

Abstract
Pluripotent embryonic stem (ES) cells have been used to produce genetically modified mice as experimental models of human genetic diseases. Increasingly, human ES cells are being considered for their potential in the treatment of injury and disease. Here we have shown that mutation in murine ES cells, heterozygous at the selectable Aprt locus, differs from that in embryonic somatic cells. The mutation frequency in ES cells is significantly lower than that in mouse embryonic fibroblasts, which is similar to that in adult cells in vivo. The distribution of spontaneous mutagenic events is remarkably different between the two cell types. Although loss of the functional allele is the predominant mutation type in both cases, representing about 80% of all events, mitotic recombination accounted for all loss of heterozygosity events detected in somatic cells. In contrast, mitotic recombination in ES cells appeared to be suppressed and chromosome loss/reduplication, leading to uniparental disomy (UPD), represented more than half of the loss of heterozygosity events. Extended culture of ES cells led to accumulation of cells with adenine phosphoribosyltransferase deficiency and UPD. Because UPD leads to reduction to homozygosity at multiple recessive disease loci, including tumor suppressor loci, in the affected chromosome, the increased risk of tumor formation after stem cell therapy should be viewed with concern.
The frequency and types of spontaneous mutation that occur in pluripotent stem cells may have an impact on their future clinical uses. Embryonic stem (ES) cells are intrinsically different from somatic cells in that they retain the capacity to differentiate into multiple cell types (1, 2). Maintenance of genomic stability in these cells is essential for their potential to serve as donor cells for tissue transplants. Here we have examined spontaneous and induced mutagenic events in ES cells. We have taken advantage of a recently described murine model that utilizes mice heterozygous at the selectable marker encoding the purine salvage enzyme adenine phosphoribosyltransferase (APRT) to examine this issue (3–5).
The Aprt gene, resident on mouse chromosome 8, is an endogenous marker that supports selection for cells that are APRT deficient and, conversely, for cells that have APRT activity (6). The model uses cells derived from 129SvEv × C3H/HeJ mice or embryos in which the strain 129SvEv Aprt allele has been interrupted by a targeted neo insertion (7). These cells are therefore heterozygous at Aprt, with the neo insertion allowing for rapid discrimination between the targeted and untargeted alleles. Polymorphic microsatellites that span the length of chromosome 8 can be used to confirm loss of heterozygosity (LOH) resulting from either mitotic recombination, or multilocus deletion. Centromeres in mouse strains 129SvEv and C3H/HeJ are different sizes. Hence, the parental origin of copies of chromosome 8 can be determined, providing visual determination of uniparental disomy (UPD), defined as the presence of homologous chromosomes of same parental origin.
In this paper, we demonstrate that early embryonic cells and somatic cells differ with respect to maintaining genomic stability. Mutation was less frequent in ES cells than somatic cells, and the predominant mechanism of mutation in ES cells was the formation of UPD rather than LOH mediated by mitotic recombination as described (8–10). These findings raise a concern regarding the use of stem cells that have been maintained in long-term culture for therapeutic purposes.
Methods
Derivation of 129SvEv × C3H/HeJ Aprt+/neo Mouse ES Cells.
The derivation of 129SvEv Aprtneo/neo mice has been described (7). These mice were crossed to wild-type C3H/HeJ mice purchased from The Jackson Laboratory. The 129SvEv × C3H Aprt+/neo F1 blastocysts were isolated at 3.5 days post coitus ES cells were derived from male blastocysts as described (11) and maintained on mouse embryonic fibroblast feeder layers.
Measurement of Mutation Rate.
Preexisting APRT and hypoxanthine phosphoribosyltransferase (HPRT) mutants were eliminated by culture in the presence of either alanosine/adenine, or hypoxanthine/aminopterin/thymidine, respectively. When cells were treated with either ethyl methanesulfonate (EMS) or N-ethyl-N-nitrosourea (ENU), the treatment was for 5 h for EMS and 30 min for ENU, after which the cells were washed in media. Cell survival at 300 μg/ml and 600 μg/ml EMS was 50% and 5%, respectively. Cell survival at 0.5 and 1 mg/ml ENU was 35% and 3%, respectively. Cells were plated on two sets of 100-mm dishes. At that time, either 2 μg/ml 2-fluoroadenine (FA) or 1 μg/ml 6-thioguanine (6TG) was added to one set of plates. After a specified number of population doublings, FA or 6TG was added to the second set of plates. The FA-resistant (FAr) or 6TG-resistant colonies were fixed with Carnoy's solution, stained with crystal violet, and counted. Mutation rate was calculated by the P0 method as described (12, 13) and corrected for colony forming efficiency. Rates are presented per cell per generation.
Molecular and Cytogenetic Characterization of FAr Colonies.
All FAr colonies were assayed for APRT activity as described (8). PCR was used to determine whether FA resistance was caused physical loss of the wild-type Aprt. A polymerase with proofreading activity was used to minimize artifactual mutations and maintain the integrity of the amplified sequence. Cells that had lost the wild-type Aprt allele were characterized for LOH of flanking simple sequence repeat markers (Research Genetics, Huntsville, AL), and for UPD by fluorescence in situ hybridization with either chromosome-8 paints (Vysis, Downers Grove, IL) or a chromosome 8 locus-specific probe (Applied Genetics, Melbourne, FL). The Aprt gene from colonies that had retained the untargeted Aprt allele was PCR amplified and sequenced (8–10).
Results
The Spontaneous Mutation Frequency and Rate in ES Cells Are Much Lower than in Somatic Cells.
To determine the rate and frequency of mutation in ES cells, we produced two ES cell lines (clones 3C4 and 2B5) and mouse embryo fibroblasts (MEFs) from 129SvEv × C3H F1 embryos. A third ES cell line (1837) was derived from a strain 129SvEv blastocyst. All ES cells and MEFs were heterozygous at Aprt and hemizygous for Hprt. To rid the cultures of preexisting cells lacking either APRT or HPRT activity, cells were first placed in medium containing either alanosine/adenine or hypoxanthine/aminopterin/thymidine. After 48 h, the cells were placed in nonselective medium for a specified time and then placed in medium containing either FA or 6-thioguanine to select APRT- and HPRT-deficient colonies, respectively.
The frequency of APRT-deficient colonies was 100-fold lower in ES cells (clone 3C4) than in MEFs (Fig. 1a). The difference in mutation rate was even greater; MEFs generated APRT-deficient colonies nearly 400 times faster than ES cells (Fig. 1b). Although they arose at a relatively low rate, mutant ES cells accumulated as a function of time, and observed mutant frequencies were consistent with those expected based on mutation rate (Fig. 1c).
Figure 1.

The mutant frequency and mutation rate are lower in ES cells than in MEFs. (a) ES cells and MEFs heterozygous at the Aprt locus were either treated with the indicated concentration of ethyl methanesulfonate (EMS, μg/ml) for a period of 5 h to measure EMS-induced mutant frequency or remained untreated to measure spontaneous mutant frequency. Mutant frequency is corrected for colony-forming efficiency. SEM is indicated. Solid bars indicate Aprt mutant frequency, and hatched bars represent Hprt mutant frequency. The asterisk indicates that no spontaneously arising Hprt mutants were detected (mutation frequency <10−8). (b) Preexisting APRT and HPRT mutants were eliminated in ES cell and MEF cultures, which were then treated with EMS or left untreated as described in a. Mutation rate was calculated as previously described, but adapted for mammalian cells (13) and corrected for colony-forming efficiency. Solid bars indicate Aprt mutation rate and hatched bars represent Hprt mutation rate. The asterisk indicates that no spontaneously arising Hprt mutants were detected (mutation rate < 10−9). The differences between Aprt and Hprt mutant frequency and mutation rate were analyzed by Student's t test (P = 0.002). Differences in Aprt mutation frequency and rate between ES cells and MEFs were also statistically significant as analyzed by t test (P = 0.0004). (c) Mutant frequency at Aprt in ES cells increases with number of population doublings (PD). Solid bars indicated observed mutant frequency; hatched bars indicated predicted mutant frequency based on mutation rate. Asterisk indicates that no expected measurement is determined.
MEFs and ES cells differed even more dramatically when mutation rates were compared at the hemizygous Hprt locus (Fig. 1b), indicating that the low rate of mutation in ES cells is not locus specific. In fact, no spontaneous Hprt ES cell mutants were detected in these experiments, although HPRT-deficient ES cells were obtained after treatment with EMS, demonstrating that the selection method was appropriate (Fig. 1b). Similar results were obtained with ES cell clones 2B5 and 1837, showing that the results with clone 3C4 were not a peculiarity of a single cell clone or of a particular genetic background (Table 1).
Table 1.
Mutation at Aprt
| Cell line | Mutation frequency at Aprt | Mutation rate for Aprt | Percent of Aprt− colonies caused by mitotic recombination | Percent of Aprt− colonies caused by nondisjunction |
|---|---|---|---|---|
| 3C5 | 2.2 × 10−7 | 8.3 × 10−8 | 26 | 53 |
| 2B4 | 2.8 × 10−7 | 8.8 × 10−8 | 28 | 52 |
| 1837 | 4.2 × 10−7 | 1.0 × 10−7 | NA | NA |
The mutation frequencies and rates for Aprt were determined for each of the three ES cell clones as described in Methods for Aprt. The clones 3C5 and 2B4 were derived from a cross between mouse strains 129SvEv and C3H/HeJ. The 1837 clone was solely of 129SvEv origins. All Aprt colonies manifesting mitotic recombination or nondisjunction events had lost the untargeted Aprt allele. These colonies arising as a consequence of mitotic recombination had lost polymorphic markers flanking Aprt but had retained heterozygosity at proximal markers. Aprt− colonies arising as a consequence of nondisjunction had lost heterozygosity at all chromosome-8 markers. Nondisjunction was verified in colonies derived from clone 3C5 by fluorescence in situ hybridization analysis and centromere size. Mitotic recombination and nondisjunction events could not be analyzed in colonies derived from clone 1837 because it is homozygous throughout except at the Aprt locus. NA, not applicable.
That mutation at Hprt is very low or not detectable in ES cells has been reported by other laboratories (14–16). However, ES cells clearly are not refractory to mutation because treatment with the known alkylating agents EMS and ENU led to dose-dependent increases in mutation at both Aprt and Hprt (Figs. 1 and 2) (15, 16). The higher mutation frequency at Aprt compared with Hprt is consistent with data reported for in vivo mutation in adult mouse skin fibroblasts (8) and splenic T cells (10). In these somatic cell types, the higher level of mutation at Aprt is caused by elevated LOH mediated by mitotic recombination at the autosomal locus (5, 8). Because of its X-linkage and hemizygous status, Hprt cannot undergo mitotic recombination and cannot accommodate mutations involving large chromosomal changes such as large multilocus deletions and chromosome loss. Hence, mutation at Hprt is restricted to intragenic events.
Figure 2.

ES cell mutation rate at Aprt and Hprt both increase after exposure to ENU. ES cells heterozygous at the Aprt locus were either treated with the indicated concentration of ENU (μg/ml) for a period of 5 h to measure ENU-induced mutant frequency or remained untreated to measure spontaneous mutant frequency. Mutation rate was calculated as previously described (13) and corrected for colony-forming efficiency. SEM is indicated. Solid bars indicate Aprt mutation rate, and hatched bars represent Hprt mutation rate. The asterisk indicates that no spontaneously arising Hprt mutants were detected (mutation rate <10−9).
LOH Accounts for the Majority of Spontaneous Mutational Events in both ES Cells and MEFs.
Cells lacking APRT activity fell into two classes when the Aprt locus was analyzed by PCR. Class I cells, which constituted approximately 80% of the APRT-deficient clones, lacked the wild-type allele (Fig. 3a). Class II cells retained the Aprt allele and had presumably sustained an intragenic Aprt mutation. To verify that this was the case, the Aprt genes in 20 class II cell lines were sequenced. In all 20 cases examined, the Aprt allele had sustained a nonsilent mutation in the coding region. The spectrum of intragenic mutation was similar in ES cells and MEFs, with transversions representing about 60% of the events. After mutagenesis with EMS, the mutation spectrum shifted drastically (Fig. 4). Of 10 mutants sequenced, all showed G-to-A transitions, as would be expected after EMS exposure.
Figure 3.

ES cells and MEFs have distinctly different spectra of mutation. (a) ES cell and MEF variants were designated as class I (loss of the untargeted Aprt allele) or class II (retention of the untargeted Aprt allele) based on allele-specific PCR. PCR of the untargeted Aprt allele yields a 700-bp product; the targeted allele yields a 300-bp product. The +/+, −/−, and +/− controls were derived from wild type, null, and heterozygous (parental) ES cells, respectively. Class I variants and class II variants are designated “I” and “II” below their respective lanes. (b) PCR of informative microsatellite markers was used to define the mechanism of LOH in class I variants. D8Mit155 (1 centimorgan) and D8Mit 56 (73 centimorgan) are markers at the most centromeric and telomeric regions of mouse chromosome 8. 129SvEv × C3H: parental control; C3H: C3H genomic DNA control; a–h: representative FAr class I variants. (c) Distribution of mutation types in MEFs. (d) Distribution of mutation types in ES cells. Dashed line indicates mutagenic mechanisms contributing to loss of the untargeted Aprt allele. □, Point mutation/epigenetic inactivation; ■, chromosome loss and reduplication (nondisjunction); ▨, mitotic recombination; ▩, gene conversion/multilocus deletion/double crossover.
Figure 4.

Distribution of spontaneous (A) and EMS-induced (B) mutational events in ES cells. □, Intragenic mutation; ▨, mitotic recombination; ▩, chromosome loss/reduplication (nondisjunction); □, double crossover/gene conversion/deletion.
Chromosome Loss Is the Predominant Mechanism of Spontaneous Mutation in ES Cells but Not in MEFs.
In both MEFs and ES cells, about 80% of the APRT-deficient cells had suffered LOH at the Aprt locus. To determine whether LOH at Aprt had been caused by mitotic recombination, microsatellites linked to Aprt were analyzed by the PCR, which revealed that all of the LOH observed in class I MEF colonies occurred as a consequence of mitotic recombination (Fig. 3c). Hence, in the MEFs, the cause of LOH mimicked that reported for adult primary skin fibroblasts and splenic T cells from mice (8, 10, 17). In contrast, mitotic recombination accounted for only 41% of the LOH events seen in class I ES cells (Fig. 3d). Multilocus deletion, two recombination events, or a very large gene conversion event accounted for 2% of events and were assigned based on LOH at Aprt and retention of heterozygosity at flanking markers. The remaining 57% of cells that had lost the nontargeted Aprt allele also had lost heterozygosity at all alleles linked to this gene.
Cells that exhibited LOH at all chromosome-8 loci were further characterized by fluorescence in situ hybridization to determine whether these cells were isodisomic with respect to chromosome 8. Painting with chromosome-8-specific probes combined with and analysis of centromeric size identified the parental origin of each painted chromosome 8. Metaphase chromosomes from Aprt heterozygous cells (Fig. 5a) showed two copies of chromosome 8, one with a large centromere (C3H origin containing the wild-type Aprt allele) and one with a small centromere (129SvEv origin containing the targeted allele). In contrast, in many metaphase spreads from class I Aprt-deficient ES cell mutants, both copies of chromosome 8 had a small centromere (129SvEv origin), indicative of UPD (Fig. 5b). The mechanism by which UPD arises is most likely by sequential nondisjunction events, producing first a trisomic cell followed by a reduction to isodisomy. Indeed, fluorescence in situ hybridization of ES cells heterozygous for Aprt detected cells trisomic for chromosome 8 (Fig. 5c).
Figure 5.
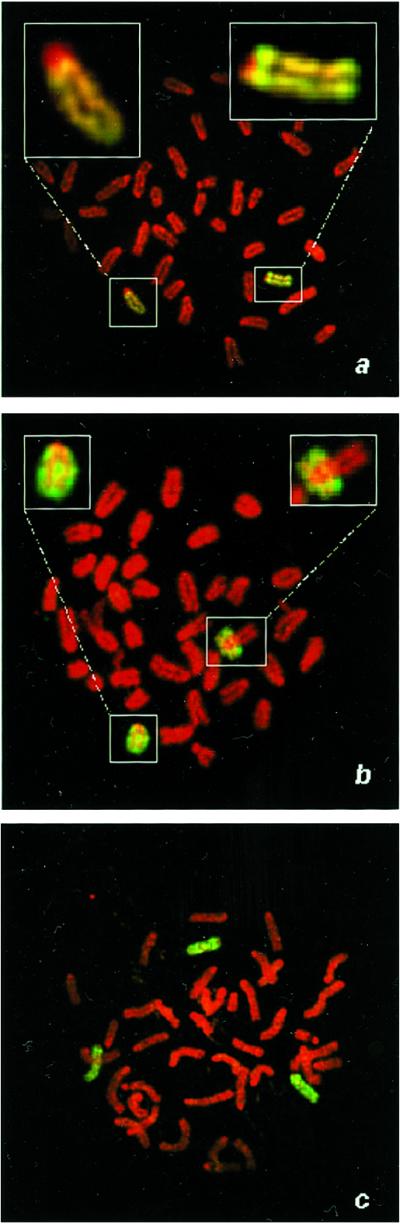
UPD is the major mechanism of mutation in ES cells. (a) A metaphase spread from a parental 129SvEv × C3H ES cell heterozygous at Aprt was hybridized with a paint specific for chromosome 8. Two chromosomes 8 are pictured, one with a large centromere (C3H parental origin, Left Inset) and one with small centromere (129SvEv parental, Right Inset). (b) Class I ES cell variant exhibiting UPD possesses two chromosomes 8 of 129SvEv parental origin (Insets). (c) Class I ES cell variant exhibiting trisomy 8.
Discussion
ES cells lost APRT function much less often than MEFs, which would suggest that stem cells are more stable genetically. However, loss of APRT occurred predominantly by chromosome loss, which was an event not detected in MEFs. The finding that the predominant spontaneous mutation in ES cells is LOH as a consequence of chromosome loss/reduplication is previously unreported and represents a pathway that is not commonly observed in other normal somatic cells. Others have reported epigenetic instability (18, 19) and karyotypic instability (20, 21), including involvement of chromosome 8 (20), with continued ES cell culture. Lefebvre et al. (22) demonstrated that large segments of four chromosomes containing a neo marker become isodisomic under high G418 selection, but this study could not formally distinguish between proximal mitotic recombination and nondisjunction events. We have quantified the frequency of chromosome loss in ES cells without the potential influence of long-term culture and demonstrated that chromosome loss in ES cells is frequently associated with reduplication of the remaining chromosome resulting in apparent euploidy. The incidence of chromosome loss/reduplication in most somatic cells is either very rare and undetectable or the preponderance of other mutational events reduces the likelihood of its detection. In pluripotent ES cells, however, where the frequency of mutational events common in somatic cells is greatly reduced, chromosome loss/reduplication emerges as the predominant mechanism of mutation.
The low frequency of mutation at Aprt in ES cells could be accounted for, in part, by suppression of mitotic recombination, which is the predominant mutational pathway in somatic cells (8). However, suppression of mitotic recombination cannot be ascribed to a deficiency in homologous recombination because genes are frequently inactivated in ES cells through targeted homologous recombination (23). Gene targeting (24) and intrachromosomal recombination (25) in ES cells are known to be strongly dependent on sequence identity, and this reliance on sequence homology is relieved when mismatch repair is absent. Similarly, we have shown that mitotic recombination is also highly dependent on DNA homology, and is much more so than is meiotic recombination (26). Further, the absence of mismatch repair alleviates a suppression of gene amplification in human cells (27), a process mediated by double-strand DNA breaks, and also renders mammalian cells hypersensitive to camptothecin, a drug that inhibits the DNA breakage-reunion activity of topoisomerase I (28). Thus, it is likely that mismatch repair modulates mitotic recombination and allows it to occur more frequently in MEFs than in ES cells. Alternatively, mitotic recombination might be observed less frequently in ES cells because ES cells that incur a double-strand break as an initiating event for mitotic recombination may survive poorly. ES cells appear to be acutely sensitive to agents that cause double-strand breaks such as mitomycin C (data not shown) and ionizing radiation (29, 30), but are far less sensitive to alkylating agents (31). Thus, the production of double-strand DNA breaks from endogenous sources such as oxidative stress may lead to recombination in most MEFs, but in relatively few ES cells. Chromosome loss and the generation of UPD are not a consequence of double-strand breaks, and therefore would not be diminished by such a hypothetical mechanism. The reduction in intragenic mutation in ES cells, observed both at Aprt and Hprt, may also be associated with mismatch repair activity. This observation would be consistent with elevated levels of point mutations that occur in mice lacking MLH1 or PMS2 (32).
The finding that mutation frequency in ES cells is lower than that in somatic cells raises hopes for the utility of stem cells in clinical applications. However, the propensity for ES cells to undergo UPD is worrisome. A single such event has the potential to generate a cell with multiple genetic defects because all of the recessive alleles on the affected chromosome are unmasked. The deleterious effects of UPD in humans manifest as congenital diseases, including certain cases of cystic fibrosis (human chromosome 7 maternal) and hemophilia A (human X chromosome paternal), and have been thoroughly reviewed (33). Similarly, UPD can alter the pattern of differential imprinting as is seen in Prader–Willi syndrome (human chromosome 15 maternal) and Angelman syndrome (human chromosome 15 paternal). UPD in ES cells may be associated with reduced p53 function and the absence of a fully functional G1 checkpoint because of the inefficient nuclear localization of p53 after DNA damage (29). Loss of p53 activity contributes to chromosome instability and increases the likelihood of nondisjunction (34, 35). This instability is reflected in the altered distribution of in vivo mutational events in skin fibroblasts from p53-deficient mice, which show a marked increase of isodisomy (9).
These data show that UPD can arise spontaneously in pluripotent mitotic stem cells as a predominant mutational mechanism. UPD could affect all chromosomes (20, 21), in which case the overall rate of UPD could be 10- to 20-fold higher than that measured for chromosome 8 alone. A previous report has argued that aneuploidy involving chromosome 8 is more common than missegregation involving other chromosomes (20), in which case the frequencies we report may not be indicative of those for other chromosomes. However, another report suggested that isodisomy occurred with similar frequency for each of four chromosomes analyzed (22). Therefore, the possibility that ES cells suffer UPD involving multiple chromosomes should be of concern. Because UPD allows all recessive loci on a given chromosome to manifest, including alleles encoding tumor suppressors, the accumulation of UPD in cultured stem cells raises a concern regarding clinical use of stem cells continuously maintained in culture. This concern does not constitute an argument against the therapeutic use of stem cells but rather indicates the need for screening such cultures to ensure the absence of UPD.
Acknowledgments
This work was supported by National Institutes of Health Grants PO1 ESO5652, DK 38185, and P30 ESO6096. R.B.C. was supported in part by training Grant T32 ESO7250 and an individual National Research Service Award.
Abbreviations
- ES
embryonic stem
- APRT
adenine phosphoribosyltransferase
- LOH
loss of heterozygosity
- UPD
uniparental disomy
- HPRT
hypoxanthine phosphoribosyltransferase
- EMS
ethyl methanesulfonate
- ENU
N-ethyl-N-nitrosourea
- FA
2-fluoroadenine
- MEF
mouse embryo fibroblast
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder J C. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brook F A, Gardner R L. Proc Natl Acad Sci USA. 1997;94:5709–5712. doi: 10.1073/pnas.94.11.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vrieling H, Wijnhoven S, van Sloun P, Kool H, Giphart-Gassler M, van Zeeland A. Environ Mol Mutagen. 1999;34:84–89. [PubMed] [Google Scholar]
- 4.Tischfield J A. Am J Hum Genet. 1997;61:995–999. doi: 10.1086/301617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stambrook P J, Shao C, Stockelman M, Boivin G, Engle S J, Tischfield J A. Environ Mol Mutagen. 1996;28:471–482. doi: 10.1002/(SICI)1098-2280(1996)28:4<471::AID-EM25>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 6.Tischfield J A, Ruddle F H. Proc Natl Acad Sci USA. 1974;71:45–49. doi: 10.1073/pnas.71.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engle S J, Stockelman M G, Chen J, Boivin G, Yum M N, Davies P M, Ying M Y, Sahota A, Simmonds H A, Stambrook P J, Tischfield J A. Proc Natl Acad Sci USA. 1996;93:5307–5312. doi: 10.1073/pnas.93.11.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao C, Deng L, Henegariu O, Liang L, Raikwar N, Sahota A, Stambrook P J, Tischfield J A. Proc Natl Acad Sci USA. 1999;96:9230–9235. doi: 10.1073/pnas.96.16.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao C, Deng L, Henegariu O, Liang L, Stambrook P J, Tischfield J A. Proc Natl Acad Sci USA. 2000;97:7405–7410. doi: 10.1073/pnas.97.13.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang L, Deng L, Shao C, Stambrook P J, Tischfield J A. Environ Mol Mutagen. 2000;35:150–157. [PubMed] [Google Scholar]
- 11.Martin G. Science. 1980;209:768–776. doi: 10.1126/science.6250214. [DOI] [PubMed] [Google Scholar]
- 12.Lea D E, Coulson C A. J Genet. 1949;119:264–284. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 13.Li I C, Chu E H. Mutat Res. 1987;190:281–287. doi: 10.1016/0165-7992(87)90010-8. [DOI] [PubMed] [Google Scholar]
- 14.Chen R Z, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. Nature (London) 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Yee D, Dains K, Chatterjee A, Cavalcoli J, Schneider E, Om J, Woychik R P, Magnuson T. Nat Genet. 2000;24:314–317. doi: 10.1038/73557. [DOI] [PubMed] [Google Scholar]
- 16.Munroe R J, Bergstrom R A, Zheng Q Y, Libby B, Smith R, John S W, Schimenti K J, Browning V L, Schimenti J C. Nat Genet. 2000;24:318–321. doi: 10.1038/73563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Sloun P P, Wijnhoven S W, Kool H J, Slater R, Weeda G, van Zeeland A A, Lohman P H, Vrieling H. Nucleic Acids Res. 1998;26:4888–4894. doi: 10.1093/nar/26.21.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphries D, Eggan K, Akutsu H, Hochedlinger K, Rideout W M, 3rd, Biniszkiewicz D, Yanagimachi R, Jaenisch R. Science. 2001;293:95–97. doi: 10.1126/science.1061402. [DOI] [PubMed] [Google Scholar]
- 19.Ohgane J, Wakayama T, Kogo Y, Senda S, Mattori N, Tanaka S, Yanagimachi R, Shiota K. Genesis. 2001;30:45–50. doi: 10.1002/gene.1031. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Wu H, Loring J, Hormuzdi S, Disteche C M, Bornstein P, Jaenisch R. Dev Dyn. 1997;209:85–91. doi: 10.1002/(SICI)1097-0177(199705)209:1<85::AID-AJA8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 21.Longo L, Bygrave A, Grosveld F G, Pandolfi P P. Trans Res. 1997;6:321–328. doi: 10.1023/a:1018418914106. [DOI] [PubMed] [Google Scholar]
- 22.Lefebvre L, Dionne N, Karaskova J, Squire J A, Nagy A. Nat Genet. 2001;27:257–258. doi: 10.1038/85808. [DOI] [PubMed] [Google Scholar]
- 23.Gossler A, Doetschman T, Korn R, Serfling E, Kemler R. Proc Natl Acad Sci USA. 1986;83:9065–9069. doi: 10.1073/pnas.83.23.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.deWind N, Decker M, Berns A, Radman M, le Riehl H. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 25.Elliott B, Jasin M. Mol Cell Biol. 2001;21:2671–2682. doi: 10.1128/MCB.21.8.2671-2682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao C, Stambrook P J, Tischfield J A. Nat Genet. 2001;28:169–172. doi: 10.1038/88897. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Bigner S H, Modrich P. Proc Natl Acad Sci USA. 2001;98:13802–13807. doi: 10.1073/pnas.241508098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pichierri P, Franchitto A, Piergentili R, Colussi C, Palitti F. Carcinogenesis. 2001;22:1781–1787. doi: 10.1093/carcin/22.11.1781. [DOI] [PubMed] [Google Scholar]
- 29.Aladjem M I, Spike B T, Rodewald L W, Hope T J, Klemm M, Jaenisch R, Wahl G M. Curr Biol. 1998;8:145–155. doi: 10.1016/s0960-9822(98)70061-2. [DOI] [PubMed] [Google Scholar]
- 30.Corbet S W, Clarke A R, Gledhill S, Wyllie A H. Oncogene. 1999;18:1537–1544. doi: 10.1038/sj.onc.1202436. [DOI] [PubMed] [Google Scholar]
- 31.Van Sloun P P, Jansen J G, Weeda G, Mullenders L H, van Zeeland A A, Lohman P H, Vrieling H. Nucleic Acids Res. 1999;27:3276–3282. doi: 10.1093/nar/27.16.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baross-Francis A, Makhani N, Liskay R M, Jivik F R. Oncogene. 2001;20:619–625. doi: 10.1038/sj.onc.1204138. [DOI] [PubMed] [Google Scholar]
- 33.Robinson W P. BioEssays. 2000;22:452–459. doi: 10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 34.Bouffler S D, Kemp C J, Balmain A, Cox R. Cancer Res. 1995;55:3883–3889. [PubMed] [Google Scholar]
- 35.Fukasawa K, Choi T, Kuriyama R, Rulong S, Van de Woude G F. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]