

Abstract
Blue and green sunlight become available for photosynthetic energy conversion through the light-harvesting (LH) function of carotenoids, which involves transfer of carotenoid singlet excited states to nearby (bacterio)chlorophylls (BChls). The excited-state manifold of carotenoids usually is described in terms of two singlet states, S1 and S2, of which only the latter can be populated from the ground state by the absorption of one photon. Both states are capable of energy transfer to (B)Chl. We recently showed that in the LH1 complex of the purple bacterium Rhodospirillum rubrum, which is rather inefficient in carotenoid-to-BChl energy transfer, a third additional carotenoid excited singlet state is formed. This state, which we termed S*, was found to be a precursor on an ultrafast fission reaction pathway to carotenoid triplet state formation. Here we present evidence that S* is formed with significant yield in the LH2 complex of Rhodobacter sphaeroides, which has a highly efficient carotenoid LH function. We demonstrate that S* is actively involved in the energy transfer process to BChl and thus have uncovered an alternative pathway of carotenoid-to-BChl energy transfer. In competition with energy transfer to BChl, fission occurs from S*, leading to ultrafast formation of carotenoid triplets. Analysis in terms of a kinetic model indicates that energy transfer through S* accounts for 10–15% of the total energy transfer to BChl, and that inclusion of this pathway is necessary to obtain a highly efficient LH function of carotenoids.
Carotenoids are a class of photoactive biomolecules that are widespread in nature and play a vital role in the photochemistry of photosynthesis (1, 2). They are found in most photosynthetic organisms where they serve a number of functions. In the light-harvesting (LH) proteins, carotenoids absorb the blue-green sunlight that is not absorbed by the (bacterio)chlorophyll (BChl) molecules, a property vital for organisms living in environments that are poor in light. Carotenoids transfer their excitation energy to the (B)Chl molecules, which in turn funnel it to a unit called the reaction center, to trigger a sequence of electron-transfer processes that take place on time scales of up to microseconds (3, 4). The presence of carotenoids is crucial for the survival of the photosynthetic organisms. During the photochemical processes that follow LH by carotenoids and (B)Chls, a (B)Chl singlet excitation can undergo intersystem crossing to a triplet state. This process takes place in a few nanoseconds and therefore is not of high yield because it competes with faster processes. However, the (B)Chl triplet excited species can promote ground-state oxygen molecules to their highly reactive and hazardous singlet excited state. Carotenoids accept and safely dissipate the (B)Chl triplet energy or scavenge the excited oxygen, thereby protecting the organism (1).
The essential structural element of all carotenoids is a chain of alternating carbon double bonds augmented with diverse end groups and substitutions. This polyene backbone accounts for many of the properties of their excited states. Knowledge of these properties is important in understanding the function of carotenoids in the LH complexes, because they determine the interactions between the different pigments. The currently established picture of the low-lying excited electronic states of the carotenoids involves two singlet states, referred to as S1 and S2 (2A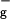 and 1B
and 1B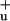 in ideal C2h symmetry notation). Because of the symmetry selection rules, only the latter can be populated by the absorption of one photon from the ground state S0, which has an Ag symmetry (2). This electronic transition accounts for the strong absorption observed around 450–550 nm that gives carotenoids their characteristic color. Excitation to S2 is followed by fast internal conversion (IC) to S1 (typically in the order of 150 fs) and then by IC from S1 to S0 with time constants ranging from 1 to 10s of ps, mainly depending on the length of the polyene chain (5). In the presence of potential excitation energy acceptors such as the BChl molecules in the LH complexes of photosynthetic bacteria (LH1 and LH2), the ultrafast IC competes with the excitation energy-transfer (EET) processes. In general, EET is known to occur from either or both of the S2 and S1 states depending on the species and the carotenoid contained therein (2). The carotenoid-to-(B)Chl EET processes are governed by the Coulombic coupling and spectral overlap between donor and acceptor states (2), and the overall efficiency can vary from as low as 35% in the LH1 complex of Rhodospirillum (Rs.) rubrum (6) and the PSII core complex of oxygenic photosynthesis (7) to as much as 90% or more in the LHCII of plants (8), the PSI complex of cyanobacteria (9), and various LH2 antennae of purple bacteria (1).
in ideal C2h symmetry notation). Because of the symmetry selection rules, only the latter can be populated by the absorption of one photon from the ground state S0, which has an Ag symmetry (2). This electronic transition accounts for the strong absorption observed around 450–550 nm that gives carotenoids their characteristic color. Excitation to S2 is followed by fast internal conversion (IC) to S1 (typically in the order of 150 fs) and then by IC from S1 to S0 with time constants ranging from 1 to 10s of ps, mainly depending on the length of the polyene chain (5). In the presence of potential excitation energy acceptors such as the BChl molecules in the LH complexes of photosynthetic bacteria (LH1 and LH2), the ultrafast IC competes with the excitation energy-transfer (EET) processes. In general, EET is known to occur from either or both of the S2 and S1 states depending on the species and the carotenoid contained therein (2). The carotenoid-to-(B)Chl EET processes are governed by the Coulombic coupling and spectral overlap between donor and acceptor states (2), and the overall efficiency can vary from as low as 35% in the LH1 complex of Rhodospirillum (Rs.) rubrum (6) and the PSII core complex of oxygenic photosynthesis (7) to as much as 90% or more in the LHCII of plants (8), the PSI complex of cyanobacteria (9), and various LH2 antennae of purple bacteria (1).
With our recent work on the LH1 complex of the purple photosynthetic bacterium Rs. rubrum (10) we clarified long-standing questions (6, 11) about the pathway of excitation energy deactivation in this specific antenna system, where the carotenoid spirilloxanthin shows a particularly high triplet yield on direct excitation (30%) and a low carotenoid-BChl EET efficiency of 35%. We have shown that on spirilloxanthin, an excited state besides the S1 is formed on optical excitation, which we tentatively referred to as S*. When bound to the LH1 complex of Rs. rubrum, S* is the precursor state in the reaction pathway toward triplet formation, which occurs within a few picoseconds through an intramolecular homofission mechanism. For spirilloxanthin in solution, S* returns to the singlet ground state on a picosecond time scale. We asserted that S* very likely corresponds to the 1B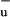 state, which in theoretical work on polyenes has been predicted to lie energetically below S2 (12). Koyama and coworkers were the first to present experimental evidence concerning the existence of 1B
state, which in theoretical work on polyenes has been predicted to lie energetically below S2 (12). Koyama and coworkers were the first to present experimental evidence concerning the existence of 1B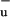 in carotenoids by carrying out Raman excitation profile measurements (13).
in carotenoids by carrying out Raman excitation profile measurements (13).
To investigate the occurrence of S* in other carotenoid-containing antennae and its possible involvement in photosynthetic LH, we studied an antenna complex with a highly efficient carotenoid LH function, the LH2 antenna of Rhodobacter (Rb.) sphaeroides 2.4.1. This complex is a membrane-bound protein that consists of a circular aggregate of 8 or 9 α,β-polypeptides. The overall structure of the complex is similar to the LH2 complex of Rhodopseudomonas acidophila, of which the atomic structure is known (14). Each α,β-polypeptide binds two BChls at the periplasmic side, giving rise to an absorption band at 850 nm (accordingly labeled B850), one BChl at the cytoplasmic side that absorbs at 800 nm (B800), and one spheroidene spanning the membrane, absorbing around 500 nm. EET among the BChls has been well studied and is characterized by very fast equilibration among the B850s in ≈100 fs (4) and rapid EET from B800 to B850 in 700 fs (4).
In the LH2 complex of Rb. sphaeroides, spheroidene is more than 90% efficient in transferring excitations to BChl (15). We will show that a state similar to S* of spirilloxanthin exists in the excited-state manifold of spheroidene bound to the LH2 antenna, and that it is involved actively in the EET processes to BChl, thereby constituting an alternative mechanism of EET in photosynthetic LH.
Materials and Methods
Sample Preparation.
The LH2 antenna complexes were isolated and purified from cells of Rb. sphaeroides 2.4.1 as described in ref. 16 and suspended in a 20 mM Tris/0.1% lauryl dimethyl amine oxide buffer. The absorption spectrum of the LH2 complex of Rb. sphaeroides is shown in Fig. 1.
Figure 1.

Absorption spectrum of the LH2 antenna of R. sphaeroides 2.4.1. The absorption of the S2 state of spheroidene is resolved clearly in the 440–520-nm region, whereas the BChl absorption appears at 590 (Qx), 800, and 850 (Qy) nm.
Laser Spectroscopy.
Femtosecond transient absorption measurements in the visible region were carried out with a Ti:sapphire laser system as described earlier (8). In brief, part of the output of a 1-kHz amplified Ti:sapphire laser system (Coherent-BMI α1000, Coherent Radiation, Palo Alto, CA) was used to drive a home-built noncollinear optical parametric amplifier. In the experiments presented here, the excitation wavelength was tuned to 475 nm (17-nm full-width half-maximum) so as to selectively excite S2. The pulse width was 80 fs, and the energy was 100 nJ. A white light continuum that was generated by focusing amplified 800-nm light on a 1-mm sapphire crystal was split into probe and reference. The beams were focused in a 1-mm path-length quartz cuvette at a spot of 150-μm diameter. To avoid sample degradation and exposure of the sample to multiple laser shots, the cuvette was mounted on a shaker. The polarization between the pump and the probe was set to magic angle (54.7°). After passing through the sample, the probe and reference beams were dispersed by a polychromator and projected on a home-built double diode array detector. Femtosecond time delays between pump and probe were controlled by a delay line covering delays of up to 2 ns. We also performed experiments in the near-IR region to probe the changes in absorption of the BChl Qy bands. It was crucial to use the lowest pump energy possible to avoid excitation annihilation effects on the LH2 ring that would mask other processes occurring simultaneously. We therefore used a 125-kHz amplified Ti:sapphire system (Coherent Mira-Rega), which in combination with lock-in detection allowed us to maintain annihilation-free conditions with a high signal-to-noise ratio. An optical parametric amplifier provided the 490- and 590-nm light for excitation of the spheroidene S2 and the BChl Qx states, whereas a portion of the 800-nm light focused in a 1-mm sapphire plate created a white light continuum, out of which the probe wavelengths (840, 860, and 880 nm) were selected with interference filters. For these experiments the sample was placed in a 1-mm path-spinning cell, the two beams were focused at a spot of ≈150-μm diameter, and the pump pulse energy was 4.8 nJ. All measurements were done at room temperature.
Data Analysis.
The recorded time-resolved spectra (covering the visible region) and kinetic traces (in the IR region) were analyzed simultaneously with a global analysis program using a kinetic model (17, 18) consisting of sequentially interconverting species, i.e., 1 → 2 → 3 → … The arrows indicate successive monoexponential decays of increasing time constants, which can be regarded as the lifetime of each species. Associated with each species is a lifetime and a difference spectrum called the species-associated difference spectrum (SADS). The first SADS corresponds to the time zero difference spectrum. In general the SADS may well correspond to mixtures of excited states and should not be considered to portray spectra of pure states. This procedure enables us to visualize clearly the evolution of the (excited) states of the system. The instrument response function was fit to a Gaussian (120-fs full-width half-maximum) and the group velocity dispersion of the probing white light was fit to a third-order polynomial. To illustrate our observations optimally we present them by means of showing the SADS instead of raw time-resolved spectra. For selected detection wavelengths we present raw data by showing the measured kinetic traces including the fit.
Results and Discussion
Rb. sphaeroides 2.4.1 LH2 Dynamics.
We investigated the excited-state dynamics of carotenoids and BChls in the LH2 complex of Rb. sphaeroides 2.4.1 by performing femtosecond transient-absorption experiments with excitation of spheroidene and broad-band white light probing. We probed in the visible and near-IR regions of the spectrum where the excited species of spheroidene and BChl absorb, respectively. Simultaneous analysis of the data indicated that six sequential components were required to achieve a satisfactory description. The resulting SADS are portrayed in Fig. 2 together with the corresponding lifetimes, and selected kinetic traces at 535, 555, and 860 nm are presented in Fig. 3.
Figure 2.

Sequential SADS and associated lifetimes for spheroidene (Left) and BChl (Right) in the LH2 antenna of Rb. sphaeroides 2.4.1 on spheroidene excitation. Note the different baseline and scale. a.u., arbitrary units.
Figure 3.

Kinetic traces measured at 535 (A), 555 (B), and 860 nm (C) on excitation of spheroidene in the LH2 antenna of Rb. sphaeroides 2.4.1 at 475 or 490 nm. The black lines denote the fitted curves as determined by the global analysis sequential model, of which the SADS are displayed in Fig. 2. (D) Kinetic trace measured at 860 nm on direct excitation of BChl at 590 nm of the LH2 antenna of Rb. sphaeroides 2.4.1. The black line denotes a fit with exponential functions with an instantaneous and a 700-fs rise component (see Results and Discussion). a.u., arbitrary units.
The instantaneously (i.e., limited by the instrument response) appearing SADS (thin black line) has a lifetime of 90 fs. It features the sharp bleach of the red-most peak of the S0 → S2 transition at 512 nm and a broad, negative signal caused by stimulated emission (SE) on the red part of the visible spectrum. It is straightforward to assign this spectrum to the S2 state of spheroidene. The decay of S2 is followed by the appearance of the second SADS (red), which has a lifetime of 320 fs. It contains a broad excited-state absorption (ESA) that peaks at 555 nm and has a shoulder at 535 nm, whereas the bleach at 512 nm has decreased. The 555 nm ESA peak is characteristic of the spheroidene S1 state, indicating that IC from S2 to S1 has occurred. The evolution that follows, from the second to the third SADS (blue), consists of the significant loss of bleach at 512 nm and ESA above 570 nm, whereas the peak and the blue wing of the ESA essentially remain the same. The spectral characteristics of this process are attributed to vibrational cooling of S1 (19), which therefore is characterized by the time constant of 320 fs. The third SADS, which has a lifetime of 1.5 ps, depicts the relaxed S1 state of spheroidene. The spectral changes after going to the fourth SADS (green) indicate that this step corresponds to the depopulation of S1. The observed lifetime of 1.5 ps is significantly shorter than that of S1 of spheroidene in solution, 9 ps (20, 21), and because the IC rate from S1 to the ground state does not depend on the properties of the solvent or protein environment (22), this effect demonstrates that the S1 state of spheroidene is an efficient donor of energy to BChl. It is important also to note that the 320-fs process is not conservative, because a significant amount of spheroidene bleach and ESA is lost after going from the second to the third SADS, indicating that EET takes place also from the “hot” S1 state to BChl.
The fourth SADS (green) has a lifetime of 7 ps and represents a significant ESA feature with a peak at 535 nm. The spectral and temporal characteristics of this SADS are clearly distinct from those of S1. It describes an excited state of spheroidene that bears close resemblance to S* of spirilloxanthin in the LH1 complex of Rs. rubrum. Notably, a feature at 535 nm was present already in the ESA after the decay of S2 (see second SADS), suggesting that this new state is populated directly from S2, in parallel with S1, as we demonstrated previously for S* in Rs. rubrum (10). These observations strongly indicate that the fourth SADS actually corresponds to the S* state, which, judging from its amplitude, is formed with a significant yield in the LH2 complex of Rb. sphaeroides. An estimate on the basis of a kinetic model would indicate that ≈20% of S2 decays directly into S* (see below). The fifth SADS (magenta) that is formed by the decay of S* follows the transient absorption features of the spheroidene triplet as determined by microsecond triplet-singlet spectroscopy (data not shown), with a small bleach at 512 nm and an ESA peak at 530 nm. This observation indicates that spheroidene triplets are created directly from S* on a picosecond time scale, as we reported for spirilloxanthin in the LH1 antenna of Rs. rubrum (10). The ESA feature that we ascribe to the triplet state remains present for times longer than those we can measure with the femtosecond setup. However, as seen in Fig. 2, significant changes take place after several 10s of ps, with the fifth SADS evolving into the sixth SADS (black), which represents the “final” state of the system. Relaxation of the spheroidene triplet as well as the ground-state band shift signal induced by excited BChl (23) may play a role in these dynamics. Triplet–triplet annihilation is a spin-forbidden process, because the triplet pair is generated with an overall singlet spin and therefore is not expected to occur on this time scale.
After our previous work on spirilloxanthin, we assert that S* is a singlet excited state and not a triplet state. S* acts as a precursor to the formation of triplet states of the LH1 complex of Rs. rubrum, and it returns to the (singlet) ground state on a picosecond time scale in spirilloxanthin in solution (10). The latter phenomenon indicates beyond doubt that S* is a singlet state.
The transient characteristics of S1 and S* are illustrated by the kinetic traces measured at the peaks of their ESA at 555 and 535 nm, respectively (Fig. 3). The 555-nm trace has a major decay component of 1.5 ps, and a slower phase with small amplitude corresponds to the red tail of the ESA that originates from S* (Fig. 2, fourth SADS). The 535-nm trace decays much slower with 7 ps and does not relax to zero, indicating the formation of the triplet state. We note that the carotenoid ground-state bleaching region (<510 nm) is not a good measure for the dynamics of the various excited states in the carotenoid, because it is insensitive to which excited state is populated and is contaminated by ESA from S* (10).
After having established that S1 and S* have distinctly different temporal characters, we are in a position to discriminate their possible contributions to the EET process to BChl by measuring the rates at which the excitations arrive on B850. To this end, we have measured the rise of the transient absorption signal at 860 nm on spheroidene excitation at 490 nm. At 860 nm, we probe the bleaching and SE of the B850 band (24). It is important to note beforehand that the rise of the B850 signal is partly associated with EET from B800 to B850, because spheroidene is expected to transfer a fraction of its energy to B800 (21). B800–B850 EET occurs with a time constant of 700 fs (4, 24), which implies that on this time scale, the rise signals at 860 nm are a convolution of spheroidene-BChl and B800–B850 EET. Thereafter, the rise exclusively reflects spheroidene-BChl EET. The result is shown in Fig. 3C. The rise of the signal is clearly multiphasic, comprising a very fast initial rise in ≈200 fs followed by slower components on the time scale of picoseconds. It is noticeable that the signal keeps rising well beyond 5 ps, indicating that excitation energy is being transferred to BChl long after 1.5 ps, which is the lifetime of S1. Fitting this trace independently of the time-resolved data in the visible region with three rise components yielded a 350-fs time constant followed by a 1.5-ps and finally a significant 6-ps component, carrying 11% of the amplitude. An independent two-component fit yielded lifetimes of 1.2 and 4.5 ps, but the quality of the fit was reduced considerably. These independent fit results are presented in Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org. The observation of a rise component on a 6-ps time scale implies that spheroidene states other than S1 and S2 participate actively in the EET process to BChl. Given its 7-ps lifetime, the newly discovered S* state is a very likely candidate. The correlation between the disappearance of S* and the arrival of excitations on BChl is demonstrated in the SADS of Fig. 2, where the right side describes the BChl dynamics in the near-IR region. Three wavelengths of detection are shown, 840, 860, and 880 nm, where B850 shows ESA, bleaching/SE, and pure SE, respectively (24). The instantaneous SADS (squares) reflects the BChl in its ground state with zero amplitude at 840 and 860 nm, whereas at 880 nm it contains a small ESA feature that may arise from S2 of spheroidene or Qx of B850. The amplitudes of the SADS in the near-IR increase on the time scales associated with the S2 and S1 states of spheroidene and the B800–B850 EET process, i.e., 90 fs (squares to open circles), 320 fs (open circles to diamonds), and 1.5 ps (diamonds to solid circles), as expected from previous studies (21, 24, 25). The 7-ps component, which in the visible part of the spectrum denotes the decay of S*, represents the final rise step of the BChl signal (solid circles to down triangles), accounting for ≈10% of the final amplitude.
It can be argued that the existence of a slow phase in the rise of the signal at 860 nm might originate from internal BChl dynamic processes. The spectroscopic properties of the BChls of this LH2 antenna have been studied extensively in the past (3, 4, 24), and no EET occurring on the time scale of picoseconds has been reported. To verify these previous results, we performed a control experiment by exciting directly the BChl of the LH2 antenna at 590 nm (Qx) and measuring the transient absorption signal at 860 nm under exactly the same conditions as in the measurements described above. The trace that depicts the rise of the bleach and the SE of B850 is shown in Fig. 3D. The rise is rapid, comprising an instantaneous and a 700-fs component. By comparing it to the trace recorded after 490-nm excitation, it becomes evident that a slow phase is now absent and that after 1 ps, all excitations reside on the Qy level of the B850. These two components agree with the established values of 100 and 700 fs, corresponding to Qx → Qy IC and B800 → B850 EET times, respectively (4, 20, 24).
Our observations provide strong evidence that the observed S* state is indeed a donor state in the EET process from spheroidene to BChl.
Target Analysis, a Compartmental Model Approach.
To model the different processes that take place simultaneously we propose that the excitation of the S2 state of spheroidene in the LH2 complex of Rb. sphaeroides 2.4.1 is followed by the general kinetic scheme shown in Fig. 4. It is important to realize that our model depicts the established picture about the role and properties of S1 and S2 of spheroidene and the two types of BChl (4), with the only addition being S* and the two paths by which it decays, i.e., EET to BChl and singlet fission to the spheroidene triplet state. In our previous study on spirilloxanthin, we showed that S1 and S* evolve independently in vivo as well as in vitro (10). The spectral evolution of spheroidene in Fig. 2 shows a very similar pattern, and therefore we did not incorporate a communication path between S1 and S*.
Figure 4.

Proposed kinetic scheme for excitation transfer and decay pathways in the LH2 antenna of R. sphaeroides 2.4.1 describing the processes occurring in the first 10s of ps after excitation of spheroidene. S2, S1, S*, and T denote the singlet and triplet excited states of spheroidene, and BChl denotes collectively the singlet excited states of bacteriochlorophyll that comprise the Qx and Qy states of B800 and B850. Note that the actual model used in the target analysis comprises nine compartments (see Target Analysis and Table 2, which is published as supporting information on the PNAS web site), that for clarity reasons are not displayed here.
The reaction scheme that we used in the so-called target analysis (18) to distinguish the contribution of the spheroidene excited states to the transient absorption spectra comprises nine compartments (for a complete description see Table 2). In this analysis, by adjusting the branching ratios we can judge the applicability of the model on the basis of plausibility of the resulting SADS and yields. The cornerstones of our modeling are the following assumptions: (i) S2 decay occurs in parallel via four competing channels, the unrelaxed hot S1, S*, B800, and the Qx of B850, (ii) the hot S1, the S1, and the S* states transfer energy to the Qy level of both the B800 and B850 bands, and (iii) the triplet state of spheroidene is formed via S* on a picosecond time scale. To limit the degrees of freedom of the fitting routine, we applied the spectral constraint that in the ground-state bleach region up to 535 nm, the SADS of S2, hot S1, and S1 should be identical. This assumption is valid provided the SE and ESA bands of these states do not overlap with the ground-state bleaching. We note that S* cannot be included in this procedure, because it has a significant ESA in the ground-state bleaching region (10). The B800 → B850 EET time has been fixed to 700 fs (20, 24) and the S1–S0 IC time to 9 ps (20, 21), whereas the Qx level of B850 is treated as a short-living intermediate with a fixed lifetime of 100 fs in the EET pathway from S2 to the final acceptor Qy (2), which has a fixed lifetime of 1 ns.
After fitting the visible spectra and kinetic trace at 860 nm to this model, we were able to estimate the SADS shown in Fig. 5. We note that the fitted lifetimes of the various excited states of spheroidene in the context of this particular kinetic model do not necessarily exactly match those derived from the sequential analysis shown in Figs. 2 and 3. The instantaneous SADS (thin black line) with a lifetime of 65 fs describes the S2 state of spheroidene and is identical to that of the sequential model in Fig. 2, as expected. The red SADS belongs to the vibrationally hot S1 state and is characterized by a decay constant of 320 fs. The blue SADS has a lifetime of 1.5 ps and corresponds to the relaxed S1 state, with the relaxation process visualized spectrally by the complete loss of the ESA wing on the low-energy side. It is important to note that the shoulder on the high-energy side of the ESA at 535 nm that was present in the sequential model (Fig. 2, red and blue SADS) is now absent, indicating that the spectral separation of the spectrum of S1 from a coexisting species (S*, see below) has been successful, as in spirilloxanthin (10). At the same time, the decay of S2 gives rise to the green SADS, which peaks at 535 nm and is characterized by a lifetime of 5 ps; it is ascribed to S*, which is well separated from S1 both spectrally and temporally. The magenta SADS follows, and its features correspond to those of the spheroidene triplet state. The apparent decay in the induced absorption around 530 nm that occurs on the time scale of several 10s of ps (see Fig. 2) is not considered further here. In the IR region (data not shown), the processes describe solely the BChl Qx and Qy excited states with lifetimes of 100 fs and 1 ns, respectively. The bleach of the BChl Qx band at 590 nm is present also in the visible region, where because of the complex dynamics it has been impossible to separate it from the spheroidene ESA, and it appears on the red wing of the SADS. Table 1 summarizes the results of the target analysis that are relevant to the role of the singlet excited states of spheroidene in the EET to BChl, and Table 2 includes the yields and the branching ratios of each compartment. The Qy level of B850 receives 91% of the total energy, whereas the spheroidene triplet yield is 5%. The spectra of the excited states of the system that are shown in Fig. 5 correspond to values for the branching from S2 to the four compartments that imply that the direct EET from S2 to BChl accounts for 57 ± 10% of the total. The S1 state has an overall EET efficiency of 87%, with the loss channel via IC to the spheroidene ground state accounting for 4% of the total energy. The S* state is formed with an efficiency of 18.5 ± 5%, and including the S* → BChl EET in the model is essential for reaching a high total EET efficiency, pointing out the importance of this pathway that accounts for the transfer of 13 ± 4% of the total energy. This fraction is consistent with the amplitude of the slow rise component in the 860-nm trace (Fig. 6).
Figure 5.

SADS and respective ascription to pure states of spheroidene as determined by the target analysis modeling. The SADS below 535 nm is common for S2, hot S1, and S1.
Table 1.
The lifetimes of the spheroidene singlet excited states and the contributions that they make to the 91% total EET efficiency to BChl
| Donor state | S2 | Hot S1 | S1 | S* |
|---|---|---|---|---|
| Lifetime | 65 fs | 320 fs | 1.5 ps | 5 ps |
| EET to BChl, % | 57 | 5 | 16 | 13 |
These parameters were determined by the target analysis as described in the text and correspond to the SADS shown in Fig. 5.
Discussion.
The results presented here show that an additional singlet excited state other than S1 and S2 of spheroidene may be involved in photosynthetic LH. It becomes clear that these molecules possess a complicated manifold of singlet excited states that are inaccessible by one-photon absorption by the ground state but can be used by the organism for efficient LH. Our measurements on the LH2 complex of Rb. sphaeroides are in good agreement with the previous work concerning the S1 state (2, 21, 25). However, we report the observation of an additional singlet state in spheroidene, which we refer to as S* following similar observations in spirilloxanthin in the LH1 complex of Rs. rubrum (10). We present a complete view of the dynamics of this state as they are reflected in the visible region, and we resolve its main function in the LH2 antenna. We conclude that S* is populated directly from S2 after excitation and constitutes an additional channel of energy flow toward the BChl, thereby carrying an obvious biological significance. The spheroidene triplets that we observe a few picoseconds after excitation are most likely a by-product of S*. They do not play an apparent biological role and very likely are created via the homofission mechanism as we have demonstrated recently for spirilloxanthin in the LH1 antenna of Rs. rubrum (10).
Kingma has performed optical experiments in magnetic fields on the LH2 complex of Rb. sphaeroides 2.4.1 (26) and showed that there is a correlation between the triplet yield and the emission of BChl. Kingma observed that a magnetic field of 250 G increases the spheroidene triplet yield and at the same time decreases the BChl fluorescence yield. On the grounds of the newly discovered S* state, this observation indicates that S* may well be on a pathway of energy flow to both the BChl and spheroidene triplet.
The existence of a low-lying carotenoid singlet excited state other than S1 and S2 was pointed out for the first time experimentally by Koyama and coworkers (13), who identified a state that they labeled 1B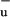 by measuring the resonance-Raman excitation profiles of a variety of carotenoids in crystalline hosts and of spheroidene in the LH2 complex of Rb. sphaeroides 2.4.1. They proposed that the 1Bu− state acts as a short-lived intermediate in the IC process from S2 to S1, which, in a weak, Fermi's golden rule-type coupling, would otherwise be inefficient because of their different symmetries (27). However, recent transient absorption experiments with sub-10-fs time resolution have shown that the formation of S1 immediately follows the decay of S2 without the involvement of an intermediate state (28). Moreover, the S2–S1 IC process in carotenoids and polyenes is very rapid and efficient irrespective of the length of the conjugated polyene chain (29). In light of this experimental observation, the involvment of 1B
by measuring the resonance-Raman excitation profiles of a variety of carotenoids in crystalline hosts and of spheroidene in the LH2 complex of Rb. sphaeroides 2.4.1. They proposed that the 1Bu− state acts as a short-lived intermediate in the IC process from S2 to S1, which, in a weak, Fermi's golden rule-type coupling, would otherwise be inefficient because of their different symmetries (27). However, recent transient absorption experiments with sub-10-fs time resolution have shown that the formation of S1 immediately follows the decay of S2 without the involvement of an intermediate state (28). Moreover, the S2–S1 IC process in carotenoids and polyenes is very rapid and efficient irrespective of the length of the conjugated polyene chain (29). In light of this experimental observation, the involvment of 1B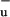 as a short-lived intermediate is hard to reconcile with theoretical calculations, which have indicated that for short polyenes 1B
as a short-lived intermediate is hard to reconcile with theoretical calculations, which have indicated that for short polyenes 1B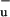 lies energetically above S2, and for long polyenes the energy of 1B
lies energetically above S2, and for long polyenes the energy of 1B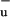 approaches that of S1 (30). Quantum chemical calculations recently indicated that the S2 and S1 surfaces in polyenes may intersect conically (29), which would explain the aforementioned properties of the S2–S1 IC process without the need to invoke 1B
approaches that of S1 (30). Quantum chemical calculations recently indicated that the S2 and S1 surfaces in polyenes may intersect conically (29), which would explain the aforementioned properties of the S2–S1 IC process without the need to invoke 1B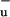 as an intermediate state. The absence of a conical intersection of the S1 and 1B
as an intermediate state. The absence of a conical intersection of the S1 and 1B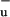 surfaces at coordinates favorable for IC would allow for a coexistence of these states, which is consistent with our experimental observation that S1 and S* evolve independently.
surfaces at coordinates favorable for IC would allow for a coexistence of these states, which is consistent with our experimental observation that S1 and S* evolve independently.
Despite unraveling the role of S* in the LH function of carotenoids in photosynthetic antennae and elucidating its spectroscopic properties, the questions concerning its nature remain unsettled. The covalent 1B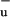 state of carotenoids bears a double triplet (TT) character (30) and therefore is a plausible assignment given the significant amount of triplets that we observe being generated via this state. In this case the singlet-fission pathway competes with EET to BChl and leads to the creation of a pair of triplets on the same carotenoid molecule. Each of the triplets resides on a different part of the long polyene chain and experiences a different protein environment or a different molecular conformation (30). These properties probably account for some of the effects observed after the triplet has been generated and which require further study. The occurrence of S* could be a consequence of distortion or twisting of the carotenoid. The possibility that it has a partial charge-transfer character cannot be excluded, and formation of a carotenoid radical seems unlikely because such a species is not expected to have ESA in the visible spectrum (e.g., see ref. 31).
state of carotenoids bears a double triplet (TT) character (30) and therefore is a plausible assignment given the significant amount of triplets that we observe being generated via this state. In this case the singlet-fission pathway competes with EET to BChl and leads to the creation of a pair of triplets on the same carotenoid molecule. Each of the triplets resides on a different part of the long polyene chain and experiences a different protein environment or a different molecular conformation (30). These properties probably account for some of the effects observed after the triplet has been generated and which require further study. The occurrence of S* could be a consequence of distortion or twisting of the carotenoid. The possibility that it has a partial charge-transfer character cannot be excluded, and formation of a carotenoid radical seems unlikely because such a species is not expected to have ESA in the visible spectrum (e.g., see ref. 31).
Conclusions
With this article we report the discovery of S*, a singlet state of spheroidene that is transferring energy to BChl in the LH2 antenna of Rb. sphaeroides. This state was observed already on spirilloxanthin of the LH1 antenna of Rs. rubrum (10). In that case it was asserted that S* may be characteristic for spirilloxanthin, and its appearance could be related to its particularly long polyene chain. In view of the differences in the structure and the carotenoid content between the two antenna systems, we can assert that the observed S* state is most probably an excited state inherently present in the carotenoids of many photosynthetic LH complexes. It is likely that the structural differences between various carotenoids and the variations within the protein environment of different complexes define the exact character and dynamics of this carotenoid excited state. Thus far we have identified two different carotenoid properties associated with S*: the direct generation of triplets in picoseconds by singlet fission and the EET process to BChl, of which the latter is of physiological importance.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Jan Amesz, Emeritus Professor of Biophysics at the University of Leiden. We recollect Jan's devotion to his students and colleagues and are grateful for his unconditioned support. Jan untimely died on January 29, 2001 (J.T.M.K. and R.v.G.). This project was supported by The Netherlands Organization for Scientific Research through the Council of Earth and Life Sciences (E.P., J.T.M.K., and R.v.G.).
Abbreviations
- LH
light harvesting
- (B)Chl
(bacterio)chlorophyll
- IC
internal conversion
- B800 and B850
BCh1 absorption band at 800 and 850 nm, respectively
- Rs.
Rhodospirillum
- Rb.
Rhodobacter
- EET
excitation energy transfer
- SADS
species-associated difference spectrum
- SE
stimulated emission
- ESA
excited-state absorption
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Frank H A, Cogdell R. In: Carotenoids in Photosynthesis. Young A, Britton G, editors. London: Chapmann & Hall; 1993. pp. 252–326. [Google Scholar]
- 2.Ritz T, Damjanovic A, Schulten K, Zhang J-P, Koyama Y. Photosynth Res. 2000;66:125–144. doi: 10.1023/A:1010750332320. [DOI] [PubMed] [Google Scholar]
- 3.van Grondelle R, Dekker J P, Gillbro T, Sundström V. Biochim Biophys Acta. 1994;1187:1–65. [Google Scholar]
- 4.Sundström V, Pullerits T, van Grondelle R. J Phys Chem B. 1999;103:2327–2346. [Google Scholar]
- 5.Frank H A, Farhoosh R, Gebhard R, Lugtenburg J, Gosztola D, Wasielewski M R. Chem Phys Lett. 1993;207:88–92. [Google Scholar]
- 6.Duysens LNM. Ph.D. thesis. Utrecht, The Netherlands: University of Utrecht; 1952. [Google Scholar]
- 7.van Dorssen R J, Breton J, Plijter J J, Satoh K, van Gorkom H J, Amesz J. Biochim Biophys Acta. 1987;893:267–274. [Google Scholar]
- 8.Gradinaru C C, van Stokkum I H M, Pascal A A, van Grondelle R, van Amerongen H. J Phys Chem B. 2000;104:9330–9342. [Google Scholar]
- 9.Kennis J T M, Gobets B, van Stokkum I H M, Dekker J P, van Grondelle R, Fleming G R. J Phys Chem B. 2001;105:4485–4494. [Google Scholar]
- 10.Gradinaru C C, Kennis J T M, Papagiannakis E, van Stokkum I H M, Cogdell R J, Fleming G R, Niederman R A, van Grondelle R. Proc Natl Acad Sci USA. 2001;98:2364–2369. doi: 10.1073/pnas.051501298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rademaker H, Hoff A J, van Grondelle R, Duysens L N M. Biochim Biophys Acta. 1980;592:240–257. doi: 10.1016/0005-2728(80)90185-1. [DOI] [PubMed] [Google Scholar]
- 12.Tavan P, Schulten K. J Chem Phys. 1986;85:6602–6609. [Google Scholar]
- 13.Sashima T, Nagae H, Kuki M, Koyama Y. Chem Phys Lett. 1999;299:187–194. [Google Scholar]
- 14.MacDermott G, Prince S M, Freer A A, Hawthornthwaite-Lawless A M, Papiz M Z, Cogdell R J, Isaacs N W. Nature (London) 1995;374:8012–8023. doi: 10.1016/s0969-2126(96)00050-0. [DOI] [PubMed] [Google Scholar]
- 15.Cogdell R J, Hipkins M F, MacDonald W, Truscott G. Biochim Biophys Acta. 1981;634:191–202. doi: 10.1016/0005-2728(81)90138-9. [DOI] [PubMed] [Google Scholar]
- 16.Hawthornethwaite-Lawless A M, Cogdell R J. In: The Chlorophylls. Scheer H, editor. Boca Raton, FL: CRC; 1991. pp. 493–528. [Google Scholar]
- 17.van Stokkum I H M, Scherer T, Brouwer A M, Verhoeven J W. J Phys Chem. 1994;98:852–866. [Google Scholar]
- 18.Holzwarth A R. In: Biophysical Techniques in Photosynthesis. Amesz J, Hoff A J, editors. Dordrecht, The Netherlands: Kluwer; 1996. pp. 75–92. [Google Scholar]
- 19.De Weerd F L, Van Stokkum I H M, Van Grondelle R. Chem Phys Lett. 2002;354:38–43. [Google Scholar]
- 20.Shreve A P, Trautman J K, Frank H A, Owens T G, Albrecht A C. Biochim Biophys Acta. 1991;1058:280–288. doi: 10.1016/s0005-2728(05)80248-8. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J-P, Fujii R, Qian P, Inaba T, Mizoguchi T, Koyama Y, Onaka K, Watanabe Y. J Phys Chem B. 2000;104:3683–3691. [Google Scholar]
- 22.Frank H A, Bautista J A, Josue J, Pendon Z, Hiller R G, Sharples F P, Gosztola D, Wasielewski M R. J Phys Chem B. 2000;104:4569–4577. [Google Scholar]
- 23.Herek J L, Polívka T, Pullerits T, Fowler G J S, Hunter C N, Sundström V. Biochemistry. 1998;37:7057–7061. doi: 10.1021/bi980118g. [DOI] [PubMed] [Google Scholar]
- 24.Kennis J T M, Streltsov A M, Vulto S I E, Aartsma T J, Nozawa T, Amesz J. J Phys Chem B. 1997;101:7827–7834. [Google Scholar]
- 25.Walla J P, Linden P A, Hsu C-P, Scholes G, Fleming G R. Proc Natl Acad Sci USA. 2000;97:10808–10813. doi: 10.1073/pnas.190230097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kingma H. Ph.D. thesis. Leiden, The Netherlands: Rijksuniversiteit; 1983. [Google Scholar]
- 27.Zhang J P, Inaba T, Watanabe Y, Koyama Y. Chem Phys Lett. 2000;332:351–358. [Google Scholar]
- 28.Cerullo G, Lanzani G, Zavelani-Rossi M, De Silvestri S. Phys Rev B. 2001;63:1–4. [Google Scholar]
- 29.Fuss W, Haas Y, Zilberg S. Chem Phys. 2000;259:273–295. [Google Scholar]
- 30.Tavan P, Schulten K. Phys Rev B. 1987;36:4337–4357. doi: 10.1103/physrevb.36.4337. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J-P, Fujii R, Koyama Y, Rondonuwu F S, Watanabe Y, Mortensen A, Skibsted L H. Chem Phys Lett. 2001;348:235–241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.