

Abstract
The heptapeptide H-Iva-Api-Iva-ATANP-Iva-Api-Iva-NHCH3 (P1a), where Iva is (S)-isovaline, Api is 4-amino-4-carboxypiperidine, and ATANP is (S)-2-amino-3-[1-(1,4,7-triazacyclononane)]propanoic acid, has been synthesized. Its conformation in aqueous solution is essentially that of a 310-helix. By connecting three copies of P1a to a functionalized Tris(2-aminoethyl)amine (Tren) platform a new peptide template, [T(P1)3], was obtained. This molecule is able to bind up to four metal ions (CuII or ZnII): one in the Tren subsite and three in the azacyclononane subunits. The binding of the metals to the Tren platform induces a change from an open to a closed conformation in which the three short, helical peptides are aligned in a parallel manner with the azacyclonane units pointing inward within the pseudocavity they define. T(P1)3 shows a peculiar behavior in the transphosphorylation of phosphate esters; the tetrazinc complex is a catalyst of the cleavage of 2-hydroxypropyl-p-nitrophenyl phosphate (HPNP), whereas the free ligand is a catalyst of the cleavage of an oligomeric RNA sequence with selectivity for pyrimidine bases. In the case of HPNP, ZnII acts as a positive allosteric effector by enhancing the catalytic efficiency of the system. In the case of the polyanionic RNA substrate, ZnII switches off the activity, thus behaving as a negative allosteric regulator. It is suggested that the opposite behavior of the catalyst induced by ZnII is associated with the change of conformation of the Tren platform, and consequently of the relative spatial disposition of the three linked peptides, that occurs after binding of the metal ion.
Typical features that characterize the catalytic site of natural enzymes are the placement of several functional groups in appropriate relative positions, the control of their solvation, and the recognition of the substrate. They are achieved via protein folding into precise secondary and tertiary conformations. Cofactors such as metal ions may provide additional, and in many instances critical, elements for performing the catalytic process and/or impart further structural rigidity or conformational control. Cooperativity is the key motif for achieving the astonishing rate accelerations observed by using the functional groups present in protein amino acids that, taken individually, show modest or no reactivity at all (1).
Although the design of peptides that fold into well defined tertiary structures mimicking native proteins has been reported recently for sequences of relatively modest size, the exploitation of these structures for the obtainment of efficient catalysts is still in its infancy (2). Rapid progress is being made, however. Notable examples taken from the most recent literature are those provided by Baltzer and coworkers (3, 4), Shogren-Knaak and Imperiali (5), Ghadiri and coworkers (6, 7), and Chmielewski and coworkers (8).
We recently reported that peptide sequences as short as 7 aa may adopt an essentially helical conformation in water (9) provided at least five Cα-tetrasubstituted α-amino acids are present. Furthermore, dinuclear ZnII heptapeptides incorporating two copies of a 1,4,7-triazacyclononane (Tacn)-functionalized amino acid showed clear evidence of cooperativity in the cleavage of RNA model substrate 2-hydroxypropyl-p-nitrophenyl phosphate (HPNP; refs. 10 and 11) and plasmid DNA (12) as well, with rate accelerations in this latter case in the order of 10 million-fold over the uncatalyzed reaction. These peptides set a clear principle: to obtain a cooperative catalyst it is not necessary to design a large molecule. However, they lack a specific recognition site, the number of cooperating functional units is limited, and there is little possibility to incorporate control units such as allosteric regulators (1, 13).
For these reasons we decided to move a step forward in the design of a de novo catalyst by increasing the complexity of the system and maintaining to a minimum the synthetic effort required. Because of our interest in synthetic phosphatases (10–12), we focused on a system incorporating catalytic units typically present in this class of enzymes (14–16), i.e., two or more functional metal centers. As an additional feature, we wanted also a regulatory metal ion to provide an allosteric control element (13) in analogy with the role played by MgII in alkaline phosphatases (17).
Materials and Methods
Details for all of the synthetic procedures, compounds characterization, and analytical techniques used are published as supporting information on the PNAS web site, www.pnas.org.
Results and Discussion
Design.
The dissection of the structural features outlined above led us to identify the following elements for the construction of our putative synthetic phosphatase: (i) a templating unit, the conformation of which could be controlled by metal ion complexation, and (ii) a conformationally stable, albeit short, peptide sequence incorporating at least a metal ion binding subunit. Several copies of this peptide connected to the templating platform would provide the binding and catalytic site of the system. As the metal ion binding platform we selected Tris(2-aminoethyl)amine (Tren), because derivatives of this tetraamine are very strong ligands (18) for transition metal ions (such as ZnII) and can be functionalized very easily. We have used this templating unit already for the preparation of peptide-based systems for which a metal ion acts as an allosteric regulator (19). The choice of Tren as the platform sets to three the number of peptides to be connected to its arms. Three is also the minimal number of structural units required to define a catalytic cavity. The peptide sequence had to be selected very carefully because the relative position of the three metal ion binding subunits had to be such to allow for their cooperativity in the catalytic process: they had to point inward within the pseudocavity. For all of the above reasons we designed the following sequence: H-Iva-Api-Iva-ATANP-Iva-Api-Iva-NHCH3 (P1a), where Iva is (S)-isovaline (20), Api is 4-amino-4-carboxypiperidine (21), and ATANP is (S)-2-amino-3-[1-(Tacn)]propanoic acid (22). Because of the presence of six Cα-tetrasubstituted α-amino acids, peptide P1a is expected to assume a 310-helical conformation in aqueous solution, which is in line with our recent findings (9). Furthermore, in this helical conformation the amino group of the two Api residues will be located on different sides of the helix with respect to the triazacyclononane of ATANP (Fig. 1a). When three copies of P1a are bound to the Tren platform via an aromatic spacer, the resulting template will likely assume, after metal ion binding, the conformation shown in Fig. 1b. The correct location of the three metal ions within the thus-defined pseudocavity should be ensured by the exposure of the side of each helix bearing the two Api lateral chains to the bulk water solution. It must be pointed out that at the pH at which we planned to run our experiments (≈7), the nitrogens of the piperidine units are protonated, whereas metal ion binding removes the protons from the three nitrogens of the azamacrocycles. Because of their coordination to the macrocycles, the ZnII ions are less hydrophilic than the ammonium ions (23).
Figure 1.
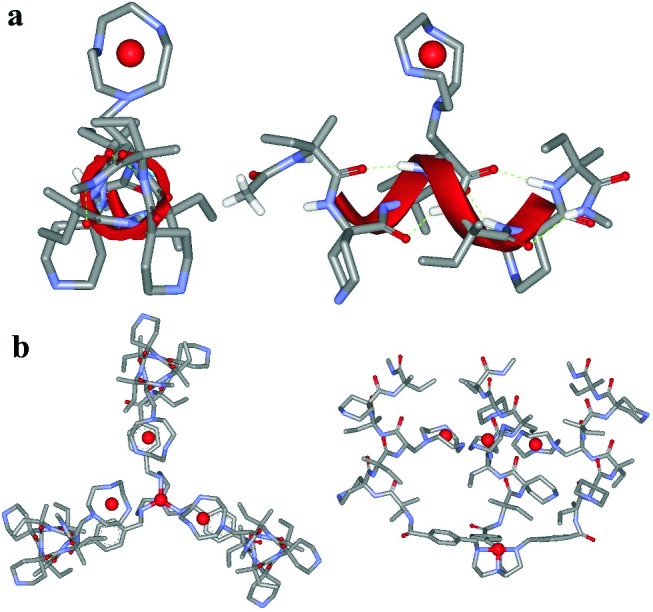
Top and side views of the molecular models of peptide P1b–ZnII complex (a) and the tetrazinc complex of the peptide template T(P1)3 (b). The peptide backbone is in the canonical 310-helical conformation. The template has been obtained by docking three copies of peptide P1a–ZnII to the ZnII complex of the Tren platform in the conformation obtained from the x-ray crystallographic analysis of an analogous complex (43).
Synthesis.
The synthesis of the Tren templating unit T1 (R = OH) was performed as reported (24). Peptide P1a was prepared by solution synthesis and fully characterized (see supporting Materials and Methods for details). The solid-phase approach is of limited use for Cα-tetrasubstituted α-amino acids such as Iva and Api because of their sluggish coupling reactivity. Regrettably, the synthetic shortcut by directly coupling Z-Iva-Api(Boc)-Iva-OH (Z, benzyloxycarbonyl; Boc, tert-butyloxycarbonyl) to H-ATANP(Boc)2-Iva-Api(Boc)-Iva-R gave a mixture of products with only traces of the desired heptapeptide. The connection of three copies of peptide P1a to the platform T1 (R = OH) was performed by using EDC [N-ethyl, N′-(3-dimethylaminopropyl)-carbodiimide] and HOAt (1-hydroxy-7-aza-1,2,3-benzotriazole) as coupling agents. Deprotection led eventually to the desired tripodal derivative T(P1)3. For comparison purposes in the subsequent kinetic studies, the Tren trisamide derivative T1 (R = NHCH3) and Ac-(S)-ATANP-NHCH3 have been prepared also. The structures of the compounds synthesized are shown in Scheme S1.
Scheme 1.

Structures of the compounds synthesized and acronyms used for their identification.
Conformational Studies.
Peptides P1b and templates T(P1)3 are fully soluble in aqueous solution in the pH interval 2.0–11.0. Consequently, they were studied in this medium by circular dichroism and, in the case of P1b, by bidimensional NMR too. The circular dichroism spectra of peptide P1b and its ZnII complex are reported in Fig. 2 (Upper). The free peptide shows the typical profile of a right-handed 310-helical conformation characterized by a negative Cotton band at 207 nm with a shoulder at 222 nm (24–26). The ratio between the molar ellipticity at the two wavelengths (R = θ222/θ207) is 0.6, slightly larger than what has been reported for peptides adopting a 310-helical conformation. This finding might suggest a modest deviation of the canonical 310-helical conformation toward an α-helix. After metal ion binding, almost 50% of the helicity is lost, suggesting that the interaction with ZnII has an adverse effect on the stability of the helix.
Figure 2.

Circular dichroism spectra of peptide P1b (Upper) and template T(P1)3 (Lower) in a pH 6.3 aqueous solution. Trace a is the system without metal, and trace b is in the presence of 1.5 or 6.0 equivalents of ZnII for P1b and T(P1)3, respectively.
Support to the 310-helical secondary structure comes from the rotating-frame Overhauser effect spectroscopy NMR spectrum of peptide P1b recorded in 9:1 H2O/D2O and 312 K. The spectrum highlights the connectivities between NH(i) and NH(i + 1) protons in the sequence. The strong intensities of the cross-peaks lend support to a highly structured conformation. The spectrum shows also two long-range connectivities between CαH(4) → NH(6) and CαH(4) → NH(7) (Fig. 9, which is published as supporting information on the PNAS web site). The first of the two is diagnostic of a 310-helical conformation (9), because in this helix dαN(i, i + 2), i.e., the distance between the CαH in the i position and the NH in the i + 2 position, is in the range of 3.5–3.9 Å as opposite to ≈4.5 Å in an α-helix. The second cross-peak indicates that dαN(i, i+3) should be relatively short (≈3.6 Å), and hence the torsion angle ψ should be close to −30°. Furthermore, the cross-peak between CαH(4) → NH(8), the terminal methylamide hydrogen, is missing. Such a peak would be present only with an α-helix conformation.
Once the peptides have been connected to the platform, the resulting template presents a circular dichroism spectrum similar to that of the parent oligomer in the 207-nm region but for θ values that are 3 times larger (Fig. 2, Lower). On the contrary, the 222-nm region most probably is influenced by the negative portion of the exciton band of the benzoyl substituents (27) present in the platform, the positive component of which is clearly visible above 240 nm (Fig. 2, Lower). Because of this overlapping, we believe that the conformation of each peptide in the template is still that of a 310-helix, and there is no conformational shift to an α-helix. Quite interestingly, the addition of ZnII (6 equivalents) in excess to those, 4, required for the saturation of all binding subunits led to a modest decrease (−20%) of helicity. This result suggests a more stable conformation for the clustered peptides in the template than when they are isolated. Both single peptide P1b and template T(P1)3 show an increase in helicity after increasing the pH (Fig. 10, which is published as supporting information on the PNAS web site). Again, this effect is more significant for P1b than for T(P1)3 and follows the deprotonation of the ammonium nitrogens in the side arms of the functional amino acids and the Tren platform as well [the reported (28) pKa of the parent amines are: piperidine, 11.1; Tren, 10.1, 9.5, and 8.4; Tacn, 10.4 and 6.8].
Summing up the conformational analysis confirms our predictions formulated in the design of P1b and T(P1)3 in that both systems largely adopt the conformation represented in Fig. 1.
Site Selectivity in Metal Ion Binding.
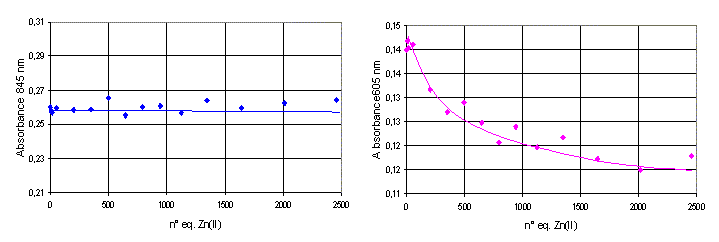
Template T(P1)3 is able to bind up to four metal ions: one in the Tren platform expected to control the relative spatial position of the three peptides and three on the azacyclononane units, one for each peptide. The Tren binding site should not be involved in catalysis, rather its role should be that of tuning the conformation of the template similar to an allosteric regulator and inducing, after metal ion binding, the formation of the putative catalytic site at which the three metal ions, bound to the triazacyclononanes, will be located. To obtain a deeper understanding of the binding sequence of the metal ions to the two different binding sites we followed the formation of the complexes with CuII ions in the 500–900-nm region. The band caused by the complex with the Tren platform is centered at 845 nm, whereas that with the triazacyclononanes is centered at 605 nm. The visible spectrum of T(P1)3–4CuII is the result of these two independent contributions, because it is almost superimposable to that obtained with three equivalents of P1b–CuII and one equivalent of T1–CuII (R = NHCH3, see Fig. 11, which is published as supporting information on the PNAS web site). This result means that there is no relevant interference between the two different binding sites. The comparison of the increase of absorbance at the wavelength of the maxima of the two different complexes when CuII is added progressively to a solution of T(P1)3 provides the distribution curve shown in Fig. 3. It shows that the metal ion first binds to the Tren site and then to the three azamacrocycles. The selectivity, however, is low with an apparent ratio between the binding constants of the two sites of ≈5. It should be pointed out that the ratio KTren/KTacn for the binding to CuII is >103 according to the literature (18, 28). However, it is known also that the functionalization of the three arms of Tren with benzyl groups leads to a substantial decrease of the binding constant (29).
Figure 3.

Distribution of CuII ions in the two different binding sites of T(P1)3 as a function of the number of equivalents of metal ion added. Filled symbols refer to the Tren site, and the open symbols refer to the triazacyclonane sites. Circles, free ligands; squares, complexed ligands.
Such a direct evaluation of the selectivity of binding for ZnII was not possible for the lack of appropriate absorption bands that could allow to discriminate between the two binding sites. We performed, however, a competition experiment with the tetracopper complex. It is known that the binding constants of the parent polyamines (Tren and Tacn) with ZnII are ≈3 orders of magnitude lower than those with CuII (18, 28). After the addition of 2.5 × 103 equivalents of ZnII, this metal ion displaces CuII from the triazacyclononane binding sites, whereas that bound to the Tren site is not affected at all (Fig. 12, which is published as supporting information on the PNAS web site). This result indicates that in the case of ZnII, KTren-site/KTacn-site < 5, i.e., lower than in the case of CuII.
The experiments described above indicate that template T(P1)3 binds up to four metal ions (CuII or ZnII) with very little preference for the binding to the Tren platform in the case of CuII and an even lower preference in the case of ZnII.
Cleavage of an RNA Model Substrate.
The major purpose of this investigation was to prepare a catalyst able to bind and subsequently hydrolyze a phosphate diester. To this aim we selected HPNP as a model substrate. Because of the presence of the secondary alcoholic function that may act as an intramolecular nucleophile (Scheme S2), this substrate is considered a good model for the cleavage of RNA, although it must be pointed out that the presence of p-nitrophenol as the leaving group renders HPNP a far more reactive phosphate than any present in an RNA sequence. By exposing HPNP (2.0 × 10−5 M) to a solution containing T(P1)3 (2.0 × 10−4 M) or T1 (R = NHCH3, 2.0 × 10−4 M) and Ac-(S)-ATANP-NHCH3 (6.0 × 10−4 M) at pH = 7.0, 40°C, and progressively increasing the amount of ZnII added, the reactivity profiles shown in Fig. 4 were obtained (independent experiments have shown that the ZnII complexes of P1b and Ac-(S)-ATANP-NHCH3 have very similar catalytic efficiencies in the cleavage of HPNP). The shape of the plot for the hydrolysis by T(P1)3 clearly is indicative of a cooperative process; as the equivalents of metal ion increase to fully saturate the peptide template binding sites, the system becomes more effective in a “nonlinear” manner (13). This phenomenon can be caused by two convergent effects: (i) allosteric control of the conformation caused by the binding of a ZnII ion to the Tren platform, and (ii) cooperativity between the metal Tacn centers once they get closer because of the conformational change associated with process i. The maximum rate acceleration over that exerted by the unassembled mixture of T1 (R = NHCH3) and 3 Ac-(S)-ATANP-NHCH3 is ≈50-fold. Thus, in the peptide template, we observe a remarkable rate acceleration without changing the intrinsic nature of the catalytic elements.
Scheme 2.

The cleavage of HPNP occurs via an intramolecular nucleophilic attack as in RNA.
Figure 4.

Observed rate constants for the cleavage of HPNP by T(P1)3 (●) and the unassembled mixture of T1 (R = NHCH3) and three equivalents of Ac-(S)-ATANP-NHCH3 (○) as a function of the number of equivalents of ZnII added. Conditions: [T(P1)3] = 2 × 10−4 M, [HPNP] = 2 × 10−5 M, 0.1 M Hepes buffer, pH 7.0, 40°C.
By running the cleavage reaction in the presence of an excess substrate, i.e., under catalytic conditions, the system is still active and does not show any inhibition of its efficiency in the presence of up to 100-fold excess of HPNP. By increasing the substrate concentration, a reactivity saturation profile was observed (Fig. 5). Michaelis–Menten analysis of the curve gave kcat = 3.9 × 10−4 s−1, Km = 3.9 × 10−3 M and k2 = kcat/Km = 0.1 M−1 s−1. The value of kcat is slightly lower than that reported recently (30–32) for calixarene-based di- and trinuclear ZnII catalysts, although in these latter cases the intrinsic reactivity of the 2,6-diaminopyridine–ZnII complex, present in the catalytic site, appears to be higher than that of the triazacyclononane–ZnII, which is at the basis of our system.
Figure 5.

Dependence of the initial rate of cleavage of HPNP by T(P1)3–4ZnII (2 × 10−5 M) as a function of the initial substrate concentration (0.1 M Hepes buffer pH 7.0, 40°C). (Inset) Dependence of the observed rate constant for the cleavage of HPNP from the pH of the solution. Conditions were as described above but [HPNP] = 2 × 10−4 M.
Interestingly enough, the tetracopper catalyst is less active than its tetrazinc counterpart. A plot of the reactivity against the [ZnII]/([CuII] + [ZnII]) mole fraction revealed that a key role is played by the metal bound to the Tren platform, because the exchange of CuII with ZnII in that site was responsible for the change of activity (Fig. 6). In fact, as shown in the same Fig. 6, the last ZnII added occupies the structural position in the templating platform. Recent results by Krämer and coworkers (33) point out the relevant role played by a metal ion in an allosteric position in determining the activity of an artificial phosphodiesterase. In that particular case the rigidity of the system was such to make it very sensitive to even little changes in the geometry of binding at the allosteric site. We don't know whether the same explanation holds true in this case too. Alternatively there might be a role played by the Tren-bound CuII in interfering with the binding of the substrate at the catalytic site, possibly by coordinating it at its free apical position where it can take less advantage of the interaction with the other three metal centers. An involvement of this metal ion in the catalytic process seems unlikely. Indeed, by comparing the rate acceleration observed with the T1 (R = NHCH3) and P1b metal complexes, a modest rate acceleration was observed with the peptide-based system, whereas the complexes of the Tren derivative proved totally inactive.
Figure 6.

Dependence of the observed rate constant for the cleavage of HPNP (●) and of the absorbance at 845 nm caused by the binding of CuII to the Tren site of T(P1)3 (○) as a function of the [ZnII]/([CuII] + [ZnII]) mole fraction. Cleavage conditions: [T(P1)3] = 2 × 10−5 M, [HPNP] = 2 × 10−4 M, 0.1 M Hepes buffer, pH 7.0, 40°C.
The above results point to a mechanism in which T(P1)3, after ZnII binding, adopts the conformation depicted in Fig. 1b, thus providing the binding pseudocavity and catalytic site for the substrate. The metal centers not only are involved in the binding but also in the catalytic process. This conclusion is supported by both the reactivity profile of Fig. 4 and the shape of the pH vs. rate plot with T(P1)3–4ZnII, which shows a break point at an approximate pH of 7.0 (Fig. 5, Inset). This behavior may be caused by direct coordination of the HPNP hydroxyl to a ZnII, resulting in a decrease of its pKa and thus allowing the formation of its conjugate base in this pH region. Alternatively, general base catalysis might be operative. In this latter case a ZnII-bound, deprotonated water molecule would act as a base for the deprotonation of the alcoholic function that then would become available for intramolecular transesterification. The two mechanisms are indistinguishable kinetically. The involvement of a water molecule coordinated to the Tren platform-bound ZnII would show a break point at a higher pH in accord with the pKa value reported for these kinds of complexes (29, 34).
RNA Cleavage.
As mentioned above, advantages of HPNP as a model substrate are its increased reactivity and the formation of nitrophenolate as a single, easily detectable product. However, it is an imperfect model as far as leaving group basicity is concerned. In addition, it lacks the polyanionic character and the sequence-specific properties of RNA. For this reason we tested our peptide-based catalysts in the cleavage of a linear 29-mer RNA substrate. To analyze the multiple hydrolysis products, a commercial sequencer served as an efficient tool (35, 36). For best resolution of all possible fragments a short DNA spacer (T10) was placed between the fluorescent dye and the RNA part of the substrate, Cy5-TTT TTT TTT TCU AGC CGA CUG CCG AUC UCG CUG ACU GAC TTT T (37). The presence of very short fragments complicating the analysis thus is avoided. Four additional deoxynucleotides (T4) were attached at the 3′ end to improve the separation between substrate and the longest degradation product.
Fig. 7 reports the amount of RNA cleaved as a function of the number of equivalents of ZnII added both for the peptide template T(P1)3 and the unassembled mixture of T1 (R = NHCH3) and three equivalents of P1b. Under the experimental conditions used (140 nM RNA, 10 μM catalyst, pH 7.0, and 3 h incubation time at 37°C), T(P1)3 is quite active in the absence of ZnII ions. Addition of the first two equivalents of ZnII causes a modest decrease of activity, whereas the third and fourth equivalents almost suppress the activity of the system. The unassembled mixture is less active and apparently not much affected by the addition of the metal ion. This last evidence indicates that the free macrocycle in the peptide is only slightly less active than its metal complex. Accordingly, we interpret the relevant loss of activity of T(P1)3 by the addition of ZnII with a conformational change associated with its complexation to the Tren platform. The closed conformation of the metallated T(P1)3 template, similar to that depicted in Fig. 1b, would prevent the interaction of the macrocycles with the oligomeric phosphates of the RNA substrate, thus inhibiting the activity of the system.
Figure 7.

Percentage of RNA cleaved by 10 μM T(P1)3 (●) and the unassembled mixture of 10 μM T1 (R = NHCH3) and 30 μM P1b (○) as a function of the number of equivalents of ZnII added. Cleavage reactions were performed in 50 mM Tris⋅HCl/0.01% SDS buffer with 140 nM Cy5-labeled DNA/RNA for 3 h at 37°C.
Cleavage of RNA by diamines (38–40) and macrocyclic polyamines (41, 42) is well documented. The catalysis takes advantage of the intramolecular acid-base cooperation of the neutral amine and ammonium ion both present at neutral pH. Although Michaelis and Kalesse (42) have shown that the catalysis by 1,4,7,10-tetraazacyclododecane (cyclen) is also inhibited by ZnII, which is at variance with ours, their system requires a large excess of metal ion. Very likely the mode of action of ZnII in their cyclen-based catalyst is different from the one we propose for the present polypeptide template. The shape of the activity profile of Fig. 7 indicates that the binding process of ZnII does not follow a linear-saturation relationship (11) such that the strength of binding does not seem to be at stake in our case.
RNA hydrolysis induced by T(P1)3 and the unassembled mixture of T1 (R = NHCH3) and three equivalents of P1b occurs specifically after pyrimidines (Fig. 8). A similar RNA cleavage pattern was observed recently with an oligopeptide–cyclen conjugate by Kalesse et al. (42). Pyridine sites in our substrate are not cleaved with equal efficiency. The preference for the 5′-terminal U seems to be a sequence-specific effect also observed with other catalysts (unpublished results).
Figure 8.
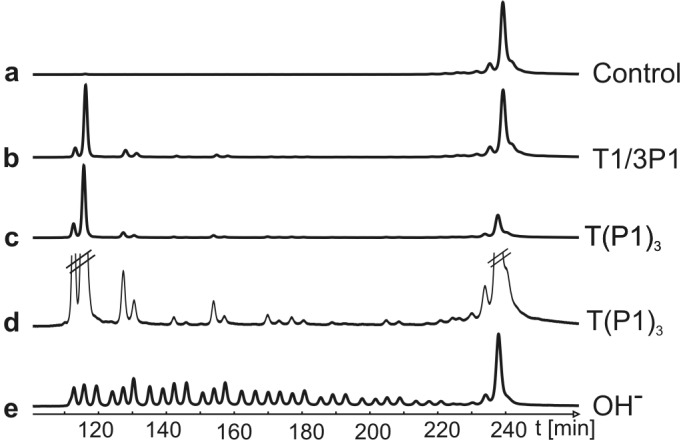
RNA cleavage pattern induced by T(P1)3 (10 μM) and a mixture of T1 (R = NHCH3, 10 μM) and P1b (30 μM). Lane e shows a base cleavage ladder of the Cy5-labeled RNA substrate. Reaction conditions were as described for Fig. 7. RNA fragmentation was analyzed by denaturing PAGE on an ALFexpress DNA sequencer. In lane d, a magnification of lane c is shown to determine the cleavage sites: Cy5-T10-CUAGCCGACUGCCGAUCUCGCUGACUGAC-T4 (cleavage sites are underlined).
Conclusions
By covalently connecting three copies of an azacrown-functionalized helical heptapeptide to a Tren platform, we succeeded in preparing a transphosphorylation catalyst, the activity of which could be controlled in opposite ways by ZnII according to the type of substrate used. In the case of HPNP, a small, monomeric phosphate diester, ZnII acts as a positive allosteric effector enhancing the catalytic efficiency of the system. In the case of an oligomeric RNA substrate, ZnII switches off the activity, thus behaving as a negative allosteric regulator. This bimodal role played by the metal ion likely is associated with the same change of conformation induced by the metal ion itself after binding to the Tren platform. In this conformation the three short helical peptides are aligned and oriented in such a way to allow the occurrence of a cooperative interaction between the ZnII ions bound to the azamacrocycles when a small substrate such as HPNP binds to them. Oligomeric RNA does not seem to be able to interact with the catalytic site in such a closed conformation, and hence the system devoid of metal ions and conformationally more flexible is more active. The higher activity with respect to the unassembled components is, in this case, likely caused by a stronger binding of T(P1)3 to polyanionic RNA because of the larger number of protonated amines present in the molecule.
This tripodal peptide provides also a striking example of selective mode of action toward two different phosphate esters and shows how, by increasing the complexity of a supramolecular catalyst, the choice of the model substrate to test its efficiency may bring about quite different conclusions and information for the future design of improved molecules. This behavior is rather similar to that often found in enzymes.
Supplementary Material
Acknowledgments
This work was supported by Italian Ministry for Education, University, and Research Grant MM03194891 and the European Union within the European Cooperation in the Field of Scientific and Technical Research D11 (D11/0009/98) and Research and Training Network (HPRN-CT1999-00008-Endevan) programs.
Abbreviations
- Tacn
1,4,7-triazacyclononane
- HPNP
2-hydroxypropyl-p-nitrophenyl phosphate
- Tren
Tris(2-aminoethyl)amine
- Iva
(S)-isovaline
- Api
4-amino-4-carboxypiperidine
- ATANP
(S)-2-amino-3-[1-(Tacn)]propanoic acid
- Z
benzyloxycarbonyl
- Boc
tert-butyloxycarbonyl
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Fersht A. Enzyme Structure and Mechanism. New York: Freeman; 1977. [Google Scholar]
- 2.Baltzer L, Nilsson J. Curr Opin Biotechnol. 2001;12:355–360. doi: 10.1016/s0958-1669(00)00227-5. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson J, Baltzer L. Chem Eur J. 2000;6:2214–2220. doi: 10.1002/1521-3765(20000616)6:12<2214::aid-chem2214>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 4.Baltzer L, Broo K S. Biopolymers. 1998;47:31–40. [Google Scholar]
- 5.Shogren-Knaak M A, Imperiali B. Bioorg Med Chem. 1999;7:1993–2002. doi: 10.1016/s0968-0896(99)00112-1. [DOI] [PubMed] [Google Scholar]
- 6.Kennan A J, Haridas V, Severin K, Lee D H, Ghadiri M R. J Am Chem Soc. 2001;123:1797–1803. doi: 10.1021/ja991266c. [DOI] [PubMed] [Google Scholar]
- 7.Saghatelian A, Yokobayashi Y, Soltani K, Ghadiri M R. Nature (London) 2001;409:797–801. doi: 10.1038/35057238. [DOI] [PubMed] [Google Scholar]
- 8.Yao S, Ghosh I, Zutshi R, Chmielewski J. Nature (London) 1998;396:447–450. doi: 10.1038/24814. [DOI] [PubMed] [Google Scholar]
- 9.Formaggio F, Crisma M, Rossi P, Scrimin P, Kaptein B, Broxterman Q B, Kamphuis J, Toniolo C. Chem Eur J. 2000;6:4498–4504. doi: 10.1002/1521-3765(20001215)6:24<4498::aid-chem4498>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 10.Rossi P, Felluga F, Tecilla P, Formaggio F, Crisma M, Toniolo C, Scrimin P. J Am Chem Soc. 1999;121:6948–6949. [Google Scholar]
- 11.Rossi P, Felluga F, Tecilla P, Formaggio F, Crisma M, Toniolo C, Scrimin P. Biopolymers. 2000;55:496–501. doi: 10.1002/1097-0282(2000)55:6<496::AID-BIP1025>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Sissi C, Rossi P, Felluga P, Formaggio F, Palumbo M, Tecilla P, Toniolo C, Scrimin P. J Am Chem Soc. 2001;123:3169–3170. doi: 10.1021/ja005675g. [DOI] [PubMed] [Google Scholar]
- 13.Shinkai S, Ikeda M, Sugasaki A, Takeuchi M. Acc Chem Res. 2001;34:494–503. doi: 10.1021/ar000177y. [DOI] [PubMed] [Google Scholar]
- 14.Sträter N, Lipscomb N, Klabunde T, Krebs B. Angew Chem Int Ed Engl. 1996;35:2024–2055. [Google Scholar]
- 15.Wilcox D E. Chem Rev (Washington, DC) 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 16.Cowan J A. Chem Rev (Washington, DC) 1998;98:1067–1087. [Google Scholar]
- 17.Reid T W, Wilson I B. The Enzymes. New York: Academic; 1971. pp. 373–416. [Google Scholar]
- 18.Canary J W, Xu J, Castagnetto J M, Rentzeperis M, Marky L A. J Am Chem Soc. 1995;117:11545–11547. [Google Scholar]
- 19.Valente S, Gobbo M, Licini G, Scarso A, Scrimin P. Angew Chem Int Ed Eng. 2001;40:3899–3902. [PubMed] [Google Scholar]
- 20.Formaggio F, Crisma M, Bonora G M, Pantano M, Valle G, Toniolo C, Aubry A, Bayeul D, Kamphuis J. Pept Res. 1995;8:6–15. [PubMed] [Google Scholar]
- 21.Wysong C L, Yokum T S, Morales G A, Gundry R L, McLaughlin M L, Hammer R P. J Org Chem. 1996;61:7650–7651. doi: 10.1021/jo961594k. [DOI] [PubMed] [Google Scholar]
- 22.Rossi P, Felluga F, Scrimin P. Tetrahedron Lett. 1998;39:7159–7162. [Google Scholar]
- 23.Scrimin P, Veronese A, Tecilla P, Tonellato U, Monaco V, Formaggio F, Crisma M, Toniolo C. J Am Chem Soc. 1996;118:2505–2506. [Google Scholar]
- 24.Polese A, Formaggio F, Crisma M, Valle G, Toniolo C, Bonora G M, Broxterman Q B, Kamphuis J. Chem Eur J. 1996;2:1104–1111. [Google Scholar]
- 25.Toniolo C, Polese A, Formaggio F, Crisma M, Kamphuis J. J Am Chem Soc. 1996;118:2744–2745. [Google Scholar]
- 26.Yoder G, Polese A, Silva R A G D, Formaggio F, Crisma M, Broxterman Q B, Kamphuis J, Toniolo C, Keiderling T A. J Am Chem Soc. 1997;119:10278–10285. [Google Scholar]
- 27.Harada N, Nakanishi K. Circular Dichroic Spectroscopy: Exciton Coupling in Organic Stereochemistry. Mill Valley, CA: Univ. Sci. Books; 1983. [Google Scholar]
- 28.Smith R M, Martell A E. Critical Stability Constants. Vol. 6. New York: Plenum; 1989. [Google Scholar]
- 29.Ichikawa K, Ibrahim M M, Shimomura N, Shiro M. Inorg Chim Acta. 2001;313:125–136. [Google Scholar]
- 30.Molenveld P, Kapsabelis S, Engbersen J F J, Reinhoudt D N. J Am Chem Soc. 1997;119:2948–2949. [Google Scholar]
- 31.Molenveld P, Stikvoort W M G, Kooijman H, Spek A L, Engbersen J F J, Reinhoudt D N. J Org Chem. 1999;64:3896–3906. [Google Scholar]
- 32.Molenveld P, Engbersen J F J, Reinhoudt D N. Chem Soc Rev. 2000;29:75–86. [Google Scholar]
- 33.Fritsky I O, Ott R, Pritzkow H, Krämer R. Chem Eur J. 2001;7:1221–1231. doi: 10.1002/1521-3765(20010316)7:6<1221::aid-chem1221>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 34.Itoh T, Fujii Y, Yoshikawa T, Hisada H. Bull Chem Soc Jpn. 1996;69:1265–1274. [Google Scholar]
- 35.Glaesner W, Merkl R, Schmidt S, Cech D, Fritz H J. Biol Chem Hoppe-Seyler. 1992;373:1223–1225. doi: 10.1515/bchm3.1992.373.2.1223. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt C, Welz R, Muller S. Nucleic Acids Res. 2000;28:886–894. doi: 10.1093/nar/28.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall J, Hüsken D, Pieles U, Moser H E, Häner R. Chem Biol. 1994;1:185–190. doi: 10.1016/1074-5521(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 38. Dalby, K. N., Kirby, A. J. & Hollfelder, F. (1993) J. Chem. Soc., Perkin Trans. 2, 1269–1281.
- 39.Komiyama M, Yoshinari K. J Org Chem. 1997;62:2155–2160. doi: 10.1021/jo961935u. [DOI] [PubMed] [Google Scholar]
- 40.Verheijen J C, Deiman B A L M, Yeheskiely E, van der Marel G A, van Boom J H. Angew Chem Int Ed Engl. 2000;39:369–372. doi: 10.1002/(sici)1521-3773(20000117)39:2<369::aid-anie369>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 41.Michaelis K, Kalesse M. Chembiochem. 2001;1:79–83. doi: 10.1002/1439-7633(20010105)2:1<79::AID-CBIC79>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 42.Michaelis K, Kalesse M. Angew Chem Int Ed Eng. 1999;38:2243–2245. [PubMed] [Google Scholar]
- 43. Scrimin, P., Tecilla, P., Tonellato, U., Valle, G. & Veronese, A. (1995) J. Chem. Soc. Chem. Commun., 1163–1164.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}