

Abstract
RNA polymerase forms competitor-resistant complexes with “forked DNA” templates that are double-stranded from the −35 promoter region through the first base pair of the −10 region, with an additional unpaired A at the 3′ end of the nontemplate strand. These types of substrates were introduced recently as model templates for the study of DNA–protein interactions occurring in the early stages of the formation of RNA polymerase–promoter open complexes. We have performed kinetic and equilibrium measurements of interactions of wild-type and mutant RNA polymerases bearing substitutions in the σ70 initiation factor, with forked DNA of wild-type and mutant sequence. Our observations reveal that formation of a competitor-resistant complex between RNA polymerase and forked DNA, similar to the formation of open complexes at promoters, is a multistep process, and some of the sequentially formed intermediates along the two pathways share common properties. This work establishes, for the forked template, progression through these intermediates in the absence of downstream DNA and validates the use of forked DNA to determine the effects of changes in promoter or RNA polymerase sequence on the process of open complex formation.
Formation of an initiation-competent or “open” complex between RNA polymerase (RNAP) and a bacterial promoter is a complicated process involving conformation changes in both the protein and the DNA. The former likely includes the “jaws” of the RNAP closing around the downstream region of the DNA, thus linking the RNAP physically to the template DNA (1, 2). The latter involves multiple DNA distortions and opening of a region of ≈14 base pairs, which includes most of the −10 hexamer sequence, and the transcription start site (+1; refs. 3 and 4). Evidence has been obtained for several intermediates on the pathway to formation of the open complex (5–7); a currently accepted pathway (8, 9) is shown below in Scheme 1.
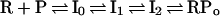 |
Scheme 1.
R is RNAP, P is the promoter, and I0, I1, and I2 are intermediates. I0 is a closed complex in which the RNAP has accomplished sequence-specific binding but no strand separation. Such a complex has been observed at the lacUV5 (10) and PRM (11) promoters, although it was not shown to be a kinetically significant intermediate at the λ PR promoter, where it would be in rapid equilibrium with more stable complexes further along on the pathway. It is only through the use of mutant RNAP that an I0-like intermediate has been observed at PR (12). I0 is sensitive to a challenge with heparin, a polyanionic competitor with DNA for binding to RNAP, and has a short footprint that extends from −50 to about +1 (10–12).
For the PR promoter, the conversion between I1 and I2 was found to be the rate-limiting process in either direction (13). Although both have “long” footprints that extend in the downstream direction to about +20 (14), only I2 is heparin-stable. Strand separation is thought to be nucleated at I1 or I2. The open complex, RPo, similar to I2, is both competitor-resistant and has the long DNaseI footprint; in addition, strand separation has occurred over a region of ≈14 base pairs including the start site (refs. 3, 4, 14, and 15; M. Raffaelle, unpublished data).
Gralla and coworkers (16–18) have pioneered the use of “forked DNA” templates (see Fig. 1) to model aspects of an RNAP–promoter complex in which the nucleation of DNA melting had occurred (I1 or I2 in Scheme 1). These templates emulate the boundary between double- and single-stranded DNA. The region from position −40, including the −35 sequence, through the first base pair of the −10 region (at position −12) is double-helical, but the 3′ end of the nontemplate strand, including the −11 A (“short Fork”) of the canonical −10 sequence, or beyond (“long Fork”), is unpaired. Forked templates have been the recent focus of structural studies (K. S. Murakami, S. Masuda, E. A. Campbell, O. Muzzin, and S. A. Darst, unpublished results). The use of these and other DNAs with altered structures in probing the mechanism of open complex formation has been reviewed recently (19).
Figure 1.

The DNA templates used in this study. The wild-type (wt) Fork is the template used for most of the experiments presented here. It is identical to that used by Gralla and coworkers (16–18). The sequence is derived from that of the PR′ promoter of bacteriophage λ. The −35 and partial −10 sequences are underlined. Fork G-33C and Fork TG have base substitutions, as indicated, that decrease the similarity to the consensus −35 region and create an extended −10 region, respectively.
Here we present equilibrium and kinetic data on the interaction of short Fork (referred to here as Fork or forked DNA) with RNAP to establish the mechanism of formation of competitor-resistant complexes between RNAP and the smallest known template that allows their formation. The results support the reaction pathway shown in Scheme 2.
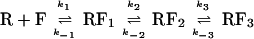 |
Scheme 2.
R again represents RNAP, F is the forked DNA, RF1 is a competitor-sensitive intermediate, and RF2 and RF3 are competitor-resistant complexes. Our findings are consistent with RF1 and RF2 being models for intermediates on the pathway to formation of open complexes and indicate that with the forked template DNA downstream of −11 is not necessary for triggering progression through these intermediates. Thus the use of forked DNA in quantitative binding assays affords a simple method for assigning the step at which mutations in DNA or RNAP, or regulators of gene expression, exert their effects to promote or attenuate open complex formation. We also show that at equilibrium a mixture of competitor-resistant and competitor-sensitive complexes coexist in solution. The distribution between these complexes is sensitive to forked DNA sequence.
Materials
Oligonucleotides were synthesized by GIBCO/BRL or Genset (La Jolla, CA). Nonradioactive nucleoside triphosphates were from Sigma, and [γ-33P]ATP, [γ-32P]ATP, and [α-32P]UTP were from New England Nuclear. DNA-modifying enzymes were purchased from either New England Biolabs or Roche Molecular Biochemicals. Escherichia coli RNAP core enzyme was purchased from Epicentre Technologies (Madison, WI). RNAP holoenzyme was purified by established methods (20, 21) and had an activity of 50% or was a gift from Epicentre Technologies.
Methods
Deoxyoligonucleotide Labeling and Annealing.
5′ end-labeled DNA oligonucleotides were generated by incubation with [γ-33P]- or [32P]ATP and T4 polynucleotide kinase (New England Biolabs) using established procedures. The two strands of forked DNA templates were reannealed at concentrations of 100 nM (5′ end-labeled nontemplate strand) and 150 nM (unlabeled template strand) in a buffer containing 25 mM Tris (pH 7.9 at 25°C) and 50 mM NaCl by heating to 90°C and slow cooling. Analysis on nondenaturing gel demonstrates that essentially all the label runs at the position of the annealed DNA.
σ70 Variants.
σ70 purification was carried out exactly as described (12, 22). The substitutions in conserved region 2.3 of σ70 have been described: the YW variant contains the Y430A and W433A substitutions; YYW and FYW additionally contain the Y425A or F427A substitutions, respectively (12, 22).
Electrophoretic Mobility Shift Assays.
Equilibrium.
All reactions contained Hepes buffer (30 mM Hepes, pH 7.5/100 mM KCl/1 mM DTT). To determine equilibrium association constants, 1 nM 5′ 32P-labeled forked DNA was incubated at 25°C with a range of concentrations of RNAP in separate reactions with final volumes of 10 μl; these were (+hep) or were not (−hep) subjected to a 10-min heparin challenge before loading onto a 5% nondenaturing gel run at room temperature. Dried gels were autoradiographed and quantified by phosphorimaging (Molecular Dynamics) to determine the fraction, θ, of the total radioactivity in each band. The data were fit as described below.
Kinetics.
To determine the rates of association of RNAP with forked DNA, ≈1 or 10 nM 32P-labeled DNA fragments were incubated with 65 nM Eσ70 at 25°C in 20 μl of Hepes buffer containing 50 μg/ml BSA for various lengths of time. Heparin challenge was with 100 μg/ml heparin for 2 min before loading onto a 5% nondenaturing polyacrylamide gel (29:1 acrylamide/bisacrylamide) run at room temperature. To determine the rate of dissociation of Eσ70–forked DNA complexes, 40 μl of solution were prepared containing 10 nM 32P-labeled forked DNA template and 65 nM Eσ70 in Hepes buffer. After a 30-min incubation at 25°C, heparin was added to 100 μg/ml, and at regular time intervals 4.5-μl aliquots were removed for analysis on a 5% nondenaturing gel as described above.
Data Analysis.
Equilibrium data.
Data analysis was carried out by using a simplified reaction scheme involving one intermediate (RF1) and a final competitor-resistant complex RFCR (where [RFCR] = [RF2] + [RF3]) as shown in Scheme 3.
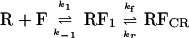 |
Scheme 3.
For the mechanism of Scheme 3, the following two equilibrium constants can be defined.
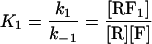 |
1 |
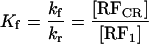 |
2 |
In the presence of a large excess of RNAP over the amount of added forked DNA, the fraction of DNA in complexes that are resistant to a challenge with heparin, θ+hep, is described by the following expression (ref. 13; R. M. Saecker, unpublished data).
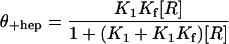 |
3 |
[R] is the concentration of unbound RNAP (uncorrected for activity), essentially equal to the total concentration of RNAP, in large excess over DNA. Because the latter condition may not be satisfied for the titrations carried out in the absence of heparin, the following equations were used to account for the reduction in the concentration of unbound RNAP caused by complex formation (up to 25% at the lowest [RNAP] used). Then θ−hep, the fractional extent of binding of DNA in the absence of heparin challenge, is given by Eqs. 4 and 5.
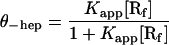 |
4 |
 |
5 |
[Rf] is the concentration of free RNAP given by Eqs. 6 and 7.
 |
6 |
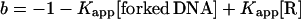 |
7 |
The θ−hep data in and of themselves are not sufficient for obtaining meaningful values of both K1 and Kf. Thus Eq. 3 was used in conjunction with Eqs. 4–7 in the simultaneous fitting of the θ+hep and θ−hep data. If only θ−hep data were available, Eqs. 4, 6, and 7 were used to obtain values for Kapp. All graphing and fitting was done with the use of SIGMA PLOT.
Kinetic data.
The rate constants for the fast and slow phases in the formation of heparin-resistant complexes between forked DNA templates and RNAP were determined by fitting kinetic data to a double-exponential function.
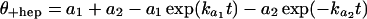 |
8 |
Here t is the time(s) of incubation of DNA and RNAP, a1 and a2 are the amplitudes, and ka1 and ka1 are the pseudo first order rate constants of the fast and slow phases of the reaction, respectively. The data for formation of complexes that were not challenged by heparin were fit to a single exponential function (similar to Eq. 8 but with a2 = 0). The kobs for the formation of heparin-resistant RF2 complexes at several [RNAP] is defined as the ka1 for the fast phase from fitting association kinetics data to the double-exponential function (Eq. 8). The products of kobs and the amplitude (a1) obtained for each concentration of RNAP were fit to Eq. 9 (13).
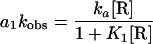 |
9 |
ka is the overall second order rate constant for the reaction, and K1 = k1/k − 1 (rate constants are as defined in Scheme 2).
The rates of dissociation were obtained by fitting the data to a two-exponential decay function.
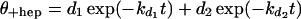 |
10 |
Here t is the time after addition of heparin, d1 and d2 are the amplitudes of the two decay processes observed, and kd1 and kd2 are the respective rate constants.
Results
Specificity of Forked DNA–RNAP Interaction.
Forked DNA with an overhanging A corresponding to the −11 A of the −10 region (see Fig. 1) is able to form a competitor-resistant complex with RNAP; this ability was shown to completely depend on the presence of a −35 region and the overhanging A (16). We have confirmed these results and extended them by demonstrating that the interaction of RNAP with forked DNA is sensitive to the length of the spacer DNA separating the −10 and −35 regions and that recognition of the first TA base pair is affected by the Q437H substitution in σ factor (L.T. and P.L.d.H., unpublished results), which is known to have the same effects on promoter DNA (23). We show below that the interaction is sensitive also to the presence of appropriately positioned TG sequences upstream of the −10 region (24, 25) and to the identity of the G-C base pair at position −33 in the −35 region. These experiments conclusively demonstrate the specific recognition of forked DNA by RNAP.
Equilibrium Binding.
wt RNAP and wt forked DNA.
Quantitative binding data were obtained by titrating the labeled wt Fork with RNAP to obtain binding isotherms. Curves for wt RNAP interacting with the wt Fork with and without a heparin challenge are shown in Fig. 2. Only a subset of the complexes is heparin-resistant, as seen from the differences in the plateau levels; the fractions of DNA in the unchallenged and challenged complexes are ≈1.0 and 0.6, respectively. The data were fit simultaneously to Eqs. 3 and 4–7 to obtain values for K1 and Kf. The curves drawn through the data reflect the fit (see also Table 1). The binding of the wt Fork to wt RNAP is seen to be quite tight under the conditions of the experiment: K1 = (2.8 ± 0.1) × 108 M−1. However, the reaction does not go to completion, with similar amounts of RF1 and RF2 accumulating at equilibrium as deduced from the value of Kf (1.0 ± 0.1).
Figure 2.

Determination of equilibrium affinities by titration of wt Fork with RNAP. The reactions contained 1 nM wt Fork and variable amounts of RNAP as shown and were analyzed by electrophoretic mobility shift assay immediately (●; data shown are averages of three independent experiments) or after a challenge with 50 μg/ml heparin for 10 min (▴; data shown are averages of four independent experiments). The curves shown reflect the simultaneous error-weighted fits of the data to Eqs. 3 and 4–7. The parameters are shown in Table 1 (line 1).
Table 1.
| Fork | RNAP | K1*, 108 M−1 | Kf* | Kapp†, 108 M−1 | |
|---|---|---|---|---|---|
| 1 | wt | wt | 2.8 ± 0.1‡ | 1.1 ± 0.1 | 3.9 ± 0.4 |
| 2 | TG | wt | 1.7 ± 0.1‡ | 3.2 ± 0.1 | 5.6 ± 0.1 |
| 3 | TG/G-33C | wt | 0.29 ± 0.01‡ | 0.33 ± 0.02 | 0.38 ± 0.04 |
| 4 | G-33C | wt | — | — | 0.18 ± 0.03§ |
| 5 | wt | Y430A | 1.77 ± 0.04¶ | 0.41 ± 0.05 | 2.9 ± 0.1 |
| 6 | wt | W433A | 1.94 ± 0.03¶ | 0.44 ± 0.04 | 2.7 ± 0.1 |
| 7 | wt | YW | 0.92 ± 0.03¶ | 0.18 ± 0.03 | 1.07 ± 0.04 |
| 8 | wt | YYW | 0.34 ± 0.05‡ | 0.21 ± 0.01 | 0.43 ± 0.01 |
| 9 | wt | FYW | — | — | 0.65 ± 0.01¶ |
| 10 | TG | FYW | — | — | 2.2 ± 0.1¶ |
| 11 | TG/G-33C | FYW | — | — | 0.13 ± 0.01¶ |
Values for K1 and Kf are from simultaneous weighted fits of the binding data obtained with and without a heparin challenge to Eqs. 3 and 4–7. Weighting was by 1/error2.
Kapp was determined from data obtained in the absence of heparin challenge by fitting to Eqs. 4, 6, and 7.
Values are based on three independent determinations (standard error shown).
Values are based on four independent determinations (standard error shown).
Values are based on two independent determinations (error is ½ difference in values).
Effects of forked DNA sequence.
The introduction of two adjacent TG sequences in the correct position with respect to the −10 region (Fork TG) is seen to not affect binding affinity significantly (compare Table 1, lines 1 and 2) but to increase the extent of conversion to RFCR, as reflected in a 3.5-fold increase in the equilibrium constant Kf. This result is consistent with the observation that TG sequences at equivalent positions of promoter DNA increase the extent of open complex formation by affecting a step subsequent to the formation of the closed complex (25). The substitution of the consensus G at −33 by a C was reported to be detrimental to promoter function (26, 27). It leads to a severely reduced ability of the wt Fork to bind RNAP in the absence of heparin and undetectable binding in its presence (data not shown). Thus Kf (Scheme 3 and Eq. 2) is small (<0.05), and the data obtained in the absence of heparin must reflect the sequence-specific formation of just RF1. The effect of the G-33C substitution could be quantified only in the context of the TG element (Fork TG/G-33C in Fig. 1), which increases the fraction of DNA bound by RNAP. Consistent with the large effect of the G-33C substitution in the context of the wt Fork (see above), it is seen to be very deleterious indeed, lowering Kf and K1 by factors of 10 and 6, respectively (compare Table 1, lines 2 and 3).
Effects of amino acid substitutions in σ70.
Substitutions of alanine for aromatic amino acids in region 2.3 of σ70 were shown to adversely affect formation of open complexes at promoters (12, 22). Consistent with these observations, we show that the Y430A and W433A substitutions both reduce Kf by 2.5-fold and K1 to a lesser extent (Table 1, compare line 1 with lines 5 and 6). Together (in YW) these two substitutions affect Kf 6-fold and K1 3-fold (Table 1, line 7). No significant formation of RFCR could be detected for the FYW mutant, which additionally bears the F427A substitution (data not shown), but almost all the wt Fork could be bound in the absence of a heparin challenge. This indicates that Kf is very small (<0.05), in good agreement with the effects of the FYW mutation on promoter binding (12). For comparison, all data for unchallenged reactions were fit to Eqs. 4, 6, and 7 to get values of Kapp (see Table 1). As for the FYW RNAP, Kf < 0.05, it is expected that Kapp ≈ K1 (Eq. 5). Indeed the Kapp values in lines 9–11 of Table 1 are in fair agreement with those for K1 in lines 1–3. Thus the FYW substitutions in RNAP have a larger effect on DNA melting than on DNA binding. Our previous studies (12) had demonstrated a somewhat less drastic effect of the YYW substitutions in σ70 on the ability of RNAP to melt promoter DNA. In agreement with these findings, DNA binding to YYW RNAP could be detected even after a heparin challenge. The fits to the data reveal a reduction in K1 as compared with the YW mutant (Table 1, compare lines 7 and 8) and 5- and 8-fold reductions in Kf and K1, as compared with wt RNAP (Table 1, compare lines 1 and 8). In addition to being consistent with the greatly impaired ability of this mutant to effect strand opening at promoters, these results indicate that DNA binding is affected also by the substitutions.
Initial experiments have been carried out with forked DNA that had an unpaired region of the sequence ATAAT (16), on which the left-most A is the −11 A, also unpaired in the short Fork (Fig. 1). For wt RNAP, K1 = 1.9 ± 0.1 × 108 M−1 and Kf = 11.2 ± 0.1, and for YYW RNAP, K1 = 2.2 ± 0.1 × 108 M−1 and Kf = 1.7 ± 0.1. Compared with the short Fork, K1 for binding wt RNAP is similar, but Kf is increased greatly for the binding of the long Fork, which accounts for the overall much tighter binding of long Fork to RNAP (22). Comparing the interaction of wt and YYW RNAP with the long Fork, both have a similar K1, but Kf is 7-fold greater for the wt RNAP, again consistent with the poor formation of open complexes by YYW RNAP. It is unclear why different K1 values are observed for the interaction of the two RNAPs with the short but not the long Fork.
RNAP–Forked DNA Interaction: Kinetics of Association.
Support for the occurrence of the intermediates indicated in Scheme 2 is derived from kinetic studies shown in Fig. 3. In the absence of a heparin challenge, complex formation occurs with a rate that is too fast to measure accurately with the manual mixing techniques used. The first few data points already are almost at the plateau level. A minimal estimate for the observed rate constant, kobs, is 0.1 ± 0.02 s−1. Also shown in Fig. 3 are the kinetics of formation of heparin-resistant complexes (RFCR), where a 2-min challenge with heparin preceded loading of the mixture onto the gel. Formation of heparin-resistant complexes is slower than that of unchallenged complexes, which is expected if a heparin-sensitive complex were an intermediate on the pathway. Both single- and double-exponential fits (Eq. 8) to the data for formation of the heparin-resistant complexes are shown in Fig. 3, with the latter giving a much better fit. The fitted values of the rate constants are shown in the legend for Fig. 3. The fast and slow phases of RFCR formation were defined as yielding RF2 and RF3, respectively (see Scheme 2). Thus RF2 is formed with a ka1 = 0.044 ± 0.002 s−1 (Eq. 8), which is significantly smaller than the lower limit of kobs for the formation of unchallenged complexes. RF3 formation has a rate constant that is smaller by a factor of 90. It is detectable, because the shift of the equilibrium to the right causes a further increase in the fraction of DNA in a heparin-resistant complex.
Figure 3.

Kinetics of complex formation. RNAP (65 nM) and wt forked DNA (1 nM) were incubated for various time intervals, and then complex formation was determined immediately (−heparin) or after a 10-min challenge with 100 μg/ml heparin (+heparin). The −heparin data (▪) were fit (error-weighted) with Eq. 8 with a2 = 0 (ka− = 0, 10 ± 0.01 s−1) and the +heparin data (▴) with both single (ka+ = 0.036 ± 0.004 s−1; thin line) and double-exponential (ka1 = 0.044 ± 0.002 s−1; ka2 = (5 ± 3) × 10−4 s−1; thick line) equations.
Rates of Binding Reach a Plateau at High [RNAP].
To probe the details of the reaction mechanism for formation of the RFCR complex, the kinetics were determined as a function of [RNAP]. These experiments were carried out at a DNA concentration of 1 rather than 10 nM as used for the experiment shown in Fig. 3 to maintain pseudo first order kinetics at RNAP concentrations of 10 nM. However, the curves were very similar to those shown in Fig. 3, again exhibiting two kinetic phases. The ka1 (= kobs) and amplitude, a1, for the fast phase of the association reaction (i.e., the formation of RF2) were determined by fitting to Eq. 8. To correct the values of kobs for the extent to which each reaction proceeded to completion, the product a1kobs was determined and plotted versus [RNAP] (13) as shown in Fig. 4. Saturation behavior is observed, in agreement with formation of RF2 involving a preequilibrium (i.e., the formation of RF1), followed by a rate-limiting step. A hyperbolic fit (Eq. 9) of the data yields K1 = (4.1 ± 0.3) × 107 M−1, ka = (4.9 ± 0.3) × 106 M−1 s−1, and k2 = ka/K1 = 0.09 ± 0.02 s−1. Fig. 4 Inset shows a double-reciprocal plot [analogous to a “τ plot” (28) for RNAP–promoter interaction]; the parameters obtained from the linear fit are K1 = 8 × 107 M−1, ka = 6 × 106 M−1 s−1 and k2 = 0.08 s−1. Omission of the outlying point at 50 nM RNAP improves the fits but only has a small effect on the values of the parameters. The above K1 values are only in order of magnitude agreement with that obtained by equilibrium titrations, likely because of the lack of kinetic measurements at nanomolar [RNAP].
Figure 4.

The formation of RF2 displays saturable kinetics. The product of the amplitude (a1) and the pseudo first order rate constant for the fast phase of the reaction (ka1 = kobs) is plotted as a function of [RNAP]. The curve represents the fit to Eq. 9 of all the data points with K1 = 4.4 ± 0.1 × 107 M−1 and k2 = 0.09 ± 0.02 s−1. (Inset) Double reciprocal plot of the data (see text).
Kinetics of Interaction with Mutant RNAP.
As shown in Fig. 5, in addition to association kinetics for the wt RNAP and wt Fork, we also obtained data for the binding of YYW RNAP to the wt Fork. The fitted values for the rate constants are shown in the legend for Fig. 5. The observed rate constant for the slow phase in the formation of the heparin-resistant complex with the YYW RNAP is in reasonable agreement with those determined for the wt RNAP. To determine dissociation rates, RNAP and the wt Fork were incubated at 25°C for 30 min before the addition of heparin. Under these conditions the majority of the heparin-resistant complexes are expected to be present as RF3. The dissociation curves, reworked from experiments discussed in ref. 22, are biphasic. The fast phase (too fast for the manual mixing technique used here) corresponds to the dissociation of the heparin-sensitive complexes after the addition of heparin. In agreement with the equilibrium measurements, the fraction of sensitive complexes is significantly greater for YYW than for the wt RNAP. Because the equilibrium between RF1 and RFCR has a constant of 1.15 for wt RNAP, and the formation of RF3 is slow, one would expect the dissociation of the heparin-resistant complexes to similarly be slow. This is indeed the case. The curves in Fig. 5 A and B show slow association and dissociation kinetics at times over 600 s, with rate constants ka2 = 5 × 10−4 s−1 and kd2 = 1 × 10−4 s−1 for wt RNAP, and ka2 = 1 × 10−3 s−1 and kd2 = 6 × 10−4 s−1 for YYW. These values should be considered approximate because of the small changes in extents of binding detected for the slow phase over the duration of our observations (30 min). All the same, comparison of the ratios of rate constants indicates that the RF3 formed with wt RNAP is 3-fold more stable than that containing YYW RNAP, which is qualitatively consistent with the results presented in Table 1 and elsewhere (12, 22).
Figure 5.

Comparison of the kinetics for formation and dissociation of competitor-resistant complexes between RNAP and wt Fork. Association data (▪) were obtained as described in the text and the legend for Fig. 3 except the concentration of forked DNA was 10 nM. Dissociation kinetics (●) were obtained by challenging a mixture of 65 nM RNAP and 10 nM forked DNA that had been incubated for 30 min with 100 μg/ml heparin. The curves represent double-exponential fits of the data to Eq. 10. (A) wt RNAP. The observed association rate constants are shown in the legend to Fig. 3; for the slow phase of the dissociation of the wt Fork–wt RNAP complex, kd2 = (1.3 ± 0.2) × 10−4 s−1. (B) YYW RNAP. The slow phase of the association reaction has a ka2 = (1.1 ± 0.3) × 10−3 s−1; the slow phase of the dissociation reaction, a kd2 = (6 ± 1) × 10−4 s−1.
Decreased Stability of an Early Intermediate.
We also followed the kinetics of dissociation after reducing the time of preincubation of wt RNAP and wt Fork before the addition of heparin to 30 s, which should predominantly lead to the formation of RF2 and little RF3. The dissociation kinetics again were observed to be biphasic, but now the slow phase is characterized by a rate constant, kd2, of 1 × 10−3 s−1, which is 10 times larger than that for the slow process observed after a preincubation of 30 min and interpreted above as the dissociation of RF3. This result provides additional evidence for the formation of two competitor-resistant complexes (i.e., RF2 and RF3), further justifying the use of a double-exponential equation (Eq. 8) for fitting the association data. A 2-min heparin challenge was used for the determination of the kinetics of complex formation (Fig. 3). It can be estimated that the fraction of the RF2 complexes that dissociated during this challenge is 0.12, leading to a slight underestimation of the extent of complex formation during the early phase of the association reaction.
Discussion
Evidence for Two Intermediates.
The results presented here establish that the formation of the final competitor-resistant complex between RNAP and forked DNA proceeds through heparin-sensitive and heparin-resistant intermediates, as indicated in Scheme 2. Two lines of evidence support the contention that the heparin-sensitive intermediate is RF1 on the pathway to formation of the heparin-resistant forms. First, it forms with faster kinetics than the heparin-resistant ones. Second, it is sequence-specific, inconsistent with its being a complex between the RNAP and a randomly positioned forked DNA. However, we cannot rigorously exclude the possibility that a fraction of the heparin-sensitive complexes is not RF1 but an off-pathway complex. Support for an additional heparin-resistant intermediate (RF2) is derived from the observation of biphasic kinetics in the formation of heparin-resistant complexes. We propose that these two intermediates reflect those that are encountered on the pathway to formation of an open RNAP–promoter complex.
RF1 has properties characteristic of a closed RNAP–promoter complex; it is the first demonstrable intermediate heparin-sensitive complex and the main intermediate formed by the FYW and YYW RNAP mutants, the ability of which to proceed beyond the closed complex in the formation of an RNAP–promoter complex is impaired greatly. K1, the equilibrium constant for the formation of this RF1 intermediate, is (at ≈108 M−1) somewhat larger than that measured for the interaction of RNAP and the PR promoter to form a closed complex (5) but within the range observed for other strong promoters (29). This comparison should be made cautiously, because the values obtained here may be higher than those for promoters solely because of the fact that the solutions used here did not contain Mg2+, whereas the promoter studies used buffers containing 10 mM Mg2+ (see ref. 30 for a discussion of Mg2+ effects).
Comparison of Equilibrium and Kinetic Data.
The rate constant for the conversion of RF1 to RF2, k2, is ≈0.1 s−1, which is within the range found for the composite rate constant (often referred to as kf) for the concentration-independent steps in open complex formation at strong naturally occurring promoters (29). The value of ka for this process (4 × 106 M−1 s−1) is also in line with that for formation of open complexes at such promoters, again with the caveat that the buffers used here did not contain added Mg2+. Although the second step in the three-step mechanism is rate-limiting in both directions for the RNAP–PR interaction, our findings indicate that in the interaction of RNAP with forked DNA, the last step, formation of RF3, is rate-limiting in both directions. Formation of RF3 is slow, with a rate constant comparable to that for formation of open complexes at weak promoters (29). We consider two possible interpretations of the differences in the location of the rate-limiting steps in formation of RF3 and open promoter complexes. It is assumed that the rate-limiting isomerizations between intermediates involve conformational changes in the RNAP, because no DNA melting would take place with the forked templates, and that these may be affected by the lack of downstream DNA in the forked template. The conformation of RNAP in RF2 may be similar to that of I2 (or the open complex). Any subsequent isomerization of RF2 to RF3 then would be the result of the lack of downstream DNA and would not be relevant to complexes formed at promoters. Alternatively, the conformation of RNAP in RF2 may be similar to that in I1. Then the conversion of RF2 to RF3 may involve the same additional conformational changes in RNAP that occur when I1 isomerizes to I2, but they would proceed much more slowly with the forked DNA because of the absence of the downstream DNA. We favor the first alternative (i.e., RF2 would model I2), because RF2 and I2 are both heparin-resistant and are formed with similar kinetics.
Correlation Between Forked DNA Binding and Open Complex Formation.
In published reports (16–18, 22), the relative ability of mutationally altered RNAP to yield heparin-resistant complexes with forked DNAs of various sequences generally has been determined at just one concentration of RNAP. Even so, major differences in extents of complex formation that correlated well with the ability to form open complexes at promoters were evident (22). The results presented here establish the basis for the observed correlation. For measurements carried out at sufficiently high concentrations of RNAP, the extents of binding will only reflect differences in Kf (Scheme 2 and Eq. 2) and not small differences in K1, because the binding step (formation of RF1) will be saturated. The actual concentration of RNAP then does not affect the interpretation of the experiment, because the process governed by Kf is concentration-independent. Thus interaction of RNAP with forked DNA is a fast and convenient means for quantitatively determining the extents to which changes in promoter or RNAP sequence or even added protein factors would affect the process of open complex formation.
Acknowledgments
We thank Drs. Vernon Anderson and M. Thomas Record, Jr. for helpful discussion and Dr. Timothy Lohman for reading a draft of the manuscript. We are grateful to Dr. Jeffrey Roberts for providing cells overexpressing histidine-tagged σ70 containing the Q437H substitution, and we thank Epicentre Technologies (Madison, WI) for a generous gift of RNAP holoenzyme. This work was supported by National Institutes of Health Grant GM 31808 (to P.L.d.H.). O.V.T. was supported by National Institutes of Health Grant GM23467 to Dr. M. T. Record, Jr.
Abbreviations
- RNAP
RNA polymerase
- wt
wild type
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Zhang G, Campbell E A, Minakhin L, Richter C, Severinov K, Darst S A. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- 2.Landick R. Cell. 2001;105:567–570. doi: 10.1016/s0092-8674(01)00381-6. [DOI] [PubMed] [Google Scholar]
- 3.Naryshkin N, Revyakin A, Kim Y, Mekler V, Ebright R H. Cell. 2000;101:601–611. doi: 10.1016/s0092-8674(00)80872-7. [DOI] [PubMed] [Google Scholar]
- 4.Siebenlist U, Simpson R B, Gilbert W. Cell. 1980;20:269–281. doi: 10.1016/0092-8674(80)90613-3. [DOI] [PubMed] [Google Scholar]
- 5.Roe J H, Burgess R R, Record M T., Jr J Mol Biol. 1984;176:495–521. doi: 10.1016/0022-2836(84)90174-8. [DOI] [PubMed] [Google Scholar]
- 6.Buc H, McClure W R. Biochemistry. 1985;24:2712–2723. doi: 10.1021/bi00332a018. [DOI] [PubMed] [Google Scholar]
- 7.Johnson R S, Chester R E. J Mol Biol. 1998;283:353–370. doi: 10.1006/jmbi.1998.2101. [DOI] [PubMed] [Google Scholar]
- 8.Record M T, Jr, Reznikoff W S, Craig M L, McQuade K L, Schlax P J. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt FC, editor. Washington, DC: Am. Soc. Microbiol.; 1996. , 792–820. [Google Scholar]
- 9.deHaseth P L, Zupancic M, Record M T., Jr J Bacteriol. 1998;180:3019–3025. doi: 10.1128/jb.180.12.3019-3025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacic R T. J Biol Chem. 1987;262:13654–13661. [PubMed] [Google Scholar]
- 11.Li X-Y, McClure W R. J Biol Chem. 1998;273:23549–23557. doi: 10.1074/jbc.273.36.23549. [DOI] [PubMed] [Google Scholar]
- 12.Panaghie G, Aiyar S E, Bobb K L, Hayward R S, deHaseth P L. J Mol Biol. 2000;299:1217–1230. doi: 10.1006/jmbi.2000.3808. [DOI] [PubMed] [Google Scholar]
- 13.Tsodikov O V, Record M T., Jr Biophys J. 1999;76:1320–1329. doi: 10.1016/S0006-3495(99)77294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Craig M L, Tsodikov O V, McQuade K L, Jr, Schax P E, Jr, Capp M W, Saecker R M, Record M T., Jr J Mol Biol. 1998;283:741–756. doi: 10.1006/jmbi.1998.2129. [DOI] [PubMed] [Google Scholar]
- 15.Tsodikov O V, Craig M L, Saecker R M, Record M T., Jr J Mol Biol. 1998;283:757–769. doi: 10.1006/jmbi.1998.2130. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Gralla J D. Proc Natl Acad Sci USA. 1998;95:11655–11660. doi: 10.1073/pnas.95.20.11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenton M S, Lee S J, Gralla J D. EMBO J. 2000;19:1130–1137. doi: 10.1093/emboj/19.5.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fenton M S, Gralla J D. Proc Natl Acad Sci USA. 2001;98:9020–9025. doi: 10.1073/pnas.161085798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helmann J D, deHaseth P L. Biochemistry. 1999;37:5959–5967. doi: 10.1021/bi990206g. [DOI] [PubMed] [Google Scholar]
- 20.Burgess R R, Jendrisak J J. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- 21.Gonzales N, Wiggs J, Chamberlin M J. Arch Biochem Biophys. 1977;182:404–408. doi: 10.1016/0003-9861(77)90521-5. [DOI] [PubMed] [Google Scholar]
- 22.Tomsic M, Tsujikawa L, Panaghie G, Wang Y, Azok J, deHaseth P L. J Biol Chem. 2001;276:31891–31896. doi: 10.1074/jbc.M105027200. [DOI] [PubMed] [Google Scholar]
- 23.Waldburger C, Gardella T, Wong R, Susskind M M. J Mol Biol. 1990;215:267–276. doi: 10.1016/s0022-2836(05)80345-6. [DOI] [PubMed] [Google Scholar]
- 24.Keilty S, Rosenberg M. J Biol Chem. 1987;262:1015–1022. [PubMed] [Google Scholar]
- 25.Burr T, Mitchell J, Kolb A, Minchin S, Busby S. Nucleic Acids Res. 2000;28:1864–1870. doi: 10.1093/nar/28.9.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi M, Nagata K, Ishihama A. Nucleic Acids Res. 1990;18:7367–7372. doi: 10.1093/nar/18.24.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moyle H, Waldburger C, Susskind M M. J Bacteriol. 1991;173:1944–1950. doi: 10.1128/jb.173.6.1944-1950.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClure W R. Proc Natl Acad Sci USA. 1980;77:5634–5638. doi: 10.1073/pnas.77.10.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClure W R. Annu Rev Biochem. 1985;54:171–204. doi: 10.1146/annurev.bi.54.070185.001131. [DOI] [PubMed] [Google Scholar]
- 30.Record M T, Jr, deHaseth P L, Lohman T M. Biochemistry. 1977;16:4791–4796. doi: 10.1021/bi00641a005. [DOI] [PubMed] [Google Scholar]