

Abstract
Cerebral ischemia stimulates increased activity of polyamine oxidase, a ubiquitous enzyme that catabolizes polyamines to produce 3-aminopropanal. 3-Aminopropanal is a reactive aldehyde that mediates progressive neuronal necrosis and glial apoptosis. Here we report that increased levels of 3-aminopropanal-modified protein levels in humans after aneurysmal subarachnoid hemorrhage correlate with the degree of cerebral injury as measured by admission Hunt/Hess grade. In vitro screening of clinically approved drugs reveals that N-2-mercaptopropionyl glycine (N-2-MPG), an agent clinically approved for prevention of renal stones in patients with cysteinuria, significantly inhibits the cytotoxicity of 3-aminopropanal. N-2-MPG reacts with 3-aminopropanal to yield a nontoxic thioacetal adduct, as confirmed by electrospray ionization mass spectroscopy. Administration of N-2-MPG in clinically relevant doses to rats significantly reduces cerebral 3-aminopropanal-modified protein immunoreactivity and infarct volume in a standardized model of middle cerebral artery occlusion, even when the agent is administered after the onset of ischemia. These results implicate 3-aminopropanal as a therapeutic target for cerebral ischemia.
Brain cell death during cerebral ischemia is mediated by the progressive release of cytotoxins. The spread of cytotoxins to brain areas outside the primary ischemic focus causes significant “bystander” cell death. The resultant area of secondary cell death, termed the ischemic penumbra, represents a major component of the total damage caused by ischemia in the central nervous system. We recently identified a toxic product of polyamine metabolism, 3-aminopropanal, as a cytotoxic mediator of cerebral ischemia (1, 2). 3-Aminopropanal is produced by polyamine oxidase (PAO), an enzyme activity that is up-regulated during cerebral ischemia and mediates the oxidation of spermine and spermidine (1). Brain PAO activity is significantly increased within 2 h after the onset of ischemia, resulting in the accumulation of cytotoxic levels of 3-aminopropanal (1, 2). Brain 3-aminopropanal levels continue to increase for at least 25 h after the onset of ischemia, a time frame that corresponds to the development of spreading brain cell death. Structurally distinct inhibitors of PAO attenuate the activity of brain PAO, prevent the production of 3-aminopropanal, and significantly protect against the development of ischemic brain damage in rat models of focal cerebral ischemia and head trauma (1, 3–5).
3-Aminopropanal is a highly reactive aldehyde that can form covalent derivatives by binding to amino and sulfhydryl groups on proteins (6, 7). 3-Aminopropanal causes necrotic death of neurons and caspase-1 dependent apoptosis in glial cell cultures (1, 8). The LD50 for 3-aminopropanal in neuronal cultures is 90 ± 20 μM, levels that are achieved in vivo after cerebral ischemia (1). PAO activity and 3-aminopropanal formation normally are localized to peroxisomes, where the potential cytotoxic activity of 3-aminopropanal is isolated to prevent inadvertent cell damage (9, 10). Cell damage from 3-aminopropanal also can be prevented by aldehyde dehydrogenase (11). During cerebral ischemia, however, there is a loss of normal cellular compartmentalization; release of 3-aminopropanal damages cells by binding to structural membrane proteins and altering other critical functional proteins (1, 12). Together these data implicate 3-aminopropanal as a therapeutic target in experimental cerebral ischemia in animals, but data from human samples were previously lacking.
Here we produced antibodies that recognize 3-aminopropanal-modified proteins and report that 3-aminopropanal-modified protein levels increase significantly in the cerebrospinal fluid (CSF) of patients with cerebral ischemia. To inhibit the cytotoxic effects of 3-aminopropanal, we screened a panel of clinically approved drugs, selected by the presence of structural groups that can react with 3-aminopropanal. A lead compound, N-2-mercaptopropionyl glycine (N-2-MPG), formed a nontoxic thioacetal adduct with 3-aminopropanal and prevented 3-aminopropanal-mediated cytotoxicity. Administration of N-2-MPG to rats significantly reduced cerebral 3-aminopropanal-modified protein immunoreactivity and protected against cerebral infarction in a standard model of middle cerebral artery occlusion (MCAO).
Materials and Methods
Tissue Culture.
The glial cell line (HTB14) and the neuronal cell line (HTB11) were obtained from the American Type Culture Collection and cultured in DMEM (GIBCO/BRL) containing 10% FBS (HyClone), sodium pyruvate (1 mM; Sigma), penicillin/streptomycin (0.5%; Sigma) in a humidified atmosphere (5% CO2; 37°C). For all experiments involving exposure to 3-aminopropanal, cells were grown in 6- or in 96-well micro titer plates to 90–95% confluence, and the medium was replaced with fresh serum-free medium (Opti-MEM I; GIBCO/BRL) to prevent nonspecific interaction of 3-aminopropanal with serum proteins and to mimic the very low protein concentrations present in cerebral interstitial fluids. 3-Aminopropanal was chemically prepared by hydrolysis of 3-aminopriopionaldehyde diethyl acetal, as described (1). Dilutions of 3-aminopropanal (50–2000 μM) or vehicle, and the test compounds of choice (Sigma-Aldrich; 1 mM) or control media, were premixed at the concentrations indicated and added to cells in 96-well plates at 120 μl/well. After an overnight incubation, cell viability was determined by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) assay. The absorbance of the untreated cells was considered 100% viability and the results were expressed as % viability with comparison to untreated cells. LD50 values designate the amount of 3-aminopropanal necessary to kill 50% of the cells in the presence of 1 mM of the test compound. Data are expressed as mean ± SE, n = 3–6 wells per condition.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Staining of the Glial Cell Line HTB14.
Cells were treated with 3-aminopropanal (160 μM) or vehicle, and N-2-MPG (1 mM) or medium alone, as described above. At 5 h postexposure, the cells were harvested and centrifuged (1,500 rpm for 5 min). The pellets were fixed with 1× ORTHO Permeafix (Ortho Diagnostics) at room temperature for 40 min. After washing with Dulbecco's PBS containing 1% BSA (PBS-BSA), cells were stained by the TUNEL method with the Oncor Apop Tag Direct Fluorescein kit. Negative controls were performed by using a reaction mixture devoid of terminal deoxynucleotidyltransferase. A Becton Dickinson FACScan was used for all analyses; 5,000–10,000 events (ungated) were collected by using a single-color histogram for FITC.
Animal Model of Permanent MCAO.
All procedures involving animals were conducted in conformity with institutional guidelines and under the approval of the Animal Care and Use Committee of North Shore University Hospital. MCAO is a widely used model of cerebral ischemia that is responsive to experimental therapeutics. This model was selected in this study because it is characterized by development of a spreading ischemic cascade in brain that is reproducible and readily measured. Mediators of cerebral ischemia in this model are implicated in the pathogenesis of human cerebral ischemia caused by subarachnoid hemorrhage (SAH). MCAO was performed as described (2, 13). Briefly, the ipsilateral common carotid artery was ligated and divided, the middle cerebral artery was coagulated and divided distal to the lenticulostriate branch, and the contralateral common carotid artery was occluded for 1 h. The onset of ischemia in these experiments was defined as the time the middle cerebral artery was cut. N-2-MPG (100 mg/kg, i.p.) was administered 15 min postocclusion of the middle cerebral artery, and a second administration was performed at 3 h (50 mg/kg, i.p.). Vehicle controls were performed for all experiments. For measurements of infarct volume, the animals were killed at the times indicated and fresh brain sections were prepared (1 mm), immersed in 2,3,5-triphenyltetrazolium chloride in 154 mM NaCl for 30 min at 37°C, and total cerebral infarct volume was measured by computerized quantitative planimetry as described (2, 13, 14). To determine whether N-2-MPG treatment affected any physiological variables known to influence infarct size, animals (n = 3) were subjected to common carotid artery cannulation, and the drug was administered at the doses and time points specified above. Blood pressure, heart rate, respiratory rate, and body temperature were recorded at 30-min intervals for a total of 5 h.
Generation of α-3-Aminopropanal Antibodies.
For generation of antibodies against 3-aminopropanal, a 200-μM solution of the aldehyde in water was incubated overnight with 5 mg/ml keyhole limpet hemocyanin (Sigma) in the presence of the reducing reagent cyanoborohydride (200 μM). The rationale behind the use of this antigen rested on (i) the need to conjugate the small hapten, 3-aminopropanal, to an immunogenic carrier and (ii) the observation that the carbonyl group of 3-aminopropanal can react irreversibly with sulfhydryl, and reversibly with amine, groups on proteins, thus accumulating in vivo in both a free and a protein-bound form. The modified protein complex was washed of any unreacted reagent by ultrafiltration and three subsequent washes in 1× PBS using Centricon-10 membranes (Amicon). The antigen was inoculated in rabbits at Biosynthesis (Lewisville, TX). Immunoreactive rabbit antibodies for 3-aminopropanal-modified protein were screened by dot blotting against standards of 3-aminopropanal-modified BSA (GIBCO/BRL) prepared under the same reaction conditions, as established for 3-aminopropanal-modified keyhole limpet hemocyanin. To establish specificity, antibodies were additionally screened and found to be negative against acrolein- or malonylaldehyde-modified BSA.
Dot Blot Analysis of 3-Aminopropanal Levels in Human Patients.
CSF of patients was collected by means of lumbar puncture or ventriculostomy at New York Presbyterian Medical Center. SAH and traumatic brain-injured patients frequently develop cerebral ischemia secondary to increased intracranial pressure, low cerebral perfusion pressure, or direct injury to regional blood cerebral arteries. CSF from these patients is routinely collected by the treating physicians, because ventriculostomy with external drainage of CSF is used to monitor and treat raised intracranial pressure. [Note: CSF is not routinely collected from patients with cerebrovascular accident (stroke).] CSF also was collected from patients with neuropathy, hydrocephalus, or cerebral neoplasm who did not have any evidence of cerebral ischemia. In the present study, CSF samples were collected at the time that the ventriculostomy was placed, within 48 h from the time of onset. Antibodies generated against 3-aminopropanal-modified keyhole limpet hemocyanin were then used to determine 3-aminopropanal levels in CSF by dot blotting. Values were compared with a standard curve generated for 3-aminopropanal-albumin complexes in arbitrary units. For preparation of standards, 7 mg/ml BSA were incubated overnight with 500 μM 3-aminopropanal and unreacted material was washed off by Centricon-10 ultrafiltration, as described above. A linear standard curve was established with the 3-aminopropanal-modified BSA in the range of 0.025 to 25 mg/ml. CSF or standards (5-μl volumes) were analyzed by dot blotting on a poly(vinylidene difluoride) membrane (Bio-Rad) following the manufacturer's instructions, probing with either immune or control preimmune rabbit serum (1:500 dilution). Development of the dot blots was performed with the ECL Plus kit (Amersham Pharmacia). After chemiluminescent exposure, the films were scanned with an image scanner (Silverscanner II, Lacie Limited, Beaverton, OR), and the relative band intensity was quantified by using National Institutes of Health program IMAGE 1.59.
Immunofluorescent Detection of 3-Aminopropanal-Modified Proteins.
Rats were subjected to MCAO, and N-2-MPG was administered where indicated, as described above (n = 3–4 animals/group). At 24 h, the brains were removed, and a section of the brain corresponding to the area of MCAO, 4 mm thick, located 3 mm dorsal to the frontal poles, was excised. The brain tissue was frozen on dry ice and cut into 10-μM thick sections. Glass slides with the sections were fixed with 4% paraformaldehyde, then subjected to immunocytochemistry staining using either rabbit anti-3-aminopronal-protein serum or prebleed serum for negative control (1:100 dilution). The sections were probed with secondary goat anti-rabbit IgG-FITC conjugate (Sigma; 1:200 dilution). The sections were covered with Vectashield (Vector Laboratories). Pictures were taken by using ×10 magnification on an Olympus Fluoview confocal laser scanning microscope.
Results
3-Aminopropanal Levels Are Elevated in Human CSF in Association with Severity of Ischemic Cerebral Injury.
To determine whether 3-aminopropanal-modified proteins accumulate in clinical samples, human CSF was collected from patients by means of lumbar puncture or ventriculostomy. Diagnoses included penetrating head trauma, intracranial hemorrhage, SAH, neuropathy, hydrocephalus, and cerebral neoplasm. Patients with good neurological prognosis had significantly lower 3-aminopropanal levels (1.7 ± 0.4 mg/ml 3-aminopropanal protein equivalents; n = 16), as compared with patients with poor neurological diagnosis (7.2 ± 1.0 mg/ml 3-aminopropanal protein equivalents; n = 19), P < 0.0001 (Fig. 1). 3-Aminopropanal levels in poor grade SAH patients (Hunt/Hess IV-V) were significantly higher as compared with SAH grade I-II patients (grades I-II: 1.2 ± 0.3 n = 3; grade III: 5.5 ± 1.7, n = 5; grades IV/V: 7.4 ± 1.3, n = 5, P < 0.05 vs. grades I-II). There was significant positive correlation between Hunt/Hess score and 3-aminopropanal levels by the Spearman rank correlation coefficient (rs = 0.6). These observations indicate that elevated 3-aminopropanal-modified proteins accumulate in human CSF and that the highest 3-aminopropanal levels occur in association with the most severe ischemic brain disease.
Figure 1.

3-Aminopropanal (3-AP)-protein adducts are elevated in CSF of patients with severe acute neurological injury and poor grade SAH patients, as compared with good-grade patients. Levels of 3-aminopropanal-protein adducts were estimated immunochemically by using dot blots as described in Materials and Methods. P < 0.0001 good prognosis vs. poor prognosis; P < 0.05 Hunt/Hess grade I/II vs. Hunt/Hess grade IV/V.
Identification of Compounds with 3-Aminopropanal-Neutralizing Activity in Vitro.
In earlier work we found that pharmacological inhibition of PAO activity prevented the formation of 3-aminopropanal and significantly decreased infarct size in animal models of focal cerebral ischemia and head injury (refs. 1, 2, and 13 and unpublished observation). Based on the detection of 3-aminopropanal-modified proteins in human CSF, we considered that it might be possible to directly neutralize the cytotoxicity of 3-aminopropanal with clinically approved reactive thiol-containing drugs. To identify FDA-approved agents capable of neutralizing 3-aminopropanal, we screened a library of compounds for reaction with 3-aminopropanal that inhibits 3-aminopropanal-mediated cytotoxicity in glial and neuronal cell cultures (Table 1). Sulfhydryl compounds with -SH groups on primary carbon atoms significantly neutralized 3-aminopropanal cytotoxicity (N-2-MPG, N-acetyl-l-cysteine, cysteine, glutathione, 2-mercaptoethylamine hydrochloride), but thiol compounds with –SH groups on secondary, tertiary, or aromatic carbon atoms, as well as compounds with amino groups, showed weak or no protection (aminoguanidine hydrochloride, penicillamine, 2-mercapto-1-methyl-imidazole, thiamine hydrochloride 2-mercaptopyridine, 2-mercaptoimidazole).
Table 1.
Effect of test compounds on 3-aminopropanal-mediated glial cytotoxicity
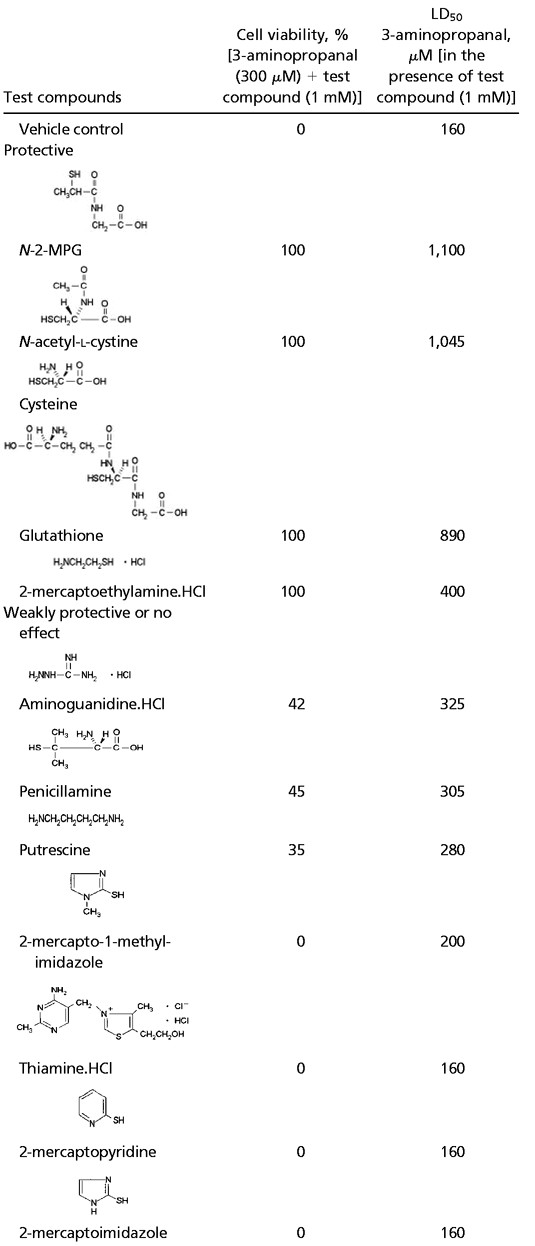 |
After an overnight incubation with 3-aminopropanal and the various compounds, cell viability was determined by using 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide reduction as described in Materials and Methods. Data are expressed as mean ± S.E.; n = 3–6 wells per condition.
Of the compounds tested, N-2-MPG, an agent used for the
treatment of cysteinuria (15), was found to be highly effective in
neutralizing 3-aminopropanal-induced glial and neuronal cytotoxicity
in vitro in a dose-dependent manner. N-2-MPG
protected neuronal cell cultures against an LD100
of 3-aminopropanal (90 μM) with an EC50 =
105 ± 10 μM (Fig. 2
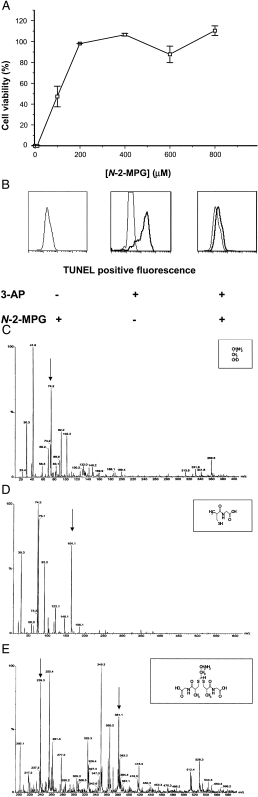 A).
N-2-MPG also protected glial cell cultures against an
LD100 of 3-aminopropanal (200 μM) with an
EC50 = 170 ± 12 μM) (not shown),
indicating that the protective effects could be extended to both glial
and neuronal cells. N-2-MPG significantly attenuated the
development of 3-aminopropanal-induced apoptotic cell death in
glial cell cultures (Fig. 2B). Coincubation of
3-aminopropanal with free radical scavengers, either ascorbic acid (16,
17) or butylated hydroxytoluene (18), failed to confer cytoprotection
(not shown), indicating that the cytoprotection of N-2-MPG
depended on direct 3-aminopropanal quenching, and not on scavenging of
free radicals. To confirm that N-2-MPG neutralized
3-aminonopropanal by direct reaction, we studied the reaction products
of 3-aminopropanal and N-2-MPG. Electron ionization mass
spectroscopy showed the expected mass ions for 3-aminopropanal and
N-2-MPG, respectively m/z 74 (hydrated form, as
3-aminopropanal in solution is found by NMR to be 90–95% hydrated;
Fig. 2C) and m/z 164 (Fig. 2D).
Reaction of 3-aminopropanal with N-2-MPG resulted in
thioacetal adduct formation between the carbonyl function of
3-aminopropanal and the thiol group of N-2-MPG (mass ion
peak m/z 381.1) (Fig. 2E), a stable reaction
product. Addition of this reaction product to cultured neurons was not
toxic, indicating that adduct formation between 3-aminopropanal and
N-2-MPG neutralized 3-aminopropanal-mediated
cytotoxicity.
A).
N-2-MPG also protected glial cell cultures against an
LD100 of 3-aminopropanal (200 μM) with an
EC50 = 170 ± 12 μM) (not shown),
indicating that the protective effects could be extended to both glial
and neuronal cells. N-2-MPG significantly attenuated the
development of 3-aminopropanal-induced apoptotic cell death in
glial cell cultures (Fig. 2B). Coincubation of
3-aminopropanal with free radical scavengers, either ascorbic acid (16,
17) or butylated hydroxytoluene (18), failed to confer cytoprotection
(not shown), indicating that the cytoprotection of N-2-MPG
depended on direct 3-aminopropanal quenching, and not on scavenging of
free radicals. To confirm that N-2-MPG neutralized
3-aminonopropanal by direct reaction, we studied the reaction products
of 3-aminopropanal and N-2-MPG. Electron ionization mass
spectroscopy showed the expected mass ions for 3-aminopropanal and
N-2-MPG, respectively m/z 74 (hydrated form, as
3-aminopropanal in solution is found by NMR to be 90–95% hydrated;
Fig. 2C) and m/z 164 (Fig. 2D).
Reaction of 3-aminopropanal with N-2-MPG resulted in
thioacetal adduct formation between the carbonyl function of
3-aminopropanal and the thiol group of N-2-MPG (mass ion
peak m/z 381.1) (Fig. 2E), a stable reaction
product. Addition of this reaction product to cultured neurons was not
toxic, indicating that adduct formation between 3-aminopropanal and
N-2-MPG neutralized 3-aminopropanal-mediated
cytotoxicity.
Figure 2.
N-2-MPG neutralizes 3-aminopropanal cytotoxicity by thioacetal formation. (A) Prevention of 3-aminopropanal-induced neuronal cytotoxicity by N-2-MPG. The glial cell line (HTB14) was exposed to 160 μM 3-aminopropanal and increasing concentrations of N-2-MPG as described in the legend to Table 1 and in Materials and Methods. Cell viability was determined after an overnight exposure by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (B) Prevention of 3-aminopropanal (3-AP)-induced glial apoptosis by N-2-MPG. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining of the glial cell line HTB14 5 h after treatment with 3-aminopropanal (160 μM) or vehicle, and N-2-MPG (1 mM) or medium alone, as described in Materials and Methods. A Becton Dickinson FACScan was used for all analyses; 5,000–10,000 events (ungated) were collected by using a single-color histogram for FITC. (C) Electrospray ionization mass spectrum of water solutions of 3-aminopropanal (300 μM), (D) N-2-MPG (1 mM), and (E) both compounds mixed together (resulting in a thioacetal adduct formation). The arrows indicate the expected molecular ions.
N-2-MPG Treatment Attenuates the Development of Cerebral Ischemic Cytotoxicity and Decreases Brain Levels of 3-Aminopropanal.
To determine whether N-2-MPG can neutralize 3-aminopropanal-mediated cytotoxicity in vivo, we measured infarct volume in rats subjected to MCAO. Administration of N-2-MPG significantly reduced infarct size as compared with vehicle-treated rats, even when the first dose of N-2-MPG was given 15 min after the onset of MCAO (Table 2). Physiological parameters including respiratory and heart rate, body temperature, and mean arterial blood pressure (recorded continuously every 30 min for 5 min during 5 h) were not influenced by N-2-MPG treatment (not shown). This finding indicated that the observed cerebroprotection by N-2-MPG was not attributable to effects on physiological variables known to influence severity of ischemic tissue damage. To obtain direct evidence that administration of N-2-MPG inhibited 3-aminopropanal-mediated cytotoxicity during cerebral ischemia in vivo, the brains of rats subjected to MCAO in the presence or absence of drug treatment were sectioned and stained by immunohistochemistry by using antibodies against 3-aminopropanal-modified proteins. Immunofluorescence for 3-aminopropanal-modified proteins was observed in the untreated animals in the anatomic region of cerebral ischemia, but was not observed in the opposite hemisphere (Fig. 3 A and B). The ischemic core revealed intense immunofluorescence, whereas 3-aminopropanal fluorescence in the adjacent ischemic penumbra showed a scattered pattern. Treatment with N-2-MPG after the onset of cerebral ischemia significantly reduced both the overall area and the intensity of 3-aminopronal-modified protein staining (Fig. 3 C and D). These results confirm that administration of N-2-MPG reduced 3-aminopropanal levels and prevented the spreading cytotoxicity in cerebral ischemia.
Table 2.
N-2-MPG attenuates cerebral ischemic damage in vivo
| Treatment | Mean infarct size, % ischemic hemisphere | SE | Mean infarct size, % vehicle treatment |
|---|---|---|---|
| Vehicle | 9.07 | 0.94 | 100 |
| N-2-MPG | 4.11* | 0.52 | 45* |
Experimental MCAOs were performed as described in Materials and Methods. For measurements of infarct volume, the animals were killed at 24 h, fresh coronal brain sections were prepared (1 mm), and infarct size was determined by using 2,3,5-triphenyltetrazolium chloride as described in Materials and Methods. Total cerebral infarct volume was measured by computerized quantitative planimetry as described (2, 13, 14). Data shown are mean ± S.E.
, P < 0.001 versus vehicle.
Figure 3.

N-2-MPG reduces 3-aminopropanal production during cerebral ischemia. Experimental MCAO, and N-2-MPG treatment where indicated, were performed as described in Materials and Methods. At 24 h, the brains were removed, and a section of the brain corresponding to the area of MCAO was sectioned and stained by immunofluorescence using antibodies against 3-aminopropanal-modified proteins as described in Materials and Methods. Pictures taken are representative of observations from 3–4 animals/group. All pictures were taken at ×10 magnification. (A) Detection of intense immunofluorescence for 3-aminopropanal-modified proteins in the anatomic region of cerebral ischemia in the ischemic hemisphere. The arrow shows intracellular 3-aminopropanal-modified protein fluorescence of scattered pattern in the ischemic penumbra. (B) 3-Aminopropanal-protein immunofluorescence was not detected in the opposite, nonischemic brain hemisphere. (C) N-2-MPG treatment reduces the overall area and intensity of 3-aminopropanal-modified protein immunofluorescence. (D) 3-Aminopropanal-protein immunofluorescence was not detected in the opposite, nonischemic brain hemisphere of N-2-MPG treated animals. Schematic diagrams designate in green the approximate area and location of the fluorescent pictures shown, and the hatched areas represent the approximate area of intense immunofluorescence for 3-aminopropanal-modified proteins.
Discussion
It now appears that 3-aminopropanal may participate as a cytotoxin in human cerebral ischemia, and that this pathway might be targeted therapeutically with a clinically approved agent (N-2-MPG). Putrescine, a terminal product of PAO, is produced contemporaneously with 3-aminopropanal, and brain putrescine levels increase within minutes after the onset of ischemia (19). Approximately 70% of putrescine formed in mammalian brain is derived from PAO-catalyzed degradation of spermine and spermidine (the remaining 30% is formed by de novo synthesis from ornithine). Stochastic analysis indicates that 2 mol of 3-aminopropanal are produced for every mol of putrescine generated by PAO. It is likely that the correlation between increased brain putrescine levels and cerebral damage is caused by the cytotoxicity of 3-aminopropanal. It has been observed that spermine and spermidine levels decrease during cerebral ischemia and putrescine levels increase in correlation with the volume of cerebral infarct (20, 21). Theoretically, putrescine can participate in the mediation of cerebral edema but, unlike 3-aminopropanal, putrescine is not cytotoxic (1, 22). Hydrogen peroxide produced by the oxidation of polyamines concomitantly with 3-aminopropanal does not participate significantly in PAO-mediated cytotoxicity (23, 24). Acetylation of the aminopropyl group of spermidine or both aminopropyl groups of spermine abrogates cytotoxicity, even though the acetylated polyamines yield H2O2 at a rate greater than the parent compounds (25). The relative contribution of 3-aminopropanal to the development of cytotoxicity together with other aldehydes formed during polyamine catabolism and with other known pathogenic mediators of cerebral ischemia (e.g., glutamate, tumor necrosis factor) can now be further addressed.
Here we show that the levels of 3-aminopropanal-modified proteins in patients with cerebral ischemia correlate to clinical disease severity. In animal models, 3-aminopropanal levels are significantly elevated within 2 h after ischemia or head trauma and increase during the subsequent period of progressive brain damage (ref. 2 and unpublished observations). To develop experimental strategies for neutralizing 3-aminopropanal-mediated cytotoxicity, we investigated an approach for direct pharmacological inhibition of 3-aminopropanal. As a result of screening Food and Drug Administration-approved drugs with structural features that can react with aldehydes, we found that N-2-MPG can effectively neutralize 3-aminopropanal in vitro. N-2-MPG is a nontoxic clinically approved drug that is used to prevent kidney stone formation in patients with cysteinuria (26). The drug has not been previously investigated in the treatment of cerebral ischemia. N-2-MPG reacts with 3-aminopropanal to form stable thioacetal derivatives, which prevents 3-aminopropanal mediation of glial and neuronal cell death. Administration of N-2-MPG markedly reduces 3-aminopropanal-modified protein levels and confers significant cerebroprotection in vivo, even when the drug is administered after the onset of brain ischemia in experimental animals subjected to MCAO. For instance, it is possible that N-2-MPG may neutralize the cytotoxicity of other aldehydes in addition to 3-aminopropanal (27–30). N-2-MPG may also confer some neuroprotective effects by scavenging free radicals and replenishing glutathione supplies (31, 32). A free radical cytoprotective mechanism is unlikely, however, because 3-aminopropanal cytotoxicity to glial cell cultures in vitro is not attenuated by other known free radical scavengers such as ascorbate and butylated hydroxytoluene (16, 33, 34). In addition to direct inactivation of 3-aminopropanal, replenishment of reduced glutathione levels by N-2-MPG remains a plausible potential mechanism for the cytoprotective effects of this drug (31). It is also plausible that our observations provide an additional mechanism for the neuroprotective effect of thioredoxin overexpression, which significantly protects against focal cerebral ischemia (35), because increased brain thiol content produced by thioredoxin can directly neutralize 3-aminopropanal-mediated cytotoxicity.
3-Aminopropanal is a normal product of cellular metabolism that is produced in discrete cellular compartments: this compound normally is detoxified in peroxisomes during the process of catabolic turnover of the polyamines spermine and spermidine by PAO. It is perhaps not surprising that loss of compartmentalization caused by cell death during cerebral ischemia or trauma leads to unrestrained production of 3-aminopropanal that initiates cytotoxic cascades. Together, our results now define a potential role for this cytotoxin as a therapeutic target in human cerebral ischemia and closed head injury. The availability of nontoxic clinically approved antagonists (N-2-MPG) of 3-aminopropanal indicates that it may be possible to develop a pharmacological strategy for neutralizing 3-aminopropanal as treatment of cerebral ischemia.
Acknowledgments
This work was supported in part by National Institutes of Health Grants GM57226 and GM62508.
Abbreviations
- PAO
polyamine oxidase
- SAH
subarachnoid hemorrhage
- N-2-MPG
N-2-mercaptopropionyl glycine
- MCAO
middle cerebral artery occlusion
- CSF
cerebrospinal fluid
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ivanova S, Botchkina G I, Al-Abed Y, Meistrell M, Batliwalla F, Dubinsky J M, Iadecola C, Wang H, Gregersen P K, Eaton J W, Tracey K J. J Exp Med. 1998;188:327–340. doi: 10.1084/jem.188.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimmerman G A, Meistrell M, Bloom O, Cockroft K M, Bianchi M, Risucci D, Broome J, Farmer P, Cerami A, Vlassara H. Proc Natl Acad Sci USA. 1995;92:3744–3748. doi: 10.1073/pnas.92.9.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao A M, Hatcher J F, Dogan A, Dempsey R J. J Neurochem. 2000;74:1106–1111. doi: 10.1046/j.1471-4159.2000.741106.x. [DOI] [PubMed] [Google Scholar]
- 4.Dogan A, Rao A M, Baskaya M K, Hatcher J, Temiz C, Rao V L, Dempsey R J. J Neurosurg. 1999;90:1078–1082. doi: 10.3171/jns.1999.90.6.1078. [DOI] [PubMed] [Google Scholar]
- 5.Dogan A, Rao A M, Hatcher J, Rao V L, Baskaya M K, Dempsey R J. J Neurochem. 1999;72:765–770. doi: 10.1046/j.1471-4159.1999.0720765.x. [DOI] [PubMed] [Google Scholar]
- 6.Lo T W, Westwood M E, McLellan A C, Selwood T, Thornalley P J. J Biol Chem. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 7.Ichihashi K, Osawa T, Toyokuni S, Uchida K. J Biol Chem. 2001;276:23903–23913. doi: 10.1074/jbc.M101947200. [DOI] [PubMed] [Google Scholar]
- 8.Parchment R E, Pierce G B. Cancer Res. 1989;49:6680–6686. [PubMed] [Google Scholar]
- 9.Beard M E, Baker R, Conomos P, Pugatch D, Holtzman E. J Histochem Cytochem. 1985;33:460–464. doi: 10.1177/33.5.3921604. [DOI] [PubMed] [Google Scholar]
- 10.Holtta E. Biochemistry. 1977;16:91–100. doi: 10.1021/bi00620a015. [DOI] [PubMed] [Google Scholar]
- 11.Averill-Bates D A, Agostinelli E, Przybytkowski E, Mondovi B. Biochem Cell Biol. 1994;72:36–42. doi: 10.1139/o94-006. [DOI] [PubMed] [Google Scholar]
- 12.Dewey D. Polyamines in Biomedical Research. Chichester, U.K.: Wiley; 1980. [Google Scholar]
- 13.Cockroft K M, Meistrell M, Zimmerman G A, Risucci D, Bloom O, Cerami A, Tracey K J. Stroke. 1996;27:1393–1398. doi: 10.1161/01.str.27.8.1393. [DOI] [PubMed] [Google Scholar]
- 14.Bederson J B, Pitts L H, Germano S M, Nishimura M C, Davis R L, Bartkowski H M. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 15.Eaton P, Li J M, Hearse D J, Shattock M J. Am J Physiol. 1999;276:H935–H943. doi: 10.1152/ajpheart.1999.276.3.H935. [DOI] [PubMed] [Google Scholar]
- 16.May J M, Qu Z, Morrow J D, Cobb C E. Biochem Pharmacol. 2000;60:47–53. doi: 10.1016/s0006-2952(00)00312-9. [DOI] [PubMed] [Google Scholar]
- 17.Riemersma R A, Carruthers K F, Elton R A, Fox K A. Am J Clin Nutr. 2000;71:1181–1186. doi: 10.1093/ajcn/71.5.1181. [DOI] [PubMed] [Google Scholar]
- 18.Yesilkaya A, Yegin A. Gen Pharmacol. 1998;30:495–498. doi: 10.1016/s0306-3623(97)00285-1. [DOI] [PubMed] [Google Scholar]
- 19.Rohn G, Kocher M, Oschlies U, Hossmann K A, Paschen W. Exp Neurol. 1990;107:249–255. doi: 10.1016/0014-4886(90)90142-f. [DOI] [PubMed] [Google Scholar]
- 20.Bolkenius F N, Seiler N. Int J Dev Neurosci. 1986;4:217–224. doi: 10.1016/0736-5748(86)90061-4. [DOI] [PubMed] [Google Scholar]
- 21.Seiler N, Bolkenius F N. Neurochem Res. 1985;10:529–544. doi: 10.1007/BF00964656. [DOI] [PubMed] [Google Scholar]
- 22.Shohami E, Nates J L, Glantz L, Trembovler V, Shapira Y, Bachrach U. Exp Neurol. 1992;117:189–195. doi: 10.1016/0014-4886(92)90126-b. [DOI] [PubMed] [Google Scholar]
- 23.Bishop C T, Mirza Z, Crapo J D, Freeman B A. In Vitro Cell Dev Biol. 1985;21:229–236. doi: 10.1007/BF02620934. [DOI] [PubMed] [Google Scholar]
- 24.Ferrante A, Maxwell G M, Rencis V O, Allison A C, Morgan D M. Int J Immunopharmacol. 1986;8:411–417. doi: 10.1016/0192-0561(86)90125-6. [DOI] [PubMed] [Google Scholar]
- 25.Morgan D M. Biochem J. 1987;242:347–352. doi: 10.1042/bj2420347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow G K, Streem S B. J Urol. 1996;156:1576–1578. doi: 10.1016/s0022-5347(01)65451-x. [DOI] [PubMed] [Google Scholar]
- 27.Quash G, Ripoll H, Gazzolo L, Doutheau A, Saba A, Gore J. Biochimie. 1987;69:101–108. doi: 10.1016/0300-9084(87)90241-0. [DOI] [PubMed] [Google Scholar]
- 28.Morgan D M, Bachrach U, Assaraf Y G, Harari E, Golenser J. Biochem J. 1986;236:97–101. doi: 10.1042/bj2360097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrante A, Rzepczyk C M, Allison A C. Trans R Soc Trop Med Hyg. 1983;77:789–791. doi: 10.1016/0035-9203(83)90290-0. [DOI] [PubMed] [Google Scholar]
- 30.Casero R A J, Pegg A E. FASEB J. 1993;7:653–661. [PubMed] [Google Scholar]
- 31.Coles B, Meyer D J, Ketterer B, Stanton C A, Garner R C. Carcinogenesis. 1985;6:693–697. doi: 10.1093/carcin/6.5.693. [DOI] [PubMed] [Google Scholar]
- 32.Lammel B, Pilz S, Freisleben H J, Schraven E, Zimmer G. Free Radical Res Commun. 1987;3:331–335. doi: 10.3109/10715768709088073. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Pinzon M A, Mumford P L, Rosenthal M, Sick T J. Brain Res. 1997;754:163–170. doi: 10.1016/s0006-8993(97)00066-8. [DOI] [PubMed] [Google Scholar]
- 34.Block F, Schwarz M. Pharmacol Biochem Behav. 1997;56:755–761. doi: 10.1016/s0091-3057(96)00484-4. [DOI] [PubMed] [Google Scholar]
- 35.Takagi Y, Mitsui A, Nishiyama A, Nozaki K, Sono H, Gon Y, Hashimoto N, Yodoi J. Pros Natl Acad Sci USA. 1999;96:4131–4136. doi: 10.1073/pnas.96.7.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]