

Abstract
In addition to its role in calcium and phosphorous homeostasis, 1α,25-dihydroxyvitamin D3 [1,25-(OH)2D3] appears to be a modulator of the immune system. Administration of 1,25-(OH)2D3 prevents disease in several autoimmune animal models, including experimental autoimmune encephalomyelitis (EAE). The vitamin D receptor is believed to mediate this activity. Among cells of the immune system, CD8+ T cells have the highest levels of the vitamin D receptor. Because CD8+ T cells have been implicated as both suppressors and effectors of the inflammation associated with multiple sclerosis and EAE, we examined the question of whether the 1,25-(OH)2D3 suppression of EAE occurs through a CD8+ T cell-dependent mechanism. To test this hypothesis, mice that are homozygous knockouts for the α chain of the CD8 receptor and have been characterized as lacking functional CD8+ T cells (CD8+ −/−) were provided 1,25-(OH)2D3 in their diet before EAE induction. Although CD8+ −/− mice fed the same diet lacking 1,25-(OH)2D3 have a high incidence of EAE, EAE did not occur in CD8+ −/− mice fed the diet containing 1,25-(OH)2D3. We conclude that CD8+ T cells neither are needed nor do they play a role in the prevention of EAE by 1,25-(OH)2D3.
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system that is the leading cause of disease-associated neurological disability in western countries (1). This disease is characterized by destruction of the myelin sheaths that surround axons in the central nervous system and ultimately results in numerous neurological defects that manifest in symptoms including fatigue, limb spasticity, bladder dysfunction, and muscle weakness so severe that death is sometimes the result (2, 3). This inflammation is caused by either a breakdown in immunological self tolerance and/or an infection that triggers central nervous system-specific T cells to form inflammatory lesions. Although very little is known about the triggering event for MS, it is clear that both environmental and genetic factors are involved (4). In examining environmental factors, the geographical distribution of MS must be considered. A north-to-south gradient of higher-to-lower MS incidence in the northern hemisphere has been demonstrated in epidemiology studies, whereas a similar gradient running south to north has been observed in the southern hemisphere (5, 6). In 1974, P. Goldberg noted that this MS incidence gradient correlated with the incidence of vitamin D deficiency in temperate regions, relative to equatorial regions (7). Further studies demonstrated a lower MS incidence in temperate areas where vitamin D is abundant because of increased sun exposure, high altitudes, or diets rich in fish oils (8–11).
Insights into the disease course of MS have been found by using the animal model experimental autoimmune encephalomyelitis (EAE). EAE is a CD4+ T cell-mediated inflammatory demylenating disease that shares many similarities with MS and is induced by injecting an antigen such as myelin basic protein (MBP) in an adjuvant into a susceptible strain of rodent. It has been demonstrated that pharmacological doses of the functional metabolite of vitamin D, 1α,25-dihydroxyvitamin D3 [1,25-(OH)2D3], can greatly reduce or eliminate the incidence and progression of EAE in mice (12, 13). In addition, 1,25-(OH)2D3 has been shown to be an inhibitor of activated T cells both in vivo and in vitro and a modulator of antigen-presenting cells (14–16).
All known biological activities of 1,25-(OH)2D3 occur through the vitamin D receptor (VDR), a nuclear hormone receptor that binds to specific promoter elements in target genes and influences their transcription rates in the presence of the 1,25-(OH)2D3 ligand. Besides being expressed in tissues involved in regulating calcium and phosphorus metabolism, VDR is expressed in other tissues including components of the immune system. VDR protein is found in peripheral blood monocytes, macrophages, and lymphocytes of human and mouse origin (17–19). In a recent study of VDR protein concentrations in CD4+ T cells, CD8+ T cells, B cells, and macrophages, our lab observed the highest concentration of VDR in activated CD8+ T cells (17). Although it is well established that CD8+ T cells act as cytotoxic killer cells against virally infected cells by recognizing viral antigens bound to class I MHC protein complexes, its role in inflammatory processes is not clear. Some studies suggest that CD8+ T cells may be effector cells in inflammation. CD8+ T cell clones specific to myelin antigen have been isolated from the periphery of MS patients and are found as part of the inflammatory infiltrate in central nervous system lesions (20–23). Autoreactive CD8+ T cells have also been implicated as potent effector cells in several animal models of autoimmune diseases including EAE. Adoptive transfer studies of CD8+ T cells isolated from animals with EAE have successfully propagated the disease to naïve recipients (24, 25). Mixed results have been reported in EAE studies in animals lacking CD8+ T cells. EAE was observed to cause lower mortality but more relapses in the CD8+ T cell knockout mouse model as well as in a mouse model where CD8+ T cells were removed with anti-CD8+ monoclonal antibodies (26, 27). These results suggest both an effector and a suppressor role. However, these studies were contradicted by studies performed on rats that showed the presence of CD8+ T cells had no influence on relapse or recovery in EAE (28, 29). In addition, beneficial effects of CD8+ T cells have been reported in studies where feeding of myelin antigens leads to the generation of CD8+ T cells that suppress EAE induction (30). There have been reports of isolation of a CD8+ T cell suppressor cell line that reduces EAE severity in mice and rats by targeting and destroying CD4+ effector cells (31, 32).
Because activated CD8+ T cells have high levels of VDR and have been implicated as effectors or suppressors of EAE, we decided to investigate whether 1,25-(OH)2D3 prevents EAE induction through a mechanism involving CD8+ T cells. To do this, we obtained mice that have been genetically engineered not to express the α chain of the CD8 marker and are characterized as lacking functional CD8+ T cells (33). They were backcrossed onto a B10.PL background that is susceptible to EAE induction by MBP immunization (34). In this study, we show induction of EAE is possible in mice lacking CD8+ T cells and that 1,25-(OH)2D3 is fully able to prevent the induction of EAE in these mice.
Materials and Methods
Animals and Diet.
B6.129S2-Cd8atm1Mak mice were obtained that are homozygous for a targeted deletion of the CD8 α chain and are characterized as lacking CD8+ T cells while having normal development and function of CD4+ T cells (The Jackson Laboratory). These mice were backcrossed in our breeding facility for seven generations onto a B10.PL-H2u(73NS)/Sn background. Genotyping was performed by PCR using by primers specific for neomycin (forward: CTTGGGTGGAGAGGCTATTC and reverse: AGGTGAGATGACAGGAGATC), the CD8 α chain (forward: TCCAGCTCCAAACTCCCC and reverse: TCTGAAGGACTGGCACGAC) and the class II MHC alleles H-2 I-Au and H2 I-Ab as described previously (35). Only mice with the EAE-susceptible MHC allele H-2 I-Au were selected for further breeding. After seven generations of backcrossing, CD8+/+ and CD8−/− breeding lines that were homozygous for the MHC allele H-2 I-Au were established from the same heterozygous breeders. All animals were maintained on Purina Chow until 5 weeks of age. In the first study, they were continued on Chow diet. For the second study, litters were then divided into different experimental groups and were given a 0.87% Ca2+-purified diet in agar form that was replaced three times a week for the duration of the study, as described previously (36). Control diets contained no 1,25-(OH)2D3, whereas experimental diets were supplemented to provide 100 ng per day of 1,25-(OH)2D3 based on a 4-g/mouse/day consumption. EAE was induced in mice 10 days after they were switched to the experimental diets. At the end of the study, mice were killed, serum collected for calcium analysis, and spleens collected for flow cytometry analysis.
EAE Disease Induction.
MBP was isolated from guinea pig spinal cords as described in Deibler et al. (37). MBP was lyophilized and stored at −80°C. For immunizations, MBP was dissolved in 0.1 M acetic acid at a concentration of 8 mg/ml. Mice were s.c. injected with 0.1 ml of Freund's complete adjuvant (Difco) containing 400 μg of MBP and 40 mg of killed Mycobacterium tuberculosis (Difco). An i.p. injection of 200 ng of pertussis toxin (List Biological Laboratories, Campbell, CA) in saline was also given at the time of immunization followed by a duplicate injection 48 h later. Development of EAE was monitored by scoring the mice daily by using the following disease index: (i) limp tail; (ii) paraparesis with a clumsy gait; (iii) hind-limb paralysis; (iv) hind- and fore-limb paralysis; and (v) moribund. Intermediate scores of ±0.5 were used for mice with symptoms between any two scores.
Serum Calcium.
Serum calcium values were determined by using a Perkin–Elmer atomic absorption spectrometer. Serum was diluted in 0.1% LaCl3 for this determination.
Antibodies and Flow Cytometry.
Spleens were pushed through a stainless steel wire mesh, and lymphocytes were purified at 800 × g on a Histopaque 1083 gradient (Sigma). Biotin rat anti-mouse CD8 α monoclonal antibody, biotin rat IgG2aκ, and streptavidin–FITC were purchased from BD PharMingen, and CD8 staining was performed on the purified lymphocytes following the protocols of the supplier. Flow cytometry was performed by using the FACScan machine at the Flow Cytometry Facility in the University of Wisconsin Comprehensive Cancer Center, and the data were analyzed by using cellquest (BD Immunocytometry Systems, San Jose, CA) software.
Statistics.
Statistical analysis was performed by using a statistics program for the Macintosh computer (graphpad prism). The two-tailed Fisher exact probability test was performed on incidence rates, and the unpaired two-group Student's t test was used on other measurements. Values of P < 0.05 were considered statistically significant.
Results
EAE Is Inducible in CD8+ −/− Mice Backcrossed onto the B10.PL Background.
As indicated in Table 1, EAE was inducible in both CD8+ −/− and CD8+ +/+ mice. The CD8+ −/− mice had an incidence rate of 93.8%, whereas the wild-type CD8 mice had a lower incidence rate of 55.6%. This incidence rate was similar to incidence rates observed in the B10.PL strain immunized with the same batch of MBP (data not shown). Although the CD8+ −/− mice had a higher incidence rate, the results were not significant according to the two-tailed Fisher exact test (P = 0.053065). Mean peak severity was calculated for mice that developed EAE by taking the highest score observed for each mouse in the 40-day observation period and averaging this value with other values from mice in the same group. The mean day of onset was calculated in a similar manner by using the first day of observance of EAE as the value. There was no statistical difference between the two groups in these two categories by using the unpaired Student's t test.
Table 1.
EAE incidence and progression for CD8+ −/− and +/+
| Incidence | Mean peak severity | Mean day of onset | |
|---|---|---|---|
| CD8+ +/+ | 55.6% (n = 4 m, 7 f) | 2.9 ± 1.1 | 20 ± 7 |
| CD8+ −/− | 93.8% (n = 9 m, 7 f) | 3.6 ± 1.1 | 14 ± 3 |
At 6 weeks of age, CD8+ −/− and +/+ mice (B10.PL background) were s.c. injected with 400 μg of MBP along with an i.p. injection of 200 ng of pertussis toxin. A second pertussis toxin injection was given 48 h later. Mice were scored for EAE once per day for 40 days. For mice that developed an EAE score of 2 or greater, the incidence, mean peak severity ± SD, and mean day of onset ±SD were calculated. Values adjacent to the incidence rates indicate the number of male and female mice in the group. Mice were fed a commercial chow (Purina) for the duration of the experiment.
1,25-(OH)2D3 Prevents CD8+ −/− Mice from Developing EAE.
To determine whether the presence of CD8+ lymphocytes was necessary for 1,25-(OH)2D3 to prevent EAE, CD8+ −/− and CD8+ +/+ were fed a diet containing 100 ng of 1,25-(OH)2D3 or a control diet containing no supplemental 1,25-(OH)2D3 for 10 days before immunization with MBP. The mice were kept on these diets through the 40-day observation period after EAE induction with MBP. In Fig. 1, the daily mean score of EAE for each group is plotted. Both wild-type and CD8+ −/− mice fed the control diet develop a progressive form of EAE, whereas the wild-type and CD8+ −/− mice fed the diet containing 100 ng of 1,25-(OH)2D3 prevented all EAE symptoms. In Table 2, the results from this study are summarized. 1,25-(OH)2D3 was effective for suppressing EAE in CD8+ −/− (P < 0.005) and +/+ (P < 0.05) animals. Of the two groups of mice fed a diet containing no supplemental 1,25-(OH)2D3, there was no significant difference in mean peak severity or mean day of onset observed between the two groups. The calcium concentration in the serum of the mice was measured at the end of the study as a measure of the physiological effect of 1,25-(OH)2D3 treatment. The mean serum calcium concentration of the two groups of mice fed a control diet were in the normal range of 9–11 mg of Ca2+/dl. The two groups of mice fed a diet containing 1,25-(OH)2D3 had significantly elevated serum calcium concentration (P < 0.005 for both groups as compared with their respective genotype controls). To confirm that the mice in this study genotyped as CD8+ −/− were not expressing the α chain of the CD8 marker, flow cytometry was performed on the mice used in this study. In Fig. 2 are the data collected by flow cytometry that show the lymphocytes stained with a rat monoclonal antibody specific for the α chain of the CD8 marker or the Ig isotype control. In this example, CD8 α was detected in the lymphocytes collected from a CD8+ +/+ mouse but not in lymphocytes collected from CD8+ −/− mouse. The flow cytometry results from the 30 mice used in this study matched the predicted results from genotyping.
Figure 1.

1 25-(OH)2D3 prevents EAE in mice lacking functional CD8+ T cells. CD8+ −/− and +/+ mice were switched at 5 weeks of age to a 0.87% Ca2+-purified diet containing 0 ng/day or 100 ng/day of 1,25-(OH)2D3. Ten days later, 400 μg of MBP was injected s.c. along with an i.p. injection of 200 ng of pertussis toxin. A second pertussis toxin injection was given 48 h later. Mice were scored for EAE once per day for 40 days. For each group, the mean EAE score was calculated and plotted for each day.
Table 2.
EAE incidence and progression for CD8+ −/− and +/+ on 0.87% Ca2+ diets with or without 100 ng of 1,25-(OH)2D3 per day
| Incidence | Mean severity | Mean day of onset | Serum calcium, mg % | |
|---|---|---|---|---|
| CD8+ +/+ | 62.5% (n = 4 m, 4 f) | 2.0 ± 0 | 20 ± 4 | 10.0 ± 0.6 |
| CD8+ −/− | 87.5% (n = 4 m, 4 f) | 2.9 ± 1.1 | 18 ± 4 | 10.6 ± 0.8 |
| CD8+ +/+, + 1,25 D3 | 0%* (n = 3 m, 4 f) | — | — | 14.1 ± 0.8†− |
| CD8+ −/−, + 1,25 D3 | 0%† (n = 4 m, 3 f) | — | — | 12.8 ± 1.3†− |
CD8+ −/− and +/+ mice were switched at 5 weeks of age to a 0.87% Ca2+-purified diet containing 0 ng/day or 100 ng/day of 1,25-(OH)2D3. Ten days later, 400 μg of MBP was injected s.c. along with an i.p. injection of 200 ng of pertussis. A second pertussis toxin injection was given 48 h later. Mice were scored for EAE once per day for 40 days. For mice that developed an EAE score of 2 or greater, the incidence, mean peak severity ± SD, and mean day of onset ± SD were calculated. Values adjacent to incidence rates indicate the number of male and female mice in the group. At the end of study, mice were killed and serum calcium concentrations determined by atomic spectrometry. (*, P < 0.05;
, P < 0.005)
Figure 2.
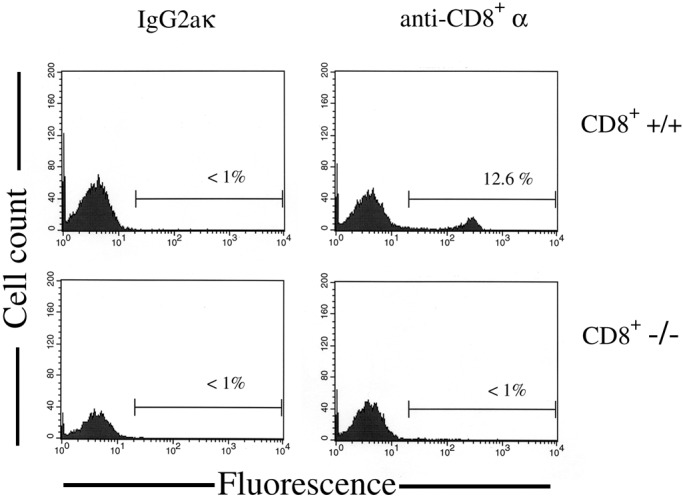
CD8+ α chain is not expressed in mice genotyped as CD8+ −/−. Splenocytes were collected from CD8+ +/+ and CD8+ −/− mice and were stained for CD8 α chain expression or with the IgG2aκ Ig isotype control. A FACScan analysis was performed and histograms were generated from data gated to include lymphocytes. Regions and percentages indicate the subset of gated cells that stained positive. Results were typical for the 30 mice screened.
Discussion
To investigate whether the immunosuppressive ability of 1,25-(OH)2D3 is acting through a mechanism dependent on CD8+ T cells, we induced EAE in mice lacking functional CD8+ T cells. The results show that 1,25-(OH)2D3 is fully able to suppress EAE induction in the absence of CD8+ T cells. There was also a general trend of a higher incidence rate in mice lacking CD8+ T cells, which may be attributed to several factors. In the study summarized in Table 1, the male-to-female ratio was higher in the knockout group than in the wild-type group. Males are more likely to develop EAE (36), which was observed in the wild-type group of the first study with three females remaining disease free compared with one male. However, in the second study, equal numbers of male and female mice in the knockout and wild-type groups were fed a control diet (Table 2), and a higher incidence rate was still observed. This trend in the data suggests CD8+ T cells may have a suppressor role in this model of EAE. Although this study clearly demonstrates that 1,25-(OH)2D3 is fully able to prevent EAE in the absence of CD8+ T cells, it does not examine a possible effect of 1,25-(OH)2D3 on CD8+ T cells involved in the inflammatory process.
By eliminating the necessity of CD8+ T cells in the suppression of EAE by 1,25-(OH)2D3, the possible target cells are further narrowed. These cells include antigen-presenting cells whose ability to present antigen to CD4+ T cells may be diminished by the hormone, and the CD4+ effector cells whose activation or cytokine expression may be altered in the presence of 1,25-(OH)2D3 (16, 38). Although these are not mutually exclusive, several lines of evidence argue that 1,25-(OH)2D3 is acting on CD4+ T cells in prevention of EAE. Many in vitro studies demonstrate the ability of 1,25-(OH)2D3 to suppress the activation and cytokine release of T cells (39–43). Studies done in vivo show that 1,25-(OH)2D3 up-regulates expression of the antiinflammatory cytokines transforming growth factor-β1 and IL-4 and is a potent inhibitor of the proinflammatory Th1 CD4+ T cell subset (35, 44–46). Clearly in the hands of more than one investigator, 1,25-(OH)2D3 arrests the development of EAE after the disease course has moved from the activation phase caused by antigen-presenting cells (13, 47). Recently, Nashold et al. demonstrated that 1,25-(OH)2D3 is unable to prevent EAE in transgenic recombination-activating gene-1 (RAG-1−/−) mice that are genetically engineered to express only T cells specific for MBP (48). They propose 1,25-(OH)2D3 acts through RAG-1-dependent cell in suppressing EAE. RAG-1 enzyme catalyzes V(D)J recombination events in B cells and T cells and is known only to be expressed in these cells. Because there are no detectable amounts of VDR in B cells (17) and our results eliminate a necessary role for CD8+ T cells, the CD4+ T cell is likely to be the key target in the immunosuppression ability of 1,25-(OH)2D3.
Acknowledgments
This work was supported in part by funds from the National Foundation for Cancer Research and the Wisconsin Alumni Research Foundation.
Abbreviations
- 1,25-(OH)2D3
1,25-dihydroxyvitamin D3
- EAE
experimental autoimmune encephalomyelitis
- VDR
vitamin D receptor
- MS
multiple sclerosis
- MBP
myelin basic protein
References
- 1.Kuby J, editor. Immunology. 3rd Ed. New York: Freeman; 1992. p. 490. [Google Scholar]
- 2.Metz L M, Patten S B, McGowan D. Biomed Pharmacother. 1999;53:371–379. doi: 10.1016/s0753-3322(99)80108-5. [DOI] [PubMed] [Google Scholar]
- 3.Gallien P, Robineau S. Biomed Pharmacother. 1999;53:380–385. doi: 10.1016/s0753-3322(99)80109-7. [DOI] [PubMed] [Google Scholar]
- 4.Kahana E. Biomed Pharmacother. 2000;54:100–102. doi: 10.1016/S0753-3322(00)88859-9. [DOI] [PubMed] [Google Scholar]
- 5.Alter M, Morariu M. Lancet. 1973;1:1126–1127. doi: 10.1016/s0140-6736(73)90443-1. [DOI] [PubMed] [Google Scholar]
- 6.Miller D H, Hammond S R, McLeod J G, Purdie G, Skegg D C. J Neurol Neurosurg Psychiatry. 1990;53:903–905. doi: 10.1136/jnnp.53.10.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg P. Int J Environ Studies. 1974;6:19–27. [Google Scholar]
- 8.Prestus J. Acta Psychiatr Neurol Scand. 1960;35:88–92. [Google Scholar]
- 9.Westlund K. Acta Neurol Scand. 1970;46:455–483. doi: 10.1111/j.1600-0404.1970.tb05806.x. [DOI] [PubMed] [Google Scholar]
- 10.Kurtzke J F. Acta Neurol Scand. 1967;43:257–282. doi: 10.1111/j.1600-0404.1967.tb05733.x. [DOI] [PubMed] [Google Scholar]
- 11.Freedman D M, Dosemeci M, Alavanja M C. Occup Environ Med. 2000;57:418–421. doi: 10.1136/oem.57.6.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemire J M, Archer D C. J Clin Invest. 1991;87:1103–1107. doi: 10.1172/JCI115072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cantorna M T, Hayes C E, DeLuca H F. Proc Natl Acad Sci USA. 1996;93:7861–7864. doi: 10.1073/pnas.93.15.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsoukas C D, Provvedini D M, Manolagas S C. Science. 1984;224:1438–1440. doi: 10.1126/science.6427926. [DOI] [PubMed] [Google Scholar]
- 15.Bargman J M, Silverman E D, Klein M H. Miner Electrolyte Metab. 1989;15:359–364. [PubMed] [Google Scholar]
- 16.Griffin M D, Lutz W H, Phan V A, Bachman L A, McKean D J, Kumar R. Biochem Biophys Res Commun. 2000;270:701–708. doi: 10.1006/bbrc.2000.2490. [DOI] [PubMed] [Google Scholar]
- 17.Veldman C M, Cantorna M T, DeLuca H F. Arch Biochem Biophys. 2000;374:334–338. doi: 10.1006/abbi.1999.1605. [DOI] [PubMed] [Google Scholar]
- 18.Kreutz M, Andreesen R, Krause S W, Szabo A, Ritz E, Reichel H. Blood. 1993;82:1300–1307. [PubMed] [Google Scholar]
- 19.Reichel H, Koeffler H P, Tobler A, Norman A W. Proc Natl Acad Sci USA. 1987;84:3385–3389. doi: 10.1073/pnas.84.10.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monteiro J, Hingorani R, Peroglizzi R, Apatoff B, Gregersen P K. Autoimmunity. 1996;23:127–138. doi: 10.3109/08916939608995336. [DOI] [PubMed] [Google Scholar]
- 21.Dressel A, Chin J L, Sette A, Gausling R, Hollsberg P, Hafler D A. J Immunol. 1997;159:4943–4951. [PubMed] [Google Scholar]
- 22.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, et al. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vos A F, Dick A D, Klooster J, Broersma L, McMenamin P G, Kijlstra A. Invest Ophthalmol Visual Sci. 2000;41:3001–3010. [PubMed] [Google Scholar]
- 24.Huseby E S, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. J Exp Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun D, Whitaker J N, Huang Z, Liu D, Coleclough C, Wekerle H, Raine C S. J Immunol. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 26.Jiang H, Zhang S I, Pernis B. Science. 1992;256:1213–1215. doi: 10.1126/science.256.5060.1213. [DOI] [PubMed] [Google Scholar]
- 27.Koh D R, Fung-Leung W P, Ho A, Gray D, Acha-Orbea H, Mak T W. Science. 1992;256:1210–1213. doi: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- 28.Sedgwick J D. Eur J Immunol. 1988;18:495–502. doi: 10.1002/eji.1830180402. [DOI] [PubMed] [Google Scholar]
- 29.Lohse A W, Schwerdt A, Herkel J, Spahn T, Meyer zum Buschenfelde K H. J Autoimmun. 1995;8:395–404. doi: 10.1006/jaut.1995.0031. [DOI] [PubMed] [Google Scholar]
- 30.Miller A, Lider O, Roberts A B, Sporn M B, Weiner H L. Proc Natl Acad Sci USA. 1992;89:421–425. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ofosu-Appiah W, Mokhtarian F. Cell Immunol. 1991;135:143–153. doi: 10.1016/0008-8749(91)90261-9. [DOI] [PubMed] [Google Scholar]
- 32.Jiang H, Braunstein N S, Yu B, Winchester R, Chess L. Proc Natl Acad Sci USA. 2001;98:6301–6306. doi: 10.1073/pnas.101123098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fung-Leung W P, Schilham M W, Rahemtulla A, Kundig T M, Vollenweider M, Potter J, van Ewijk W, Mak T W. Cell. 1991;65:443–449. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- 34.Zamvil S S, Mitchell D J, Moore A C, Schwarz A J, Stiefel W, Nelson P A, Rothbard J B, Steinman L. J Immunol. 1987;139:1075–1079. [PubMed] [Google Scholar]
- 35.Cantorna M T, Humpal-Winter J, DeLuca H F. Arch Biochem Biophys. 2000;377:135–138. doi: 10.1006/abbi.2000.1765. [DOI] [PubMed] [Google Scholar]
- 36.Cantorna M T, Humpal-Winter J, DeLuca H F. J Nutr. 1999;129:1966–1971. doi: 10.1093/jn/129.11.1966. [DOI] [PubMed] [Google Scholar]
- 37.Deibler G E, Martenson R E, Kies M W. Prep Biochem. 1972;2:139–165. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 38.Deluca H F, Cantorna M T. FASEB J. 2001;15:2579–2585. doi: 10.1096/fj.01-0433rev. [DOI] [PubMed] [Google Scholar]
- 39.Nonnecke B J, Franklin S T, Reinhardt T A, Horst R L. Vet Immunol Immunopathol. 1993;38:75–89. doi: 10.1016/0165-2427(93)90114-j. [DOI] [PubMed] [Google Scholar]
- 40.Provvedini D M, Manolagas S C. J Clin Endocrinol Metab. 1989;68:774–779. doi: 10.1210/jcem-68-4-774. [DOI] [PubMed] [Google Scholar]
- 41.Vanham G, Ceuppens J L, Bouillon R. Cell Immunol. 1989;124:320–333. doi: 10.1016/0008-8749(89)90134-2. [DOI] [PubMed] [Google Scholar]
- 42.Jordan S C, Toyoda M, Prehn J, Lemire J M, Sakai R, Adams J S. Mol Immunol. 1989;26:979–984. doi: 10.1016/0161-5890(89)90116-8. [DOI] [PubMed] [Google Scholar]
- 43.Willheim M, Thien R, Schrattbauer K, Bajna E, Holub M, Gruber R, Baier K, Pietschmann P, Reinisch W, Scheiner O, Peterlik M. J Clin Endocrinol Metab. 1999;84:3739–3744. doi: 10.1210/jcem.84.10.6054. [DOI] [PubMed] [Google Scholar]
- 44.Cantorna M T, Woodward W D, Hayes C E, DeLuca H F. J Immunol. 1998;160:5314–5319. [PubMed] [Google Scholar]
- 45.Lemire J M, Archer D C, Beck L, Spiegelberg H L. J Nutr. 1995;125:1704.S–1708S. doi: 10.1093/jn/125.suppl_6.1704S. [DOI] [PubMed] [Google Scholar]
- 46.Mattner F, Smiroldo S, Galbiati F, Muller M, Di Lucia P, Poliani P L, Martino G, Panina-Bordignon P, Adorini L. Eur J Immunol. 2000;30:498–508. doi: 10.1002/1521-4141(200002)30:2<498::AID-IMMU498>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 47.Nashold F E, Miller D J, Hayes C E. J Neuroimmunol. 2000;103:171–179. doi: 10.1016/s0165-5728(99)00247-7. [DOI] [PubMed] [Google Scholar]
- 48.Nashold F E, Hoag K A, Goverman J, Hayes C E. J Neuroimmunol. 2001;119:16–29. doi: 10.1016/s0165-5728(01)00360-5. [DOI] [PubMed] [Google Scholar]