

Abstract
It has been argued that genetic instability is required to generate the myriad mutations that fuel tumor initiation and progression and, in fact, patients with heritable cancer susceptibility syndromes harbor defects in specific genes that normally maintain DNA integrity. However, the vast majority of human cancers arise sporadically, in the absence of deficiencies in known “mutator” genes. We used a cII-based mutation detection assay to show that the mean frequency of forward mutations in primary mammary adenocarcinomas arising in mouse mammary tumor virus-c-erbB2 transgenic mice harboring multiple copies of the λ bacteriophage genome was significantly higher than in aged-matched, wild-type mammary tissue. Analysis of the cII mutational spectrum within the mammary tumor genomic DNA demonstrated a >6-fold elevation in transversion mutation frequency, resulting in a highly unusual inversion of the transition/transversion ratio characteristic of normal epithelium; frameshift mutation frequencies were unaltered. Arising oncogenic point mutations within the c-erbB2 transgene of such tumors were predominantly transversions as well. Data from this model system support the notion that elaboration of a mutator phenotype is a consequential event in breast cancer and suggest that a novel DNA replication/repair gene is a relatively early mutational target in c-erbB2-induced mammary tumorigenesis.
From the genetic perspective, cancer represents the phenotypic consequence of the accumulation of a minimal number of mutations in genes controlling cell growth, differentiation, and/or survival. Because this minimal number is likely too large to be accounted for by the relatively low mutation rate characteristic of normal somatic cells, it has been hypothesized that cancer must develop through manifestation of a genetically unstable state, or a “mutator” phenotype (1, 2). Such a state could be achieved through early mutations in genetic stability genes, spawning subsequent mutations throughout the genome over time. This concept has been strengthened by the detection of cancer cell-specific alterations in the lengths of tandem repetitive nucleotide sequences, or microsatellites, infrequently in sporadic and frequently in familial human colorectal tumors (reviewed in ref. 3). The suggestion that microsatellite instability associated with familial human disorders, such as hereditary nonpolyposis colorectal cancer (HNPCC), was the consequence of defects in mismatch repair genes (4) was realized by the discovery in HNPCC kindreds of inactivating germ-line mutations in hMSH2 and hMLH1, the homologs of the bacterial mismatch repair genes mutS and mutL, respectively (5–8). Subsequently, mice harboring germ-line null mutations in Msh2, Msh6, Pms2, or Mlh1 were shown to be cancer prone, demonstrating various tumor spectra (reviewed in ref. 3). Thymic lymphomas, but not nonthymic tumors, arising in Msh2-deficient mice carrying a transgenic lacI gene serving as a screen for subtle sequence changes demonstrated a mutant frequency that was elevated an average of about 8-fold compared with normal thymi (9, 10). However, the lacI mutational spectrum in thymic lymphomas was not significantly altered relative to wild-type controls.
It is now appreciated that other cancer susceptibility syndromes, such as ataxia telangiectasia, Fanconi's anemia, Bloom's syndrome, xeroderma pigmentosum, and Li-Fraumeni syndrome, are associated with genetic instability attributable to defects in DNA damage response (reviewed in refs. 11 and 12). These heritable instabilities can occur at the nucleotide level, as when defects involve nucleotide excision and mismatch repair genes, as well as at the chromosome level, leading to changes in substantial portions of, or even whole, chromosomes. However, the vast majority of cancers arise with no obvious family history and without germ-line mutations in known DNA replication/repair genes. In these sporadic tumors, the role of genetic instability and the identity of genetic defects responsible for the creation of such states is largely unknown.
ErbB2, also known as Neu or HER2, is a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases, together with the EGFR (ErbB1/HER1), ErbB3 (HER3), and ErbB4 (HER4) (reviewed in refs. 13 and 14). Although originally discovered as a highly oncogenic mutant in carcinogen-induced rat tumors (15), this activated form is rarely found in human cancer. Instead, wild-type c-erbB2 is overexpressed and/or amplified in 10–30% of breast cancers (16, 17), where it correlates with chemo-resistance and poor patient prognosis (reviewed in refs. 18 and 19). Notably, Herceptin, a mAb against ErbB2, has been found to be an effective treatment for a subset of patients with advanced breast cancer (20, 21). Mouse models of breast cancer have been created through mouse mammary tumor virus (MMTV)-targeted expression of both wild-type and mutationally activated c-erbB2, confirming their oncogenic role in mammary carcinogenesis (22, 23). Here we use a transgenic mouse model, relevant to human breast cancer by virtue of targeted aberrant overexpression of c-erbB2, to determine whether genetic instability is associated with mammary tumorigenesis in vivo in the absence of heritable defects in known DNA maintenance genes.
Materials and Methods
Escherichia coli Strains.
The E. coli host strain G1250 (Stratagene) used in this study, which carries mutant hflA and hflB genes that facilitate the lysogenic response by increasing the stability of the cII protein, has been described in detail (24).
Mouse Strains.
The ErbB2/Big Blue bitransgenic mice used in these studies were generated by mating female mice carrying the MMTV-c-erbB2 transgene on a FVB/N background (23) with male mice carrying a transgene consisting of multiple copies of the λ transgene (25) on a C57BL/6 background (Big Blue Mouse; Stratagene). Crossing the FVB/N-based ErbB2 transgenic mice onto C57BL/6 lengthened mammary tumor latency in the F1 generation to more than 18 months, as described (26). All female mice experienced 2–4 pregnancies, a practice that stimulated the development of mammary tumors. Control mice were generated by mating wild-type FVB/N and Big Blue transgenic mice. The genotypes of the F1 generation were determined by PCR analysis of tail genomic DNA. Primers specific for the λ transgene were 5′-GTTATGACGAAGAAGAACGG-3′ and 5′-GCTCAAGAGCGATGTTAATT-3′. All animals were cared for and maintained in accordance with National Institutes of Health animal care guidelines.
λ cII Mutagenesis Assay.
Genomic DNA was isolated from Big Blue mouse tissues with a RecoverEase DNA Isolation Kit (Stratagene). λ cII mutagenesis assays were performed as described (24). The λLIZ shuttle vector was rescued from genomic DNA by using Transpack packaging extract (Stratagene). The packaged phages were adsorbed to G1250 and plated on TB1 plates according to instructions for the λ Select-cII Mutation Detection System for Big Blue Rodents (Stratagene). To determine the total titer of packaged sample, λ phage-infected G1250 cultures were incubated overnight at 37°C—the nonselective condition. To select for phage containing mutant cII genes, λ phage-infected G1250 cultures were plated and incubated at 24°C for 40–48 h, the conditions under which only phage containing a mutant cII gene commit to the lytic pathway and form plaques. The frequency of λ cII mutants was then determined by dividing the plaque number on the selecting plates by the plaque number on the nonselecting plates. The mutant frequency is expressed as the mean ± SEM. Differences among mutant frequencies were tested for statistical significance by using a Student's t test.
DNA Sequence Analysis.
λ cII mutant plaques were picked at random and eluted into SM buffer (50 mM Tris⋅HCl, pH 7.5/10 mM MgSO4/0.01% gelatin/0.2 NaCl). The entire 294-bp cII gene was PCR-amplified with Vent polymerase (New England Biolabs) under conditions designed to minimize mutations introduced by PCR. Sequencing was performed by using an Applied Biosystems model 310 automated DNA sequencer. Primer pairs used in PCR amplification and sequencing reactions were 5′-CTTGCTCAATTGTTATCAGC-3′ and 5′-GTCATAATGACTCCTGTTGA-3′, and 5′-ACCACACCTATGGTGTATGCA-3′ and 5′-GTCATAATGACTCCTGTTGA-3′, respectively.
Tumor Transplantation.
Portions of mammary adenocarcinomas arising in multiparous ErbB2/Big Blue bitransgenic mice (FVB/N × C57BL/6 F1 generation) were immediately snap-frozen for use in mutation analyses. For the generation of metastases, a piece of the primary tumor was either implanted s.c. onto the backs of athymic nude mice (NCR-NU) or smashed through a filter to make a single cell suspension, 1 × 105 to 106 cells of which were immediately injected into the tail vein of nude mice without prior in vitro culturing. The resulting pulmonary metastases were then subjected to the λ cII mutagenesis assay.
Detection of cII Mutations in Vitro.
Primary low-passage BBM1 fibroblasts established from the Big Blue mouse harboring multiple tandom copies of the λLIZ shuttle vector were used for transfection of pJ4Ω-based expression vectors. Fibroblasts were cotransfected with pcDNA and either the pJ4Ω vector alone, the pJ4Ω vector expressing wild-type ErbB2 (pJ4Ω-Neu), or the pJ4Ω vector expressing mutationally activated ErbB2 (pJ4Ω-NT), and exposed to G418. After 2 weeks, the resistant pool and several individual clones were established. The cII mutant frequencies in these selected cells were determined as above. ErbB2 expression in these cells was assessed by using Western blotting. Confluent cells were lysed in RIPA-B buffer (20 mM sodium phosphate, pH 7.4/150 mM NaCl/100 mM sodium fluoride/2 mM PMSF/2 mM sodium orthovanadate/protease inhibitor, Roche Molecular Biochemicals), and protein concentrations were determined by using the Bio-Rad protein assay. Fifty micrograms of protein of each sample was loaded onto Novex 4–20% Tris-glycine gels, subjected to electrophoresis, and transferred to poly(vinylidene difluoride) membranes. The p185 ErbB2 protein was visualized by using the mAb Ab-3 (Oncogene Research Products, Boston) and the ECL detection system (Amersham Pharmacia). The ErbB2-overexpressing cell line SKBR3 was used as a positive control.
Results
ErbB2/Big Blue bitransgenic mice were generated carrying both a transgene expressing wild-type c-erbB2 by virtue of an MMTV promoter (23) and the λ bacteriophage genome (25). Mammary adenocarcinomas arising in multiparous ErbB2/Big Blue bitransgenic mice between 14 and 25 months of age (mean age of 20.8 months) were subjected to mutational analysis using a now well-used transgenic shuttle phage assay that selects for phage containing forward mutations in the λ cII gene (24). Mammary glands from female mice of an identical FVB/N × C57BL/6 F1 genetic background harboring the λ transgene alone failed to develop tumors and served as control tissue.
Table 1 shows that the mean mutant frequency in 11 primary mammary adenocarcinomas (designated neu-1 through neu-11) was 29.8 ± 3.3 × 10−5 (mean ± SE), significantly greater (P < 0.005) than the corresponding mean mutant frequency in wild-type mammary epithelium (from Big Blue mice without the ErbB2 transgene, designated wt-1 through wt-7) of 13.4 ± 1.0 × 10−5. Individual tumors had mutant frequencies up to 3.4-fold greater than mean wild-type controls. The tumor mutant frequency was also significantly higher (P < 0.05) than that detected in contralateral mammary glands within the same animals, 19.5 ± 2.1 × 10−5 (n = 7). Six fragments from a single tumor (neu-2) each were subjected to cII analysis and found to possess equivalently high mutant frequencies (mean of 34.8 ± 0.8 × 10−5), statistically indistinguishable from the original neu-2 test fragment (38.3 × 10−5). This result suggested that the observed elevation in mutant frequency was the consequence of an early genetic event in tumorigenesis, occurring before tumor cell clonal expansion. To determine whether further mutagenic enhancement was associated with late-stage tumor progression, we examined the cII mutant frequency in pulmonary metastases derived from a primary mammary adenocarcinoma (neu-10). The mean mutant frequency of 10 pulmonary metastases, 26.3 ± 2.0 × 10−5, was not significantly different from that of the original primary mammary tumor (18.0 × 10−5) or from s.c. transplants of that primary tumor (23.0 × 10−5).
Table 1.
λ cII mutant frequencies in mammary glands and tumors
| Mouse ID | Age (months) | Mammary glands
|
Mammary tumors
|
||||
|---|---|---|---|---|---|---|---|
| Total pfu × 103 | Mutant plaques | Mutant frequency × 10−5 | Total pfu × 103 | Mutant plaques | Mutant frequency × 10−5 | ||
| neu-1 | 19 | 381 | 56 | 14.7 | 280 | 98 | 35.0 |
| neu-2 | 19 | 274 | 42 | 15.3 | 188 | 72 | 38.3 |
| neu-3 | 20 | 220 | 60 | 27.3 | 288 | 123 | 42.7 |
| neu-4 | 20 | 263 | 63 | 24.0 | |||
| neu-5 | 23 | 272 | 54 | 19.8 | 285 | 98 | 34.4 |
| neu-6 | 23 | 178 | 82 | 46.1 | |||
| neu-7 | 17 | 184 | 25 | 13.6 | |||
| neu-8 | 23 | 233 | 50 | 21.7 | 164 | 180 | 31.7 |
| neu-9 | 24 | 223 | 38 | 17.0 | 192 | 56 | 29.2 |
| neu-10 | 25 | 234 | 42 | 18.0 | |||
| neu-11 | 25 | 102 | 21 | 20.7 | 443 | 64 | 14.4 |
| Mean ± SEM | 19.5 ± 2.1 | 29.8 ± 3.3 | |||||
| wt-1 | 6.0 | 144 | 21 | 14.6 | |||
| wt-2 | 4.5 | 523 | 98 | 18.7 | |||
| wt-3 | 4.0 | 220 | 29 | 13.2 | |||
| wt-4 | 4.0 | 243 | 26 | 10.7 | |||
| wt-5 | 5.5 | 157 | 17 | 10.8 | |||
| wt-6 | 4.0 | 148 | 19 | 12.8 | |||
| wt-7 | 5.5 | 142 | 18 | 12.7 | |||
| Mean ± SEM | 13.4 ± 1.0 | ||||||
Tissues are derived from: (neu-1 through neu-11) ErbB2/Big Blue bitransgenic mice carrying both the MMTV-c-erbB2 transgene and a second transgene consisting of multiple copies of the λ bacteriophage genome; or (wt-1 through wt-7) phenotypically wild-type Big Blue single transgenic mice crossed once to FVB/N mice. Note that although the wild-type group was substantially younger than the bitransgenic group, contralateral “normal” mammary tissues from seven tumor-bearing ErbB2/Big Blue bitransgenic mice were subjected to cII mutation analyses and their collective mutant frequency was significantly lower than the frank tumors (P < 0.05).
These data are consistent with the hypothesis that primary mammary neoplasms arise in mice expressing the c-erbB2 transgene in the context of genetic instability, negatively impacting the ability of a cell to repair spontaneous DNA damage. To determine the nature of this genetic instability, mutant cII genes in tumor cells were scrutinized for any shift in mutational spectrum relative to wild-type tissue. The entire 294-bp cII gene was sequenced in 41 λ cII-mutants derived from various mammary glands taken from single transgenic λ mice, 33 from the neu-3 tumor, and 41 from the neu-6 tumor. Upon amplification, all plaques gave full-length PCR products, indicating that there were no large cII deletions. Approximately 80% of the samples sequenced contained detectable mutations in cII, all of which altered the amino acid sequence; the remainder were presumed to be mutations elsewhere in λ that enhances λ PR transcription (24, 27). No sample was found to harbor more than one mutation in the λ cII gene. To ensure that only independent mutational events were being scored, mutations at the same nucleotide position occurring more than once in any tumor were excluded. Table 2 shows that within a collection of normal mammary tissues most cII mutations (66%) were transitions, predominantly of the G/C to A/T variety; the vast majority of these (95%) occurred at CpG islands. Only 29% of the cII mutations in normal tissue were transversions and 6% were frameshifts, consistent with other published data for the background mutational spectrum measured in mouse tissue (25, 28–30). Detailed analysis of a contralateral mammary gland from one of the oldest tumor-bearing animals revealed a typical transition-dominated mutation spectrum, indicating the absence of any overt age-dependent effect on mutagenesis (Table 2). In striking contrast, transversions were the predominant mutational type in two independent tumors, neu-3 and neu-6, representing 57% and 56% of all cII mutations analyzed, respectively. G/C to T/A mutations represented approximately half of the total number of transversions in the tumors. Transitions in the tumor DNA were poorly represented relative to nontumorous tissue (Table 2). The transversion mutation frequency was elevated 6.3- and 6.6-fold in the neu-3 and neu-6 tumors, respectively, relative to normal tissue; the transition mutation frequency was increased only about 1.8-fold. Frameshift mutations were not significantly altered, consistent with mismatch repair proficiency (Table 2).
Table 2.
λ cII mutational spectrum is shifted in ErbB2-induced mammary tumors
| Wild-type mammary glands | Aged contralateral mammary gland | Mammary tumor | Mammary tumor | |
|---|---|---|---|---|
| Mouse ID | Multiple glands | neu-11 | neu-6 | neu-3 |
| Mutants sequenced | 41 | 109 | 41 | 33 |
| Individual mutants | 35 (85%) | 32 (29%) | 27 (66%) | 21 (64%) |
| Transition | 23 (66%) | 18 (56%) | 9 (33%) | 8 (38%) |
| G/C → A/T | 21 (60%) | 18 (56%) | 7 (26%) | 7 (33%) |
| (% at CpG) | 20 (95%) | 12 (67%) | 5 (71%) | 4 (57%) |
| A/T → G/C | 2 (6%) | 0 (0%) | 2 (7%) | 1 (5%) |
| Transversion | 10 (29%) | 8 (25%) | 15 (56%) | 12 (57%) |
| G/C → T/A | 5 (14%) | 3 (9%) | 8 (30%) | 6 (29%) |
| G/C → C/G | 3 (9%) | 1 (3%) | 4 (15%) | 3 (14%) |
| A/T → T/A | 2 (6%) | 3 (9%) | 1 (4%) | 1 (5%) |
| A/T → C/G | 0 | 1 (3%) | 2 (7%) | 2 (10%) |
| Frameshift | 2 (6%) | 6 (19%) | 3 (11%) | 1 (5%) |
| +1 | 1 (3%) | 2 (6%) | 2 (7%) | 1 (5%) |
| −1 | 1 (3%) | 4 (13%) | 1 (4%) | 0 |
Identical mutations appearing at the same site in more than one phage were tabulated as a single mutational event.
Mammary tumorigenesis in MMTV-c-erbB2 transgenic mice has already been shown to occur in association with activating in-frame deletions in the extracellular region proximal to the transmembrane domain of the transgene itself, resembling alternatively spliced forms of ErbB2 detected in some human breast tumors (31). Further sequence analysis of primary mammary adenocarcinomas arising in these transgenic mice revealed the frequent occurrence of single nucleotide substitutions that invariably affected cysteine residues between amino acids 628 and 647 of ErbB2 (Table 3). The majority of these mutations were transforming in nature (data not shown). Notably, 60% of the independent tumors characterized by c-erbB2 substitutional mutations contained transversions, indicating that the unusually high incidence of transversions detected by cII analysis was not caused by an artifact associated with the ectopic placement of λ bacteriophage DNA in transgenic mouse cells, but could be found in bona fide mammalian sequences as well.
Table 3.
Point mutations detected in transgenic ErbB2
 |
The first line shows the wild-type ErbB2 (Neu) amino acid sequence for the cysteine-rich region proximal to the juxtatransmembrane domain. The five cysteine amino acids are indicated in bold. All point mutations were isolated from independent mammary tumors (BT) or from a lung metastasis (Lg) of MMTV-c-erbB2 transgenic mice (N202). The altered amino acid change is shown in bold and is underlined. Note that all mutations either add or delete a cysteine residue. The corresponding nucleotide point mutations shown on the right are underlined and bold.
The fact that eight of 11 primary mammary tumors arising in ErbB2/Big Blue bitransgenic mice demonstrated an enhanced mutant frequency of similar proportion raised the possibility that overexpression of c-erbB2 was sufficient to induce genetic instability. To test this hypothesis, vectors encoding either wild-type or activated c-ErbB2 were transfected into fibroblasts isolated from the λ-bearing Big Blue mouse. ErbB2 expression in stable clones of this transfection was found to be comparable to SKBR3, a human breast cancer cell line characterized by c-ERBB2 amplification/overexpression, but somewhat lower than in the mammary tumors arising in MMTV-c-erbB2 transgenic mice (Fig. 1). Analysis of cII mutations in representative stable clones grown over many cellular generations showed no general effect of either wild-type or activated c-erbB2 overexpression on mutagenesis, although one clone (Neu2) did exhibit an enhanced rate of cII mutation in culture (Fig. 2). These data suggest that c-erbB2 overexpression may be necessary to induce this unusual genetic instability, but it is not sufficient.
Figure 1.

Ectopic ErbB2 expression in (A) tissues from MMTV-c-erbB2/Big Blue bitransgenic mice and (B) stably transfected fibroblast cell lines derived from the Big Blue mouse. (A) Western blot of proteins extracted from mammary glands from either a Big Blue transgenic mouse (wt-7 gland) or ErbB2/Big Blue bitransgenic mouse (neu-12 gland), or from mammary adenocarcinomas from ErbB2/Big Blue bitransgenic mice (neu-12, neu-7 and neu-3 tumors). Blots were probed with the anti-ErbB2 mAb Ab-3, and then anti-β-actin as a loading control. The ErbB2-overexpressing cell line SKBR3 was included as a positive control. (B) Western blot of proteins extracted from Big Blue fibroblast cell lines stably transfected with vectors expressing either wild-type ErbB2 (NEU2, NEU3, and NEU pool) or mutationally activated ErbB2 (NT2 and NT4), or vector alone. The cell lines were from passage five (NEU2), seven (NEU3) or 13 (NT2 and NT4). Blots were probed with anti-ErbB2 antibody. Fifty micrograms of protein was loaded for each sample.
Figure 2.

Analysis of mutation rates in the cII gene of Big Blue fibroblasts ectopically expressing either wild-type ErbB2 [NEU2 (■), NEU3 (□), NEU pool (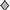 )], activated ErbB2 [NT4 (▴)], or harboring vector alone (
)], activated ErbB2 [NT4 (▴)], or harboring vector alone ( ). Only a single clone, NEU2, shows a consistent increase in mutant frequency.The expression was pJ4Ω (53).
). Only a single clone, NEU2, shows a consistent increase in mutant frequency.The expression was pJ4Ω (53).
Discussion
Acquisition of the full arsenal of mutations required to successfully build a cancer cell can be facilitated through deficiencies in genetic stability genes. A number of such genes have now been identified, mostly through their association with heritable cancer susceptibility syndromes, which function in DNA maintenance or cell cycle control (reviewed in refs. 3, 11, and 12). However, the identity of mutator genes that are not heritably deficient and their potential role in spontaneous tumorigenesis in vivo is largely unknown. In this study we used a cII-based mutation detection assay to examine subtle sequence genetic instability in cancer in vivo and show that the majority of sporadic mammary tumors arising spontaneously in mice carrying an MMTV-c-erbB2 transgene at a mean onset of 20 months possessed a significantly elevated frequency of forward mutations relative to wild-type mammary tissue. Notably, in contrast to recessive cancer susceptibility syndromes caused by defects in mismatch repair or nucleotide excision repair genes (3, 11, 12), spontaneous tumors arising in this transgenic breast cancer model were associated with an enhanced incidence of transversion base substitutions, which are recovered less often than transitions in repair competent cells; frameshift mutations characteristic of mismatch repair deficiencies were rarely detected. Mismatch repair-associated microsatellite instability, common in patients with familial colorectal tumors such as hereditary nonpolyposis colorectal cancer (5–8), is thought to be relatively uncommon in breast cancer (32). Moreover, Watanabe et al. (33) recently showed that single nucleotide mutations are frequently detected in human breast cancer cell lines in the absence of microsatellite instability. Interestingly, a relatively high incidence of transversions was recently reported for both a lacI transgene and the endogenous hprt gene in mammary carcinoma cell lines derived from PhIP-treated rats; two primary tumors from these rats had a similar mutation profile (34). Together, these data suggest the existence of a novel type of genetic instability in mammary tumorigenesis under conditions of mismatch repair proficiency. Such tissue-specific mutagenesis could be fueled, for example, by oxidative damage associated with the generation of estrogen metabolites in the breast (reviewed in refs. 35 and 36).
This genetic instability was not common to all mouse mammary carcinomas. We previously reported the absence of any overt mutator phenotype in mammary adenocarcinomas arising in MMTV-polyoma middle T transgenic mice (24). Moreover, the unusual transversion-dominated mutation profile was not a consequence of aging, which has been shown to affect somatic mutations in specific mouse tissues (37–40), because aged normal contralateral mammary glands possessed a typical mutation spectrum dominated by transitions. In fact, our finding that all pieces from a single tumor had the same elevated mutant frequency suggested that this mutator phenotype arose as a relatively early event in tumorigenesis. These findings raise the possibility that the novel genetic instability observed in this mouse model was a direct consequence of chronic c-erbB2 overexpression. It is known that c-erbB2 overexpression, a relatively early event in human breast cancer pathogenesis, disrupts cell cycle checkpoints and facilitates evasion of drug-induced apoptosis (41, 42). However, we could obtain no compelling evidence in cultured fibroblasts that forced c-erbB2 expression directly elevated cII mutations. Although we cannot exclude the possibility that ectopic c-erbB2 overexpression has a direct effect on mutagenesis in mammary epithelial cells, which express the ErbB2 signaling partners epidermal growth factor receptor and ErbB3, we believe it is more likely that through induction of mammary epithelial cell hyperplasia, early expression of the MMTV-c-erbB2 transgene creates an expanded population of mutational targets from which the genetic instability phenotype arises.
The mutator gene driving this novel genetic instability is not known. Our analysis indicates that transversions arising in the MMTV-c-erbB2 mammary tumors are predominantly of the G/C to T/A type. Interestingly, mutations in a small number of DNA repair genes that cause such transversions have been identified in single-cell organisms. For example, MutY/MutM complexes act to prevent G/C to T/A transversions, and MutY mutations can induce this mutator phenotype in E. coli and Schizosaccharomyces pombe (43–45). Similarly, deficiencies in Saccharomyces cerevisiae Rad6/Rad18 complexes are associated with G/C to T/A mutations (46, 47). These complexes all are involved in repair of oxidative DNA damage. Mammalian homologs of members of these complexes also have been cloned (48–51), and point mutations exhibiting dominant negative activity have been identified (45, 52). Together, these findings predict that a single early dominant mutation in a specific DNA repair/replication gene has the potential to create genetic instability in vivo, resulting in an accumulation of a transversion-dominated spectrum of mutations throughout tumor latency and/or in the context of a hypoxic tumor environment. It is anticipated that the identification of the transversion mutator gene(s) associated with ErbB2-induced mammary tumorigenesis, and the analysis of single nucleotide mutations in additional breast cancer mouse models, will provide critical insight into the molecular pathogenesis of breast cancer in general, and early initiating/promoting events in particular. Such knowledge may aid in devising new diagnostic and therapeutic weapons in the war against this deadly disease.
Acknowledgments
We are indebted to Drs. Shelley Berger and David Stern for their critical review of this manuscript and Elizabeth Snyderwine and the Merlino lab for useful discussions.
Abbreviation
- MMTV
mouse mammary tumor virus
Footnotes
See commentary on page 3368.
References
- 1.Loeb L A, Springgate C F, Battula N. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 2.Nowell P C. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 3.Buermeyer A B, Deschenes S M, Baker S M, Liskay R M. Annu Rev Genet. 1999;33:533–564. doi: 10.1146/annurev.genet.33.1.533. [DOI] [PubMed] [Google Scholar]
- 4.Strand M, Prolla T A, Liskay R M, Petes T D. Nature (London) 1993;365:274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- 5.Fishel R, Lescoe M K, Rao M R, Copeland N G, Jenkins N A, Garber J, Kane M, Kolodner R. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 6.Leach F S, Nicolaides N C, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen L A, Nystrom-Lahti M, et al. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 7.Papadopoulos N, Nicolaides N C, Wei Y F, Ruben S M, Carter K C, Rosen C A, Haseltine W A, Fleischmann R D, Fraser C M, Adams M D, et al. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 8.Bronner C E, Baker S M, Morrison P T, Warren G, Smith L G, Lescoe M K, Kane M, Earabino C, Lipford J, Lindbloom A, et al. Nature (London) 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 9.Baross-Francis A, Andrew S E, Penney J E, Jirik F R. Proc Natl Acad Sci USA. 1998;95:8739–4873. doi: 10.1073/pnas.95.15.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baross-Francis A, Milhausen M K, Andrew S E, Jevon G, Jirik F R. Carcinogenesis. 2000;21:1259–1262. [PubMed] [Google Scholar]
- 11.Auerbach A D, Verlander P C. Curr Opin Pediatr. 1997;9:600–616. doi: 10.1097/00008480-199712000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Lengauer C, Kinzler K W, Vogelstein B. Nature (London) 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 13.Stern D F. Breast Cancer Res. 2000;2:176–183. doi: 10.1186/bcr51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yarden Y, Sliwkowski M X. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 15.Schechter A L, Stern D F, Vaidyanathan L, Decker S J, Drebin J A, Greene M I, Weinberg R A. Nature (London) 1984;312:513–516. doi: 10.1038/312513a0. [DOI] [PubMed] [Google Scholar]
- 16.Slamon D J, Clark G M, Wong S G, Levin W J, Ullrich A, McGuire W L. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 17.Slamon D J, Godolphin W, Jones L A, Holt J A, Wong S G, Keith D E, Levin W J, Stuart S G, Udove J, Ullrich A, et al. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 18.Hynes N E, Stern D F. Biochim Biophys Acta. 1994;1198:165–184. doi: 10.1016/0304-419x(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 19.Ross J S, Fletcher J A. Am J Clin Pathol. 1999;112, Suppl. 1:S53–S67. [PubMed] [Google Scholar]
- 20.Vogel C L, Cobleigh M A, Tripathy D, Gutheil J C, Harris L N, Fehrenbacher L, Slamon D J, Murphy M, Novotny W, F, Burchmore M, et al. Oncology. 2001;61,Suppl. S2:37–42. doi: 10.1159/000055400. [DOI] [PubMed] [Google Scholar]
- 21.Slamon D, Pegram M. Semin Oncol. 2001;28, Suppl. 3:13–19. doi: 10.1016/s0093-7754(01)90188-5. [DOI] [PubMed] [Google Scholar]
- 22.Muller W J, Sinn E, Pattengale P K, Wallace R, Leder P. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 23.Guy C T, Webster M A, Schaller M, Parsons T J, Cardiff R D, Muller W J. Proc Natl Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jakubczak J L, Merlino G, French J E, Muller W J, Paul B, Adhya S, Garges S. Proc Natl Acad Sci USA. 1996;93:9073–9078. doi: 10.1073/pnas.93.17.9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohler S W, Provost G S, Fieck A, Kretz P L, Bullock W O, Putman D L, Sorge J A, Short J M. Environ Mol Mutagen. 1991;18:316–321. doi: 10.1002/em.2850180421. [DOI] [PubMed] [Google Scholar]
- 26.Rowse G J, Ritland S R, Gendler S J. Cancer Res. 1998;58:2675–2679. [PubMed] [Google Scholar]
- 27.Obuchowski M, Shotland Y, Koby S, Giladi H, Gabig M, Wegrzyn G, Oppenheim A B. J Bacteriol. 1997;179:5987–5991. doi: 10.1128/jb.179.19.5987-5991.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Provost G S, Kretz P L, Hamner R T, Matthews C D, Rogers B J, Lundberg K S, Dycaico M J, Short J M. Mutat Res. 1993;288:133–149. doi: 10.1016/0027-5107(93)90215-2. [DOI] [PubMed] [Google Scholar]
- 29.Mirsalis J C. Toxicol Lett. 1995;82–83:131–134. doi: 10.1016/0378-4274(95)03472-2. [DOI] [PubMed] [Google Scholar]
- 30.Watson D E, Cunningham M L, Tindall K R. Mutagenesis. 1998;13:487–497. doi: 10.1093/mutage/13.5.487. [DOI] [PubMed] [Google Scholar]
- 31.Siegel P M, Dankort D L, Hardy W R, Muller W J. Mol Cell Biol. 1994;14:7068–7077. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anbazhagan R, Fujii H, Gabrielson E. Clin Cancer Res. 1999;5:839–844. [PubMed] [Google Scholar]
- 33.Watanabe N, Okochi E, Mochizuki M, Sugimura T, Ushijima T. Cancer Res. 2001;61:7739–7742. [PubMed] [Google Scholar]
- 34.Watanabe N, Okochi E, Hirayama Y, Shimada Y, Yanagihara K, Yoshida M C, Takahashi S, Mochizuki M, Sugimura T, Nagao M, et al. Cancer Res. 2001;61:2632–2640. [PubMed] [Google Scholar]
- 35.Bradlow L, Telang N T, Osborn M P. In: Biological Reactive Intermediates V. Snyder R, editor. New York: Plenum; 1996. pp. 285–296. [Google Scholar]
- 36.Jackson A L, Loeb L A. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 37.Dollé M E, Giese H, Hopkins C L, Martus H-J, Hausdorff J M, Vijg J. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 38.Dolle M E, Snyder W K, Gossen J A, Lohman P H, Vijg J. Proc Natl Acad Sci USA. 2000;97:8403–8408. doi: 10.1073/pnas.97.15.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono T, Ikehata H, Nakamura S, Saito Y, Hosoi Y, Takai Y, Yamada S, Onodera J, Yamamoto K. Mutat Res. 2000;447:165–177. doi: 10.1016/s0027-5107(99)00200-6. [DOI] [PubMed] [Google Scholar]
- 40.Stuart G R, Oda Y, de Boer J G, Glickman B W. Genetics. 2000;154:1291–1300. doi: 10.1093/genetics/154.3.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alaoui-Jamali M A, Paterson J, Al Moustafa A E, Yen L. Biochem Cell Biol. 1997;75:315–325. [PubMed] [Google Scholar]
- 42.Harari D, Yarden Y. Oncogene. 2000;19:6102–6114. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 43.Nghiem Y, Cabrera M, Cupples C G, Miller J H. Proc Natl Acad Sci USA. 1988;85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michaels M L, Cruz C, Grollman A P, Miller J H. Proc Natl Acad Sci USA. 1992;89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang D Y, Gu Y, Lu A L. Mol Genet Genomics. 2001;266:336–342. doi: 10.1007/s004380100567. [DOI] [PubMed] [Google Scholar]
- 46.Kunz B A, Kang X L, Kohalmi L. Mol Cell Biol. 1991;11:218–225. doi: 10.1128/mcb.11.1.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang X L, Yadao F, Gietz R D, Kunz B A. Genetics. 1992;130:285–294. doi: 10.1093/genetics/130.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider R, Eckerskorn C, Lottspeich F, Schweiger M. EMBO J. 1990;9:1431–1435. doi: 10.1002/j.1460-2075.1990.tb08259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arai K, Morishita K, Shinmura K, Kohno T, Kim S R, Nohmi T, Taniwaki M, Ohwada S, Yokota J. Oncogene. 1997;14:2857–2861. doi: 10.1038/sj.onc.1201139. [DOI] [PubMed] [Google Scholar]
- 50.Radicella J P, Dherin C, Desmaze C, Fox M S, Boiteux S. Proc Natl Acad Sci USA. 1997;94:8010–8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xin H, Lin W, Sumanasekera W, Zhang Y, Wu X, Wang Z. Nucleic Acids Res. 2000;28:2847–2854. doi: 10.1093/nar/28.14.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fousteri M I, Lihmann A R. EMBO J. 2000;19:1691–1702. doi: 10.1093/emboj/19.7.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morgenstern J P, Land H. Nucleic Acids Res. 1990;18:1068. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]