

Abstract
Rod photoreceptors are highly compartmentalized sensory neurons that maintain strict ultrastructural and molecular polarity. Structural subdivisions include the outer segment, inner segment, cell body, and synaptic terminal. The visual pigment rhodopsin is found predominantly in membranes of the rod cell outer segment but becomes mislocalized, appearing throughout the plasma membrane of the cell in many retinal diseases and injuries. Currently, there is no known link between rhodopsin redistribution and rod cell death. We propose that activation of mislocalized rhodopsin kills rod cells by stimulating normally inaccessible signaling pathways. This hypothesis was tested in primary retinal cell cultures, which contain photoreceptors. In rod photoreceptors, opsin immunofluorescence occurred throughout the rod cell plasma membrane. Activation of this mislocalized opsin by photostimulation after formation of isorhodopsin or by incubation with β-ionone (opsin agonist) killed 19–30% of rod cells. Rod cell death was apoptotic, as indicated by marked chromatin condensation and the requirement for caspase-3 activation. Rod cell death could be induced by forskolin (adenylate cyclase agonist), and conversely, β-ionone-induced cell death could be blocked by cotreatment with SQ22536 (an adenylate cyclase inhibitor). Pertussis toxin (a G protein inhibitor) also blocked β-ionone-induced cell death. The data support a mechanism by which activation of mislocalized opsin initiates apoptotic rod cell death through G protein stimulation of adenylate cyclase.
The first step in vision is the absorption of light by rod and cone photoreceptors of the retina. Many degenerative and age-related retinal diseases that lead to blindness or visual disability show an early loss of rod cells followed by a loss of cone cells (1). Efforts to understand the process of rod cell death in hereditary retinal diseases and in mechanical or photic injuries indicate that rod cells die through an apoptotic mechanism (2–5). In addition, several lines of evidence suggest that the visual pigment rhodopsin plays a major role in apoptotic rod cell death (3, 6). However, the connections between rhodopsin and apoptotic cell death are completely unknown.
Rhodopsin is a light sensitive, G protein (transducin)-coupled receptor localized predominantly in the membranes of rod cell outer segments. In transgenic mice lacking the last five amino acids of opsin's C terminus (Q344ter), a mutation also found in human autosomal-dominant retinitis pigmentosa (RP), defective delivery of opsin to the outer segment leads to rod cell apoptosis and retinal degenerative disease (3, 7). Concomitant with this defect in trafficking, opsin becomes mislocalized to the plasma membrane of the inner segment, cell body, and synaptic terminal. Similar changes in opsin localization have been reported for many other diseases and injuries where rod cell death occurs, including human RP, light-induced retinal degeneration, retinal detachment, rd mice, rds mice, RCS rats, and prcd dogs (8–15). More recently, studies using Rpe65 knockout mice, a deletion that results in complete loss of rhodopsin activity, showed protection against light-induced rod cell death (6). Conversely, in arrestin knockout mice, the inability to terminate light-stimulated rhodopsin signaling killed rod cells (16). Thus, the evidence linking rhodopsin to retinal disease suggests that its localization and activity are significant components of the mechanisms that cause rod cell death.
Based on rhodopsin's ability to transduce light into biochemical signals, we propose that activation of mislocalized rhodopsin generates signals through normally unavailable targets that initiate rod cell apoptosis in retinal disease. In the present study, we tested whether activation of mislocalized opsin can cause apoptotic rod cell death and whether cell death occurs through stimulation of specific intracellular signaling pathways.
Methods
Animals.
Data were obtained from 38 adult, aquatic-phase tiger salamanders (Ambystoma tigrinum, 17–28 cm in length). Animals were maintained at 5°C on a 12 h light/12 h dark cycle.
Materials.
β-Ionone, 9-cis-retinal, all trans-retinal, phosphatidylcholine type-V, forskolin, SQ22536, and pertussis toxin were purchased from Sigma. DEVD-fluoromethylketone was purchased from CLONTECH. Before use, 9-cis-, all trans-retinal, and β-ionone stock solutions were scanned for the presence of impurities or breakdown products with a DU-64 spectrophotometer (Beckman Coulter). None were detected. Calculation of retinoid concentration was based on the following λ max and extinction coefficients in ethanol: β-ionone (295 nm, ɛ = 8,700 M−1 cm−1), 9-cis-retinal (373 nm, ɛ = 36,200 M−1 cm−1) and all trans-retinal (383 nm, ɛ = 43,143 M−1 cm−1).
Immunolabeling for Rod Opsin.
For frozen sections, eyes were fixed in 4% (wt/vol) paraformaldehyde/0.1 M phosphate buffer (PB), pH 7.4 for 30 min. After removal of the anterior segment, the eyecup was placed in fixative overnight at 4°C. Eyecups were rinsed in 0.1 M PB with 5% (wt/vol) sucrose, equilibrated in 30% (wt/vol) sucrose/PB and frozen in Tissue-Tek OCT compound. Tissue sections 10-μm thick were placed on gelatin-subbed slides and rinsed with wash buffer (450 mM NaCl/20 mM sodium phosphate buffer, pH 7.4). For cultured cells, cells were fixed with 4% (wt/vol) paraformaldehyde in 0.1 M PB (pH 7.4) for 1 h at room temperature and then rinsed with wash buffer. Immunofluorescent labeling was performed as described (17). Antirod opsin 4D2 was kindly provided by Robert Molday (University of British Columbia, Vancouver).
Cell Culture.
Dissociation of adult salamander retina was performed as described (17). Briefly, light-adapted animals were decapitated, pithed, and enucleated. The anterior segment of the eye was removed, and the neural retina was scooped from the eyecup and placed in Ringer's solution (17). Isolated retinas were transferred to a solution containing 14 units/ml of papain (Worthington) and digested for 45 min. After trituration with a 3-mm wide-bore glass pipette, cell suspensions were plated onto glass coverslips coated with Sal-1 antibody substrate (18) (generously provided by P. R. MacLeish, Morehouse School of Medicine, Atlanta) and grown in serum-free medium containing 90% Ringer's solution, 7% medium 199, 1× MEM vitamin mix, 0.1× MEM essential amino acids, 0.1× MEM nonessential amino acids, 2 mM glutamine, 2 μg/ml bovine insulin, 1 μg/ml transferrin, 5 mM taurine, 0.8 μg/ml thyroxine, 10 μg/ml gentamicin, and 1 mg/ml BSA. Cells were maintained in a humidified chamber at 10°C in air. Because pertussis toxin activity is reduced at low temperatures, experiments testing G protein signaling were carried out at room temperature.
Photoactivation of Isorhodopsin.
Cultured rod cells were made photosensitive by forming isorhodopsin with 10 μM 9-cis-retinal in phosphatidylcholine vesicles (19). White light from a Nikon MKII fiber optic light was filtered through a 488 nm ± 2 nm (isorhodopsin λ max = 487 nm) narrow bandpass filter (Edmund Industrial Optics, Barrington, NJ) held within a Uniblitz Electronic Shutter (Vincent Associates, Rochester, NY). Exposure times and intervals were set by using an electronic shutter driver with timer (JML Optical Industries, Rochester, NY). Cultures were maintained at 10°C during the light treatment with a Peltier plate (Acopian, Easton, PA). Cultures were exposed to 5 × 103 photons per μm2 of 488 nm light for 200 ms every 30 s for 3 h. After light treatment, the retinal solution was replaced with retinal-free medium, and the cells were grown in the dark for 3 additional days. In some experiments, cell death was analyzed immediately after the 3-h light treatment.
Survival Assay.
We had previously determined that salamander photoreceptors are stable in culture; cell death is not prominent until at least 3 weeks after plating the cells (E.T.-A., J. W. Mandell, and P. R. MacLeish, unpublished results). To assess viability after various treatments, cultured cells were immunofluorescently labeled with antirod opsin 4D2, and the total number of rod cells was counted blind in 20–25 fields at 20× or 40× by using conventional fluorescence microscopy. Cone cells were identified through phase-contrast microscopy by their pear-shaped morphology, the presence of an ellipsoid, and the absence of fluorescent staining. All other cell types were classified as nonphotoreceptors.
To determine relative changes in cell number, one control (ctrl) and one experimental (exp) culture dish from the same animal composed an experiment. For each experiment, the percent change in cell number was calculated as follows: number of rod cells (exp) − number of rod cells (ctrl)/number of rod cells (ctrl) × 100. The average percent change of 5–11 experiments from at least two animals was reported. Statistical significance was determined by using a two-tailed t test for a single population, where t = average % change/SEM. To determine if any systematic error occurred during cell plating, counts of 2 h-old untreated cultures were performed. In dishes prepared from the same animal, no statistically significant change in rod, cone, or nonphotoreceptor cell numbers between dishes was detected [In n = 6 experiments from two animals, control and experimental dishes were assigned arbitrarily. Percent differences in plating (means ± SEM): rods 6 ± 8%, cones .3 ± 4%, and nonphotoreceptors 2 ± 4%]. To study the time course of β-ionone-induced cell death, cell counts were performed after 30 min, 3 h, 6 h, 12 h, or 24 h of treatment.
Time-Lapse Photography.
To induce cell death by calcium overload, cultures were treated with high K+ Ringer's solution (54 mM NaCl/50 mM KCl/1.0 mM NaHCO3/0.5 mM NaH2PO4/1 mM sodium pyruvate/17 mM glucose/0.5 mM MgSO4/2 mM Hepes/1.8 mM CaCl2, pH 7.7). Rod cells viewed with Nomarski optics were photographed periodically with a charge-coupled device camera or recorded on videotape. Video images were recorded at 4-s intervals after averaging 16 frames by using an Argus-10 image processor (Hamamatsu Photonics, Hamamatsu City, Japan).
Cell Death Assay.
Rod cell death was characterized by using the fluorescent DNA-binding dyes acridine orange (AO, 1 μg/ml) and ethidium bromide (EtdBr, 10 μg/ml) as described (20) in conjunction with morphological identification of cell-type by using phase-contrast optics. To quantitate cell death, the total number of early and late apoptotic rod cells was counted in 40 fields at 40×. The data were analyzed and reported as described above.
Results
Opsin Protein Becomes Mislocalized in Cultured Rod Cells.
Rod photoreceptors are structurally subdivided into an outer segment, inner segment, cell body with nucleus, and synaptic terminal (Fig. 1A). Opsin, the protein component of the light-receptive molecule rhodopsin, is synthesized in the inner segment and then incorporated and maintained with high fidelity in the outer segment. Immunofluorescent labeling of the intact tiger salamander retina showed opsin labeling predominantly in rod cell outer segments (Fig. 1B). Immunofluorescent labeling of cultured rod cells showed opsin labeling throughout the entire plasma membrane of the inner segment, cell body, and synaptic terminal, when present (Fig. 1C). Mislocalized opsin was seen as early as 2 h after plating the cells and could be detected throughout the culture period. The pattern of opsin labeling in cultured rods, therefore, mimicked the mislocalization of opsin seen in retinal disease. The rapid appearance of mislocalized opsin most likely represented both newly synthesized and preexisting opsin. Existing opsin can diffuse from outer segment membranes to the plasma membrane of the inner segment, cell body, and synaptic terminal after retinal isolation because, under these circumstances, there is rapid fusion of outer segment to inner segment plasma membranes (21). In the isolated retina, fusion occurs in ≈28% of rod cells; in culture, all rod cells with an outer segment show fusion after plating.
Figure 1.

Activation of mislocalized opsin kills rod cells. (A) Structural subdivisions of rod cells viewed with Nomarski optics. Rod cells in culture exist with or without their outer segment. The ellipsoid (*), an accumulation of mitochondria, lies within the inner segment. OS, outer segment; IS, inner segment; N, nucleus; T, synaptic terminal. (Bar = 10 μm.) [Reproduced from ref. 47, p. 111 by courtesy of Marcel Dekker, Inc.)]. (B) Conventional fluorescence image of a 10-μm frozen section of the salamander retina labeled with a monoclonal antibody to rod opsin (4D2) and a rhodamine-conjugated secondary antibody. Intense opsin staining was present in rod cell outer segments (arrows) but not in inner segments indicated by (*) or cell bodies, located in the ONL. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. (Bar = 10 μm.) (C) One-μm optical sections of isolated rod cells obtained by confocal microscopy showed opsin labeling throughout the plasma membrane of the inner segment and cell body. Opsin labeling also was seen within the Golgi apparatus. OS, outer segment; IS, inner segment; N, nucleus; G, Golgi apparatus; *, ellipsoid. (Bar = 10 μm.) (D) Percent change in rod cell number after photoactivation of reconstituted isorhodopsin. Light plus 10 μM 9-cis-retinal killed 19% of rod cells in culture compared with light-treated phospholipid controls. When maintained in the dark, rod cell number was unchanged by 9-cis-retinal treatment. In n = 9 experiments from three animals, a total of 1,874 rod cells was counted; *, P < 0.01, means ± SEM. (E) Percent change in cell number after treatment with 2.5 μM β-ionone. Rod cell number decreased 20% in β-ionone-treated cultures compared with ethanol controls. Cone and nonphotoreceptor cell numbers did not change. In n = 6 experiments from two animals, a total of 1,425 rod, 1,335 cone, and 3,735 nonphotoreceptor cells was counted; *, P < 0.01, means ± SEM. (F) The effect of β-ionone on rod cell death was blocked by cotreatment with 10 μM 9-cis-retinal in the dark. In n = 5 experiments from two animals, a total of 671 rod cells was counted; *, P < 0.02 compared with control; **, P < 0.05 compared with β-ionone treated; unpaired t test, means ± SEM.
Activation of Mislocalized Opsin Kills Rod Cells.
Photosensitive rhodopsin-like pigments can be generated in vitro by supplying exogenous chromophores (22). Three days after plating the cells, isorhodopsin was formed, and the cells were exposed to 488-nm light. The cells were grown in the dark for 3 additional days, immunofluorescently labeled for rod opsin, and counted for viability in 25 fields at 20×. Treatment with light and 9-cis-retinal killed 19% ± 5% of rod cells, compared with light-treated phospholipid controls (Fig. 1D). Cultures supplied with 10 μM 9-cis-retinal but maintained in the dark showed no rod cell loss. Up to 100 μg/ml of phosphatidylcholine vesicles alone had no effect on rod cell viability (data not shown).
Absorption of light by 9-cis-retinal initiates its isomerization to all trans-retinal and its subsequent release from the opsin protein. Although all trans-retinal does not activate opsin in cultured salamander rod cells (23), it could affect rod cell survival through another mechanism. To determine the effect of all trans-retinal on rod cell death, 3 day-old cultures were treated with 10 μM all trans-retinal for 3 additional days. All trans-retinal did not kill rod cells (data not shown).
To develop a second and more efficient approach for activating mislocalized opsin in culture, a retinal analogue, β-ionone, was used. Exogenous application of β-ionone has been shown to activate opsin and its associated G protein, transducin (Gt), in isolated salamander rod cells (24). In our paradigm, cells were cultured for 2 h before receiving 2.5 μM β-ionone in 0.08% ethanol for an additional 24 h. Viability of rod, cone, and nonphotoreceptor cells was assessed in 20 fields at 40×. β-ionone treatment killed 20 ± 4% of rod cells compared with ethanol controls but had no effect on either cone or nonphotoreceptor cells (Fig. 1E). Because β-ionone has been shown to down-regulate phototransduction in cone cells (25), induction of cell death was not expected. Ethanol alone at 0.08% had no effect on any retinal cell-type (data not shown). The requirement for opsin was indicated by the fact that β-ionone-induced cell death could be blocked by cotreatment with 10 μM 9-cis-retinal in the dark (Fig. 1F). Ten micromolars 9-cis-retinal alone had no effect on rod cell number (data not shown). Finally, to determine the kinetics of β-ionone-induced cell death, a time-course study was performed. It was found that rod cell death occurred between 30 min and 3 h after treating with β-ionone (In n = 6 experiments from two animals per time point).
Because some rod cells retained their outer segment in culture, it was possible that β-ionone or 9-cis-retinal exerted their effect through activation of opsin in the outer segment. To ensure that rod cells died through activation of mislocalized opsin, we analyzed separately the death of cells, cultured in 2.5 μM β-ionone, with and without their outer segments. Rod cells without an outer segment comprised 86% of the total number of rod cells and accounted for 84% of the total number of dead cells (data from 17 experiments). Rod cells with an outer segment comprised 14% of the total number of rod cells and accounted for 16% of the total number of dead cells (data from 17 experiments). Because the majority of rod cells did not have an outer segment, and because death of each cell-type mirrored its percentage in the population, the conclusion that activation of mislocalized opsin kills rod cells was confirmed.
Rod Cell Death Occurs Through an Apoptotic Mechanism.
To determine if the mechanism of rod cell death after activation of mislocalized opsin was consistent with that seen in retinal disease, we tested the nature of light- and β-ionone-induced cell death. For both treatments, cultures were first grown for 2 h in medium only. Based on the time course of β-ionone-induced death, cell death was analyzed after 3 h of treatment. By using AO/EtdBr staining and phase-contrast optics, live rod cells were identified by a teardrop shape, the presence of an ellipsoid, and bright green (from AO) homogeneously stained chromatin (Fig. 2A). Early apoptotic rod cells showed the same morphology as live cells but contained bright green, highly condensed chromatin (Fig. 2B). Late apoptotic and necrotic rod cells had bright red (from EtdBr) chromatin because of the presence of permeable membranes. However, these rod cells were difficult to identify by using the morphological criteria above because of the loss of membrane integrity. To learn to identify these cell types, cultures were treated with high K+ Ringer's solution to induce cell death by calcium overload; individual cells were followed by using time-lapse photography. Rod cells remained distinguishable from other cell types by the presence of a rectangular, elongated ellipsoid (Fig. 2C); cone cells retained a round- or oval-shaped ellipsoid (not shown). Thus, late apoptotic and necrotic rod cells were identified by using these morphological criteria in conjunction with bright red (EtBr), highly condensed chromatin (Fig. 2D) or bright red, homogeneously stained chromatin, respectively.
Figure 2.
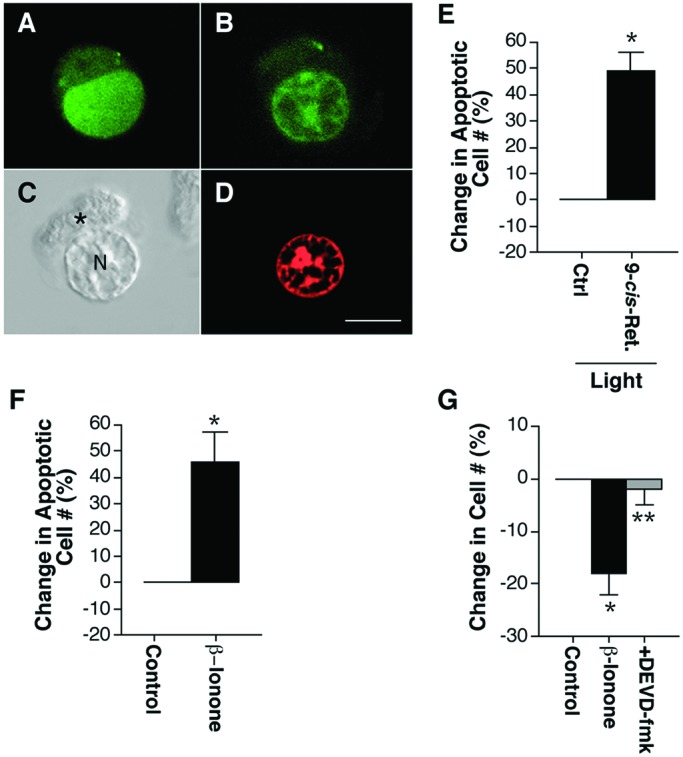
Characterization of rod cell death. (A, B, D) One-μm optical sections of isolated rod cells fluorescently labeled with AO and EtdBr obtained by confocal microscopy. (C) Nomarski optics image of an isolated rod cell treated with high K+ Ringer's solution demonstrating rod cell morphology after loss of membrane integrity. (A) Live rod cells had green (AO), homogeneously stained chromatin. (B) Early apoptotic rod cells had green (AO), condensed chromatin. (C) Rod cells that had lost membrane integrity could be identified by their rectangular, elongated ellipsoid. *, ellipsoid; N, nucleus. (D) Late apoptotic cells with permeant membranes had red EtdBr, condensed chromatin. (Bar = 10 μm.) (E) Percent change in the number of apoptotic rod cells after treatment with light and 10 μM 9-cis-retinal. Photoactivation of reconstituted isorhodopsin increased apoptotic rod cell death by 49%. In n = 5 experiments from two animals, a total of 83 apoptotic rod cells was counted; *, P < 0.01, means ± SEM. (F) Percent change in the number of apoptotic rod cells after treatment with 2.5 μM β-ionone. β-ionone treatment increased apoptotic rod cell death by 46%. In n = 9 experiments from three animals, a total of 120 apoptotic rod cells was counted; *, P < 0.01, means ± SEM. (G) The effect of β-ionone on rod cell death was blocked by preincubation and cotreatment with 1 μM DEVD-fmk. In n = 11 experiments from four animals, a total of 4,417 rod cells was counted; *, P < 0.01 compared with control; **, P < 0.01 compared with β-ionone treated; unpaired t test, means ± SEM.
Light plus 9-cis-retinal increased the number of apoptotic rod cells by 49 ± 7% compared with light-treated phospholipid controls (Fig. 2E). Treatment with β-ionone increased apoptotic rod cell death 46 ± 11% compared with ethanol-treated controls (Fig. 2F). Necrotic rod cells were not detected in either light- or β-ionone treated cultures.
Inhibition of the proteolytic enzyme caspase-3 has been shown to reduce apoptotic rod cell death in N-methyl-N-nitrosourea-, lead-, calcium-, and ischemia-induced retinal degenerations, as well as in a transgenic rat model of RP (26–29). To confirm that rod cells died through an apoptotic mechanism, cells were preincubated with an irreversible caspase-3 inhibitor, DEVD-fmk, for 2 h and then cotreated with 2.5 μM β-ionone for 3 h. Cells were immunofluorescently labeled with antirod opsin, and the total number of rod cells was counted in 20 fields at 20×. β-ionone-induced rod cell death was not blocked by 1 nM DEVD-fmk (data not shown) but was completely inhibited by 1 μM DEVD-fmk (Fig. 2G). One micromolar DEVD-fmk alone had no effect on rod cell number (data not shown).
Activation of Mislocalized Opsin Kills Rod Cells by Stimulating G Protein and Adenylate Cyclase Activity.
In rod cell outer segments, absorption of light by rhodopsin isomerizes 11-cis-retinal and activates its associated G protein, transducin (Gt). Activated Gt then binds to and activates a cGMP-dependent phosphodiesterase (PDE) that hydrolyzes cGMP to 5′-GMP and closes a cGMP-gated ion channel in the outer segment plasma membrane. When rhodopsin becomes mislocalized to the membranes of the inner segment, cell body, and synaptic terminal, stimulation of normally unavailable targets may generate signals that ultimately kill the cell. In support of this possibility, light-stimulation of rhodopsin in cell-free systems can enhance adenylate cyclase activity in a transducin-dependent manner (30, 31). In addition, baseline cAMP levels are higher in photoreceptors from animal models of RP in which opsin has become mislocalized (32), and a cAMP analogue, 8-Cl-cAMP, has been shown to cause apoptosis in Y79 retinoblastoma cells (33). Therefore, we examined whether activation of mislocalized opsin can kill rod cells through G protein activation and stimulation of adenylate cyclase.
To test the role of adenylate cyclase in rod cell death, cells were grown for 2 h in medium and then treated for 3 h with 10 μM forskolin (adenylate cyclase agonist) in 0.1% DMSO. Cells were immunofluorescently labeled with antirod opsin, and the total number of rod cells was counted in 20 fields at 20×. Forskolin treatment killed 22 ± 4% of rod cells as compared with 0.1% DMSO-treated controls (In n = 6 experiments from two animals, a total of 1,523 rod cells was counted; P < 0.01, means ± SEM). DMSO alone had no effect on rod cell survival (data not shown). To test the role of adenylate cyclase in β-ionone-induced rod cell death, cells were preincubated for 2 h in 100 μM SQ22536 (an adenylate cyclase inhibitor) then cotreated with 2.5 μM β-ionone for an additional 3 h. SQ22536 completely blocked β-ionone-induced rod cell death (Fig. 3A); 100 μM SQ22536 alone had no effect (data not shown). To test for the possible involvement of Gt, cells were preincubated with 100 ng/ml of pertussis toxin (inhibits Gi, Go, and Gt activation through ADP-ribosylation) for 24 h and then cotreated with 2.5 μM β-ionone for an additional 3 h. Treatment with pertussis toxin blocked a significant amount of β-ionone-induced cell death, but the effect was not complete (Fig. 3B). Pertussis toxin alone at 100 ng/ml had no effect on rod cell number. The data supports a mechanism for rod cell death in which activation of mislocalized opsin kills rod cells through G protein, possibly transducin stimulation of adenylate cyclase activity (Fig. 4). The fact that pertussis toxin did not completely block β-ionone-induced rod cell death suggests that other G proteins are also involved, a suggestion forwarded (32) and supported by the known promiscuity between receptors and G proteins.
Figure 3.
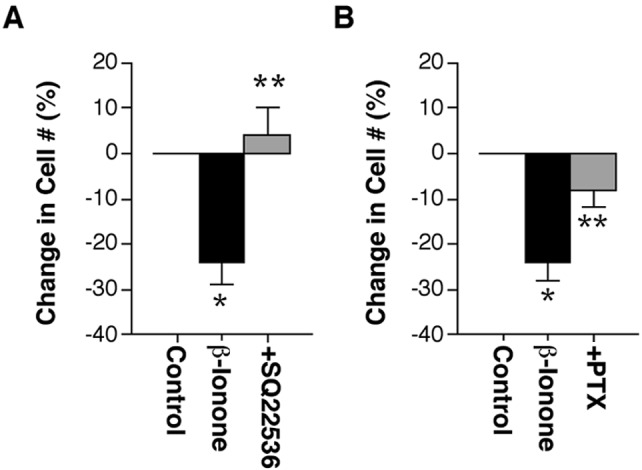
Mislocalized opsin activity stimulates normally inaccessible signaling pathways. (A) Inhibition of adenylate cyclase with 100 μM SQ22536 blocked β-ionone-induced cell death. In n = 6 experiments from two animals, a total of 855 rod cells was counted in 20 fields at 20×; *, P < 0.01 compared to control; **, P < 0.005 compared with β-ionone treated; unpaired t test, means ± SEM. (B) Preincubation and cotreatment with 100 ng/ml of pertussis toxin blocked β-ionone-induced rod cell death. In n = 6 experiments from two animals, a total of 1,226 rod cells was counted in 40 fields at 40×; *, P < 0.01 compared with control; **, P < 0.03 compared with β-ionone treated; unpaired t test, means ± SEM.
Figure 4.
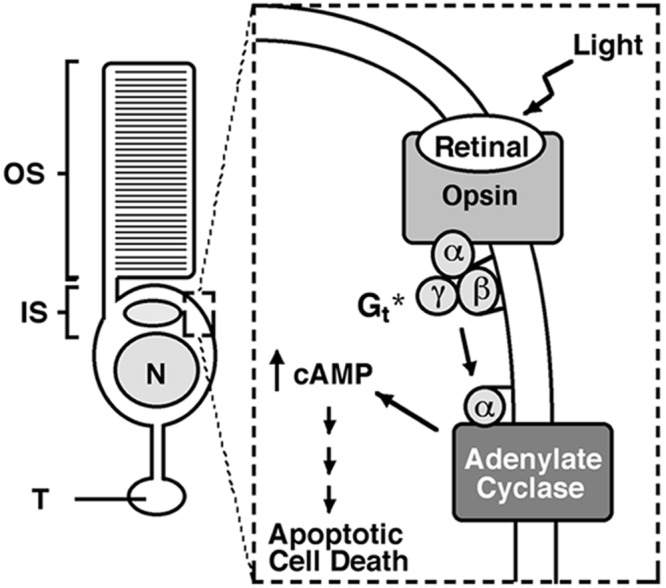
Proposed mechanism of rod cell death. During retinal degenerative disease, opsin becomes mislocalized to the plasma membrane of the inner segment, cell body, and synaptic terminal. Photostimulation of mislocalized rhodopsin activates G proteins such as transducin. In the absence of its normal target, PDE, transducin binds to and activates adenylate cyclase. The subsequent increase in cAMP levels initiates signals within the rod cell that eventually lead to caspase-3 activation and apoptotic cell death. *, photoactivation of mislocalized opsin may kill rod cells through the stimulation of other G proteins. OS, outer segment; IS, inner segment; N, nucleus; T, synaptic terminal.
Discussion
In the present study, we have demonstrated that opsin activity, when mislocalized, can kill rod cells by stimulating G protein signaling and adenylate cyclase activity. In addition, we have shown that rod cell death occurs through an apoptotic mechanism involving the proteolytic enzyme caspase-3. These data are consistent with previous work identifying apoptosis as the mode (2–5) and rhodopsin activity (6, 16) and localization (3, 7) as key components of rod cell death in disease states. Because opsin redistribution is observed in so many retinal diseases and injuries, we propose that activation of mislocalized rhodopsin may be a mechanism of rod cell death with broad applicability to retinal disorders.
Light exposure has been shown to both cause degeneration and to accelerate some forms of genetic retinal degenerative disease (34, 35). To suggest that rod cells in the intact retina die after photoactivation of mislocalized rhodopsin requires that the chromophore, 11-cis-retinal, and the signaling components, such as transducin and adenylate cyclase, be available to mislocalized opsin. Current evidence suggests that rhodopsin can form in locations other than the outer segment. Addition of 11-cis-retinal during biosynthesis of rhodopsin has been reported at the level of the endoplasmic reticulum (36), located within the inner segment. In addition, interstitial-retinoid binding protein, the protein that transports 11-cis-retinal from retinal pigmented epithelial cells to photoreceptors, is found throughout the interphotoreceptor matrix (37). Because this region extends from the tip of the outer segment to the external limiting membrane (located between the juncture of the inner segment and cell body), it indicates that rhodopsin can form in the plasma membrane of the inner segment when opsin becomes mislocalized. Finally, the enzyme adenylate cyclase and the G protein transducin have been detected in rod cell inner segments (38, 39). Collectively, this work supports the idea that activation of mislocalized opsin can kill rod cells in the intact retina.
In seeming contradiction, dark-rearing seems to have little protective effect on the time course of rod cell death in certain retinal degenerations, including rds mice and RCS and Ser334ter rats (40–42). However, light is not the only way to activate opsin signaling. Opsin without bound 11-cis-retinal, for example, can stimulate the phototransduction cascade (43). Because a measurable amount of chromophore-free opsin has been found during postnatal development in the rat (44), it is possible that spontaneous activation of mislocalized opsin may lead to rod cell death in diseases where opsin mislocalization occurs during development. This possibility may explain in part the lack of protection against cell death in dark-reared rds, RCS, or Ser334ter retinas. In addition, excessive stimulation of rod cell adenylate cyclase through other mechanisms such as decreased dopamine signaling, increased adenosine signaling, or elevation of intracellular calcium levels (all of which occur in the dark) also would be expected to contribute to rod cell death in dark-reared animals.
In summary, we have identified a previously uncharacterized mechanism of rod cell death that involves activation of mislocalized opsin and stimulation of normally inaccessible signaling pathways. Increasing evidence supports the idea that fidelity of protein trafficking in highly polarized cells such as neurons and epithelia is critical for their survival. For example, mislocalized Cdk5 activation has been implicated in the pathogenesis of Alzheimer's disease (45), whereas aberrant, apical Na+/K+ ATPase activity may lead to polycystic kidney disease (46). Our results suggest that survival of rod photoreceptors depends on the proper maintenance of rhodopsin polarity. Future experiments with intact retina and animal models of retinal degeneration should help clarify the role of rhodopsin mislocalization in retinal disease.
Acknowledgments
We gratefully acknowledge support from National Institutes of Health Grant EY12031.
Abbreviations
- RP
retinitis pigmentosa
- AO
acridine orange
- EtdBr
ethidium bromide
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Schmidt S Y. In: Handbook of Neurochemistry, Volume 10. Lajthe A, editor. New York: Plenum; 1985. pp. 461–507. [Google Scholar]
- 2.Li Z-Y, Milam A H. In: Degenerative Diseases of the Retina. Anderson R E, editor. New York: Plenum; 1995. pp. 1–8. [Google Scholar]
- 3.Portera-Cailliau C, Sung C-H, Nathans J, Adler R. Proc Natl Acad Sci USA. 1994;91:974–978. doi: 10.1073/pnas.91.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cook B, Lewis G P, Fisher S K, Adler R. Invest Ophthalmol Visual Sci. 1995;36:990–996. [PubMed] [Google Scholar]
- 5.Abler A S, Chang C J, Ful J, Tso M O M, Lam T T. Res Commun Mol Pathol Pharmacol. 1996;92:177–189. [PubMed] [Google Scholar]
- 6.Grimm C, Wenzel A, Hafezi F, Yu S, Redmond T M, Remé C E. Nat Genet. 2000;25:63–66. doi: 10.1038/75614. [DOI] [PubMed] [Google Scholar]
- 7.Sung C-H, Makino C, Baylor D, Nathans J. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z-Y, Kljavin I J, Milam A H. J Neurosci. 1995;15:5429–5438. doi: 10.1523/JNEUROSCI.15-08-05429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edward D P, Lim K, Sawaguchi S, Tso M O M. Graefe's Arch Clin Exp Ophthalmol. 1993;231:289–294. doi: 10.1007/BF00919107. [DOI] [PubMed] [Google Scholar]
- 10.Lewis G P, Erickson P A, Anderson D H, Fisher S K. Exp Eye Res. 1991;53:629–640. doi: 10.1016/0014-4835(91)90223-2. [DOI] [PubMed] [Google Scholar]
- 11.Bowes C, van Veen T, Farber D B. Exp Eye Res. 1988;47:369–390. doi: 10.1016/0014-4835(88)90049-8. [DOI] [PubMed] [Google Scholar]
- 12.Nir I, Agarwal N, Sagie G, Papermaster D S. Exp Eye Res. 1989;49:403–421. doi: 10.1016/0014-4835(89)90050-x. [DOI] [PubMed] [Google Scholar]
- 13.Nir I, Papermaster D S. Invest Ophthalmol Vis Sci. 1986;27:836–840. [PubMed] [Google Scholar]
- 14.Nir I, Sagie G, Papermaster D S. Invest Ophthalmol Vis Sci. 1987;28:62–69. [PubMed] [Google Scholar]
- 15.Huang J C, Chesselet M-F, Aguirre G D. Exp Eye Res. 1994;58:17–30. doi: 10.1006/exer.1994.1191. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Simon M I, Matthes M T, Yasumura D, LaVail M M. Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- 17.Nachman-Clewner M, Townes-Anderson E. J Neurocytol. 1996;25:597–613. doi: 10.1007/BF02284827. [DOI] [PubMed] [Google Scholar]
- 18.MacLeish P R, Barnstable C J, Townes-Anderson E. Proc Natl Acad Sci USA. 1983;80:7014–7018. doi: 10.1073/pnas.80.22.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones G J, Crouch R K, Wiggert B, Cornwall M C, Chader G J. Proc Natl Acad Sci USA. 1989;86:9606–9610. doi: 10.1073/pnas.86.23.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGahon A J, Martin S J, Bissonnette R P, Mahboubi A, Shi Y, Mogil R J, Nishioka W K, Green D R. Methods Cell Biol. 1995;46:153–185. doi: 10.1016/s0091-679x(08)61929-9. [DOI] [PubMed] [Google Scholar]
- 21.Townes-Anderson E. Invest Ophthalmol Vis Sci. 1995;36:1918–1933. [PubMed] [Google Scholar]
- 22.Pepperberg D R. Methods Enzymol. 1982;81:452–459. doi: 10.1016/s0076-6879(82)81063-x. [DOI] [PubMed] [Google Scholar]
- 23.Corson D W, Cornwall M C, MacNichol E F, Jin J, Johnson R, Derguini F, Crouch R K, Nakanishi K. Proc Natl Acad Sci USA. 1990;87:6823–6827. doi: 10.1073/pnas.87.17.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kefalov V J, Cornwall M C, Crouch R K. J Gen Physiol. 1999;113:491–503. doi: 10.1085/jgp.113.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornwall M C, Matthews H R, Crouch R K, Fain G L. J Gen Physiol. 1995;106:543–557. doi: 10.1085/jgp.106.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshizawa K, Yang J, Senzaki H, Uemura Y, Kiyozuka Y, Shikata N, Oishi Y, Miki H, Tsubura A. Exp Eye Res. 2000;71:629–635. doi: 10.1006/exer.2000.0921. [DOI] [PubMed] [Google Scholar]
- 27.He L, Poblenz A T, Medrano C J, Fox D A. J Biol Chem. 2000;275:12175–12184. doi: 10.1074/jbc.275.16.12175. [DOI] [PubMed] [Google Scholar]
- 28.Katai N, Yoshimura N. Invest Ophthalmol Vis Sci. 1999;40:2697–2705. [PubMed] [Google Scholar]
- 29.Liu C, Li Y, Peng M, Laties A M, Wen R. J Neurosci. 1999;19:4778–4785. doi: 10.1523/JNEUROSCI.19-12-04778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bitensky M W, Wheeler M A, Rasenick M M, Yamazaki A, Stein P J, Halliday K R, Wheeler G L. Proc Natl Acad Sci USA. 1982;79:3408–3412. doi: 10.1073/pnas.79.11.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muschietti J P, Bianchini G M, Martinetto H E, Carricarte V C, Giusto N, Farber D B, Torres H N, Flawia M M. Eur J Biochem. 1989;185:205–210. doi: 10.1111/j.1432-1033.1989.tb15103.x. [DOI] [PubMed] [Google Scholar]
- 32.Weiss E R, Hao Y, Dickerson C D, Osawa S, Shi W, Zhang L, Wong F. Biochem Biophys Res Commun. 1995;216:755–761. doi: 10.1006/bbrc.1995.2686. [DOI] [PubMed] [Google Scholar]
- 33.Fassina G, Aluigi M G, Gentleman S, Wong P, Cai T, Albini A, Noonan D M. Int J Cancer. 1997;72:1088–1094. doi: 10.1002/(sici)1097-0215(19970917)72:6<1088::aid-ijc25>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 34.Noell W K, Walker V S, Kang B S, Berman S. Invest Ophthalmol Vis Sci. 1966;5:450–473. [PubMed] [Google Scholar]
- 35.Naash M L, Peachey N S, Li Z-Y, Gryczan C C, Goto Y, Blanks J, Milam A H, Ripps H. Invest Ophthalmol Vis Sci. 1996;37:775–782. [PubMed] [Google Scholar]
- 36.St. Jules R S, Wallingford J C, Smith S B, O'Brien P J. Exp Eye Res. 1989;48:653–665. doi: 10.1016/0014-4835(89)90007-9. [DOI] [PubMed] [Google Scholar]
- 37.Fong S-L, Liou G I, Landers R A, Alvarez R A, Gonzalez-Fernandez F, Glazebrook P A, Lam D M K, Bridges C D B. J Neurochem. 1984;42:1667–1676. doi: 10.1111/j.1471-4159.1984.tb12758.x. [DOI] [PubMed] [Google Scholar]
- 38.DeVries G W, Campau K M, Ferrendelli J A. J Neurochem. 1982;38:759–765. doi: 10.1111/j.1471-4159.1982.tb08696.x. [DOI] [PubMed] [Google Scholar]
- 39.Brann M R, Cohen L V. Science. 1987;235:585–587. doi: 10.1126/science.3101175. [DOI] [PubMed] [Google Scholar]
- 40.LaVail M M, Battelle B-A. Exp Eye Res. 1975;21:167–192. doi: 10.1016/0014-4835(75)90080-9. [DOI] [PubMed] [Google Scholar]
- 41.Sanyal S, Hawkins R K. Vision Res. 1986;26:1177–1185. doi: 10.1016/0042-6989(86)90099-4. [DOI] [PubMed] [Google Scholar]
- 42.Green E S, Menz M D, LaVail M M, Flannery J G. Invest Ophthalmol Vis Sci. 2000;41:1546–1553. [PubMed] [Google Scholar]
- 43.Cornwall M C, Fain G L. J Physiol. 1994;480:261–279. doi: 10.1113/jphysiol.1994.sp020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ratto G M, Robinson D W, Yan B, McNaughton P A. Nature (London) 1991;351:654–657. doi: 10.1038/351654a0. [DOI] [PubMed] [Google Scholar]
- 45.Patrick G N, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai L-H. Nature (London) 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 46.Wilson P D, Sherwood A C, Palla K, Du J, Watson R, Norman J T. Am J Physiol. 1991;260:F420–F430. doi: 10.1152/ajprenal.1991.260.3.F420. [DOI] [PubMed] [Google Scholar]
- 47.Townes-Anderson E, Nachman-Clewner M. In: Axonal Regeneration in the Central Nervous System. Ingoglia N A, Murray M, editors. New York: Dekker; 2000. p. 111. [Google Scholar]