

Abstract
Protein–protein interactions are central to most biological processes. Although much recent effort has been put into methods to identify interacting partners, there has been a limited focus on how these interactions compare with those known from three-dimensional (3D) structures. Because comparison of protein interactions often involves considering homologous, but not identical, proteins, a key issue is whether proteins that are homologous to an interacting pair will interact in the same way, or interact at all. Accordingly, we describe a method to test putative interactions on complexes of known 3D structure. Given a 3D complex and alignments of homologues of the interacting proteins, we assess the fit of any possible interacting pair on the complex by using empirical potentials. For studies of interacting protein families that show different specificities, the method provides a ranking of interacting pairs useful for prioritizing experiments. We evaluate the method on interacting families of proteins with multiple complex structures. We then consider the fibroblast growth factor/receptor system and explore the intersection between complexes of known structure and interactions proposed between yeast proteins by methods such as two-hybrids. We provide confirmation for several interactions, in addition to suggesting molecular details of how they occur.
A major goal of functional genomics is to determine protein interaction networks for whole organisms. Large-scale studies have identified hundreds of potentially interacting proteins or complexes in yeast (1–3). Computational methods have used gene fusion (4, 5), gene order (6), phyletic distribution (7), or a combination of approaches (5) to predict functional associations (and putative interactions) between thousands of proteins, and efforts are underway to catalog interaction data contained within the literature (8, 9).
Despite attempts to identify putative interactions, little attention has been paid to one of the best sources of protein interaction data: complexes of known three-dimensional (3D) structure. Decades of x-ray crystallography have produced hundreds of structures for protein complexes, and these structures provide a rich source of data for learning principles of how proteins interact and for validating interactions determined by other methods. Although the number of complexes of known 3D structure is relatively small, it is possible to expand this set by considering homologous proteins.
Any interaction, whether known from two-hybrids, crystallography, or another method, will typically involve two or more proteins that themselves are members of homologous families. A major problem in genome annotation efforts is to understand when it is possible to transfer functional information, determined by experiment, from one protein to its homologues. Recent years have seen progress in predicting whether details such as enzymatic specificity or binding sites can be extrapolated to other members of a protein family (e.g., refs. 10 and 11), although similar studies on protein–protein interactions have been limited. It is not known whether it is generally possible to say that proteins homologous to a known interacting pair will interact in the same way, or indeed interact at all. For example, cytokines in the same family can be either promiscuous or highly specific regarding the receptors they prefer (e.g., ref. 12), and some homologues do not bind receptors at all (e.g., ref. 13). Moreover, analysis of interactions within the protein databank suggests considerable variation in the interaction partners preferred by particular protein families (14). Clearly detailed studies are required to understand when it is possible to infer an interaction between proteins when one is known to occur between homologues.
Here, we present a method to model putative interactions on known 3D complexes and to assess the compatibility of a proposed protein–protein interaction with such a complex. After identifying the residues that make atomic contacts in a known crystallographic complex, we look to homologues of both interacting proteins to see whether these interactions are preserved by means of empirical potentials. This method permits us to score all possible pairs between two protein families, and say which are likely to interact. We apply the method to the fibroblast growth factor/receptor system, and explore the intersection between all complexes of known 3D structure and interactions between yeast proteins proposed by methods such as two-hybrids. We demonstrate and discuss the importance of incorporating 3D structure information into studies of protein–protein interactions.
Methods
Overview of the Methodology.
Our initial analysis of complexes of known 3D structure showed that interactions between proteins occurred through a variety of main-chain and side-chain contacts (Fig. 1) as described in part previously (15). This prompted us to derive empirical potentials for amino acids to be involved in particular side-chain to side-chain and side-chain to main-chain contacts at protein–protein interfaces (see below).
Figure 1.

Interaction types across the four classes.
For a complex of known structure, we identify residues making atomic contacts at the interface of the two proteins. We then use the potentials to score the compatibility of any homologous intra-species pair of sequences, and calculate a statistical confidence based on a comparison to scores for a background of sequences that are unlikely to make favorable complexes (see below). In the sections that follow, we refer to scores as being significant if their probability to occur by chance is ≤0.01 or ≤0.1 when compared with the background.
Database of 3D Protein Complexes.
To construct a nonredundant database of interacting protein domains, we used blast2 (16) to compare sequences from representatives of the SCOP database (17) of protein structures to those from the Pfam database of protein domain families (18). We defined links between the two databases as matches with an expectation E-value ≤0.01. We then searched for instances where different Pfam domains matched different chains that were in contact (any atom distance <5Å) in the same structure. We manually grouped the final, nonredundant set of 356 unique interacting pairs of different Pfam families into broad classes: (i) enzyme inhibitors; (ii) cytokine receptors; (iii) signaling complexes; (iv) hetero-multimers; (v) immune system complexes (i.e., antibody/antigen or MHC complexes); and (vi) other protein–protein interactions.
Derivation of Empirical Potentials for Protein–Protein Interactions.
Empirically derived potentials incorporate thermodynamic effects without explicitly having to model proteins, which makes them useful in protein fold-recognition and protein–protein docking (e.g., refs. 19 and 20). We derived main-chain to side-chain and side-chain to side-chain potentials from the complexes above. We defined interacting residues by requiring one or more of: hydrogen bonds (N—O distances ≤ 3.5 Å), salt bridges (N—O distances ≤ 5.5 Å), or van de Waals interactions (C—C distances ≤ 5 Å). We also require a relative accessibility of the unbound proteins ≥10% to exclude buried side-chains. We found a total of 4,517 side-chain to main-chain and 3,316 side-chain to side-chain contacts. We then defined the empirical potentials by using a molar-fraction random state model based on the observed tendency of residues to be on protein surfaces:
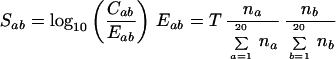 |
where na and nb are the total number of amino acids a and b and T is the total number of interacting pairs. Eab is the molar expected frequency for the a − b pair, Cab is the number of observed contacts between residues a and b and Sab the log-odds score. We assess the compatibility of a potential complex by evaluating a total score as the sum of Sab values for all interacting residue pairs identified in the crystallographic complex. Sab values are evaluated only if Cab > 0 and Eab > 5 to ensure sufficient data. Otherwise Sab is set to −0.5 and −1.0 for side-chain to main-chain and side-chain to side-chain potential, respectively, which are approximately the negative of the largest values. High scoring complexes are evaluated by this function to be more favorable.
Assigning Statistical Significance and Evaluation.
To assess whether an interface contains a sufficient number and type of contacts to be used further, we generated 1,000 random sequences for each interacting protein, based on surface residue frequencies. We scored them on the known complexes by using the empirical potentials on the interacting residues identified from the crystallographic complex. We used random sequences because we could not systematically extract information about noninteracting pairs of proteins homologous to a known complex, because such information is scant and subjective. Only when the known complex gave a score with Z ≥ 2.3 (2.3 standard deviations above the mean; significance ≤10−3) did we consider it suitable for further use. This process effectively removes complexes where there are few interactions or that are mediated mainly through main-chain to main-chain contacts.
To test the method, we extracted 59 pairs of homologous, nonidentical complexes of known structure, and scored one complex by using the other. We ignored immune system complexes as different interacting partners are expected (e.g., immunoglobulins). To ensure the integrity of the potentials we performed the test as a jack-knife (leave-one-out) procedure during this process.
Evaluating Potential Interactions.
Given a new potential interaction, we first assign the two components to one or more Pfam domains by using blast2 to see whether any complex 3D structure for this pair exists in the database described above. If one or more complexes are found, we align the new sequences to their homologues of known structure, and apply the empirical potentials and statistical significance to assess the fit of the interaction.
Data Sources and Availability.
We obtained protein 3D coordinate data from the protein databank (20) (http://www.rcsb.org) and yeast protein–protein interactions from the Munich Information Center for Protein Sequences Database (MIPS; ref. 21; http://mips.gsf.de), which collates interactions from a number of sources [including the Yeast Proteome Database, YPD (22)]. Data from two large-scale yeast two-hybrid experiments (1, 2) were taken from the web sites http://depts.washington.edu/sfields/yp_project and http://genome.c.kanazawa-u.ac.jp/Y2H/.
All data used in this study are available at http://www.russell.embl-heidelberg.de/add_info/.
Results and Discussion
Evaluation of the Method.
To evaluate the ability of the method to predict whether a pair of proteins homologous to a known complex could interact, we identified all examples of similar, but not identical, complex 3D structures. Essentially, these are examples where two or more 3D structures are known for interactions between the same homologous families. For example, structures are known for two different trypsin homologues (trypsin and proteinase B) in contact with different “Kunitz” inhibitors (tryptase and ovomucoid inhibitor). The examples were diverse, coming from 20 different protein family pairs. For clarity, we divided them into classes: cytokine/receptor, signaling, peptidase/inhibitor, and other (see Methods).
First, we found that, on average, 70% of pairs of residues in contact were common to these homologous complexes (cytokine/receptor 92%; signaling 89%; peptidase/inhibitor 59%; other 66%) and thus might, in the absence of a known structure, be identified by homology. We then used the potentials to score one complex by using the other; the results are summarized in Table 1. For cytokine/receptor, signaling, and other systems, all modeled complexes have a significant score, compared with 65% for peptidases and inhibitors. The poorer results for the peptidase/inhibitor complexes are likely because they interact via many main-chain to main-chain contacts (see ref. 15 and Fig. 1). Indeed, for 13 of 36 peptidase/inhibitor complexes, the crystal structure itself did not score significantly (see Methods), compared with only 3/18 from the three other classes. Inhibitors need to bind tightly to the target proteases, and this binding may be achieved by constrained main-chain conformations (15). Cytokine/receptor and signaling domain interactions are more transient, with lower affinities (24), and involve more side-chain contacts.
Table 1.
Accuracy of the method on interacting families of proteins with multiple complex structures
| Domain 1 | % Id range (interface) | Domain 2 | % Id range (interface) | Nc | T | P < 0.01 | P < 0.1 |
|---|---|---|---|---|---|---|---|
| Signaling | 16 | 16 | 16 | ||||
| G-α | 97 (88) | Guanylate cyclase | 99 (100) | 2 | 2 | 2 | 2 |
| RhoGAP | 99–100 (90–100) | ras | 50–100 (82–100) | 4 | 12 | 12 | 12 |
| RhoGDI | 87 (96) | ras | 51 (78) | 2 | 2 | 2 | 2 |
| Cytokines–Receptors | 12 | 12 | 12 | ||||
| FGF receptor | 71–99 (62–100) | FGF | 53–100 (60–100) | 4 | 12 | 12 | 12 |
| Peptidases–Inhibitors | 172 | 83 | 112 | ||||
| Peptidase S8 | 40–84 (35–84) | Potato inhibitor | 36–100 (38–100) | 4 | 12 | 5 | 10 |
| TIMP | 42 (36) | Peptidase M10 | 47 (50) | 2 | 2 | 2 | 2 |
| Trypsin | 27–99 (22–100) | Kunitz BPTI | 38–100 (28–100) | 10 | 70 | 21 | 37 |
| Trypsin | 12–100 (10–100) | Kazal | 27–100 (0–100) | 10 | 20 | 2 | 3 |
| Trypsin | 85 (88) | Kringle | 80 (83) | 2 | 4 | 4 | 4 |
| Trypsin | 65–100 (61–100) | Squash | 57–100 (44–100) | 6 | 24 | 12 | 19 |
| Other | 2 | 1 | 1 | ||||
| Colicin/pyocin | 56 (30) | HNH endonuclease | 47 (17) | 2 | 2 | 1 | 1 |
| Elongation factor TU | 69 (100) | Elongation factor TS | 27 (14) | 2 | N/A | N/A | N/A |
Domain 1/Domain 2 show the two interacting families. % Id range, Overall (and interface) sequence identity ranges between the proteins in the family. Nc, Number nonidentical complexes of known structure for each family. T, Number of predictions for which the template complex gave significant scores. P < 0.01 and P < 0.1, Number of pairs identified with significances of <0.01 and <0.1, respectively. N/A, Not applicable.
There is no clear correlation between the performance of the method at modeling interacting interfaces and sequence similarity (data not shown). Even at low sequence identities (see Table 1), we are able to use one complex to predict most of the correct side-chain to side-chain and side-chain to main-chain interactions that occur in another.
The evaluation above considers only instances where different pairs of proteins are known to interact in the same way. It is clearly also important to consider examples where pairs of proteins homologous to a complex structure are known definitely not to interact. A search of the literature demonstrated that there are only a few instances like this, one of which is discussed in the section below. Note that many interaction detection methods, for example two-hybrids, can provide only possible positive examples, and do not rule out interactions.
Fibroblast Growth Factors and Receptors.
We sought an example from the literature to illustrate the operation and accuracy of the method. Some of the most intensively studied interactions are those between fibroblast growth factors (FGFs) and receptors. FGFs play key roles in morphogenesis, development, angiogenesis, and wound healing. There are more than 20 human FGFs that bind to one or more of 7 FGF receptors (FGFR1c, -1b, -2c, -2b, -3c, -3b, and -4; c and b denote isoforms IIIc & IIIb formed by alternative splicing; ref. 25). Four different structures are known, involving all possible interactions between FGF-1 & FGF-2 and receptors FGFR-1c and FGFR-2c (26–28). Fig. 2 shows the identified interacting residues for the FGF-1/FGFR-1c complex. These results agree closely with those reported (26), although the method finds several others, including a hydrogen bond between Tyr-24 and Lys-163 and many hydrophobic interactions. Table 1 shows that the method successfully predicts all four complexes starting from any of the others (12 predictions in total) with significance <0.01. FGFs adopt a β-trefoil fold, and have been proposed to be ancient relatives of IL-1, a cytokine that binds to a receptor in a similar fashion to FGFs, and to proteins sharing no apparent functional similarity, such as actin bundling proteins, toxins, and protease inhibitors (29). The method correctly gives poor scores to all of these molecules when modeled onto the FGF receptors (>0.1), although, interestingly, IL-1α scores highest, possibly reflecting the similarity with FGF/receptor system as is known from the IL-1β/receptor structure (30).
Figure 2.

The FGFR system. alscript (37) alignment and molscript (38) structure figures showing the FGF (Upper)/receptor (Lower) interaction. Sequences are denoted by SWISSPROT or PDB identifiers. Residues are colored according to property conservation: hydrophobic, yellow background; small, blue background; and polar, red characters. Residues in the known structure (FGF-2/FGFR-1; PDB code 1cvs (26) participating in side-chain to side-chain contacts are shown and colored: positive, blue; negative, red; hydrophobic, yellow; and tyrosine, green. Residues participating in side-chain to main-chain contacts are boxed. Contacts between side-chains are listed.
Ornitz et al. performed a study of FGFR specificity by measuring mitogenic activity of FGFR-inducible BaF3 cell lines (12). They assayed the binding of FGFs 1–9 to all 7 receptors as a percentage relative to the affinity of FGF-1, which binds well to all of them (i.e., 100%). From this study, there are 252 different interactions that can be predicted (4 FGF/receptor structures, 7 receptors, and 9 FGFs). For 112 of these, the experimentally determined binding affinity relative to FGF-1 is <10% (which we term “low affinity”); for the other 140, the binding is ≥10% (“high”). Our method predicts 158 interactions with highly significant values (≤0.01), 105 of which are high affinity interactions. In addition, 59 of the 94 predictions that have low significance values (≥0.1) are low affinity, giving an overall prediction accuracy of 65%. There is good agreement between the predicted scores and the observed affinities, showing a correlation between molecular interaction details and macromolecular observations. This result is despite other factors that may be involved in determining the strength of the FGFR interaction (e.g., differing heparin affinities).
The interactions between FGF-2 with receptors FGFR-2b or FGFR-3b are consistently predicted to be significant despite being low affinity. The lack of affinity is thought to be the result of a deletion that changes the hydrophobic core of the receptor (27). We make no attempt to model such changes here. Further improvements may come from explicit modeling of loops or side-chain conformations.
An intriguing recent finding within the FGF family is that several members are not extracellular growth factors, but intracellular signaling molecules. FGFs-12, -13, -11, and -14 have recently been renamed as FGF homologous factors (FHF) 1–4, because, despite a high sequence identity to FGFs (up to 42%), they do not bind to FGF receptors, being instead associated with intracellular mitogen-activated protein (MAP) kinases (13). Encouragingly, our method gives poor scores to all of the interactions between FHFs and the FGF receptors. Inspection shows that substitutions in these proteins lead to the loss of many key contacts discussed above (see Fig. 2).
Interrogating the Yeast Protein Interaction Network.
The many interactions proposed between yeast proteins provide a large set to be studied by our method. Fig. 3 shows those interactions that are homologous to a known 3D complex and how they are scored by the method. Of the 2,590 interactions proposed (by two-hybrids, coimmunoprecipitation, cross-linking, etc.; see Methods) only 59 could be mapped onto our set of interacting complexes. For 23 of these, despite being homologous to a 3D complex, we found that the two interacting domains were not in direct atomic contact (see Methods). For the 36 remaining interactions, we identified domains in direct contact, thus suggesting the molecular basis for the interaction. We ignored one crystallographic structure (chaperonin-TCP1 complex) that did not score significantly itself when compared with random sequences, leaving the 35 interactions shown. Considering data from the large scale two-hybrid studies, 7 of the 1,466 interactions proposed by Uetz et al. (1) and 2 of the 841 from Ito et al. (2) could be mapped on to this set (labeled in Fig. 3). The low intersection between these interactions and known 3D structures agrees with that discussed previously (14), and also broadly agrees with that seen when comparing two-hybrid data to predictions made from gene-fusions (31).
Figure 3.

Yeast interactions mapped onto known 3D complexes. Yeast proteins are given by their YPD names, codes, and Pfam families. Thick lines denote interactions significant at ≤0.01, thin lines 0.01–0.1, and dashed lines >0.1.
Fig. 3 shows instances of interaction promiscuity where the method gives significant scores only to some of the proposed interactions. This result suggests that the approach could be used to rank interacting pairs that are to be investigated by further experimental means. Significant scores indicate those interactions that are likely to be most compatible with a known 3D structure, and thus where a model will give most accurate details regarding, for example, site-directed mutagenesis experiments. For those where the score is not significant, the interaction may either be weak, or may involve detailed loop or side-chain changes not considered here.
Most experimental and computational methods for predicting interactions are unable to discern direct physical interactions from those involving intermediate proteins. Sometimes this result can be clarified simply by mapping proposed interactions onto 3D structures. For example, a cyclin/cyclin-dependent kinase regulatory subunit (CKS) interaction was proposed by yeast two-hybrids (1). Although no cyclin/CKS 3D complex is known, separate structures have been determined for cyclin dependent kinase 2 (CDK2) in complex with cyclin A and CKS. Superimposing the kinases shows the cyclin and CKS domains to be more than 15 angstroms apart. Whereas we cannot completely rule out the presence of N- or C-terminal extensions, absent from the crystal structures, that could bridge this distance, the structures suggest that the proposed CKS/cyclin interaction may involve CDK2 as an intermediary.
Several of the interactions shown in Fig. 3 are those between kinases and cyclins, which is a system that shows considerable promiscuity. Multiple cyclins control eukaryotic cell division by regulating CDKs. However, it is unclear whether all are required for cell cycle progression, because there is evidence for considerable redundancy in function (32, 33). Differential activation of CDKs by cyclins could occur through differences in affinities, but may also be a result of differences in expression, protein associations, or subcellular localizations. We applied our method to see whether the scoring system could model the promiscuity within this system.
The structure of a human CDK2/cyclin A is the only kinase/cyclin complex known in atomic detail (explaining its absence from Table 1). However, sequence comparison permits 14 of the 22 known yeast kinase/cyclin interactions (34) to be modeled on this complex. Our method identifies all key interacting residues described previously (35) in addition to others (e.g., Lys-278-Asp-9 hydrogen bond, Arg-122-Gln-141 salt-bridge; Fig. 4). There is often little similarity between the yeast and human proteins in the known complex, considering either overall sequence identities (CDKs 38–58%, cyclins 3–32%) or only residues on the interacting surfaces (CDKs 24–78%, cyclins 10–38%). Nevertheless, we find significant scores for all 14 interactions and for 12 scores have significances ≤0.01. The kin28/ccl1 pair is the most different from the human complex (24% and 10% identity on the interacting surface for the CDK and cyclin, respectively). Although some key interactions are lost in the kin28/ccl1 complex with respect to CDK2/cyclin A (e.g., Lys-278–Asp-9 changes to Val-278–Lys-9), others show a good correlation between the substitutions in the interacting partners (e.g., Arg-122–Tyr-13 and Gln-141 in CDK2/cyclin A changes to Phe-122–Ile-13 and Ile-122 in kin28/ccl1). Despite the low degree of identity, our method is able to model the new interacting residues and correctly predicts the interaction between these two proteins with confidence.
Figure 4.

The kinase/cyclase system in yeast. Sequences are denoted by YPD or PDB codes. The known structure [1fin (35)] is that of human CDK2 (Upper)/cyclin A (Lower). Details are otherwise as for Fig. 2.
Considering the 28 putative CDK/cyclin pairs not detected by experimental methods (i.e., not in Fig. 3), all but one (cdc28/pcl1) give significant scores, with 14 significant at ≤0.01. The surprising suggestion that most kinase/cyclin combinations can interact agrees with cyclin knockouts in yeast (32) that show that deletion of all but a single cyclin yields viable strains.
Conclusions
We have presented a method to model protein interactions on 3D complexes. The method can successfully model one known protein complex on another, and predicts correct interactions within several systems. Given a known 3D complex structure and homologous sequences for each interacting protein, the method can rank all of the possible interactions between homologues of the same species. For studies of protein families that are known to show different interaction specificities, such a ranking can be used to prioritize experiments, and save laboratory time and costs. This ranking could also help to interpret results from gene-fusion studies (4, 5), where multiple homologues in one organism relative to another can create ambiguities. More generally, we have shown how 3D structures can be used to interrogate whole interaction networks to validate and infer molecular details for interactions proposed by other approaches.
There remain several questions regarding protein–protein interactions. Whereas we can predict whether proteins homologous to a complex are likely to interact in the same way, we still do not generally know how protein-interacting partners change during evolution. Some systems appear to change gradually, or via genetic events such as alternative splicing, to optimize pairings. Ultimately, these changes may lead to differences in the relative orientations of the interacting molecules, or to homologous proteins interacting with entirely different molecules. However, it is also becoming clear that nature makes use of strategies other than alteration of surfaces to ensure that the correct interactions occur in the cell, foremost among these being regulation of gene expression (e.g., ref. 36). Optimal interactions may be obtained simply by the presence or absence of proteins. Each pair of interacting protein families will need to be considered in context in order for strategies like that described here to be most useful.
Structural genomics efforts and the increasing pace of structure determination will provide knowledge of many more complexes in the future. With these data, our method will permit critical interrogation of interactions predicted by other areas of functional genomics, and provide molecular details for proposed protein interaction networks in advance of experimental structural biology.
Acknowledgments
We are grateful to I. Gelfand and A. Kister (Rutgers) for numerous helpful comments. We thank P. Bork, T. Gibson, R. Copley, E. Conti, C. Perez-Iratxeta, M. Andrade, D. Torrents & T. Sardon (EMBL) for critical reading of the manuscript.
Abbreviations
- 3D
three-dimensional
- FGF
fibroblast growth factor
- FGFR
FGF receptor
- CKS
cyclin-dependent kinase regulatory subunit
References
- 1.Uetz P, Giot L, Cagney G, Mansfield T A, Judson R S, Knight J R, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. Nature (London) 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavin A C, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick J M, Michon A M, Cruciat C M, et al. Nature (London) 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 4.Enright A J, Iliopoulos I L, Kyrpides N C, Ouzounis C A. Nature (London) 1999;402:25–26. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 5.Marcotte E M, Pellegrinin M, Thompson M J, Yeates T O, Eisenberg D. Nature (London) 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- 6.Huynen M A, Snel B, Bork P. Trends Genet. 2001;17:304–306. doi: 10.1016/s0168-9525(01)02302-2. [DOI] [PubMed] [Google Scholar]
- 7.Pellegrini M, Marcotte E M, Thompson M J, Eisenberg D, Yeates T O. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xenarios I, Rice D W, Salwinski L, Baron M K, Marcotte E M, Eisenberg D. Nucleic Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bader G D, Donaldson I, Wolting C, Ouellette B F, Pawson T, Hogue C W. Nucleic Acids Res. 2001;29:242–245. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hannenhalli S S, Russell R B. J Mol Biol. 2000;303:61–76. doi: 10.1006/jmbi.2000.4036. [DOI] [PubMed] [Google Scholar]
- 11.Aloy P, Querol E, Aviles F X, Sternberg M J E. J Mol Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. [DOI] [PubMed] [Google Scholar]
- 12.Ornitz D M, Xu J, Colvin J S, McEwen D G, MacArthur C A, Coulier F, Gao G, Goldfarb M. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 13.Schoorlemmer J, Goldfarb M. Curr Biol. 2001;11:793–797. doi: 10.1016/s0960-9822(01)00232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park J, Lappe M, Teichmann S A. J Mol Biol. 2001;307:929–938. doi: 10.1006/jmbi.2001.4526. [DOI] [PubMed] [Google Scholar]
- 15.Jackson R M. Protein Sci. 1999;8:603–613. doi: 10.1110/ps.8.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;253:389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo Conte L, Ailey B, Hubbard T J, Brenner S E, Murzin A G, Chothia C. Nucleic Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bateman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones D T, Taylor W R, Thornton J M. Nature (London) 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 20.Moont G, Gabb H A, Sternberg M J. Proteins. 1999;35:364–373. [PubMed] [Google Scholar]
- 21.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mewes H W, Frishman D, Gruber C, Geier B, Haase D, Kaps A, Lemcke K, Mannhaupt G, Pfeiffer F, Schuller C, Stocker S, Weil B. Nucleic Acids Res. 2000;28:37–40. doi: 10.1093/nar/28.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodges P E, McKee A H, Davis B P, Payne W E, Garrels J I. Nucleic Acids Res. 1999;27:69–73. doi: 10.1093/nar/27.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones S, Thornton J M. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szebenyi G, Fallon J F. Int Rev Cytol. 1999;185:45–106. doi: 10.1016/s0074-7696(08)60149-7. [DOI] [PubMed] [Google Scholar]
- 26.Plotnikov A N, Schlessinger J, Hubbard S R, Mohammadi M. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 27.Plotnikov A N, Hubbard S R, Schlessinger J, Mohammadi M. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 28.Stauber D J, DiGabriele A D, Hendrickson W A. Proc Natl Acad Sci USA. 2000;97:49–54. doi: 10.1073/pnas.97.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponting C P, Russell R B. J Mol Biol. 2000;302:1041–1047. doi: 10.1006/jmbi.2000.4087. [DOI] [PubMed] [Google Scholar]
- 30.Schreuder H, Tardif C, Trump-Kallmeyer S, Soffientini A, Sarubbi E, Akeson A, Bowlin T, Yanofsky S, Barrett R W. Nature (London) 1997;386:194–200. doi: 10.1038/386194a0. [DOI] [PubMed] [Google Scholar]
- 31. Enright, A. J. & Ouzounis, C. A. (2001) Genome Biol.2, research0034.1–research0034.7. [DOI] [PMC free article] [PubMed]
- 32.Fisher D L, Nurse P. EMBO J. 1996;15:850–860. [PMC free article] [PubMed] [Google Scholar]
- 33.Miller M E, Cross F R. J Cell Sci. 2001;114:1811–1820. doi: 10.1242/jcs.114.10.1811. [DOI] [PubMed] [Google Scholar]
- 34.Andrews B, Measday V. Trends Genet. 1998;14:66–72. doi: 10.1016/s0168-9525(97)01322-x. [DOI] [PubMed] [Google Scholar]
- 35.Jeffrey P D, Russo A A, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletiich N P. Nature (London) 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 36.Sowa M E, He W, Wensel T G, Lichtarge O. Proc Natl Acad Sci USA. 2000;97:1483–1488. doi: 10.1073/pnas.030409597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton G J. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 38.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]