

Abstract
The Escherichia coli umuDC (pol V) gene products participate in both a DNA damage checkpoint control and translesion DNA synthesis. Interactions of the two umuD gene products, the 139-aa UmuD and the 115-aa UmuD′ proteins, with components of the replicative DNA polymerase (pol III), are important for determining which biological role the umuDC gene products will play. Here we report our biochemical characterizations of the interactions of UmuD and UmuD′ with the pol III β processivity clamp. These analyses demonstrate that UmuD possesses a higher affinity for β than does UmuD′ because of the N-terminal arm of UmuD (residues 1–39), much of which is missing in UmuD′. Furthermore, we have identified specific amino acid residues of UmuD that crosslink to β with p-azidoiodoacetanilide, defining the domain of UmuD important for the interaction. We have recently proposed a model for the solution structure of UmuD2 in which the N-terminal arm of each protomer makes extensive contacts with the C-terminal globular domain of its intradimer partner, masking part of each surface. Taken together, our findings suggest that UmuD2 has a higher affinity for the β-clamp than does UmuD′2 because of the structures of its N-terminal arms. Viewed in this way, posttranslational modification of UmuD, which entails the removal of its N-terminal 24 residues to yield UmuD′, acts in part to attenuate the affinity of the umuD gene product for the β-clamp. Implications of these structure–function analyses for the checkpoint and translesion DNA synthesis functions of the umuDC gene products are discussed.
Significant insights into possible molecular mechanisms of induced mutagenesis leading to the onset of certain types of cancers were recently provided by the discovery of a family of DNA polymerases present in all three kingdoms of life. A defining characteristic of these polymerases is their ability to replicate imperfect DNA templates that cannot be replicated by other polymerases (1–4). Mutations inactivating a human member of this superfamily, the hRAD30A or XP-V gene product, leads to the UV-hypersensitive disorder xeroderma pigmentosum variant, or XP-V (5–7). This polymerase family, originally referred to as the UmuC-DinB-RAD30-REV1 superfamily of DNA polymerases (8), so named for the founding member of each of its four subfamilies, has recently been renamed the DNA polymerase Y superfamily (9). The most extensively characterized member of this superfamily is Escherichia coli UmuC protein that is encoded by the umuDC operon (4, 10). UmuC is also referred to as pol V, whether it is by itself (11) or in a complex with UmuD′ (UmuD′2C) (12).
The umuDC operon is part of the E. coli SOS response (4, 10, 13). This response consists of more than 40 unlinked genes (14) whose expression is coordinately regulated by the LexA and RecA proteins (10). LexA functions as a transcriptional repressor that acts to block transcription of the SOS-regulated genes (4, 10, 15). In response to DNA damage, single-strand DNA (ssDNA) accumulates as a result of the cell's failed attempts to replicate over DNA lesions. RecA protein, the main bacterial recombinase, binds to this ssDNA forming RecA/ssDNA nucleoprotein filaments. Direct interaction of LexA with these RecA/ssDNA nucleoprotein filaments activates the latent capacity of LexA to autodigest, thereby inactivating its repressor function leading to derepression of transcription of the SOS-regulated genes (10, 16), including the umuDC-encoded pol V (17).
The umuDC gene products participate in two temporally separated roles in response to DNA damage. First, the UmuD2C complex plays a noncatalytic role in promoting cell survival by participating in a DNA damage checkpoint control (18, 19). This checkpoint acts to regulate the rate of DNA synthesis in response to DNA damage, thereby allowing additional time for accurate repair processes, such as nucleotide excision repair, to remove lesions in the DNA before the cell's attempts to replicate its genome (18). Approximately 25 min after irradiation with 25 J/m2 of UV light (18), UmuD undergoes a RecA/ssDNA-facilitated autodigestion that is mechanistically similar to that undergone by LexA (15, 20–22). This posttranslational modification serves to remove the N-terminal 24 residues of UmuD, yielding UmuD′ (20–22). UmuD′ does not act in the DNA damage checkpoint control, but rather participates in the second role of the umuDC gene products. The UmuD′2 homodimer interacts with UmuC, forming the UmuD′2C complex (pol V), that functions as a lesion-bypass DNA polymerase to enable translesion DNA synthesis (TLS) (11, 12, 23, 24). Viewed in this way, posttranslational modification of UmuD to yield UmuD′ acts as a molecular switch to regulate temporally the two physiological roles of the umuDC gene products. In addition, specific interactions of UmuD and UmuD′ with the α (catalytic), ɛ (proofreading), and β (processivity clamp) subunits of the E. coli replicative DNA polymerase, DNA polymerase III (pol III), are believed to be important for regulating which role the umuDC gene products will play (25–28).
Our previous characterizations of the interactions of the umuD gene products with components of pol III suggested that UmuD interacts more strongly with the β processivity clamp of pol III than does UmuD′, whereas UmuD′ interacts more strongly with the α-catalytic subunit than does UmuD (25). On the basis of these findings, we have proposed that interaction of UmuD′ with α is important for TLS, perhaps as part of a polymerase-switching mechanism (4, 25, 27–29), whereas interaction of UmuD with β is important for enabling the UmuD2C-dependent checkpoint control (4, 25, 28, 29). Although genetic and biochemical analyses are consistent with the UmuD′-α interaction being important for TLS in vivo (reviewed in refs. 30 and 31), recent biochemical studies of UmuD′2C-dependent TLS have demonstrated that lesion bypass by UmuD′2C complex is facilitated by the β-clamp and γ-clamp loader complex of pol III (12, 23, 32). Therefore, in an effort to better define the biological role(s) of the interactions between β and the umuD gene products, we have further characterized them biochemically by using a variety of chemical crosslinkers. Our findings, presented in this report, indicate that the N-terminal arm of intact UmuD (comprising residues 1–39), the first 24 aa residues of which are missing in UmuD′, serves an important role in the interaction of the umuD gene product with the β-clamp. Implications of these structure–function analyses for the checkpoint and TLS functions of the umuDC gene products are discussed.
Materials and Methods
Proteins and Reagents.
UmuD, UmuD′ (33), and βHMK (34) were purified as described. βHMK bears both an N-terminal polyhistidine tag and a heart muscle kinase (HMK) motif, neither of which affect its activity (34). UmuD derivatives bearing nested deletions of N-terminal sequence were purified as described for wild-type UmuD. The plasmid constructs used for their overproduction will be described elsewhere (I.N. and G.C.W., unpublished work). Purified β lacking a tag and polyclonal antibodies specific to the β-clamp were generous gifts from C. S. McHenry, University of Colorado. Affinity-purified polyclonal antibodies specific to UmuD/D′ and UmuC have been described (35, 36). Oxidation of UmuD[C24-F94C]2 was catalyzed by copper-phenanthroline as described (33). The doubly disulfide-linked UmuD[C24-F94C]2 homodimer was purified by MonoQ chromatography (Amersham Pharmacia) as was wild-type UmuD. βHMK was radiolabeled in vitro with [γ-32P]ATP (DuPont-NEN) and HMK (Pierce), as described (25).
In Vitro Protein–Protein Crosslinking.
In vitro crosslinking of highly purified proteins with formaldehyde (Malinckrodt, Phillipsburg, NJ) or glutaraldehyde (Polysciences) was performed as described (36) with 67 pmol of homodimeric UmuD or UmuD′ with 12 pmol of homodimeric β or 32P-βHMK. Crosslinking efficiency was monitored by either Western blotting by using chemiluminescent detection (Tropix, Bedford, MA), as described (25, 37), or by PhosphorImager analysis of dried SDS-polyacrylamide gels.
Results
UmuD Interacts More Strongly with the β Processivity Clamp of DNA pol III than Does UmuD′.
By using purified proteins, we have reported that UmuD interacts more strongly with β than does UmuD′ in vitro (25). To examine whether these differences in affinity of UmuD and UmuD′ for β can be detected in a more physiologically relevant fashion, we used affinity chromatography to investigate whether UmuD or UmuD′ could interact with β present in a crude soluble extract prepared from an E. coli strain that was induced for SOS functions. Aliquots of this extract were applied to three different affinity columns corresponding to either UmuD, UmuD′, or BSA covalently coupled to Affi-Gel A15. The BSA column served as a control to distinguish between specific and nonspecific interactions. After extensive washing with buffer containing 75 mM sodium chloride, bound proteins were eluted with the same buffer containing 0.25 M followed by 1.0 M sodium chloride. We have demonstrated that interactions involving β and the UmuD and UmuD′ proteins are sensitive to salt, and hence are not hydrophobic in character (25). Aliquots of each wash and elution were then fractionated by SDS/PAGE and processed as a Western blot with anti-β-polyclonal antibodies.
As shown in Fig. 1A, the β-clamp was retained at easily detectable levels on the UmuD column; neither the UmuD′ nor the BSA columns retained detectable levels of β in the 0.25 and 1 M sodium chloride elutions; however, small amounts of β were observed in the 75 mM sodium chloride wash of the UmuD′ column (data not shown). It is possible that UmuD present in the soluble extract competed with UmuD′ that was bound to the column matrix for binding to β. Nonetheless, these results indicate that β interacts with UmuD′ very weakly under these conditions.
Figure 1.
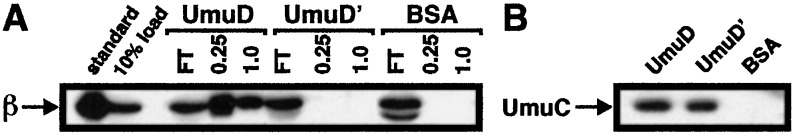
UmuD and UmuD′ affinity chromatography. Affinity chromatography was performed as described (25). Nine milliliters (corresponding to ≈4 mg of total protein) of a crude soluble fraction prepared from an E. coli strain induced for SOS functions was supplemented with 35 μg each of UmuD, UmuD′, and the α, ɛ, θ, and β subunits of pol III. (A) After applying 3 ml of this lysate to each column, aliquots of the flowthrough (FT), 0.25 and 1.0 M sodium chloride elutions were fractionated by SDS/PAGE, electroblotted to poly(vinylidene difluoride) membrane, and processed as a Western blot with anti-β-polyclonal antibodies. (B) After 1.0 M sodium chloride, columns were washed with buffer containing 4% SDS and aliquots from each wash were processed as a Western blot with affinity-purified anti-UmuC polyclonal antibodies.
To confirm that the inability of the UmuD′ column to retain easily detectable levels of the β-clamp was caused by the reduced affinity of UmuD′ for the clamp and not by a problem with the column, we probed aliquots of these same column fractions for the presence of UmuC; both UmuD and UmuD′ interact specifically with UmuC (36, 38). A previous report found that elution of UmuC from a UmuD/UmuD′ affinity column required mild denaturing conditions (38), suggesting that interactions of the umuD gene products with UmuC were hydrophobic in nature. Consistent with this report, only barely detectable quantities of UmuC were detected in the 0.25 M and 1.0 M sodium chloride wash fractions (data not shown). In contrast, significantly higher levels of UmuC were detected in SDS wash fractions from both the UmuD and the UmuD′ column but not the BSA column (Fig. 1B). Taken together, these results (i) confirm the specificity of the UmuD and UmuD′ affinity columns, and (ii) clearly indicate that UmuD possesses a comparatively higher affinity for the β-clamp of pol III than does UmuD′.
Characterization of the UmuD-β Interaction by Solution Crosslinking in Vitro.
Our attempts to characterize the interactions between β and the umuD gene products by gel filtration (25) or native-PAGE (data not shown) were unsuccessful, suggesting that the interactions are weak in vitro. We therefore used solution crosslinking to trap the complexes, thus allowing their further characterization. We chose to use formaldehyde and glutaraldehyde for these experiments because they have proven useful for stabilizing UmuD and UmuD′ as both homo- and heterodimers in our analyses (35–37). Crosslinked species were identified by Western blot analysis with antibodies specific to either β or the umuD gene products.
Both formaldehyde and glutaraldehyde crosslinked UmuD to β. An example of a typical experiment with glutaraldehyde is shown in Fig. 2A. With glutaraldehyde, we routinely observed a UmuD-β complex of ≈80 kDa (Fig. 2A). Its apparent size in SDS/PAGE suggests that this complex corresponds to one UmuD2 homodimer (≈30 kDa) crosslinked to a single β monomer (41 kDa). The crosslinking efficiency of UmuD as a homodimer increased in the presence of β (Fig. 2A), possibly because of some structural rearrangement of the UmuD2 homodimer when in complex with β that increases its susceptibility to glutaraldehyde crosslinking. Likewise, the amount of β trapped as a homodimer was reduced in the presence of UmuD, suggesting that UmuD masks the side chain of the residue in β that interacts with glutaraldehyde to mediate its crosslinking as a homodimer. In addition, depending on the glutaraldehyde preparation used, we observed a second complex of >115 kDa (data not shown) that may correspond to either one UmuD2 homodimer crosslinked to one β2 homodimer, or two UmuD2 homodimers crosslinked to a single β monomer.
Figure 2.

Stabilization of the UmuD-β interaction by glutaraldehyde or formaldehyde in vitro. (A) Crosslinking was performed as described in Materials and Methods. Aliquots of each mixture were fractionated in duplicate by SDS/PAGE, electroblotted to poly(vinylidene difluoride) membrane, and processed as Western blots with the indicated antibodies. Positions of molecular weight markers (GIBCO/BRL), UmuD, β, and protein complexes are indicated. (B) Schematic representation of structures of UmuD derivatives. (C and D) Characterization of the abilities of the UmuD derivatives to interact with β by solution crosslinking in vitro. The indicated UmuD derivatives were mixed with wild-type β (C) or 32P-βHMK (D) in vitro as described in Materials and Methods. After fractionation by SDS/PAGE, mixtures treated with formaldehyde (C) were processed as a Western blot with anti-β-polyclonal antibodies, whereas mixtures treated with glutaraldehyde (D) were analyzed by using a PhosphorImager. Positions of free β and UmuD-β complexes are indicated. The percent of the total β present in the UmuD-β complex for each UmuD derivative is indicated in D.
The N-Terminal Arm of the umuD Gene Product (Residues 1–39) Serves an Important Role in Interaction with β.
Because in vitro solution crosslinking provided us with a convenient method to characterize the interaction of the umuD gene product with the β-clamp, we used it to investigate the role played by the N-terminal arm of the umuD gene product in the interaction with the β-clamp. In the context of this report, we will refer to the residues proximal to amino acid 40 of the umuD gene products as comprising their N-terminal arms. Thus, residues 1–39 of the 139-aa UmuD comprise its N-terminal arm, whereas residues 25–39 (with use of the same numbering as for intact UmuD) of the 115-aa UmuD′ comprise its N-terminal arm. Also important for the purposes of this study are the respective structures of the N-terminal arms of UmuD and UmuD′. Although the N-terminal arms of UmuD′2 are mobile in solution (39), those of UmuD2 are structured because of contact with the C-terminal domains of their intradimer partners (40, 41).
To investigate the role played by the N-terminal arm of the umuD gene products in the interaction with β, we investigated the ability of a collection of UmuD derivatives bearing nested deletions of N-terminal sequences to interact with β. Before discussing results, it is worthwhile emphasizing that the inability of a particular chemical agent to crosslink two proteins in vitro does not necessarily mean the two proteins do not interact. However, under conditions where proteins can be crosslinked, a reduction in the crosslinking efficiency for a mutant form of one of the proteins is indicative of a reduced affinity. The UmuD derivatives used in this study are described in Fig. 2B and include, in addition to UmuD and UmuD′, UmuDΔ9 (lacking residues 2–9), UmuDΔ19 (lacking residues 2–19), and UmuDΔ37 (lacking residues 2–37). On the basis of both the solution (39, 40) and the crystal structures of UmuD′ (42), and on our working model for the solution structure of the UmuD2 homodimer (40, 41), UmuDΔ37 lacks all but two of the 39 residues comprising the N-terminal arm (39, 43). All of these UmuD derivatives form homodimers in vitro, as expected (data not shown).
Formaldehyde was able to crosslink both intact UmuD and UmuDΔ9 to β. However, deletion of more than the first nine residues of the umuD gene product greatly impaired its ability to be crosslinked to β with formaldehyde, illustrating the importance of the N-terminal arm of UmuD for interaction with β (Fig. 2C). In striking contrast to the results observed with formaldehyde, glutaraldehyde was able to stabilize the interaction of all five UmuD derivatives with β, indicating that the β-clamp interacts with elements of UmuD within its C-terminal globular domain (Fig. 2D). Because we used 32P-labeled β in the glutaraldehyde crosslinking experiment shown in Fig. 2D, we were able to quantitate the crosslinking efficiency of β to each UmuD derivative by using a PhosphorImager. This analysis indicated that crosslinking efficiency was roughly proportional to the length of the N-terminal arm of UmuD. However, although crosslinking efficiency is proportional to the strength of an interaction, it is not a direct measure of affinity because it shifts the equilibrium irreversibly toward complex formation. Intact UmuD and UmuDΔ9 interacted with β similarly, trapping 7.2% and 7.3% of the total β in the form of a UmuD-β or a UmuDΔ9-β complex, respectively (Fig. 2D). UmuDΔ19 and UmuD′ also crosslinked to β similarly, trapping 5.5% and 5.8% of the total β in the complex, respectively. UmuDΔ37, which corresponds to the C-terminal globular domain of the umuD gene product, crosslinked to β with the lowest efficiency of all, trapping just 2.9% of the β in two different complexes. Taken together, these results demonstrate that both the N-terminal arm and the C-terminal globular domains of UmuD are important for its interaction with β.
We believe that the differences we observed between formaldehyde and glutaraldehyde with respect to stabilizing the interactions between the individual UmuD derivatives and β relate to the fact that glutaraldehyde is ≈5- to 10-fold more efficient than formaldehyde at crosslinking UmuD to β (data not shown). This difference in efficiency is presumably because, at least in part, glutaraldehyde reacts predominantly with primary amines (i.e., amino termini and lysine side chains), whereas formaldehyde reacts with primary amines as well as histidine, tryptophan, and cysteine side chains (44). Furthermore, formaldehyde crosslinks constituent sites by means of a methylene linkage, whereas glutaraldehyde tends to multimerize, thereby allowing crosslinking of more distant sites (44). Thus, the lack of detectable crosslinking of the shorter UmuD derivatives to β with formaldehyde is presumably the result of the reduced affinity of the UmuD derivatives for β. Consistent with this interpretation, we have observed low levels of formaldehyde-mediated crosslinking of UmuD′ (compared with those observed for UmuD) to β by using 32P-labeled β-protein (data not shown). Further evidence that positions within the N-terminal 37 residues of the umuD gene product are important for the interaction of UmuD with β and not for formaldehyde-mediated crosslinking includes the fact that the only residues within the first 37 expected to react efficiently with formaldehyde are C24 and the amino terminus at M1. Because both UmuDΔ9 (Fig. 2) and a derivative of UmuD bearing a C24A substitution (data not shown) crosslinked to β with formaldehyde nearly as well as did wild-type UmuD, the inability of the UmuDΔ19, UmuD′, and UmuDΔ37 derivatives to crosslink to β with formaldehyde cannot be caused by deletion of the residue required for formaldehyde crosslinking, but rather must be caused by a reduced affinity of these UmuD derivatives for β.
Identification of Amino Acid Positions in UmuD That Crosslink Efficiently to β with p-Azidoiodoacetanilide (AIA).
To begin to understand the UmuD-β interaction at the molecular level, we made use of our substantial collection of UmuD and UmuD′ single cysteine derivatives (33, 45) and the heterobifunctional crosslinking agent AIA (46). We have used this crosslinker to characterize interactions within the UmuD2 homodimer, as well as interaction of UmuD with the RecA/ssDNA nucleoprotein filament (47). In such analyses, we first attach the AIA to the single cysteine in each UmuD or UmuD′ derivative. Each of these derivatives contains a single cysteine residue in a different position that is known to be quite reactive (47). All of these UmuD and UmuD′ derivatives retain some biological activity with most being fully functional for SOS mutagenesis in vivo (33, 45). After purifying each AIA-conjugated UmuD or UmuD′ derivative and adding purified β, mixtures were exposed to UV light, as described (46, 47). Covalent attachment of the AIA-modified UmuD or UmuD′ derivative to β was visualized by Western blot analysis with anti-β-antibodies.
This analysis indicated that UmuD derivatives bearing cysteines at positions 24, 34, 52, and 126 crosslinked more efficiently to β with AIA than those bearing cysteines at positions 19, 57, 60, 81, 94, 112, 133, and 139 (Fig. 3A). Thus, the AIA crosslinking confirmed the results of the formaldehyde and glutaraldehyde crosslinking, demonstrating the importance of both the N-terminal arm and the C-terminal globular domain of UmuD for interaction with the β-clamp. The native form of UmuD contains a cysteine at position 24, thus wild-type UmuD crosslinked to β with AIA. Attempts to crosslink β to derivatives of UmuD′ containing single cysteines at positions 34, 44, 57, 67, 81, and 112 by using AIA were unsuccessful (data not shown), consistent with our finding that UmuD′ has a lower affinity for β than does intact UmuD (Fig. 1).
Figure 3.

Identification of amino acid residues in UmuD that are close to β in the UmuD-β complex. (A) UmuD derivatives either lacking a cysteine (C24A) or containing a single cysteine at the indicated position that was conjugated to AIA were mixed with purified β, exposed to UV light, then fractionated in SDS/PAGE, and processed as a Western blot with anti-β-polyclonal antibodies. (B and C) Single cysteine positions in UmuD that crosslinked most efficiently to β are colored red and are labeled, and those that did not are in green. One UmuD protomer is colored blue and the other is gray, except for residues 1–9 of each that are magenta. The suffixes a and b in the labels are intended to distinguish to which protomer the indicated residues belong. Residues 1–19 of each protomer are represented in stick form, and residues 20–139 are displayed as space-filled. Views shown depict the front (B) and side (C) of UmuD2. Structure figures were generated by using the structural model for UmuD2 proposed recently (41) and MOLMOL (54).
Model of the Domain of UmuD2 Involved in Interaction with the β Clamp.
Neither the solution nor the crystal structure of the uncleaved UmuD2 homodimer has been reported. However, we have recently proposed a working model for the solution structure of the UmuD2 homodimer (40, 41). In this model, the overall protein fold of the C-terminal globular domain of UmuD2 (residues 40–139) resembles that reported for the UmuD′2 crystal (42). However, the interacting α-helixes near the N terminus (residues 40–44) are restructured, presumably the result of the N-terminal arm of each UmuD protomer making extensive contacts with the C-terminal globular domain of its intradimer partner such that the cleavage site of each protomer is located nearby the active site dyad of its partner (40, 41). Residues 1–19 in UmuD2 may be unstructured (40, 41).
We have represented the results of our AIA crosslinking on our working model of the UmuD2 structure (41). Those positions that crosslinked most efficiently (residues 24, 34, 52, and 126) are in red, and those that crosslinked poorly, or not at all, are in green (Fig. 3). The most striking feature of this representation is that the residues that crosslinked to β most efficiently are located in both the N-terminal arm (residues 1–39) and the C-terminal globular domain of UmuD and cluster near the top and sides of the UmuD2 homodimer (see Fig. 3). The fact that a UmuD derivative bearing an HMK motif at its C terminus (attached to residue 139) that was radiolabeled with 32P did not crosslink to β with glutaraldehyde (data not shown) suggests that β interacts with the portion of the extended interface of UmuD2 in the vicinity of the base of the arms that is formed by the N-terminal arm of one UmuD protomer contacting the C-terminal globular domain of its intradimer partner. With the exception of position 34, the residues identified in this study that crosslinked to β are different from those identified in a previous study that identified residues of UmuD (residues 34, 57, 67, 81, 112) that crosslinked to a RecA/ssDNA nucleoprotein filament (47). Taken together, these analyses suggest that different residues in UmuD are involved in interaction with RecA/ssDNA nucleoprotein filaments and the β-clamp.
The UmuD2 Homodimer Interacts with β.
As summarized in Fig. 3B, our results to this point suggest a model in which the N-terminal arm of UmuD confers upon the homodimer a unique structure with a comparatively higher affinity than UmuD′ for the β-clamp. Based on the AIA crosslinking experiment discussed above, both residues C24 and V34, which are located within the arm, must be close to β, although neither makes a contact required for the interaction, because their conjugation to AIA, or substitution of V34 with C, did not eliminate the interaction. As a test of this model, we measured the ability of a UmuD derivative containing two cysteines (UmuD[C24-F94C]2), one at the native position of 24 located within the N-terminal arm, and the other at position 94, within the C-terminal globular domain, to interact with β. Under oxidizing conditions, this mutant readily forms two disulfide linkages between positions C24 of each UmuD protomer and positions C94 of their intradimer partners (see Fig. 4A) (41). Thus, this derivative can be covalently “locked” into a structure that is presumably similar to that of the UmuD2 homodimer under normal conditions, allowing us to test whether the interaction of UmuD with the β-clamp requires that the N-terminal arms undergo a major conformational change.
Figure 4.

The UmuD2 homodimer interacts with the β-clamp. (A) Model of UmuD2 showing the relative positions of C24 and F94 (in yellow) in each protomer. One UmuD protomer is shown in blue and the other in gray. Residues 1–9 of each protomer are colored magenta. Residues 1–37 of each protomer are represented in stick form to illustrate how the structures of the N-terminal arms form an extended interface. As in Fig. 3, labels include the suffixes a and b to distinguish between protomers. (B) SDS/PAGE analysis of untreated and copper-phenanthroline (Cu-Ph)-treated UmuD[C24-F94C], with or without subsequent treatment with β-mercaptoethanol (β-Merc). The positions of molecular weight markers, and monomeric (D) and homodimeric UmuD2 derivatives (D-D) are indicated. (C) Glutaraldehyde crosslinking was performed with the reduced (R) or copper-phenanthroline-induced oxidized (O) forms of UmuD[C24-F94C] (24–94), as described in Materials and Methods. The percent of the total β trapped in the UmuD-β complex for each UmuD derivative is indicated. NA, not applicable.
For this analysis, we purified both the reduced and the oxidized form of UmuD[C24-F94C]2 and compared their affinities for β with that of the wild-type UmuD. The crosslinking efficiency for the reduced form of the mutant UmuD[C24-F94C] protein was only modestly lower than that observed for the wild-type UmuD protein (6.2% of the total β was trapped in the UmuD[C24-F94C]-β complex, and 7.2% was trapped with UmuD), suggesting that the F94C substitution has a modest affect on the interaction of UmuD with β (Fig. 4C). However, the fact that the crosslinking efficiency for both the reduced and oxidized forms of UmuD[C24-F94C] were essentially identical (6.2% for the reduced form compared with 5.9% for the oxidized form) indicates clearly that β interacts with the UmuD2 homodimer and suggests that this interaction involves contacts of β with the extended interface in UmuD2 formed by the N-terminal arm of each protomer bending down and contacting the C-terminal globular domain of its intradimer partner.
Discussion
Our findings, discussed in this report, demonstrate clearly that intact UmuD has a greater affinity for the β processivity clamp of pol III than does the cleaved form, UmuD′, and that this higher affinity of UmuD for β is due to the extra N-terminal 24 residues it has relative to UmuD′. We have recently proposed a model for the structure of the UmuD2 homodimer in solution (40, 41). In this model, the N-terminal arm of each UmuD protomer bends down over the C-terminal globular domain of its intradimer partner, thus forming an extended interface that masks a significant portion of its solvent-exposed surface. Representing our AIA crosslinking results on this proposed structure for the UmuD2 homodimer suggests that the extended interface in UmuD2 created by the N-terminal arms contacting the C-terminal globular domains of their intradimer partner is an important determinant of the interaction of UmuD2 with the β-clamp, and likely accounts for the increased affinity of UmuD for β relative to UmuD′. Consistent with this conclusion, by using the UmuD[C24-F94C]2 derivative, which under oxidizing conditions forms two intermolecular disulfide linkages involving C24 in each arm to F94C in each globular domain, we have demonstrated that association of β with UmuD2 involves an interaction with the N-terminal arms in the extended interface rather than an interaction that requires the arms to become free as they are in UmuD′2 (39).
On the basis of our finding that intact UmuD interacts more strongly with the β-clamp of pol III than does UmuD′, we suggest that the UmuD-β interaction is an important component of the UmuD2C-dependent DNA damage checkpoint control by affecting resumption of DNA replication after DNA damage. With respect to this checkpoint control, the interaction of intact UmuD with the β-clamp is somewhat reminiscent of the interaction of the eukaryotic processivity clamp, proliferating cell nuclear antigen, with the C-terminal 22 residues of p21 (48). However, it should be stressed that although interaction of p21 with proliferating cell nuclear antigen can inhibit processive replication by pol δ in vitro (48), the role of this interaction in vivo is less clear (49). Despite the obvious similarities, important differences with respect to how UmuD and p21 interact with their respective clamp proteins exist. Specifically, the C-terminal 22 residues of p21 are disordered in solution and take on an ordered structure only in the presence of proliferating cell nuclear antigen (50). In contrast to this finding, we have recently demonstrated that a large portion of the N-terminal arms of UmuD2 are not mobile but rather form an extended interface (40, 41). In this report, we demonstrate that this structure seems to be important for interaction of UmuD2 with the β-clamp. Finally, in addition to interacting with proliferating cell nuclear antigen, p21 also serves an important role in governing the G1-to-S phase transition by regulating the activity of cyclin-CDK complexes (51). Given that the umuD gene product is ≈12-fold more abundant than the umuC gene product in vivo (52), it will be interesting to see whether UmuD and UmuD′ have functions in addition to their currently recognized roles in checkpoint control and TLS.
Despite the fact that UmuD interacts with β more strongly than does UmuD′, the β-clamp and γ-clamp loader complex of pol III have been shown to stimulate UmuD′2C-dependent TLS in vitro (12, 23, 32). We suggest that interaction of UmuD′2C with β and α (and possibly ɛ) might constitute part of a polymerase switch involving pol III and pol V important for enabling TLS in the living cell. Although E. coli pol IV (DinB) interacts with β (53), it is currently unknown whether UmuC does so. Assuming UmuC does interact physically with β (based on its amino acid similarity to pol IV), it is unclear whether both UmuD′ and UmuC must interact directly with β for pol V-dependent TLS to be enabled. It is possible that the interaction of UmuD′ with β is not physiologically relevant. However, because our previously described genetic analyses suggested that both UmuD2C and UmuD′2C interact with similar surfaces on β (27), it seems likely that UmuD′ nonetheless has some limited ability to interact with β both in vivo and in vitro. Further work will be required to better understand the UmuD′-β interaction at the molecular level, as well as the importance of this interaction to TLS in the living cell.
We have suggested that interactions of the umuD gene products with components of pol III, as well as other cellular proteins, constitute part of a higher-order regulatory system of replication fork management that acts to manage the nature and the order of events at the replication fork (25, 28, 29). Our findings discussed in this report provide further evidence for this proposal by demonstrating that structural differences between UmuD and UmuD′ translate directly into differences in how these two forms of the umuD gene product interact with at least one component of pol III, namely the β processivity clamp. Thus, by interacting differently with the β-clamp of pol III, the UmuD and UmuD′ proteins presumably help to determine whether replication is blocked by the UmuD2C-dependent checkpoint control, or whether replication over DNA lesions is facilitated by UmuD′2C-dependent TLS, thus helping to decide both the nature and the order of events when the replisome encounters a DNA lesion. Further biochemical characterization of the interactions of the two umuD gene products with the α- and ɛ-subunits of pol III, as well as with other cellular proteins, together with further characterizations of protein–protein interactions involving pol V and the other four E. coli DNA polymerases, will help to better define the molecular mechanism of this higher-order regulatory system. We anticipate that these analyses will provide insights into how other organisms regulate the actions of their multiple DNA polymerases (28).
Acknowledgments
We thank Charles McHenry, Brad Glover, and Cherie Mueller (Univ. of Colorado) for purified β protein and the anti-β-antibodies, Mike O'Donnell and Zvi Kelman (Rockefeller Univ.) for the βHMK overproducer, Tim Opperman (Massachusetts Institute of Technology) for cloning the UmuDΔ37 overexpresser, J. D. Zhang (Massachusetts Institute of Technology) for synthesis of AIA, Ann Ferentz (Harvard Univ.) and Sharotka Godzina (Massachusetts Institute of Technology) for help with generating Figs. 3 and 4, and the members of our laboratory, particularly Brad Smith, for helpful discussions. This work was supported by Public Health Service Grant CA21615 (to G.C.W.) from the National Cancer Institute. M.D.S. was supported by Fellowship 5 F32 CA79161-03 from the National Cancer Institute. I.N. was the recipient of an Atomic Energy Research Exchange Program Fellowship from the Japan Atomic Energy Research Institute and the Science and Technology Agency of Japan.
Abbreviations
- AIA
p-azidoiodoacetanilide
- pol III
DNA polymerase III holoenzyme
- pol V
DNA polymerase V
- HMK
heart muscle kinase
- ssDNA
single strand DNA
- TLS
translesion DNA synthesis
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Friedberg E C, Gerlach V L. Cell. 1999;98:413–416. doi: 10.1016/s0092-8674(00)81970-4. [DOI] [PubMed] [Google Scholar]
- 2.Woodgate R. Genes Dev. 1999;13:2191–2195. doi: 10.1101/gad.13.17.2191. [DOI] [PubMed] [Google Scholar]
- 3.Johnson R E, Washington M T, Prakash S, Prakash L. Proc Natl Acad Sci USA. 1999;96:12224–12226. doi: 10.1073/pnas.96.22.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutton M D, Smith B T, Godoy V G, Walker G C. Annu Rev Genet. 2000;34:479–497. doi: 10.1146/annurev.genet.34.1.479. [DOI] [PubMed] [Google Scholar]
- 5.Johnson R E, Kondratick C M, Prakash S, Prakash L. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 6.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature (London) 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 7.Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlach V L, Aravind L, Gotway G, Schultz R A, Koonin E V, Friedberg E C. Proc Natl Acad Sci USA. 1999;96:11922–11927. doi: 10.1073/pnas.96.21.11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohmori H, Friedberg E C, Fuchs R P, Goodman M F, Hanaoka F, Hinkle D, Kunkel T A, Lawrence C W, Livneh Z, Nohmi T, et al. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 10.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol. Press; 1995. [Google Scholar]
- 11.Reuven N B, Arad G, Maor-Shoshani A, Livneh Z. J Biol Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 12.Tang M, Shen X, Frank E G, O'Donnell M, Woodgate R, Goodman M F. Proc Natl Acad Sci USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodgate R, Levine A S. Cancer Surv. 1996;28:117–140. [PubMed] [Google Scholar]
- 14.Courcelle J, Khodursky A, Peter B, Brown P O, Hanawalt P C. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Little J W, Mount D W, Yanisch-Perron C R. Proc Natl Acad Sci USA. 1981;78:4199–4203. doi: 10.1073/pnas.78.7.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Little J W, Edmiston S H, Pacelli L Z, Mount D W. Proc Natl Acad Sci USA. 1980;77:3225–3229. doi: 10.1073/pnas.77.6.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bagg A, Kenyon C J, Walker G C. Proc Natl Acad Sci USA. 1981;78:5749–5753. doi: 10.1073/pnas.78.9.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Opperman T, Murli S, Smith B T, Walker G C. Proc Natl Acad Sci USA. 1999;96:9218–9223. doi: 10.1073/pnas.96.16.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murli S, Opperman T, Smith B T, Walker G C. J Bacteriol. 2000;182:1127–1135. doi: 10.1128/jb.182.4.1127-1135.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nohmi T, Battista J R, Dodson L A, Walker G C. Proc Natl Acad Sci USA. 1988;85:1816–1820. doi: 10.1073/pnas.85.6.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burckhardt S E, Woodgate R, Scheuermann R H, Echols H. Proc Natl Acad Sci USA. 1988;85:1811–1815. doi: 10.1073/pnas.85.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shinagawa H, Iwasaki H, Kato T, Nakata A. Proc Natl Acad Sci USA. 1988;85:1806–1810. doi: 10.1073/pnas.85.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang M, Pham P, Shen X, Taylor J-S, O'Donnell M, Woodgate M, Goodman M F. Nature (London) 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 24.Maor-Shoshani A, Reuven N B, Tomer G, Livneh Z. Proc Natl Acad Sci USA. 2000;97:565–570. doi: 10.1073/pnas.97.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutton M D, Opperman T, Walker G C. Proc Natl Acad Sci USA. 1999;96:12373–12378. doi: 10.1073/pnas.96.22.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutton M D, Murli S, Opperman T, Klein C, Walker G C. J Bacteriol. 2001;183:1085–1089. doi: 10.1128/JB.183.3.1085-1089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutton M D, Farrow M F, Burton B M, Walker G C. J Bacteriol. 2001;183:2897–2909. doi: 10.1128/JB.183.9.2897-2909.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutton M D, Walker G C. Proc Natl Acad Sci USA. 2001;98:8342–8349. doi: 10.1073/pnas.111036998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker G C, Smith B T, Sutton M D. In: Bacterial Stress Responses. Stroz G, Hengge-Aronis R, editors. Washington DC: Am. Soc. Microbiol. Press; 2000. pp. 131–144. [Google Scholar]
- 30.Bridges B. Mutat Res. 2001;485:61–67. doi: 10.1016/s0921-8777(00)00074-4. [DOI] [PubMed] [Google Scholar]
- 31.Walker G C. Mutat Res. 2001;485:69–81. doi: 10.1016/s0921-8777(00)00075-6. [DOI] [PubMed] [Google Scholar]
- 32.Pham P, Bertram J G, O'Donnell M, Woodgate R, Goodman M F. Nature (London) 2001;409:366–370. doi: 10.1038/35053116. [DOI] [PubMed] [Google Scholar]
- 33.Lee M H, Ohta T, Walker G C. J Bacteriol. 1994;176:4825–4837. doi: 10.1128/jb.176.16.4825-4837.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner J, Hingorani M M, Kelman Z, O'Donnell M. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battista J R, Ohta T, Nohmi T, Sun W, Walker G C. Proc Natl Acad Sci USA. 1990;87:7190–7194. doi: 10.1073/pnas.87.18.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sutton M D, Walker G C. J Bacteriol. 2001;183:1215–1224. doi: 10.1128/JB.183.4.1215-1224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohta T, Sutton M D, Guzzo A, Cole S, Ferentz A E, Walker G C. J Bacteriol. 1999;181:177–185. doi: 10.1128/jb.181.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodgate R, Rajagopalan M, Lu C, Echols H. Proc Natl Acad Sci USA. 1989;86:7301–7305. doi: 10.1073/pnas.86.19.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferentz A E, Opperman T, Walker G C, Wagner G. Nat Struct Biol. 1997;4:979–983. doi: 10.1038/nsb1297-979. [DOI] [PubMed] [Google Scholar]
- 40.Ferentz A E, Walker G C, Wagner G. EMBO J. 2001;20:4287–4298. doi: 10.1093/emboj/20.15.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutton M D, Guzzo A, Narumi I, Costanzo M, Altenbach C, Ferentz A E, Hubbell W, Walker G C. DNA Repair. 2002;1:77–93. doi: 10.1016/s1568-7864(01)00006-4. [DOI] [PubMed] [Google Scholar]
- 42.Peat T S, Frank E G, McDonald J P, Levine A S, Woodgate R, Hendrickson W A. Nature (London) 1996;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- 43.Peat T S, Frank E G, McDonald J P, Levine A S, Woodgate R, Hendrickson W A. Structure (London) 1996;4:1401–1412. doi: 10.1016/s0969-2126(96)00148-7. [DOI] [PubMed] [Google Scholar]
- 44.Wong S S. Chemistry of Protein Conjugation and Cross-linking. Boca Raton, FL: CRC Press; 1993. [Google Scholar]
- 45.Guzzo A, Lee M H, Oda K, Walker G C. J Bacteriol. 1996;178:7295–7303. doi: 10.1128/jb.178.24.7295-7303.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, Lee M H, Walker G C. Biochem Biophys Res Commun. 1995;217:1177–1184. doi: 10.1006/bbrc.1995.2893. [DOI] [PubMed] [Google Scholar]
- 47.Lee M H, Walker G C. J Bacteriol. 1996;178:7285–7294. doi: 10.1128/jb.178.24.7285-7294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waga S, Hannon G J, Beach D, Stillman B. Nature (London) 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 49.Warbrick E. BioEssays. 2000;22:997–1006. doi: 10.1002/1521-1878(200011)22:11<997::AID-BIES6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 50.Gulbis J M, Kelman Z, Hurwitz J, O'Donnell M, Kuriyan J. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 51.Harper J W, Elledge S J, Keyomarsi K, Dynlacht B, Tsai L H, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, et al. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woodgate R, Ennis D G. Mol Gen Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 53.Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs R P. EMBO Rep. 2000;1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]