

Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1, EC 2.4.2.30), a nuclear enzyme activated by DNA strand breaks, physiologically participates in DNA repair. Excessive activation of PARP-1 by cellular insults depletes its substrate β-nicotinamide adenine dinucleotide and ATP, leading to cell death. PARP-1-deficient (PARP-1−/−) mice are protected from several forms of inflammation. In the present study, we demonstrate in PARP-1−/− glial cells a loss of several stress-activated transcription factors as well as decreased expression of genes for cytokines and cellular adhesion molecules. We also show that augmented expression of some of these genes is independent of PARP-1 catalytic activity. These findings indicate that PARP-1 plays a pivotal role in the initial inflammatory response by modulating transcription of inflammation-linked genes.
Poly(ADP-ribose) polymerase-1 (PARP-1, EC 2.4.2.30) is a nuclear enzyme activated by DNA strand breaks, which adds branched chains of up to 200 ADP-ribose units to a variety of nuclear proteins, especially PARP-1 itself. PARP-1 participates in the DNA repair process after its activation by DNA damage. Several isozymes of PARP have been identified. However, the initially described form of the enzyme, PARP-1, is the most abundant, because PARP activity is reduced markedly in most tissues of PARP-1-deficient (PARP-1−/−) mice (1). PARP-1 plays a major role in tissue damage (2) and necrotic cell death (3), because PARP-1−/− mice are protected from cerebral ischemia (4, 5), streptozotocin-induced diabetes (6–8), myocardial ischemia (9–11), and several forms of inflammation (9, 12–14). In many instances cellular death seems related to PARP-1 overactivation and ATP depletion. Tissue insults lead to DNA damage, which can arise from the formation of nitric-oxide derivatives such as peroxynitrite (2, 15). This massive DNA damage leads to pronounced overactivation of PARP-1. Because PARP-1 is an extremely abundant enzyme, its overactivation results in depletion of its substrate β-nicotinamide adenine dinucleotide (NAD+). In efforts to resynthesize NAD+, ATP is depleted, and the cell dies from energy loss.
An alternative way in which PARP-1 may influence the stress/inflammation response involves regulation of transcription factors and associated gene transcription. PARP-1 has been reported either to activate or repress transcription activity (16). PARP-1 influences on transcription activity may involve direct protein–protein interaction with PARP-1 or the catalytic activity of the PARP-1 enzyme, which can poly(ADP-ribosyl)ate transcription factors. Transcription factors such as AP-2 (17), B-MYB (18), Oct-1 (19), YY-1 (20), and TEF-1 (21) have been shown to bind directly to PARP-1. On the other hand, transcription factors such as p53 (22), fos (23), and RNA polymerases I (24) and II (25) are poly(ADP-ribosyl)ated. NF-κB transcription activation after stress/inflammatory stimuli is reduced in PARP-1−/− cells (13, 26). PARP-1 participates in the activation of NF-κB independently of energy depletion by binding directly to NF-κB (27, 28) or poly(ADP-ribosyl)ating NF-κB (29, 30).
Stress/inflammation involves multiple transcription factors. In the present study we demonstrate a loss of several stress-activated transcription factors and decreased expression of cytokine and cellular adhesion molecule genes in PARP-1−/− glial cells. We also show that augmented expression of some of these genes is independent of PARP-1 catalytic activity.
Materials and Methods
Reagents and Materials.
An affinity-purified goat polyclonal antibody (M-20) raised against the carboxyl terminus of the IL-1β precursor of mouse origin and goat polyclonal antibody (M19) raised against intracellular adhesion molecule-1 (ICAM-1) were from Santa Cruz Biotechnology. Antibody raised against inducible nitric-oxide synthase (iNOS) was from Transduction Laboratories (Lexington, KY). Murine polyclonal antibody raised against cyclooxygenase-2 (Cox-2) was from Cayman Chemicals (Ann Arbor, MI). Recombinant mouse tumor necrosis factor-α (TNF-α, catalog no. 19321T) was from PharMingen. The specific p38MAPK inhibitor SB302085 was from Calbiochem. Antisera to PARP-1 and poly(ADP-ribose) and PARP inhibitor 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1-(2H)-isoquinolinone (DPQ) were from Alexis Biochemicals (San Diego, CA). Murine monoclonal antibody raised against β-actin and lipopolysaccharide (LPS) from Escherichia coli serotype 0111:B4 were from Sigma. [α-32P]NAD+ and [γ-32P]ATP were from NEN Life Science Products.
Isolation and Culture of Mouse Mixed Glial Cells, Peritoneal Macrophages, Thymocytes, and Bone Marrow-Derived Dendritic Cells (DCs).
The PARP-1−/− mice (31) were provided kindly by Z. Q. Wang (International Agency for Research on Cancer, Lyon, France). The whole brains of 1–3-day-old mouse pups from PARP-1 wild type (PARP-1+/+, C57BL/6 × 129/Sv) and PARP-1−/− (C57BL/6 × 129/Sv) were isolated for primary mixed glial cultures as described elsewhere (32). Primary mixed glial cells were grown in T75 flasks for at least 14 days in DMEM and 10% heat-inactivated FBS, 20 mM glutamine, and antibiotics (0.1 units/ml penicillin/0.1 μg/ml streptomycin solution). The medium was changed twice per week. After reaching confluency, the cells were trypsinized and plated on 6-well plates with 50% confluency. The resulting cultures were determined by staining with glial fibrillary acidic protein to comprise ≈70% astrocytes.
Primary macrophages were isolated from the peritoneal exudates from sex- and age- (6–9 weeks old) matched PARP-1+/+ and PARP-1−/− mice by fluxing with sterile DMEM and cultured at 1 × 106 cells per well on 6-well plates in DMEM. The medium was changed to remove floating cells 4 h later. Macrophages were cultured overnight, and the experiments were carried out on the next day.
Thymuses from sex- and age- (4–6 weeks old) matched PARP-1+/+ and PARP-1−/− mice were removed aseptically and placed into ice-cold RPMI medium 1640 containing 10% heat-inactivated FBS, 20 mM glutamine, and antibiotics (0.1 units/ml penicillin/0.1 μg/ml streptomycin solution). Single-cell suspensions were prepared by sieving the organ through a stainless wire mesh. Thymocytes were seeded in 24-well plates and cultured overnight before the experiments.
DCs were isolated from sex- and age- (4–6 weeks old) matched PARP-1+/+ and PARP-1−/− mice and cultivated in RPMI medium 1640 supplemented with granulocyte/macrophage colony-stimulating factor for 5 days before the experiments.
Primary embryonic fibroblasts (E18) were grown in DMEM and 10% heat-inactivated FBS, 20 mM glutamine, and antibiotics (0.1 unit/ml penicillin/0.1 μg/ml streptomycin solution). Only cell passages 2–5 were used for experiments.
Preparation of Cell Lysates.
Cells cultured in 6-well plates were washed with PBS and lysed in 60 μl of lysis buffer (100 mM Hepes, pH 7.0/200 mM NaCl/40 mM EDTA/4 mM EGTA/10% glycerol/1% Nonidet P-40/14 μM pepstatin A/100 μM leupeptin/100 μM phenylmethylsulfonyl fluoride/1 mM sodium pyrophosphate/2 mM orthovanadate/100 mM NaF). After incubation for 30 min on ice, cell lysates were sonicated for 15 s and centrifuged (16,000 × g, 10 min, 4°C). Protein concentrations of the recovered supernatants were determined by using the Coomassie Plus assay reagent from Pierce with BSA as a standard.
Western Blotting.
Cell lysates containing 50 μg of total protein in Laemmli buffer were separated by 4–12% SDS/PAGE gel and transferred to nitrocellulose membranes from NEN Life Science Products. The membrane was blocked with 5% nonfat milk in PBS-Tween [0.1% (vol/vol) Tween 20] and incubated with specific antibody for 2 h and peroxidase-conjugated secondary antibody for 1 h at ambient temperature. Specific bands were revealed by using the Western blot Chemiluminescence Reagent Plus from NEN Life Science Products. SeeBlue Plus2 prestained standards markers, 3–188 kDa, from Invitrogen were used as molecular mass standards.
PARP Activity Assay.
The PARP activity assay was performed as described (33) with some modifications. Primary glial cells were isolated in the absence or presence of LPS (1 μg/ml) or DPQ (10 or 20 μM). The pellet was incubated with assay buffer containing 50 mM Tris⋅HCl (pH 8.0), 28 mM KCl, 10 mM MgCl2, 0.01% digitonin in ethanol, 1 mM DTT, and [α-32P]NAD+ [0.1 μCi/nmol (1 Ci = 37 GBq)] for 20 min at 4°C. Reactions were stopped by the addition of ice-cold 10% trichloroacetic acid and centrifuged. The pellet was washed three times with cold acetone and suspended in cell lysis buffer. After sonication (3×, 10 sec), 70 μg of protein suspended in Laemmli buffer was separated by 4–12% SDS/PAGE gel and transferred to nitrocellulose membranes from NEN Life Science Products. PARP activity was visualized by autoradiography.
Electrophoretic Mobility-Shift Assay (EMSA).
Nuclear extracts for EMSA were prepared by using NE-PER nuclear and cytoplasmic extraction reagents from Pierce. Protein concentrations of the recovered nuclear extracts were determined by using the Coomassie Plus assay reagent from Pierce with BSA as a standard. EMSA was performed by incubating 3 μg of nuclear extracts with binding buffer containing 20% glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl, 50 mM Tris⋅HCl (pH 7.5), 0.25 mg/ml poly(dI-dC)⋅poly(dI-dC), and 32P-labeled consensus oligonucleotides for 20 min at room temperature. Double-stranded oligonucleotides were end-labeled with [γ-32P]ATP using T4 polynucleotide kinase. Labeled and unlabeled oligonucleotides were separated by chromatography through MicroSpin G25 columns from Amersham Pharmacia equilibrated in TE buffer (10 mM Tris⋅HCl, pH 8.0/1 mM EDTA). Bound and free DNA probes were then resolved by electrophoresis using native 6% polyacrylamide gels with TBE buffer (45 mM Tris-borate/1 mM EDTA). Dried gels were exposed to Kodak XAR-5 film at −70°C with intensifying screens. The oligonucleotide sequences used were NF-κB (AGTTGAGGGGACTTTCCCAGG), Oct-1 (TGTCCAATGCAAATCACTAGAA), AP-1 (CGCTTGATGACTCAGCCGGAA), SP-1 (ATTCGATCGGGGCGGGGCGAGC), Stat-1 (CATGTTATGCATATTCCTGTAAGTG), and YY-1 (CGCTCCGCGGCCATCTTGGCGGCTGGT). For the sequences listed, the direction is from 5′ to 3′, and only the sense strand is shown. The mutated nucleotide is underlined, and the consensus binding motif is italicized.
Microarray.
Profiles of LPS-induced gene expression from primary mixed glia of PARP-1+/+ and PARP-1−/− mice were determined by using the protocol of GEArray pathway-specific expression arrays from SuperArray (Bethesda, MD). After treatment with LPS (1 μg/ml) for 2 h, poly(A)+ RNA (8 μg) was isolated for the synthesis of cDNA probe using GEAprimer mix as reverse transcriptase primers. The resulting cDNA probes were hybridized to gene-specific cDNA fragments on the membranes. After phosphorimaging of the membrane, the relative abundance of a particular transcript was quantified by comparing its signal intensity to the signal derived from β-actin and glyceraldehyde-3-phosphate dehydrogenase.
Enzyme-Linked Immunosorbent Assay (ELISA) of TNF-α and IL-6.
All samples were assayed for immunoreactive TNF-α or IL-6 by using the OptEIA set for mouse TNF-α and IL-6 from PharMingen. TNF-α and IL-6 standards were assayed in duplicate, and samples (100 μl) were assayed in triplicate. The assay sensitivity for TNF-α and IL-6 in supernatants was 4 pg/ml.
Results
Transcription Factor Activation Is Lost in PARP-1−/− Glial Cells.
NF-κB proteins are constitutively present in the cytoplasm in an inactive form associated with inhibitory proteins, IκBs. Inflammatory stimuli typically lead to phosphorylation and degradation of IκB proteins, permitting translocation of NF-κB to the nucleus (34, 35). Hassa and Hottiger (26) and de Murcia and coworkers (13) showed that activation of nuclear NF-κB in immortalized fibroblasts and peritoneal macrophages by UV, TNF-α, etoposide, or LPS is abolished in PARP-1−/− cells. Nuclear translocation of NF-κB after TNF-α treatment was normal, but its binding to an oligonucleotide probe or NF-κB-dependent reporter gene was reduced greatly (13, 26).
In primary glial cultures from PARP-1+/+ and PARP-1−/− mice, the expression of PARP-1 protein is abolished in PARP-1−/− preparations (Fig. 1A). PARP-1 activity, measured as poly(ADP-ribosyl)ated PARP-1, is detected only in PARP-1+/+ glial cells treated with or without LPS. As expected with abolished expression of PARP-1 protein, PARP-1−/− glial cells show no PARP-1 activity (Fig. 1B). Treatment of PARP-1+/+ glial cells with LPS or TNF-α markedly augments the binding activity of NF-κB to its consensus sequences, whereas the binding activity of NF-κB is diminished greatly in PARP-1−/− glial cells (Fig. 1C). Also, basal DNA binding activity without LPS or TNF-α is reduced in PARP-1−/− glial cells. The specificity of this effect is evident in its blockade by adding a 100-fold molar excess of the cold oligonucleotide used to detect NF-κB, whereas a 100-fold molar excess of a single base mutant NF-κB oligonucleotide, which does not bind NF-κB, fails to compete for binding. NF-κB DNA binding activity is diminished also in primary cultures of nonglial cells from PARP-1−/− mice (Table 1): peritoneal macrophages stimulated with LPS or TNF-α, thymocytes stimulated by phorbol 12-myristate 13-acetate/ionophore, and fibroblasts stimulated by IL-1β or TNF-α. These results resemble those of other workers employing peritoneal macrophages from LPS-challenged mice, immortalized fibroblasts stimulated with TNF-α, and pulmonary microvascular endothelial cells stimulated with high glucose (13, 36).
Figure 1.
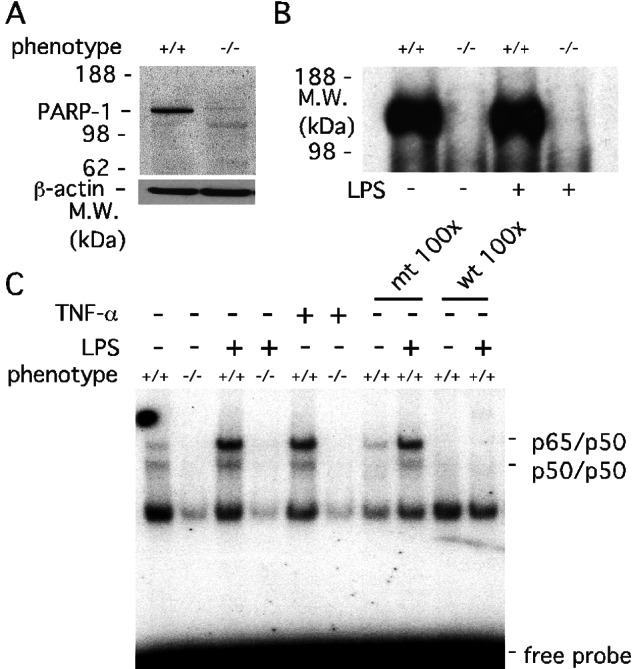
Activation of transcription factors is lost in PARP-1−/− primary mixed glial cells. (A) Protein immunoblot analysis of PARP-1 expression in PARP-1+/+ and PARP-1−/− glial cells. The PARP-1−/− lane confirmed the absence of PARP-1 protein. Total protein (50 μg) from PARP-1+/+ and PARP-1−/− glial cells was separated by 4–12% SDS/PAGE gel. β-actin was used as a protein loading control. M.W., molecular mass. (B) PARP-1 catalytic activity in PARP-1+/+ and PARP-1−/− glial cells. PARP-1 automodification by [32P]poly(ADP-ribose) was abolished in PARP-1−/− glial cells with or without LPS (1 μg/ml) for 30 min. Glial cells were labeled with [32P]NAD+ for 20 min at 4°C. Total protein (70 μg) from labeled glial cells was separated on 4–12% SDS/PAGE gel and visualized by autoradiography. (C) Defective NF-κB activation in PARP-1−/− glial cells. LPS- or TNF-α-induced DNA binding activity of NF-κB was absent in PARP-1−/− glial cells. Primary glial cells were treated with either 1 μg/ml LPS or 1 ng/ml (1,000 units/ml) TNF-α for 30 min, and the extracted nuclear fractions were analyzed for the DNA consensus binding activity of NF-κB heterodimers (p65/p50). Specificity of the DNA binding activity was assessed by using a 100-fold molar excess of unlabeled wild-type (wt) NF-κB oligonucleotides or single base mutated (mt) NF-κB oligonucleotides. Only wild type competed with labeled NF-κB oligonucleotides, indicating the specificity of the NF-κB DNA binding activity. The protein complex (p65/p50 and p50/p50 heterodimer) bound to the NF-κB element was confirmed by supershift using anti-p65 and anti-p50 antibodies (data not shown).
Table 1.
NF-κB DNA binding activity of primary cultures from PARP+/+ and PARP−/− mice
| Cell types* | Inducers† | Phenotype
|
|
|---|---|---|---|
| PARP+/+ | PARP−/− | ||
| Macrophages | LPS or TNF-α | +++ | + |
| Thymocytes | PMA/Ionophore | +++ | + |
| Fibroblasts | IL-1β or TNF-α | +++ | + |
Each type of primary cell from PARP-1+/+ and PARP-1−/− mice was isolated and cultivated as described in Materials and Methods.
Peritoneal macrophages were treated with 1 μg/ml of LPS or 1,000 units/ml of TNF-α, thymocytes with 10 ng/ml of phorbol 12-myristate 13-acetate and 400 ng/ml of ionophore, and fibroblasts with 1 ng/ml of IL-1β or 1,000 units/ml of TNF-α for 30 min.
To ascertain whether PARP-1 is linked only to the NF-κB transcription factor, we examined Oct-1, AP-1, SP-1, Stat-1, and YY-1 transcription factors that also can be activated by cytokines or LPS and can mediate transcription of stress/inflammation-related genes. DNA binding activity of these transcription factors is induced by the treatment of LPS or TNF-α; however, in PARP-1−/− glial cells, the basal DNA binding activity is lower than PARP-1+/+ glial cells, and treatment with LPS or TNF-α does not augment DNA binding activity (Table 2).
Table 2.
DNA binding activity of transcription factors in glial cells from PARP+/+ and PARP−/− mice
| PARP+/+
|
PARP−/−
|
|||
|---|---|---|---|---|
| Basal LPS or TNF-α* | Basal LPS or TNF-α | |||
| Oct-1† | ++ | +++ | + | + |
| AP-1 | + | ++ | + | + |
| SP-1 | ++ | ++ | + | + |
| Stat-1 | ++ | +++ | + | + |
| YY-1 | ++ | +++ | + | + |
Glial cells were treated with 1 μg/ml of LPS or 1,000 units/ml of TNF-α for 30 min.
Oligonucleotide sequences employed in the electrophoretic mobility-shift assays are described in Materials and Methods.
PARP-1 Regulates Efficient Gene Expression in Glia.
Transcription factors such as NF-κB, Oct-1, AP-1, SP-1, YY-1, and Stat-1 control the expression of a wide range of genes that are transcribed as a part of the stress/inflammatory process. By using a DNA microarray, we profiled the expression of mouse inflammatory cytokines and NF-κB-dependent genes in LPS-treated glial cultures of PARP-1+/+ and PARP-1−/− mice (Fig. 2A). Transcription of IL-6, IL-1β, ICAM-1, and vesicular adhesion molecule-1 is markedly induced in PARP-1+/+ glial cells but greatly reduced in PARP-1−/− glial cells. By contrast, other inflammatory cytokine and NF-κB-dependent genes are not activated dramatically, defined as less than 2-fold induction. These genes are c-myc, P- and E-selectin, G-CSF, NF-κB1, NF-κB2, c-rel, Ikk-α, Ikk-β, Ikk-γ, Ikk-i, I-κBα, iNOS, IRF-1, INF-β, MIF, Gro-1, IL-1α, IL-2, IL-4, IL-5, IL-10, IL-12A, IL-12B, IL-16, IL-17, IL-18, LT-β, MCP-1, TGF-α, TGF-β1, TGF-β2, TGF-β3, TNF-α, TNF-β, MCP-1, and NIK.
Figure 2.

PARP-1 regulates efficient gene expression in primary mixed glial cells activated with LPS. (A) Profile of LPS-induced gene expression from primary PARP-1+/+ (hatched columns) and PARP-1−/− (dotted columns) glial cells measuring mRNA formation in a microarray analysis. Transcription of IL-6, IL-1β, ICAM-1, and vesicular cell adhesion molecule-1 (VCAM-1) in PARP-1+/+ glial cells was induced markedly by treatment with LPS (1 μg/ml) for 2 h and diminished markedly in PARP-1−/− cells. The induction levels of genes were normalized by glyceraldehyde-3-phosphate dehydrogenase and β-actin expression. Forty-three genes of mouse inflammatory response cytokines and the mouse NF-κB pathway genes were analyzed. Only differences greater than 2-fold between PARP-1+/+ and PARP-1−/− were considered significant. (B) Release of IL-6 in primary glial culture. LPS-stimulated (1 μg/ml) release of IL-6 was attenuated markedly in PARP-1−/− (dotted columns) compared with PARP-1+/+ (hatched columns) cells. IL-6 release was quantified by ELISA. Values represent the means ± SE from triplicate samples. The results are representative of three independent experiments. (C) Expression of pro-IL-1β in primary glial cells. LPS-stimulated (1 μg/ml) expression of pro-IL-1β was virtually abolished in PARP-1−/− cells. Pro-IL-1β was visualized by immunoblot with 50 μg of total protein. GFAP, glial fibrillary acidic protein. (D) Expression of ICAM-1 in primary glial cells. LPS-stimulated (1 μg/ml) expression of ICAM-1 was reduced substantially in PARP-1−/− cells. ICAM-1 protein was visualized by immunoblot with 50 μg of total protein. (E) Release of TNF-α in primary glial culture. LPS-stimulated (1 μg/ml) release of TNF-α was attenuated significantly in PARP-1−/− (dotted columns) compared with PARP-1+/+ (hatched columns) cells. TNF-α release was quantified by ELISA. Values are the means ± SE from triplicate samples. (F) Expression of iNOS in primary mixed glial cells. LPS-stimulated (1 μg/ml) expression of iNOS was attenuated notably in PARP-1−/− cells. iNOS was visualized by immunoblot with 70 μg of total protein. (G) Expression of Cox-2 in primary glial cells. LPS-stimulated (1 μg/ml) expression of Cox-2 was attenuated only slightly in PARP-1−/− compared with PARP-1+/+ peritoneal macrophages at 4 and 24 h. Expression of Cox-2 was visualized by immunoblot with 70 μg of total protein.
There are several limitations of microarray experiments. Our microarray analysis was done only at a single time point after LPS treatment. Quantification of microarray results is limited such that only relatively large changes are detected readily. Accordingly, we have confirmed the microarray results by analysis of protein expression. LPS augments expression in glia markedly of IL-6, pro-IL-1β, ICAM-1, TNF-α, Cox-2, and iNOS (Fig. 2 B–G). Expression is reduced profoundly in PARP-1−/− preparations for all these except for Cox-2, which manifests an ≈40–50% reduction.
Differential Dependence of LPS-Induced Gene Expression on PARP-1 Catalytic Activity.
The rapidity of PARP-1-dependent activation of transcript factors and associated gene expression does not seem to result from ATP depletion after excess activation of PARP-1 catalytic activity. We wondered whether the gene expression we had observed required catalytic activity of PARP-1 at all. Accordingly, we examined the effects of the specific PARP inhibitor DPQ on LPS-induced expression of cytokines and cellular adhesion molecules in glial cultures. DPQ abolishes PARP-1 activity (Fig. 3A). DPQ fails to influence DNA binding activity of NF-κB and LPS-induced pro-IL-1β and ICAM-1 expression (Fig. 3 B–D). p38MAPK is a major inflammatory signal transduction mediator, activated by treatment of cells with LPS, cytokines, and stress (37, 38). SB203580, a selective p38MAPK inhibitor, treatment abolishes the LPS-induced pro-IL-1β expression in glial preparations but only marginally reduces ICAM-1 expression, suggesting differential dependence of these two mediators on the p38MAPK pathway.
Figure 3.
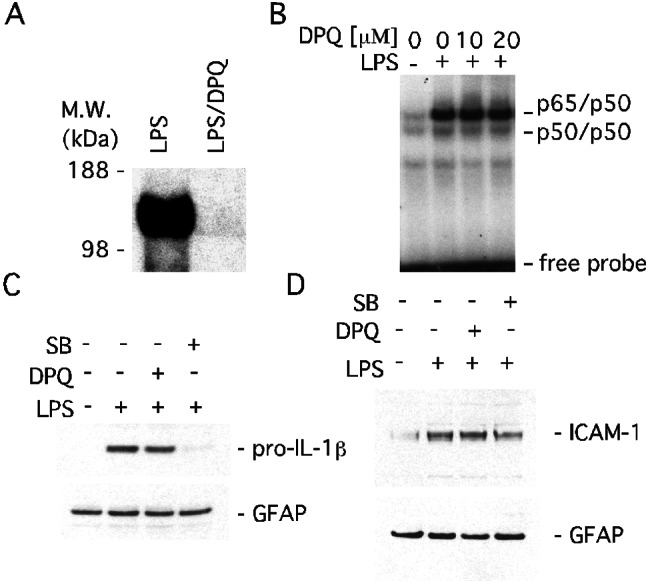
PARP-1 regulates LPS-induced gene expression independent of its catalytic activity in primary mixed glial cells. (A) Inhibition of PARP activity by DPQ. DPQ, a specific PARP inhibitor, inhibited automodification of PARP-1 in LPS-treated PARP-1+/+ glial cells. Cells were pretreated with DPQ (20 μM) for 1 h before adding LPS (1 μg/ml) for 30 min. M.W., molecular mass. (B) PARP activity-independent DNA binding of NF-κB. DPQ had no effect on LPS-induced DNA binding activity. Glial cells were pretreated with 10 or 20 μM DPQ for 1 h before adding LPS (1 μg/ml) for 30 min. (C) PARP activity-independent pro-IL-1β expression. DPQ (20 μM) had no effect on the expression of pro-IL-1β, whereas the p38MAPK inhibitor SB203580 (20 μM) abolished expression of pro-IL-1β elicited by LPS (1 μg/ml) for 24 h. GFAP, glial fibrillary acidic protein. (D) PARP activity-independent ICAM-1 expression. DPQ (20 μM) did not affect the expression of ICAM-1, and SB203580 (20 μM) slightly attenuated the expression of ICAM-1. LPS (1 μg/ml) was applied for 24 h.
PARP-1 Dependence of iNOS Expression Is Regulated in a Stimulus- and Cell/Tissue Type-Specific Manner.
Unlike our findings with pro-IL-1β and ICAM-1, the catalytic activity of PARP-1 seems to be important for iNOS expression, because DPQ treatment markedly reduces the LPS-induced augmented expression of iNOS (Fig. 4A). Interestingly, the dependence of iNOS expression on PARP's catalytic activity seems to be regulated in a stimulus- and cell/tissue type-specific manner. Cotreatment of LPS with IFN-γ induces iNOS in RAW264.7 macrophages; however, DPQ paradoxically enhances the expression of iNOS (Fig. 4B). DPQ also fails to prevent LPS-induced iNOS expression in DCs. Unexpectedly, LPS-treated PARP-1−/− DCs express iNOS protein, and DPQ stimulates iNOS expression just as in RAW264.7 macrophages (Fig. 4C). By contrast, iNOS expression is absent in basal or LPS-treated PARP-1−/− peritoneal macrophages (Fig. 4D).
Figure 4.
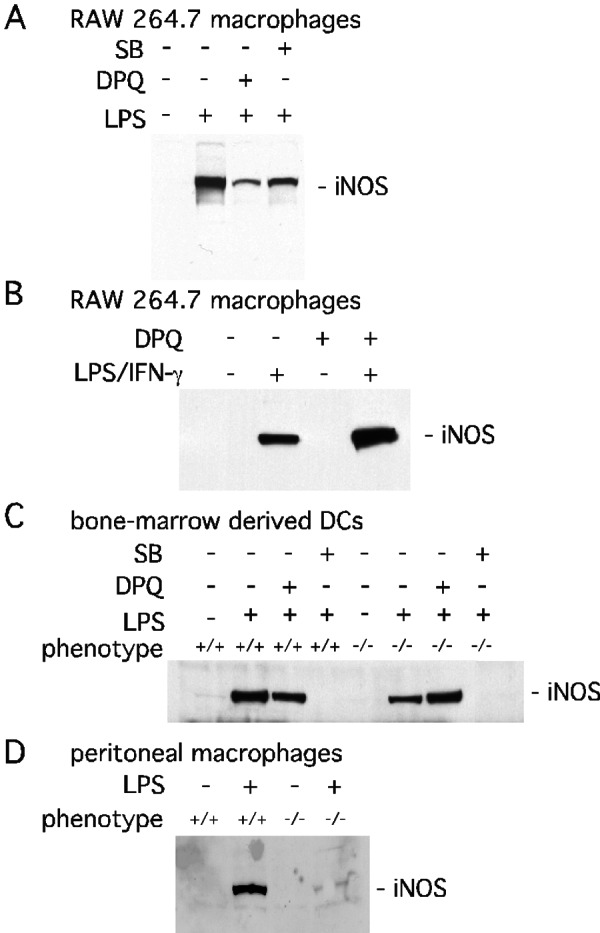
PARP-1 mediated iNOS expression is stimulus- and cell type-specific. (A) PARP activity-dependent iNOS expression induced by LPS. Treatment with LPS (1 μg/ml) for 24 h induced iNOS expression in RAW264.7 macrophages. LPS-induced iNOS expression was reduced substantially by DPQ (10 μM) and modestly by SB203580 (SB, 20 μM). (B) PARP activity-independent expression iNOS-induced by LPS/IFN-γ in RAW264.7 macrophages. DPQ (20 μM) increased iNOS expression after 24 h of treatment with LPS (1 μg/ml) and IFN-γ (400 units/ml). (C) PARP activity-independent iNOS expression in the primary DCs. LPS (1 μg/ml) for 24 h stimulated iNOS in PARP-1−/− cells but less than PARP-1+/+ cells. SB203580 (20 μM) abolished LPS-stimulated expression of iNOS in both PARP-1+/+ and PARP-1−/− cells. (D) Expression of iNOS in primary peritoneal macrophages. LPS-stimulated (1 μg/ml) expression of iNOS was attenuated significantly in PARP-1−/− peritoneal macrophages. Expression of iNOS was visualized by immunoblot with 100 μg of total protein from cells.
Even within the same cell type, iNOS regulation varies with stimulus. Thus, in RAW264.7 macrophages DPQ augments iNOS expression after induction by the combination of LPS and IFN-γ but not LPS alone (Fig. 4 A and B). SB203580 abolishes iNOS expression in cells treated with LPS and IFN-γ but only partially reduces expression in cells receiving only LPS.
Discussion
Recent reports that NF-κB activation depends on PARP-1 (13, 27, 39) as well as studies showing the direct binding of PARP-1 to related transcription factors such as Oct-1 (19) and YY-1 (20) suggest a novel role for PARP-1 as a coactivator or repressor of transcription factors that mediate the stress/inflammation response. Previously, PARP-1-dependent activation of transcription factors induced by stress/inflammatory agents had been demonstrated only for NF-κB (13, 27, 39). In the present study, we show that LPS- or TNF-α-induced activation of the stress/inflammation transcription factors AP-1, SP-1, Oct-1, YY-1, and Stat-1 also requires PARP-1.
How might PARP-1 regulate these transcription factors? One possibility is that PARP-1 ADP-ribosylates the transcription factors. There have been contradictory reports of poly(ADP-ribosyl)ation of NF-κB by PARP-1 (27–30, 39). The failure of the PARP inhibitor DPQ to reduce NF-κB DNA binding activity in our experiments and similar findings with PJ34, another PARP inhibitor (36), argue against a direct role for catalytic activity in activating NF-κB. DPQ also fails to alter expression of pro-IL-1β and ICAM-1 but does reduce expression of iNOS. Thus, the catalytic activity of PARP regulates some but not other transcription factors.
Variable effects of PARP inhibitors on gene expression may reflect influences of multiple regulatory systems. Thus, although the promoter regions of IL-1β, ICAM-1, and iNOS have NF-κB binding sites, these genes are regulated also by other transcription factors whose response to PARP catalytic activity may vary. Inhibition by DPQ of iNOS stimulated by LPS but not by LPS combined with IFN-γ may reflect IFN-γ influences on various transcription factors such IRF-1 (40), which is essential for IFN-γ action on iNOS via a binding site in the iNOS promoter (41).
PARP-1 also may act by direct binding to transcription factors as a coactivator or repressor independent of catalytic activity. PARP-1 binds directly to AP-2 (17), B-MYB (18), Oct-1 (19), YY-1 (20), and TEF-1 (21). PARP-1 binding to the p50 (27, 28) and p65 (27, 28) subunits of NF-κB is independent of catalytic activity, but the subsequent binding of the PARP-1/p65 complex to HMG-1(Y) is prevented by PARP inhibitors (28).
Our microarray analysis revealed four PARP-1-dependent genes, whereas expression of another 37 genes was not changed significantly. PARP-1-dependent LPS-induced expression of certain proteins in glia was not detected by microarray analysis. Among the genes whose protein expression was examined, TNF-α and iNOS were reduced markedly in PARP-1−/− tissue. Cox-2 expression seemed to be less dependent on PARP-1 even though there was some reduction of its expression in the PARP-1−/− glial cells. Because the time course for expression of various proteins may vary, we examined multiple time points in these experiments and found that temporal factors do not account for the differences between these proteins. Thus, it seems that the expression of some stress/inflammation genes is regulated by PARP-1, whereas the expression of others is PARP-1-independent. There are no obvious properties that differentiate the PARP-1-dependent and -independent genes.
The genes studied here are well known inflammatory mediators. They also mediate ischemic damage including myocardial infarction and cerebral ischemia (42–44). In necrotic cell death associated with myocardial and cerebral ischemia, cell death has been attributed to extensive DNA damage causing massive overactivation of PARP-1 with depletion of its substrate NAD+ and then of ATP such that cells die from energy deficit (3, 45). The present study as well as other recent work (13, 14, 27) indicates an alternative role for PARP-1 in stress-related pathophysiology. PARP-1 seems critical for activation of stress/inflammation-related genes. The initial inflammatory response associated with PARP-1-dependent gene activation does not involve massive DNA damage, overactivation of PARP, energy depletion, or cell death.
Acknowledgments
We thank Dr. Z. Q. Wang for providing the PARP-1−/− mice. We also thank Helena Ong and Loise M. Francisco for isolating peritoneal macrophages and DCs, respectively. This work was supported by U.S. Public Health Service Grants DA00266 (to S.H.S.) and Research Scientist Award DA00074 (to S.H.S.). Under an agreement between the Johns Hopkins University and Guilford, S.H.S. is entitled to a share of sales royalties related to PARP received by the University from Guilford. The University owns stock in Guilford with S.H.S. having an interest in the University Share under university policy. S.H.S. serves on the Board of Directors and the Scientific Advisory Board of Guilford. He is a consultant to the company, and he owns additional equity in Guilford. This arrangement is being managed by the university in accordance with its conflict-of-interest policies.
Abbreviations
- PARP-1
poly(ADP-ribose) polymerase-1
- ICAM-1
intracellular adhesion molecule-1
- iNOS
inducible nitric-oxide synthase
- Cox-2
cyclooxygenase-2
- TNF-α
tumor necrosis factor-α
- DPQ
3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1-(2H)-isoquinolinone
- LPS
lipopolysaccharide
- DC
bone marrow-derived dendritic cell
References
- 1.Shieh W M, Ame J C, Wilson M V, Wang Z Q, Koh D W, Jacobson M K, Jacobson E L. J Biol Chem. 1998;273:30069–30072. doi: 10.1074/jbc.273.46.30069. [DOI] [PubMed] [Google Scholar]
- 2.Szabo C, Dawson V L. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 3.Ha H C, Snyder S H. Proc Natl Acad Sci USA. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eliasson M J, Sampei K, Mandir A S, Hurn P D, Traystman R J, Bao J, Pieper A, Wang Z Q, Dawson T M, Snyder S H, Dawson V L. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 5.Endres M, Wang Z Q, Namura S, Waeber C, Moskowitz M A. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Masutani M, Suzuki H, Kamada N, Watanabe M, Ueda O, Nozaki T, Jishage K, Watanabe T, Sugimoto T, Nakagama H, Ochiya T, Sugimura T. Proc Natl Acad Sci USA. 1999;96:2301–2304. doi: 10.1073/pnas.96.5.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkart V, Wang Z Q, Radons J, Heller B, Herceg Z, Stingl L, Wagner E F, Kolb H. Nat Med. 1999;5:314–319. doi: 10.1038/6535. [DOI] [PubMed] [Google Scholar]
- 8.Pieper A A, Brat D J, Krug D K, Watkins C C, Gupta A, Blackshaw S, Verma A, Wang Z Q, Snyder S H. Proc Natl Acad Sci USA. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zingarelli B, Salzman A L, Szabo C. Circ Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z, Zingarelli B, Szabo C. Shock. 2000;13:60–66. doi: 10.1097/00024382-200013010-00011. [DOI] [PubMed] [Google Scholar]
- 11.Pieper A A, Walles T, Wei G, Clements E E, Verma A, Snyder S H, Zweier J L. Mol Med. 2000;6:271–282. [PMC free article] [PubMed] [Google Scholar]
- 12.Szabo C, Lim L H, Cuzzocrea S, Getting S J, Zingarelli B, Flower R J, Salzman A L, Perretti M. J Exp Med. 1997;186:1041–1049. doi: 10.1084/jem.186.7.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oliver F J, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet J C, de Murcia G. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhnle S, Nicotera P, Wendel A, Leist M. Biochem Biophys Res Commun. 1999;263:433–438. doi: 10.1006/bbrc.1999.1393. [DOI] [PubMed] [Google Scholar]
- 15.Pieper A A, Verma A, Zhang J, Snyder S H. Trends Pharmacol Sci. 1999;20:171–181. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 16.D'Amours D, Desnoyers S, D'Silva I, Poirier G G. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 17.Kannan P, Yu Y, Wankhade S, Tainsky M A. Nucleic Acids Res. 1999;27:866–874. doi: 10.1093/nar/27.3.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cervellera M N, Sala A. J Biol Chem. 2000;275:10692–10696. doi: 10.1074/jbc.275.14.10692. [DOI] [PubMed] [Google Scholar]
- 19.Nie J, Sakamoto S, Song D, Qu Z, Ota K, Taniguchi T. FEBS Lett. 1998;424:27–32. doi: 10.1016/s0014-5793(98)00131-8. [DOI] [PubMed] [Google Scholar]
- 20.Oei S L, Griesenbeck J, Schweiger M, Babich V, Kropotov A, Tomilin N. Biochem Biophys Res Commun. 1997;240:108–111. doi: 10.1006/bbrc.1997.7621. [DOI] [PubMed] [Google Scholar]
- 21.Butler A J, Ordahl C P. Mol Cell Biol. 1999;19:296–306. doi: 10.1128/mcb.19.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wesierska-Gadek J, Schmid G. Cell Mol Biol Lett. 2001;6:117–140. [PubMed] [Google Scholar]
- 23.Amstad P A, Krupitza G, Cerutti P A. Cancer Res. 1992;52:3952–3960. [PubMed] [Google Scholar]
- 24.Muller W E, Zahn R K. Mol Cell Biochem. 1976;12:147–159. doi: 10.1007/BF01741713. [DOI] [PubMed] [Google Scholar]
- 25.Taniguchi T, Suzuki S, Shizuta Y. Biochem Biophys Res Commun. 1985;127:526–532. doi: 10.1016/s0006-291x(85)80191-1. [DOI] [PubMed] [Google Scholar]
- 26.Hassa P O, Hottiger M O. Biol Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 27.Hassa P O, Covic M, Hasan S, Imhof R, Hottiger M O. J Biol Chem. 2001;276:45588–45597. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- 28.Ullrich O, Distel A, Eyupoglu I Y, Nitsch R. Nat Cell Biol. 2001;3:1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- 29.Kameoka M, Ota K, Tetsuka T, Tanaka Y, Itaya A, Okamoto T, Yoshihara K. Biochem J. 2000;346:641–649. [PMC free article] [PubMed] [Google Scholar]
- 30.Chang W J, Alvarez-Gonzalez R. J Biol Chem. 2001;276:47664–47670. doi: 10.1074/jbc.M104666200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z Q, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner E F. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 32.Dobrenis K. Methods. 1998;16:320–344. doi: 10.1006/meth.1998.0688. [DOI] [PubMed] [Google Scholar]
- 33.Berton G, Sorio C, Laudanna C, Menegazzi M, Carcereri De Prati A, Suzuki H. Biochim Biophys Acta. 1991;1091:101–109. doi: 10.1016/0167-4889(91)90228-p. [DOI] [PubMed] [Google Scholar]
- 34.Baeuerle P A, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 35.Baeuerle P A. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 36.Soriano F G, Virag L, Szabo C. J Mol Med. 2001;79:437–448. doi: 10.1007/s001090100236. [DOI] [PubMed] [Google Scholar]
- 37.Han J, Lee J D, Bibbs L, Ulevitch R J. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 38.Harper S J, LoGrasso P. Cell Signal. 2001;13:299–310. doi: 10.1016/s0898-6568(01)00148-6. [DOI] [PubMed] [Google Scholar]
- 39.Le Page C, Sanceau J, Drapier J C, Wietzerbin J. Biochem Biophys Res Commun. 1998;243:451–457. doi: 10.1006/bbrc.1998.8113. [DOI] [PubMed] [Google Scholar]
- 40.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh S I, Kimura T, Green S J, et al. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 41.Lowenstein C J, Alley E W, Raval P, Snowman A M, Snyder S H, Russell S W, Murphy W J. Proc Natl Acad Sci USA. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merrill J E, Benveniste E N. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 43.Kreutzberg G W. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 44.O'Neill L A, Kaltschmidt C. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 45.Ha H C, Snyder S H. Neurobiol Dis. 2000;7:225–239. doi: 10.1006/nbdi.2000.0324. [DOI] [PubMed] [Google Scholar]