

Abstract
Targeting intractable proteins remains a key challenge in drug discovery, as these proteins often lack well-defined binding pockets or possess shallow surfaces not readily addressed by traditional drug design. Covalent chemistry has emerged as a powerful solution for accessing protein sites in difficult to ligand regions. By leveraging activity-based protein profiling (ABPP) and LC-MS/MS technologies, academic groups and industry have identified cysteine-reactive ligands that enable selective targeting of challenging protein sites to modulate previously inaccessible biological pathways. Cysteines within a protein are rare, however, and developing covalent ligands that target additional residues hold great promise for further expanding the ligandable proteome. This review highlights recent advancements in targeting amino acids beyond cysteine binding with an emphasis on tyrosine- and lysine-directed covalent ligands and their applications in chemical biology and therapeutic development. We outline the process of developing covalent ligands using chemical proteomic methodology, highlighting recent successful examples and discuss considerations for future expansion to additional amino acid sites on proteins.

1. Introduction
Advancements in covalent drug discovery have alleviated the long-standing stigma that electrophilic compounds induce adverse drug events due to their promiscuous reactivity in living systems, thereby limiting their clinical applications. − The recent FDA approval of Sotorasib (Amgen, 2021) underscores the utility of covalent chemistry for developing highly targeted therapies for historically intractable protein classes. Sotorasib incorporates a strategically placed acrylamide moiety within a KRAS-targeting scaffold, enabling it to react with the mutant cysteine residue and lock KRAS G12C in its inactive state by outcompeting GTP binding. , Reversible KRAS inhibitors have proven ineffective due to the high intracellular concentrations of GTP and GDP (submillimolar range) and their strong affinities for KRAS (K d ∼ 20 pM) under physiological conditions, which have historically rendered KRAS ″undruggable″. − Covalent compounds, however, bridge this gap by targeting sites considered undruggable by reversible ligands, thereby addressing critical but challenging targets in medicinal chemistry and advancing precision medicine through amino acid-selective electrophiles that enhance target specificity.
In addition, covalent compound selectivity can be globally evaluated and derisked using ABPP, which quantifies on- and off-target interactions across the proteome via chemoproteomics coupled with tandem liquid chromatography/mass spectrometry (LC-MS/MS). − This proteome-wide assessment facilitates structure–activity relationship (SAR) studies for optimizing drug profiles and exploring novel or previously intractable targets using irreversible compounds. − Furthermore, the identification of site of binding via chemoproteomics provides valuable insights for optimizing small molecules through in silico studies. −
Numerous amino acid-selective electrophiles have been developed, including iodoacetamide (IA) for cysteines and fluorophosphonates for active-site serines. − Cysteine has been extensively targeted for chemical probe and drug discovery due to its high nucleophilicity, localization in functional pockets and demonstrated ligandability with electrophilic ligands. Several cysteine-targeting covalent ligands have been translated into drugs with Sotorasib, an FDA-approved drug for solid tumors serving as a notable example. , Ritlecitinib (Pfizer, 2023) serves as a notable example of a cysteine-targeting covalent drug for a nononcology indication. This novel therapeutic agent for alopecia areata exhibits exceptional selectivity for JAK3 compared to other JAK isoforms. , The high selectivity of Ritlecitinib is primarily due to its ability to form a covalent bond with C909, a residue uniquely present in JAK3. , Interestingly, this cysteine residue is conserved across TEC family kinases, enabling Ritlecitinib to exert dual inhibitory effects on both JAK3 and TEC family members. This distinctive dual mechanism of action, combined with its irreversible binding properties, offers significant advantages in terms of selectivity, and therapeutic efficacy, particularly for alopecia areata treatment. Recognizing its potential, the FDA approved Ritlecitinib in 2023 for the treatment of severe alopecia areata in patients aged 12 years and older. ,
Efforts to expand electrophiles targeting amino acids beyond cysteine stem from the low abundance of this residue in protein (1.7% of amino acids in the human proteome). Probes targeting other nucleophilic amino acids such as tyrosine, lysine, histidine, serine, threonine, aspartic-, and glutamic-acid have been reported. − Tyrosine is more abundant (3.2%) compared with cysteines and has recently been targeted by sulfone-based electrophiles, thus expanding the ligandable proteome. , Lysine is also of high interest due to its abundance in human proteins (6.0%) and relatively low mutation rates compared to other residues. , This highlights the potential of lysine-reactive chemistry to uncover new biological insights.
In this review, we discuss both historical and recent advances (within the past 3 years) in covalent targeting of lysine and tyrosine residues in cellular proteomes using electrophilic small molecules. Methods involving the use of photo-, enzyme-, and transition metal-catalyzed modification are beyond the scope of this review and will not be covered. Instead, we direct readers to the following comprehensive reviews. −
2. Development of Novel Covalent Ligands
2.1. Rate Constants and Mechanism of the Bond Formation
Enzyme inhibitors can function through either reversible or irreversible mechanisms, each characterized by distinct binding dynamics and reaction pathways (Figure ). Reversible noncovalent inhibitors suppress enzymatic activity by forming a transient enzyme–inhibitor complex [P + L] in a single-step reaction (Figure A). The end point of this interaction is defined by the equilibrium state, which reflects the balance between association and dissociation of the inhibitor. In contrast, irreversible inhibitors achieve complete enzymatic inactivation through covalent bond formation, rendering dissociation negligible (Figure B).
1.

Overview of the binding mechanisms between protein [P] and reversible/irreversible ligand [L]. The modes of protein-ligand interaction are generally classified as (A) one-step reversible, (B) two-step irreversible, (C) one-step irreversible, and (D) two-step covalent reversible binding. kon = noncovalent reversible association rate constant, k off = noncovalent reversible dissociation rate constant, k inact = maximum potential rate of inactivation, k chem = single turnover rate constant, k on,cov = covalent reversible association rate constant, k off,cov = covalent reversible dissociation rate constant K I = [P][L]/[P+L]; inhibition constant for reversible binding.
Unlike traditional fast-binding inhibitors, some inhibitors exhibit time-dependent or slow-binding behavior, where steady-state equilibrium or full inactivation occurs gradually. , This phenomenon is particularly common among inhibitors employing covalent binding modes, as the formation of a covalent adduct is not an instantaneous process. For reversible covalent inhibitors, the binding process involves two distinct steps: an initial rapid interaction forms a noncovalent enzyme–inhibitor [P + L] complex, followed by a slower step in which a covalent [P – L] adduct is formed. Importantly, the covalent [P – L] adduct remains in equilibrium with the noncovalent [P + L] complex due to its reversible nature (Figure D). The steady-state inhibition constant captures this overall equilibrium. Optimization of reversible covalent inhibitors typically focuses on enhancing binding affinity by reducing dissociation rates, as slower off-rates and prolonged residence times are associated with improved therapeutic efficacy. −
In contrast, irreversible inhibitors form covalent [P – L] adducts which do not revert to the starting materials. This irreversible mechanism alters how inhibitor potency is assessed, as kinetic parameters such as IC50 can vary depending on incubation time. , Many irreversible inhibitors follow a two-step mechanism: an initial noncovalent interaction forms a [P + L] complex, which subsequently transitions into a covalent [P – L] adduct. The potency of such inhibitors depends on both their noncovalent binding affinity (K I ) and the maximum rate of inactivation (k inact). The ratio k inact/K I is widely regarded as the most reliable measure of potency for two-step irreversible inhibitors. −
On the other hand, highly reactive compounds such as thiol-alkylating agents may bypass the noncovalent intermediate entirely and bind directly in a single step (Figure C). , For one-step mechanism, parameter like k chem is used to evaluate potency and efficiency.
The strategies for developing reversible and irreversible covalent inhibitors can differ substantially. For reversible covalent inhibitors, efforts focus on optimizing overall affinity and prolonging residence time to enhance therapeutic effects. On the other hand, designing irreversible covalent inhibitors requires balancing rapid covalent bond formation with minimizing off-target reactivity toward undesired proteins or endogenous nucleophiles such as glutathione (GSH). Clinically relevant ranges for k inact/K I must account for factors such as the nucleophilicity of the targeted amino acid residue and competition from endogenous molecules.
For instance, Sotorasib exhibited a k inact value of 6.24 × 10–3 s–1, and a K I value of 3.11 × 10–7 M, which is sufficient to develop as a drug. , By understanding these distinct mechanisms and optimizing key parameters, researchers can tailor inhibitor designs to achieve desired therapeutic outcomes while minimizing adverse effects.
2.2. Covalent Ligand Discovery with LC-MS/MS
ABPP coupled with LC-MS/MS is a powerful tool for developing broad-profiling probes and residue-directed covalent inhibitors. For example, competitive ABPP using cysteine-reactive covalent probes such as desthiobiotin-iodoacetamide (DBIA) and acrylamide-based ligands represent a well-established approach for covalent targeting of cysteines in the field. These probes incorporate either reporter tags directly or terminal alkynes that react with reporter-N3 reagents via click chemistry (Copper(I)-catalyzed azide–alkyne cycloaddition or CuAAC) for detection and quantification of modified protein sites.
Activity-based probes (ABPs) covalently bind a wide range of proteins and are utilized in ABPP (Figure ). This approach can monitor changes in protein binding activity under various activation stimuli. In drug discovery, competitive ABPP provides invaluable insights for medicinal chemists. First, it enables the evaluation of irreversible and reversible (under kinetically controlled conditions) ligand affinities for proteins of interest in lysates, cells and animals. Traditional drug discovery methods often rely on High-Throughput Screening (HTS) to identify ligands for protein targets; however, these approaches face significant limitations, including high cost, challenges in validating on- and off- target engagement of proteins in cells and the lack of assays to evaluate activity of target proteins without prior biological insights. − Collectively, these aforementioned factors contribute to many proteins being classified as undruggable. ,
2.

Schematic overview of (A) LC-MS/MS ABPP and isotopic labeling by SILAC (B), and TMT (C).
In contrast, LC-MS/MS facilitates the identification of proteins and their modified residues across entire proteomes, and selective ABPs serve as effective tools for functional evaluation. − Consequently, ABPP strategies can address a wider range of protein classes and probe binding in some instances can serve as surrogate activity assays for challenging targets.
Second, ABPP provides critical information on both the selectivity and potency of ligands for target proteins. Labeling techniques such as Stable Isotope Labeling by Amino acids in Cell culture (SILAC), isotopically labeled desthiobiotin azide (isoDTB), and more recently, Tandem Mass Tag (TMT) reagents utilize isotopic labeling strategies in LC-MS/MS to quantify peptides modified by ABPs. − These methods, combined with advanced data analysis software like SEQUEST, Comet, MaxQuant and Byonic, play a pivotal role in interpreting complex LC-MS/MS data. This underscores the dual importance of effective labeling strategies and robust computational tools in ABPP experiments. Additionally, pretreatment with potential covalent binders can block probe binding, allowing for the assessment of binding affinities for irreversible ligands. To maximize protein detection and minimize undetectable off-target effects caused by irreversible ligands, highly reactive and residue-directed probes are essential. The IA electrophile is a potent thiol alkylating agent and thus its alkynylated probe is highly effective as a general cysteine-reactive ABP. Counterpart ABPs are more challenging to develop and a reason why beyond cysteine ABPP is only recently emerging.
A general workflow employed by our group and others to develop electrophilic groups for protein and covalent ligand discovery by ABPP consists of the following steps: 1) Chemoselective ABP Development: Develop new global ABPs featuring tunable electrophilic warheads and analyze their binding activity including amino acid chemoselectivity in proteomes. Simplicity and high reactivity are prioritized to maximize detection of modified proteins and binding sites. Overly complex ABP structures risk fragmentation and poor LC compatibility during analysis, leading to reduced detection efficiency. 2) Ligandability assessment using Fragment Electrophiles and Competitive ABPP: Construct a library of fragment-like compounds containing electrophilic warheads for evaluating ligandability of protein sites containing residues of interest by competitive ABPP. The use of a common electrophile for ligand and ABP design can reduce the potential for missing liganded sites not detected by ABPs. 3) Prioritization of liganded sites via functional assays: Validate biochemical and cell biological effects from genetic and pharmacological perturbation of target protein site(s). 4) Targeted covalent modulator development: Translate ligandability findings into highly potent and selective tool compounds for cellular and in vivo evaluation of target protein. Conduct SAR studies to reduce off-target affinities identified in ABPP results and enhance potency against target proteins. In silico docking studies or molecular dynamics simulations can assist in fine-tuning affinities for both on- and off-targets. Additionally, pharmacokinetic optimization is required to balance intrinsic reactivity with hydrolytic and metabolic stability.
An example from our group is the development of a PTGR2 (Y100) covalent inhibitor containing a Sulfonyl-Azole (SufAz) warhead, which illustrates this workflow in practice (Figure ). , SufAz ABPs were designed as tyrosine/lysine-selective warheads with negligible detectable affinity for cysteine, which we speculate is due to the inherent instability arising from the predicted sulfonothioate (SO2–S) adduct. , Using these ABPs, quantitative ABPP methods were employed to evaluate potency and selectivity of a library of sulfonyl-azole fragment electrophiles for PTGR2 inactivation. Among these compounds, HHS-0701 emerged as a lead PTGR2 inhibitor with reasonable proteome-wide selectivity and cellular activity. Further SAR studies demonstrate that exchanging the triazole for an imidazole leaving group could further improve selectivity against common off-targets of this electrophile class.
3.

Example workflow for ‘beyond cysteine’ ABPP and protein ligand discovery. The deployment of a tunable electrophilic scaffold enables seamless development of reactive probes and corresponding fragment compounds for chemoselectivity and ligandability assessment, respectively. This approach when combined with medicinal chemistry led to discovery of RJG-3017, a targeted covalent inhibitor of the lipid enzyme PTGR2 derived from the lead hit HHS-0701. ,
Originating from the conceptual framework of activity-based protein profiling (ABPP) in drug discovery, the Kelly and Sharpless groups introduced the Inverse Drug Discovery (IDD) approach. Unlike global ABPs, which typically use broadly reactive probes, IDD employs alkynyl-substituted probes containing latent electrophilic warheads, such as aryl fluorosulfates and sulfuramidimidoyl fluorides. These probes are used for the biological evaluation of their own activity, or that of the corresponding ligand, with minimal chemical modification. Due to their weak intrinsic reactivity for covalent binding, only a limited number of conjugated proteins are identified via LC-MS/MS compared to the use of global ABPs. Although the complexity of the chemical structure may pose challenges for protein identification, this strategy can significantly reduce concerns about unexpected off-target effects when paired with an appropriately selected adduct group.
In addition, residue-directed warhead development enables selective targeting of mutants and addresses limitations associated with reversible inhibitors that exhibit short residence times due to open binding sites. Simple incorporation of residue-reactive warheads can convert reversible inhibitors into irreversible ones with enhanced therapeutic potential. ,
3. Residue-reactive Scaffolds
3.1. Cysteine-reactive Scaffolds
For drug development, cysteine is an attractive target in part because of its high nucleophilicity and low abundance, which can facilitate increased selectivity for the target proteins. − Particularly, an acrylamide electrophilic group has been extensively studied and now clinically validated for development of covalent drugs. Among acrylamide-based covalent drugs, kinases have been prominently targeted because of the ability to ligand nonconserved cysteines within the highly conserved ATP binding pocket to develop more selective drug compounds. Examples include Ibrutinib (Abbvie, 2013), − which is an acrylamide inhibitor that binds to C481 of Bruton’s tyrosine kinase (BTK) as well as series of covalent inhibitors which targeted C797 of Epidermal growth factor (EGFR) to combat T790M mutation such as Afatinib (Boehringer Ingelheim, 2013) and Osimertinib (Astrazeneca, 2015). −
Many probes with diverse cysteine-reactive warheads have also been developed. In addition to IA, many α-halogenated amide and α,β-unsaturated amides were developed as covalent probes. − In addition, halogenated heteroaromatic compounds, including simple probes and nucleoside and nucleobase mimics for RNA binding proteins (e.g., clickable electrophilic purines), undergo nucleophilic aromatic substitution (SNAr) with cysteines. − For additional information on targeting cysteines, we direct readers to recent reviews on cysteine-reactive electrophiles and their applications. −
The rationale for targeting cysteines is clear but not every protein pocket contains an available cysteine, prompting exploration of electrophilic groups that can ligand sites beyond cysteine. In the following sections, we summarize recent advancements in electrophile development for beyond cysteine targeting. We initially focus on lysine-targeting chemistry given the high abundance of lysines and their functional roles in protein biochemistry. We conclude the review with electrophiles for targeting tyrosine, which lie in between cysteine and lysine in abundance and present opportunities for disrupting protein–protein and protein-nucleic acid recognition. −
3.2. Lysine-reactive Scaffolds
3.2.1. Biological Features of Lysine
The pK a of the ε-NH3 + group of lysine is 10.4 when lysine exists as a free amino acid, meaning that most lysine residues are protonated under physiological conditions (pH = 7.4). However, the pK a of lysine can vary significantly depending on its location within a protein. For example, studies have shown a 5-log reduction in pK a for buried vs solvent-exposed lysine side-chain, thereby increasing its nucleophilicity and propensity for electrophilic modification. − García-Moreno et al. , produced 25 variants of staphylococcal nuclease (SNase) to analyze shifts in pK a of lysine side chains based on their environment. The side chain of the K92 (I92K) mutant is buried in SNase and was calculated to have a pK a of 5.2, whereas solvent exposed N118K has a pK a of 10.4 (Figure A).
4.

(A) Crystal structure of staphylococcal nuclease (SNase) with selected sites mutated to lysine (generated using Pymol) and their pK a values based on published work from García-Moreno et al. (B) Example post-translational modifications (PTMs) found on lysine residues.
Lysine undergoes various PTMs, which play critical roles in cell biology including signal transduction (Figure B). − More than 100,000 PTM sites on lysine across over 10,000 proteins have been mapped using mass spectrometry; however, many PTM functions remain elusive. , PTMs on lysine include acyl substitution (e.g., acetylation, ubiquitination) with various acyl substrates or alkylation (e.g., methyl-, dimethyl-, or trimethylation) mediated by methyltransferases. , The nucleophilicity of lysine correlates with frequency of PTMs such as acetylation. ,
Lysine is also prone to oxidative deamination to an aldehyde that is well studied and commonly observed in histones. Oxidative deamination of lysine is mediated by copper-dependent amine oxidases (CAOs).
CAOs catalyze oxidative amination using a covalently linked quinone cofactor 2,4,5-trihydroxyphenylalanine quinone (TPQ) or lysine tyrosylquinone (LTQ) that subdivide the CAO family into TPQ-dependent CAOs (i.e., CAOs) or LTQ-dependent lysyl oxidases (LOXs). − Lysine can also sequester carbon dioxide via a carboxylation process and cyanate via carbamylation. , Particularly, many PTMs on lysine are reversibly regulated by enzyme pairs such as histone acetylases (HACs) and histone deacetylases (HDACs), depending on physiological conditions. Due to the functional importance mentioned above, there are considerable research efforts focused on developing molecules that can perform PTMs such as acetylation on specific lysine residues. − Thus, the covalent modification of lysine can be influenced by its protonation state and interactions with various PTMs, as described above, making lysine an attractive residue for covalent modification.
3.2.2. Lysine-Targeted Chemistry
3.2.2.1. Natural Lysine-targeted Aldehydes and Mimetics
The formation of aldimines is commonly used in organic chemistry such as the popular reductive amination reaction that delivers primary, secondary, or tertiary amines. The use of aldehydes to produce aldimines for bioconjugation to lysine on proteins is limited by the thermodynamic instability of imines, which rapidly revert to their aldehyde and amine precursors in aqueous solutions. −
Efforts to stabilize the imine under aqueous conditions were inspired by the ability of B6 vitamers PL and PLP, the latter of which is an essential vitamin and the active B6 vitamer, to conjugate with amines in xenobiotics and biomolecules. − This topic has been comprehensively reviewed by Eliot and Kirsch so only recent literature will be covered here. PLP is known to covalently modify lysine in various proteins, which is utilized by alanine racemase, , aspartate aminotransferase, ,, serine hydroxymethyltransferase, aromatic L-amino acid decarboxylase, and other proteins to facilitate transformations of amino acids (Figure A). Using NMR, Crugeiras et al. characterized the strength of the intramolecular hydrogen bond between the imine and o-hydroxy proton of amine-conjugated pyridoxal in aqueous solution to be ∼ 3 kcal/mol (Figure B). ,
5.

(A) Scheme depicting the mechanism of amine transfer beginning with pyridoxal phosphate (PLP)-bound aspartate aminotransferase (AAT), and crystal structure of PLP bound to K258 in AAT (PDB ID: 1ARS ). (B) Schematic of the conversion of pyridoxal (PL), a B6 vitamer, to the active PLP and subsequent conversion to PLP-therapeutic conjugates. The stability of the imine is enhanced by a strong intramolecular hydrogen bond (highlighted in red). (C) Examples of therapeutics that are known to conjugate with PLP. (D) Chemical structures of ABPs mimicking PLP. The structural isomer (4) of PLP mimicking ABP 1 was used as an inactive control.
The pyridoxal-amine intramolecular hydrogen bond is more stable than i+4 hydrogen bonds in an α-helix (about 2 kcal/mol). , In E. coli, PLP binds K36 in PLP Homeostasis Protein (PLPHP or YggS) with a K D of about 1 nM and has no enzymatic function, suggesting PLPHP acts as a regulatory or PLP-transfer mechanism to modulate PLP levels or mediate PLP-dependent enzymatic reactions, respectively; however, more evidence is required to define the role of PLPHP. The ability of PLP to bind lysine in proteins warrants discussion of amine- and hydrazine-containing therapeutics that interact with PLP resulting in vitamin B6 deficiency or inactivation of the therapeutic itself. The interaction between isoniazid, the first-line treatment for tuberculosis, and PLP was one of the first drug-PLP interactions to be investigated due to induction of isoniazid neuritis in patients, i.e., isoniazid interference in vitamin B6 metabolism that is mitigated by coadministration with pyridoxine. − However, isoniazid interactions with other metabolites and cofactors, such as NAD+, may be involved in isoniazid-related toxicity issues. The kinetics of formation of the isoniazid-PLP conjugate is the most rapid compared to hydrazine and other hydrazine-containing drugs, such as carbidopa and hydralazine, and correlates with the pK a of the primary amine in each hydrazine moiety (e.g., isoniazid (pK a = 11.10) > hydrazine (pK a = 8.10) > carbidopa (pK a = 7.25) > hydralazine (pK a = 7.20)). , Carbidopa is used to treat Parkinson’s disease as it mitigates degradation of dopa by inhibiting aromatic L-amino acid decarboxylase (AAAD) in peripheral tissues, allowing more dopa to reach the brain. Decreases in concentration of PLP in serum (88%), liver (51%), muscle (18%), brain (27%), and hypothalamus (18%) was observed in rats administered 100 mg/kg carbidopa; however, clinically relevant doses, such as 10–30 mg/kg, were less effective at reducing PLP concentrations. Similar to isoniazid, coadministration of pyridoxine hydrochloride with carbidopa mitigates reductions in PLP levels. , In addition to the examples above, there are numerous drugs (e.g., hydralazine, phenelzine, cycloserine, − penicillamine, and amphetamine) capable of conjugating PLP resulting in vitamin B6 deficiency (Figure C). Sieber and colleagues synthesized PLP-mimicking ABPs (1-3) to detect PLP-dependent enzymes (PLP-DE) which are phosphorylated in situ by pyridoxal kinase and identify off-targets of PLP-DE inhibitor such as D-cycloserine. The inactive control 4, which lacks the electronic capability for PLP catalysis, was used to evaluate nonspecific reactivity of the PL scaffold. Using ABPs (1-3), a significant portion of the eukaryotic cells, Gram-positive, and -negative bacteria were successfully detected and utilized in antibiotic drug discovery (Figure D).
Similarly, Folkers et al. discovered that the sensitivity observed in individuals consuming food with high levels of monosodium glutamate (MSG) is linked to decreased PLP levels in the blood. Co-administration of MSG and PLP in the diet alleviated symptoms of MSG-related toxicity compared to a placebo group, suggesting that MSG conjugates with PLP, thereby reducing its concentration to unhealthy levels.
In 2014, Wilde et al. reported that hydralazine can covalently modify abasic (Ap) sites in double- and single-stranded DNA, which exist in equilibrium between the ring-closed hemiacetal (5) and ring-opened aldehyde (6). Hydralazine reacts with the aldehyde (6) to form hydrazone (7), which can undergo ring closure to produce hemiaminal (8), a structure prone to errors and potentially mutagenic (Figure A). In addition, aldose sugars naturally form advanced glycation end products (AGEs) with proteins or lipids (Figure B). These AGEs can accumulate in vessel walls, leading to micro- and macrovascular complications, , and are implicated in various aging- and alcohol-related diseases. , The formation of AGEs during aging and under hyperglycemic conditions, where they bind to cell surface receptors, increase reactive oxygen species (ROS), alter the extracellular matrix, modify hormone levels, and cross-link with other proteins such as collagen. Protein-aldose sugar bioconjugates are formed between lysine or arginine residues on a protein and an aldehyde on the sugar via mechanisms similar to those described above. To investigate biological events of AGE formation, Methylglyoxal (MG), which is major reactive carbonyl species serving as a precursor to AGEs, was employed to develop ABP for chemoproteomic profiling (Figure C). , Aminoguanidine is a therapeutic that covalently modifies 1,2-dicarbonyl compounds, increasing urinary excretion of orally absorbed AGEs and reducing their accumulation in the kidney and liver. Other AGE-related therapeutics such as ALT-946 and pyridoxamine follow a similar mechanism as aminoguanidine, whereas ALT-711 functions by breaking the bond between the 1,2-dicarbonyl (Figure D).
6.

(A) Mechanistic scheme illustrating the condensation of an aldose saccharide with hydralazine to form a stable hemiaminal. (B) Reaction scheme depicting the natural synthesis of Advanced Glycation End-Products (AGEs) through condensation between lysine residues on proteins and 1,2-dicarbonyl-containing glycans. (C) Profiling metabolites and AGE proteomes with Methylglyoxal ABP. (D) Chemical structures of AGE-related therapeutics.
3.2.2.2. Synthetic Aldehyde Warheads
As mentioned in the previous section, the inherent stability and selectivity of salicylaldehyde for reversible covalent adduction of amines inspired synthetic small molecules targeting catalytic lysines. , ORIN1001, an inhibitor of inositol-requiring enzyme 1 (IRE1), has entered clinical trials as an antitumor agent, originating from a salicylaldehyde screening hit compound 13 (Figure A). , The covalent reversible Bcr-Abl kinase inhibitor 15 was derived from the reversible inhibitor PPY-A through the introduction of aldehyde warheads (Figure B). The covalent reversible inhibitors were designed to target the catalytic lysine K271, based on the X-ray cocrystal structure of PPY-A with ABL1 kinase (Figure C, PDB ID: 2QOH). ,
7.

SAR studies for aldehyde covalent reversible inhibitor (A) ORIN1001, and (B) compound 15. (C) The X-ray cocrystal structure of the ABL1 kinase T310I mutant bound to the reversible ABL1 inhibitor PPY-A, highlighting the position of K271. (PDB ID: 2QOH ) (D) The mode of action for 2-hydroxy-1-naphthalenealdehyde derivatives (16) as PPI inhibitors.
The reversible covalent inhibitor 14, containing a carbonyl boronic acid capable of stabilizing imine intermediates, demonstrated high potency in vitro; however, its activity significantly decreased in living cells, which could be due to metabolism of the aldehyde functional group within this scaffold. , Structural modification of carbonyl boronic acid to salicylaldehyde enhanced antiproliferative effects in situ with increasing inhibitory activity to ABL, and high kinase selectivity was confirmed using ABPP with an alkynyl probe corresponding to compound 15.
Notably, solvent-exposed noncatalytic lysine residues are generally not preferred as targets for aldehyde electrophiles due to their reversible nature and high possibility to exist in the protonated form. , However, 2-hydroxy-1-naphthalenealdehydes (16) could bind specifically to K720 of KRIT1 which has inherently low nucleophilicity. K720 is a surface residue located within the HEG1 binding domain of KRIT1; thus, 16 can function as a Protein–protein Interaction (PPI) inhibitor (Figure D).
Recently, Cravan et al. modified the clinical reversible pan-AKT1 allosteric inhibitor ARQ092 into an irreversible AKT1(E17K) mutant selective inhibitor 17 by introducing salicylaldehyde warheads (Figure ). This selectivity was achieved through the recruitment of endogenous zinc ions, which stabilized the aldimine product formed between the E17K mutant and the salicylaldehyde warhead. Pan-AKT1 inhibitors have been associated with hyperglycemia as a side effect in clinical studies, − but the newly developed irreversible AKT1(E17K) mutant selective compound 17 did not induce hyperglycemia.
8.

Development of an irreversible AKT1(E17K) mutant selective inhibitor from the reversible pan-AKT inhibitor through the introduction of a salicylaldehyde warhead.
The Gao group designed boronic acids with extended residence times compared to 2-formyl or 2-keto aryl boronic acids. , By linking a 2-formylanilino group to the o-benzylic position, they observed a dramatic increase in the half-life (t 1/2 ) for covalent modification. Lysine forms an imine that coordinates with boron similarly to 2-formylboronic acids; however, the anilino nitrogen also coordinates with boron, resulting in the formation of a bicyclic ring structure (Figure ).
9.
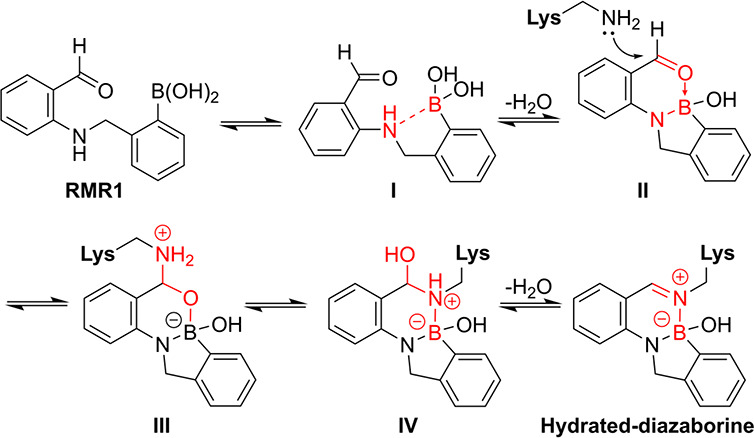
Proposed mechanism of RMR1 conjugation with lysine for the formation of a hydrated diazaborine.
The covalent reversible properties of salicylaldehyde have recently spurred exploration into diverse aldehyde-containing ABPs, which often require reduction with agents such as NaBH4 for detection. , Yang et al. developed lysine-targeted ABPs based on residence time (Figure A). While benzaldehyde (18) and salicylaldehyde probes (19-20) detected a similar number of kinases, probe 18 exhibited a short half-life following washout (Figure B). In contrast, salicylaldehydes stabilized imine covalent products, significantly increasing the half-life of reversible covalent probes after washout (Figure C). Interestingly, different dissociation rates were observed among kinases after washout, enabling ABPs to selectively bind to kinases such as Aurora Kinase A (AURKA) K162 in both in vitro and in vivo systems. Screening aminophilic compounds using ABPP with LC-MS/MS identified dicarboxaldehyde fragments, including Jensenone (21) and 22, as potential lysine- targeted scout molecules (Figure D). Building on these findings, phloroglucinol meroterpenoids and their corresponding probes were synthesized to map lysine-phloroglucinol meroterpenoid interactions in live cells (Figure D).
10.

(A) Chemical structures of the residence time-based ABPs containing a salicylaldehyde scaffold. (B) The half-life (t 1/2 ) and (C) remaining percentage of the probes for each target protein after wash-out. (D) The synthesized chemical structures of Phloroglucinol Meroterpenoids and the corresponding probes based on the dicarboxaldehyde fragments discovered from an aminophilic compound library.
Recently, Chen et al. applied a 2-ethynylbenzaldehyde (2-EBA) warhead for developing catalytic lysine-targeted ligands. Unlike other aldehyde warheads, 2-EBA irreversibly binds to the N-terminus of peptides or lysine residues, forming a stable isoquinolinium product (Figure A). Due to the low intrinsic reactivity of the 2-EBA warhead, 27 exhibited negligible reactivity with GSH. Based on the reported structure of a reversible inhibitor, 2-EBA and sulfonylfluoride derivatives were investigated (Figure B). The study demonstrated the superior efficacy and lysine-selectivity of 2-EBA derivatives. Additionally, using the 2-EBA warhead, successful modification of N-terminal amines over lysine residues was achieved under slightly acidic conditions (pH = 6.5).
11.

(A) The proposed irreversible binding mechanism of the 2-EBA warhead with lysine. (B) Chemical structures of derivatives containing the 2-EBA warhead and their corresponding reversible or sulfonylfluoride ligands.
3.2.2.3. Michael Acceptor Warheads
Although Michael acceptors such as acrylamide, vinylsulfone, and vinylsulfonamide primarily target cysteine residues, preferentially oriented Michael acceptors can also bind to lysine residues. , PX-866 (Sonolisib), an investigational drug and orally available pan-PI3K inhibitor, is a modified derivative of the natural product Wortmannin. The substituted-acrylamide of PX-866 covalently binds to and inhibits catalytic K802 in p100α and K883 in p110γ.
Through structural drug design approaches, the reversible Cyclin D kinase 2 (CDK2) inhibitor NU6155 was converted into an irreversible inhibitor NU6300 by introducing a vinylsulfonyl group targeting K87, a residue not well-conserved across the protein kinase family (Figure A). , Similarly, a reversible non-nucleoside inhibitor of HIV reverse transcriptase (33) was converted into an irreversible inhibitor (34-35) by introducing an acrylamide group near catalytic residues such as K102 and Y181C (Figure B). ,
12.

(A) Conversion of the CDK2 reversible inhibitor NU6155 into the irreversible inhibitor analog NU6300 and its X-ray cocrystal structure with CDK2 (PDB ID: 5CYI ). (B) Development strategy for HIV-1 RT irreversible inhibitors and X-ray cocrystal structure containing compound 33 (PDB ID: 4H4M ). Electrophiles were introduced at different positions on compound 33 depending on the catalytic residues of interest, such as K102, Y181C and Y181.
Additionally, studies comparing the reactivity of various Michael acceptors with GSH and N-α-acetyl-l-lysine revealed that vinylsulfone and sulfonamide react more rapidly with N-α-acetyl-l-lysine than with GSH. This observation can be attributed to the hard nucleophilic nature of N-α-acetyl-l-lysine, which interacts more effectively with hard electrophiles like vinylsulfonamides compared to softer acrylamide electrophiles. These findings provide valuable insights for optimizing lysine-targeted Michael acceptor compounds in terms of potency and stability (Figure ).
13.

Half-lives of electrophiles evaluated by their reaction with N-α-acetyl-l-lysine as a nucleophile at pH 10.2.
3.2.2.4. Activated Ester Warheads
N-hydroxysuccinimide (NHS) esters are representative lysine-reactive activated esters. One of the most widely used NHS esters, TMT reagents effectively label N-terminal free NH2 groups and lysine residues (Figure A). However, NHS esters are highly susceptible to hydrolysis, and their residue specificity can deteriorate due to their high reactivity.
14.

(A) Chemical structures and reaction mechanism of TMT reagents. 13C and 15N isotopes were used to manipulate mass differences; red asterisks indicate isotope positions for each TMT2 labeling reagent in TMT proteomics. (B, C) Chemical structures of lysine-reactive ABPs and their residue specificity profiles. (D) Rate constants for reactions depending on the structures of activated esters.
Numerous studies have attempted to minimize over labeling caused by NHS-ester-based TMT reagents, which exhibit promiscuity toward other residues such as serine, threonine, and tyrosine. , Ward et al. investigated the residue selectivity of an NHS-ester-alkyne ABP (37) in the mouse liver proteome using isotopic Tandem Orthogonal Proteolysis-Activity Based Protein Profiling (isoTOP-ABPP) platform. Their study identified 3,372 modified peptides in total, with 49% of the labeled sites corresponding to lysine residues (Figure B). Hacker et al. synthesized a variety of activated ester ABPs, including NHS, nitro-substituted phenyl, and fluoro substituted phenyl esters. Among these, the Sulfotetrafluorophenyl (STP)-alkyne ABP (38) demonstrated strong proteomic reactivity and high selectivity for lysine residues. The most frequently modified alternative sites were serine residues (∼8% of labeled peptides), with less than 5% modification observed for other residues (Figure C). Notably, lysine modification can interfere with trypsin digestion at cleavage sites, potentially leading to misinterpretation of modified sites detected by LC-MS/MS.
Additionally, using ABP 38, more than 30 aminophilic chemotypes were screened in cancer cell proteomes, revealing that 77% of the liganded proteins were non-DrugBank proteins. This finding suggests that the discovered aminophilic fragments could serve as scout molecules for targeting previously intractable proteins. Wan et al. synthesized a series of activated esters to systematically tune reactivity using lysine-containing peptides. Among these tested esters, triazine ester 42 exhibited the highest reactivity and lysine-selectivity in vitro (Figure D). Using activated ester warheads, reversible selective PI3Kδ inhibitors GSK2292767 and GSK2269557 (∼1,000-fold selectivity over PI3Kα, β, γ) were successfully converted into irreversible inhibitor 46 by modification of a sulfonamide pharmacophore interacting with K779 (Figure A). , Additionally, ATP and ADP-desthiobiotin probes have been widely used as ABPs to profile protein kinases by targeting conserved lysine residues in ATP-binding sites as well as various nucleotide-binding proteins such as ATPases and heat shock proteins. − (Figure B). Two highly conserved lysine residues are commonly found across protein kinases: one localized in subdomain II (except in the WNK family) and another in subdomain VIB within the HRDxKxxN motif (Figure C). −
15.

(A) Example of introducing an activated ester to convert a reversible inhibitor into an irreversible inhibitor 46. (B) The mechanism of tagging lysine with ATP-desthiobiotin ABP. (C) The proximity of highly conserved lysines in protein kinases to the gamma phosphate of ATP where the acyl phosphate warhead of ATP-desthiobiotin ABP is located (PDB ID: 1ATP ).
3.2.2.5. N-Acyl-N-alkylsulfonamide (NASA) Warheads
NASA moieties have been widely utilized as reactive species in safety-catch linkers for solid-phase peptide synthesis. − The NASA scaffold functions as an acyl donor that is lysine-reactive, and the Hamachi group optimized this functional group as an electrophilic warhead in ABPs (Figure A). − N-acyl-N-arylsulfonamide (ArNASA) derivatives (51) demonstrated significantly prolonged half-lives compared to NASA itself, along with increased selectivity (Figure B). Additionally, ligand-directed NASA-containing inhibitors (52-53) showed improved potency compared to their parent compounds when targeting lysine residues in proteins such as C481S Bruton’s tyrosine kinase (BTK) and Heat Shock Protein 90 (Hsp90) in live cells (Figure C). Binding of NASA warheads to N-terminal α-amines and tyrosine residues was also observed in the human double minute 2 (HDM2) protein, where they acted as PPI inhibitors. Tivon et al. further developed NASA-containing aptamers to label targeted lysine residues. In this approach, oligonucleotides facilitated proximity between the target protein and the NASA warhead, enabling lysine residues to attack the NASA electrophile and label the protein effectively. Ryan et al. reported N-acetyl-N-alkylsulfonamide derivatives capable of transferring acetyl groups site-specifically to lysine residues in human cells. Furthermore, Teng et al. developed a potential targeted degrader that combined a CDK2 ligand with a CRBN-recruiting ligand using a NASA warhead. Although the study confirmed the formation of a CDK2-CRBN complex, degradation of CDK2 was not observed.
16.

(A) Development of NASA warheads for applications in drug discovery. (B) Half-lives of various NASA derivatives depending on their chemical structures. (C) Chemical structures of NASA analogs derived from parent small molecules.
3.2.2.6. Squarate, Squaramate, and Squaramide Warheads
Squaramides are bioisosteres of ureas, thioureas, guanidines, and cyanoguanidines that are commonly employed in medicinal chemistry. , Additionally, squaramides and squaric acids have been incorporated into nucleosides as isosteres of phosphate groups. − Several methods have been developed to synthesize squarate- and squaramide-containing building blocks such as amino acids, peptides, − proteins, polymers, − nucleosides, fluorescent probes, and luminescent probes that exploit the high reactivity of squarate esters with free amines.
Kiessling and co-workers compared the reaction rates of squarate esters and squaramates with other lysine-reactive electrophiles such as NHS esters. The second consecutive reaction rate for squaramates was much slower than that for NHS esters, demonstrating their mild reactivity (>104-fold slower than NHS esters; Figure A). Subsequently, a squaramate moiety was introduced at the terminal phosphate of UDP as a substrate mimic for glycotransferase GlfT2. This compound selectively inhibited a catalytic lysine residue while dichlorotriazine analogs modified eight lysines in GlfT2. Ohara et al. designed a glycan-BODIPY-squaramate conjugate 59 that bound Human UGGT1 (HUGT1), an N-glycosylating enzyme recognizing aglycons. Irreversible modification of K1424 via reaction with squaramate 59 provided insights into the aglycon binding site and folding-sensing mechanism of HUGT1 (Figure B).
17.

(A) Comparison of reaction rates for squarate ester, squaramate, and NHS-ester. (B) Chemical structure of the HUGT1 squaramate probe (59) mimicking glycoproteins containing aglycon. (C) Broadly lysine-reactive squaramates (60-61) discovered through screening an aminophilic compound library. (D) Applications of squaramates as linkers between biomolecules and in DNA fluorescence labeling.
Lysine-specific squaramates were further explored by Abbasov et al., leading to compounds 60 and 61 as broader lysine-reactive squaramates than other sterically hindered squaramates (Figure C). Notably, 61 covalently modified K351 in Ku70 (XRCC6), inhibiting its dimerization with Ku80 (XRCC5). This process disrupts nonhomologous end-joining critical for repairing DNA double-strand breaks in human cells.
The unique properties of squaramates such as their two consecutive reactive sites and mild reactivity, have made them extensively useful in biomolecular conjugation studies. Fonvielle et al. prepared RNA-derivatized squaramates for conjugation with peptides and proteins to identify or covalently modify tRNA-enzyme binding sites. Ivancová et al. linked squarates to deoxycytidine nucleotides to create squaramate-linked dCTPs (dCESQTPs, 62), which were incorporated into DNA by DNA polymerase. These synthesized DNA probes (DNA_CESQ) efficiently reacted with DNA-binding proteins but showed limited reactivity with peptides lacking DNA-binding affinity (Figure D). As demonstrated above, lysine-specific and mild electrophilic squaramates stand out for their versatility in discovering undruggable proteins such as DNA-binding proteins through various bioconjugation strategies.
3.2.2.7. Sulfur(VI)-Fluoride Warheads: Sulfonyl Fluoride (-SO2F) and Fluorosulfate (-OSO2F)
Sulfonyl fluoride was historically regarded as an unsuitable electrophile due to its combination of kinetic and thermodynamic stability, which limited its reactivity with nucleophiles. However, Baker first demonstrated that sulfonyl fluoride could rapidly form covalent bonds if the compound first binds reversibly to a protein, facilitating access for covalent bond formation. This insight has since attracted significant interest in both drug discovery and chemoproteomics. Sulfonyl fluoride can react with residues such as tyrosine, lysine, serine, threonine, and histidine. ,, Its hydrolytic stability, which can be easily tuned, provides a valuable alternative to many unstable irreversible ligands (Figure A). , However, the affinity of the electrophile for proteins does not necessarily correlate with its intrinsic reactivity because the rate of reversible interactions between the compound and the binding site significantly influences covalent bond formation.
18.

(A) Intrinsic reactivity and hydrolytic stability of S(IV)-F electrophiles (63-71) depending on substituents. Half-lives of compounds in PBS buffer (pH = 8) are shown; values in parentheses represent half-lives at pH = 10. (B) The chemical structure of XO-44. (C) Kinome phylogenetic tree plotted using KinMap showing protein kinases detected by XO-44 (gray) in Jurkat cells and those liganded by Dasatinib (red). (D) Kinases inhibited by XO-44 based on purified kinase assays.
Zhao et al. developed XO-44, a highly selective ABP for protein kinases, which detected 133 endogenous kinases in live Jurkat cells (Figure B). XO-44 also demonstrated its potential to identify off-targets of kinase inhibitors through competitive ABPP studies with Dasatinib, a tyrosine kinase inhibitor (Figure C). Furthermore, among the 375 tested purified protein kinases, XO-44 exhibited IC50 values below 1 μM for 219 kinases (Figure D). XO-44 has also been utilized in studies identifying resistance mechanisms for the multityrosine kinase inhibitor Lenvatinib. These findings suggest that XO-44 can be leveraged to evaluate kinase inhibitor profiles and may serve as a promising scaffold for developing kinase inhibitors. ,
The Cheeseman group designed covalent inhibitors for Hsp72 by mimicking reversible nucleotide-competitive inhibitors such as 8-N-benzyladenosine through the introduction of electrophilic warheads. , For instance, introducing an acrylamide warhead converted a reversible inhibitor into an irreversible analog targeting K56. However, this modification resulted in low k inact due to unfavorable conformations between the warhead and proximal residues that hindered covalent bond formation. Based on a systematic analysis of X-ray cocrystal structures of Hsp72, a rationally designed arylsulfonylfluoride was introduced, improving the kinetic parameter (k inact /K I ) by 108-fold compared to the acrylamide derivative.
Recently, fluorosulfates (-OSO2F), which are more stable and latent aminophiles than sulfonyl fluorides, have emerged as electrophilic warheads for developing irreversible ligands. − Tang et al. introduced fluorosulfate into the first-generation EGFR inhibitor Erlotinib to target catalytically conserved K745 (Figure A). The resulting irreversible inhibitor 72 displayed comparable cellular activity to Erlotinib while demonstrating a favorable pharmacokinetic profile both in vitro and in vivo. Tang et al. also synthesized fluorosulfate derivatives of XO-44 and analyzed their profiles across the human lysine kinome while assessing their stability and reactivity (Figure B). Among these derivatives, ABP 73 detected more kinases than XO-44 and was used in combination with VX-680 to develop irreversible Aurora kinase inhibitors. The newly synthesized irreversible inhibitor 74 exhibited comparable potency against AURKA but showed higher selectivity than VX-680.
19.

(A) Chemical structures of the first-generation EGFR drug Erlotinib and its corresponding irreversible inhibitor 72, which contains a fluorosulfate electrophile. (B) Development of the fluorosulfate probe 73 and the irreversible pan-Aurora kinase inhibitor 74 containing a fluorosulfate. The Venn diagram represents the number of kinases identified by 73 and XO44 in Jurkat cells.
3.2.2.8. Sulfonyl-Azole (SufAz) Warheads
An apparent difference between Sulfur(IV)-fluoride and SufAz warheads is the choice of leaving group at the sulfonyl electrophilic center, and SufAz compounds have the advantage of additional tunability via modification of the azole leaving groups. Particularly, our group introduced sulfur-triazole exchange (SuTEx) chemistry for chemoproteomics and protein ligand discovery (Figure ). ,, 1,2,4-sulfonyl-triazoles modify tyrosine and lysine residues with a preference for tyrosine. , Of relevance to this section, the lysine binding activity of SuTEx chemistry can be augmented toward lysine residues by switching to a 1,2,3-triazole leaving group or by positioning the electrophile near catalytic lysines using kinase-binding scaffolds. ,− The latter was demonstrated using a SuTEx warhead-containing XO44 derivative, KY-26, for identifying site of binding on kinases (∼50% of the modified peptides was detected as lysine) using a modified LC-MS/MS protocol.
20.
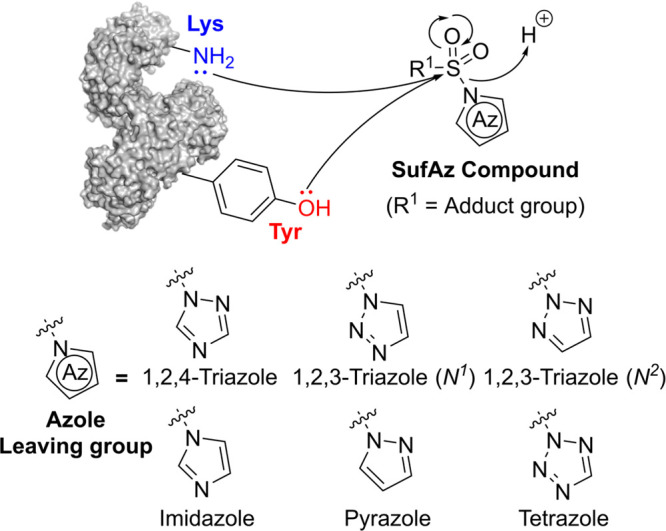
SufAz developed as Tyr/Lys-reactive warheads and chemical structures of synthesized azoles in SufAz derivatives for proteomic research.
3.3. Tyrosine-reactive Scaffolds
3.3.1. Biological Features of Tyrosine
Tyrosine, despite having a slightly lower pK a (∼10) than a protonated lysine, has been more challenging to target using covalent ligands. This can be attributed to the structural characteristics of its side chain, which is more rigid and sterically hindered by the phenolic hydroxyl group. These factors pose challenges in achieving the optimal geometries required for effective covalent binding.
Nevertheless, tyrosine-targeting electrophiles have proven valuable in studying PPIs. Tyrosine is notably abundant at protein–protein interfaces, where its unique properties contribute significantly to molecular recognition. − Its amphipathic nature allows for diverse interactions, while its rigidity enhances binding affinity and specificity. The limited conformational flexibility of tyrosine reduces entropic penalties during binding, thereby minimizing nonspecific interactions. This specificity is particularly critical in tyrosine-rich interfaces, which are inherently less accommodating to nonspecific binding events.
Additionally, PTMs such as phosphorylation of tyrosine residues plays a pivotal role in regulating PPIs that are essential for biological processes. − Targeting tyrosines subject to phosphorylation could therefore serve as a different strategy for modulating signaling pathways. − Developing tyrosine-reactive warheads may enable precise regulation of these critical residues and provide deeper insights into signaling mechanisms. − Thus, the development of novel tyrosine-reactive warheads holds great potential to advance our understanding of protein signaling networks and discover irreversible small molecule modulators with unprecedented mechanisms.
3.3.2. Tyrosine-targeted Chemistry
3.3.2.1. Mannich Type Warheads
Francis and co-workers developed a three-component Mannich-type tyrosine conjugation strategy. − Utilizing the Betti variant of the Mannich reaction, they performed an in situ imine condensation between an electron-rich aniline and formaldehyde, followed by the formation of a C–C bond with tyrosine (Figure ). Although this reaction proceeds under mild conditions, it requires an extended reaction time and an optimal pH. − Moreover, it also leads to the formation of adducts with tryptophan and cysteine, limiting its chemoselectivity.
21.

Mechanism of three-component Mannich-type tyrosine conjugation.
To address these limitations, Tanaka and co-workers introduced a two-component Mannich-type tyrosine bioconjugation strategy. , In this approach, a stable cyclic imine was presynthesized and subsequently reacted with tyrosine. This method offers several advantages: it is versatile across a wide range of pH values, prevents enamine formation during the reaction, and exhibits improved resistance to hydrolysis. These features make Mannich-type conjugation more applicable in biological systems for selective tyrosine targeting. Although the cyclic imine showed limited activity for global tyrosine labeling, it demonstrated significant potential for targeted covalent labeling and inhibition.
Building on this tyrosine bioconjugation strategy, Wang et al. synthesized cyclic imine warheads as targeted covalent inhibitors. Among the cyclic imines evaluated, cyclohexylimino-lactones and -lactams exhibited reactivity toward Ac-Tyr-NHMe in PBS buffer (pH 7, 37 °C). While lactones showed high reactivity, this electrophile was labile and subject to hydrolysis. Conversely, cyclohexyliminolactams exhibited stability under GSH treatment and in acidic and basic conditions. For the reversible dual CBX2 and CBX8 inhibitor SW2_110A (75), a cyclic imine was introduced to selectively target the nonconserved Y39 of CBX8, thereby enhancing selectivity over CBX2 (Figure A). Design of this covalent inhibitor was enabled by a cocrystal structure of CBX8 bound to an oligopeptide UNC3866 (PDB ID: 5EQ0, Figure B). Biochemical assays and LC-MS/MS analysis revealed that cyclic imine 77 improved selectivity through a targeted covalent binding strategy (Figure C).
22.

(A) A sequence alignment of CBX isoforms. (B) X-ray cocrystal structure of CBX2 including UNC3866 (PDB ID: 5EQ0 ). (C) Chemical structures of the synthesized CBX8 inhibitors.
3.3.2.2. Triazolidione (TAD) Warheads
The tyrosine conjugation potential of cyclic TAD warheads was first reported by Barbas and co-workers. Due to their high selectivity for tyrosine and tryptophan, as well as the stability of the resulting adducts, TAD warheads have been widely studied (Figure ). − However, their poor aqueous stability poses challenges for pharmaceutical development. Additionally, TAD hydrolysis generates isocyanates that can nonspecifically bind to residues such as lysine, although this can be mitigated by using Tris buffer to scavenge isocyanate byproducts. − Decoene et al. further demonstrated that the Tyr/Trp selectivity of TAD can be modulated by pH, with acidic conditions favoring tryptophan modification. Moinpour et al. quantified TAD labeling on surface tyrosine residues, offering insights into conformational changes in proteins, which are critical for understanding PPI.
23.

Mechanism of TAD conjugation to tyrosine and tryptophan.
3.3.2.3. Sulfur(VI)-Fluoride Warheads: Sulfonyl Fluoride (-SO2F) and Fluorosulfate (-OSO2F)
As discussed in Section , sulfonyl fluoride warheads exhibit significant reactivity toward tyrosine as well as lysine residues. Scammells et al. transformed a reversible A1 Adenosine Receptor (A1AR) antagonist, DPCPX, into an irreversible antagonist, FSCPX, through a traditional SAR study. Covalent inactivation of FSCPX was achieved by introducing an arylsulfonyl fluoride warhead at either the 8- or N 3-position. Next, an optimization strategy for improving metabolic stability resulted in discovery of DU172; X-ray cocrystal structures with human A1AR (hA1AR) confirmed covalent binding between the ligand and Y2657.36 of hA1AR (Figure A). In addition, an irreversible partial hA1AR agonist and ABPs containing sulfonyl fluoride were developed and used to visualize the receptor in live cells, mimicking the reversible hA1AR partial agonist, Capadenoson. ,
24.

(A) SAR studies for the development of irreversible hA1AR antagonist DU172. (B) An introduction of sulfonylfluoride electrophiles for hA3AR covalent inhibition through homology models with hA1AR and the further development for hA3AR ABP.
Homology modeling of hA1AR with DU172 further inspired Yang et al. to design novel irreversible human A3 Adenosine Receptor (hA3AR) antagonists. Since most existing hA3AR modulators are reversible ligands, developing irreversible ligands provide additional chemical probes to expand our understanding of hA3AR. − The simple incorporation of an electrophile into DU172 resulted in covalent interaction with Y2717.36 of hA3AR. This modification was further developed into ABPs with terminal alkyne groups for profiling endogenous hA3AR expression (Figure B).
Jones and co-workers designed ABPs and irreversible inhibitors for RNA binding of DcpS. , X-ray cocrystal structures of DcpS with D153249 (78) identified three nucleophilic residues (Y113, Y143, K142) near the benzyl group, motivating the introduction of sulfonyl fluoride at ortho-, meta-, or para-positions of the benzyl substituent. As expected, SF-p1 (79) modified Y143, while SF-m1 (80) and SF-o1 (81) bound with Y113 (Figure ). Additionally, alkynylation of 80 facilitated its use as a tool for evaluating DcpS target engagement.
25.

Alterations to the targeted tyrosine residue based on positioning of the sulfonylfluoride warhead.
Chen et al. reported arylfluorosulfate ABPs that selectively modify conserved tyrosine residues in the substrate-binding site of the intracellular lipid-binding protein (iLBP) family. Among these, biphenylarylfluorosulfate exhibited selective labeling of CRABP2 in living cells. The selectivity was attributed to a lowered pK a (∼7.6) of CRABP2 Y134, influenced by adjacent arginine residues. Olsen and co-workers further advanced the field by developing irreversible SIRT5-selective fluorosulfate inhibitors and ABPs. Their earlier mechanism-based reversible SIRT5 inhibitor 82, which incorporated a thiourea moiety, suffered from issues related to cell permeability and serum stability. To address these limitations, bioisostere and prodrug strategies were explored but proved insufficient for in vivo applications. − By replacing the warhead with a fluorosulfate group and incorporating a guanidine moiety to increase aqueous solubility, they successfully developed SIRT5 ABP 83, which exhibited functionality in vivo (Figure ).
26.

Chemical structures of reversible SIRT5 inhibitor and its corresponding irreversible sulfonylfluoride ABP.
3.3.2.4. Sulfonyl-Azole (SufAz) Warheads
The SufAz electrophile is one of the most tyrosine-selective scaffolds available, offering sufficient stability and tunability for chemoproteomic applications. , SuTEx ligands have been shown to disrupt enzymatic sites by liganding catalytic and noncatalytic tyrosines on proteins, and perturb protein–protein or protein-RNA complexes involved in biomolecular condensate formation and dissolution. ,, Hahm et al. synthesized a series of SuTEx compounds, among which HHS-465 (84) and HHS-475 (85) were frequently used in ABPP studies in lysates and live cells (Figure ). Notably, HHS-482 (86) exhibited moderate reactivity but superior tyrosine selectivity (Y/K ratio ∼ 5).
27.

Chemical structures of SuTEx ABPs.
Brulet et al. investigated the tunability of SuTEx compounds by modifying adduct groups and substituents on leaving groups in both organic solutions and proteomes. Aryl substituents as adduct groups demonstrated superior reactivity, whereas cyclopropyl-substituted adduct groups reduced electrophilicity. Additionally, the reactivity of SuTEx compounds correlated strongly with the electronic properties of their substituents (Figure ).
28.

Half-lives of SuTEx compounds are dependent on alterations to adduct and leaving group modifications. p-Cresol and tetramethylguanidine in DMSO/ACN (1:9) were used to compare reactivity toward tyrosine.
Huang et al. combined the RF001 fragment of the serotonin receptor antagonist Ritanserin with SuTEx warheads (Figure ). Ritanserin exhibited secondary activity against metabolic kinases such as diacylglycerol kinase (DGK) alpha, prompting the introduction of the RF001 binding element for developing kinase-targeted ABPs. The ABP design installed the structurally complex RF001 moiety in the leaving group to mediate recognition without complicating the resulting peptide-probe adduct to enhance LC-MS/MS detectability of modified sites. The resulting SuTEx ABP, TH211, identified tyrosine residues located in the catalytic or regulatory domains of over 50 protein and metabolic kinases in live cells. Additionally, TH211 was used to capture sites in predicted binding pockets of DGKs to guide future inhibitor development.
29.

Chemical structures of kinase ABP TH211 mimicking Ritanserin.
3.4. Stereoselective Recognition in Covalent Ligand Design
A traditional ligand screening for evaluation of cellular activity can be easily altered by physicochemical properties of ligands, which can increase complexity of SAR anlaysis. − Also, many proteins utilize stereochemical properties as one of the key factors to distinguish substrates. − Thus, the concept of stereoprobe pairs was proposed as an innovative solution to conduct systemic SAR analysis, minimizing the concerns of physicochemical properties. − Stereoprobe pairs serve as ideally matched controls to assess on-target versus nonspecific binding and cellular activity of electrophilic ligands. The initial proteomic mapping using enantioprobes was focused on targeting cysteines with Michael acceptor electrophiles or to broadly target the proteome in a protein residue-independent manner using photoreactive diazirine groups (Figure ). −
30.

(A) Development of enantioprobe pairs for covalent ligand discovery. Quantification of stereoselectively modified peptides and proteins were determined by ABPP analysis as shown in Figure . (B) The chemical structures of tryptoline acrylamide stereoprobe pairs possessing a terminal alkyne at R1, R2, or tryptoline core.
Recently, the Taunton group synthesized enantiomeric ABP pairs of tyrosine- and lysine- reactive sulfonyl fluorides, demonstrating that stereochemistry influences binding preferences on protein residues beyond cysteine (Figure ). By screening ten enantiomeric pairs (96-105), the authors detected 634 stereoselectively modified sites (513 Tyr, 121 Lys) out of 2,343 total modified sites (1,728 Tyr, 615 Lys) in living cells. These enantiomeric pairs, which provide valuable insights into stereochemical preferences at protein binding sites, can not only guide the design of small molecules to target novel proteins but also serve as useful tools for connecting on- versus off-target binding site engagement with phenotypes observed in compound-treated cells.
31.

Chemical structures of enantioprobe pairs with tyrosine- and lysine- reactive sulfonyl fluoride as an electrophile.
4. Conclusions and Future Outlook
Most of the human proteome (∼85%) lack well-defined binding pockets making them challenging targets for traditional drug design. , Examples of such proteins include p53 and c-Myc, which often rely on PPIs or intrinsically disordered regions for their function. − To overcome these challenges, covalent chemistry and ABPP have become indispensable tools in drug discovery, streamlining steps from target identification to lead optimization. The emergence of ABPP technologies combined with LC-MS/MS has transformed drug discovery by enabling a tractable path toward discovery of amino acid-directed covalent warheads and subsequent optimization of highly targeted covalent ligands. Additionally, ABPP has expanded the scope of ligandable proteins previously considered intractable.
While numerous cysteine-directed covalent inhibitors have been developed, targeting cysteine alone covers only a small portion of the proteome. Therefore, there is a growing demand for covalent chemistries that target a broader range of amino acids on proteins. Among these residues, lysine and tyrosine have emerged as an attractive target for covalent modulator development.
To date, various lysine-reactive warheads have been discovered and utilized for protein profiling and the development of covalent inhibitors. Furthermore, some studies have focused on PTMs at lysine residues and have applied site-selective lysine chemistry to diverse biological applications such as antibody-drug conjugation (ADC) and human serum albumin (HSA)-conjugated strategies. − Despite the development of numerous tyrosine- and lysine-reactive warheads, each scaffold has limited coverage when considering the vast diversity of potential ligandable sites in the human proteome. ,, In fact, the proximity-driven environment within the residues strongly influences the reactivity of warheads, which depends on their electronic and steric properties. − This underscores the need for continued development of diverse scaffolds to target hard to ligand proteins.
In particular, tyrosine- and lysine-reactive warheads that avoid reactivity with cysteine such as sulfonyl electrophiles, are crucial for achieving selective modification of novel targets. Thiols on cysteine and GSH are primary nucleophiles in cells that readily forms covalent bonds with electrophiles. Thus, electrophiles designed to target noncysteine residues must avoid or minimize thiol cross-reactivity in biological systems. The discovery of novel electrophiles and their precise integration into noncovalent recognition scaffolds is essential for advancing covalent inhibitors and chemoproteomic probes targeting lysine as well as other nucleophilic residues such as tyrosine, noncatalytic serine/threonine, and histidine. Tyrosines are particularly interesting because they are more abundant than cysteine but not as prevalent as lysine to expand protein targeting opportunities without compromising selectivity. Also, tyrosines are known to be enriched in protein binding surfaces, suggesting that targeting tyrosine has a high potential for developing PPI inhibitors.
In recent years, the IDD approach has emerged as an important strategy for the discovery of novel covalent ligands, including enantiomeric ABP pairs. While chirality is commonly incorporated into the adduct group of enantiomeric pairs, the inclusion of chirality within the electrophilic center itself also represents a promising and underexplored avenue for covalent ligand development.
Progress in chemoproteomic probe design combined with multiplexed quantitative mass spectrometry will further expand the scope of functional protein pockets amenable to covalent targeting. This advancement will provide new fundamental insights into protein biochemistry and biology and in the process drive innovation in therapeutic development.
Acknowledgments
This work was supported by the National Institutes of Health grant nos. GM144472 (K.-L.H.), DA043571 (K.-L.H.), AI169412 (K.-L.H.), the Mark Foundation for Cancer Research (Emerging Leader Award to K.-L.H.), Recruitment of Rising Stars Award from CPRIT (RR220063 to K.-L.H.), the National Science Foundation (CHE-2422750 to K.-L.H.), a Research Grant Award from The Welch Foundation (F-2143-20230405 to K.-L.H.), and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (RS-2024-00415392 to G. K.).
Glossary
Abbreviations
- BODIPY
Borondipyrromethene
- CBX
Chromobox protein
- CRABP2
Cellular retinoic acid-binding protein 2
- CRBN
Cereblon protein
- DBP
DNA binding protein
- DcpS
Decapping scavenger enzyme
- DPCPX
Dipropylcyclopentylxanthine
- FDA
Food and drug administration
- FSCPX
8-Cyclopentyl-3-(3-((4-(fluorosulfonyl)benzoyl)oxy)propyl)-1-propylxanthine
- GDP
Guanosine diphosphate
- GTP
Guanosine triphosphate
- GlfT2
Galactofuranosyltransferase 2
- HEG1
Heart development protein with EGF-like domains 1
- HIV
Human immunodeficiency virus
- JAK
Janus kinase
- KRIT1
Krev interaction trapped protein 1
- NAD
Nicotinamide adenine dinucleotide
- PI3K
Phosphatidylinositol-3 kinase
- SIRT5
Sirtuin 5
- TEC
Tyrosine kinase expressed in hepatocellular carcinoma
- UDP
Uridine diphosphate
- UGGT1
UDP-glucose:glycoprotein glucosyltransferase
- WNK
With-no-lysine kinases
- XRCC
X-ray repair cross-complementing protein
Biographies
Gibae Kim graduated from Seoul National University in 2018 and received his Ph.D. in Medicinal Chemistry in 2024 from the College of Pharmacy at Seoul National University. During his Ph.D. studies, he investigated modified nucleoside chemistry and SAR studies of their derivatives under the guidance of Professor Lak Shin Jeong. He then joined the Hsu Lab as a postdoctoral fellow in the Department of Chemistry at the University of Texas at Austin, where he focused on discovering novel targets using LC-MS/MS and developing new electrophiles.
R. Justin Grams received his B.S. in Biochemistry from Virginia Tech in 2012 and his Ph.D. in Organic Chemistry from Virginia Tech in 2021 under the advisorship of Prof. Webster Santos. Justin worked as a Post-Doctoral Research Associate in the Hsu Lab at the University of Texas at Austin and a Visiting Scholar at the University of Virginia until 2024 and transitioned to a Research Scientist in the Hsu Lab at UT Austin. Justin’s postdoctoral research involved designing and synthesizing sulfonyl-triazole electrophiles that covalently modify functional tyrosines in oncogene proteins towards oncology.
Ku-Lung (Ken) Hsu is currently a Stephen F. and Fay Evans Martin Endowed Associate Professor and CPRIT Scholar in the Department of Chemistry at the University of Texas at Austin. His group develops enabling covalent chemistry and chemical proteomic technologies to create novel molecules that drive chemical biology and therapeutic discovery and uncover new insights into protein and lipid biology relevant to human disease. Ken received his PhD in Chemistry and Biochemistry from the University of Texas at Austin and pursued further training in chemical biology at The Scripps Research Institute (TSRI) in La Jolla as a Hewitt Foundation for Medical Research Postdoctoral Fellow.
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. CRediT: Gibae Kim conceptualization, visualization, writing - original draft, writing - review & editing; Robert Justin Grams writing - review & editing; Ku-Lung Hsu conceptualization, funding acquisition, supervision, writing - original draft, writing - review & editing.
The authors declare the following competing financial interest(s): K.-L.H. is a founder and scientific advisory board member of Hyku Biosciences.
Published as part of Chemical Reviews special issue “Drugging the Undruggable”.
References
- Patricelli M. P., Janes M. R., Li L.-S., Hansen R., Peters U., Kessler L. V., Chen Y., Kucharski J. M., Feng J., Ely T.. et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery. 2016;6(3):316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- Janes M. R., Zhang J., Li L.-S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S. J., Darjania L.. et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172(3):578–589. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Canon J., Rex K., Saiki A. Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C. G., Koppada N.. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Kim D., Xue J. Y., Lito P.. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell. 2020;183(4):850–859. doi: 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A., Yoo E.. Sotorasib: a KRASG12C inhibitor for non-small cell lung cancer. Trends Pharmacol. Sci. 2022;43(6):536–537. doi: 10.1016/j.tips.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Haga R., Tamura S., Shimada I., Nishida N.. Real-time monitoring of the reaction of KRAS G12C mutant specific covalent inhibitor by in vitro and in-cell NMR spectroscopy. Sci. Rep. 2023;13(1):19253. doi: 10.1038/s41598-023-46623-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A. R., Rosenberg S. C., McCormick F., Malek S.. RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discovery. 2020;19(8):533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D., Gmachl M., Mantoulidis A., Martin L. J., Zoephel A., Mayer M., Gollner A., Covini D., Fischer S., Gerstberger T.. et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. U. S. A. 2019;116(32):15823–15829. doi: 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Guo Z., Wang F., Fu L.. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021;6(1):386. doi: 10.1038/s41392-021-00780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grams R. J., Hsu K.-L.. Reactive chemistry for covalent probe and therapeutic development. Trends Pharmacol. Sci. 2022;43(3):249–262. doi: 10.1016/j.tips.2021.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Esbroeck A. C. M., Janssen A. P. A., Cognetta A. B., Ogasawara D., Shpak G., van der Kroeg M., Kantae V., Baggelaar M. P., de Vrij F. M. S., Deng H.. et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10–2474. Science. 2017;356(6342):1084–1087. doi: 10.1126/science.aaf7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhong B., Xu C., Zhan D., Zhao S., Wu H., Liu M., Lan X., Cai D., Ding Q.. et al. Global profiling of AMG510 modified proteins identified tumor suppressor KEAP1 as an off-target. iScience. 2023;26(2):106080. doi: 10.1016/j.isci.2023.106080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen S., Dix M. M., Barbas S., Potter Z. E., Lu S., Brodsky O., Planken S., Behenna D., Almaden C., Gajiwala K. S.. et al. Proteome-wide Map of Targets of T790M-EGFR-Directed Covalent Inhibitors. Cell Chem. Biol. 2017;24(11):1388–1400. doi: 10.1016/j.chembiol.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P.-Y., Liu K., Ngai M. H., Lear M. J., Wenk M. R., Yao S. Q.. Activity-Based Proteome Profiling of Potential Cellular Targets of Orlistat - An FDA-Approved Drug with Anti-Tumor Activities. J. Am. Chem. Soc. 2010;132(2):656–666. doi: 10.1021/ja907716f. [DOI] [PubMed] [Google Scholar]
- Ward C. C., Kleinman J. I., Nomura D. K.. NHS-Esters As Versatile Reactivity-Based Probes for Mapping Proteome-Wide Ligandable Hotspots. ACS Chem. Biol. 2017;12(6):1478–1483. doi: 10.1021/acschembio.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boike L., Henning N. J., Nomura D. K.. Advances in covalent drug discovery. Nat. Rev. Drug Discovery. 2022;21(12):881–898. doi: 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counihan J. L., Ford B., Nomura D. K.. Mapping proteome-wide interactions of reactive chemicals using chemoproteomic platforms. Curr. Opin. Chem. Biol. 2016;30:68–76. doi: 10.1016/j.cbpa.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T., Wang J., Liang R., Chen W., Chen Y., Ma M., He A., Du Y., Zhou W., Zhang Z.. et al. Selective inhibition reveals the regulatory function of DYRK2 in protein synthesis and calcium entry. eLife. 2022;11:e77696. doi: 10.7554/eLife.77696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Wang Y., Ma N., Tian J., Shao Y., Zhu B., Wong Y. K., Liang Z., Zou C., Wang J.. Target identification of natural medicine with chemical proteomics approach: probe synthesis, target fishing and protein identification. Signal Transduct. Target. Ther. 2020;5(1):72. doi: 10.1038/s41392-020-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimster N. P., Connelly S., Baranczak A., Dong J., Krasnova L. B., Sharpless K. B., Powers E. T., Wilson I. A., Kelly J. W.. Aromatic Sulfonyl Fluorides Covalently Kinetically Stabilize Transthyretin to Prevent Amyloidogenesis while Affording a Fluorescent Conjugate. J. Am. Chem. Soc. 2013;135(15):5656–5668. doi: 10.1021/ja311729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y., Zhang H., Wang Y., Zong Z., Bogyo M., Chen S.. DNA encoded peptide library for SARS-CoV-2 3CL protease covalent inhibitor discovery and profiling. RSC Chem. Biol. 2024;5(7):691–702. doi: 10.1039/D4CB00097H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner M., Lentz C. S., Jamieson S. A., Brewster J. L., Chen L., Bogyo M., Mace P. D.. Structural Basis for the Inhibitor and Substrate Specificity of the Unique Fph Serine Hydrolases of Staphylococcus aureus. ACS Infect. Dis. 2020;6(10):2771–2782. doi: 10.1021/acsinfecdis.0c00503. [DOI] [PubMed] [Google Scholar]
- White M. E. H., Gil J., Tate E. W.. Proteome-wide structural analysis identifies warhead- and coverage-specific biases in cysteine-focused chemoproteomics. Cell Chem. Biol. 2023;30(7):828–838. doi: 10.1016/j.chembiol.2023.06.021. [DOI] [PubMed] [Google Scholar]
- Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B. D., Bachovchin D. A., Mowen K., Baker D., Cravatt B. F.. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468(7325):790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abo M., Li C., Weerapana E.. Isotopically-Labeled Iodoacetamide-Alkyne Probes for Quantitative Cysteine-Reactivity Profiling. Mol. Pharmaceutics. 2018;15(3):743–749. doi: 10.1021/acs.molpharmaceut.7b00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Patricelli M. P., Cravatt B. F.. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1999;96(26):14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun D. P., Ritchie J., Jung Y., Holewinski R., Kim H.-R., Tagirasa R., Ivanic J., Weekley C. M., Parker M. W., Andresson T.. et al. Covalent Inhibition by a Natural Product-Inspired Latent Electrophile. J. Am. Chem. Soc. 2023;145(20):11097–11109. doi: 10.1021/jacs.3c00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M., Ware T. B., Hsu K.-L.. DAGL-Beta Functions as a PUFA-Specific Triacylglycerol Lipase in Macrophages. Cell Chem. Biol. 2020;27(3):314–321. doi: 10.1016/j.chembiol.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A., Zhang X., Lei X.. Covalent Probe Finds Carboxylic Acid. Cell Chem. Biol. 2017;24(5):537–539. doi: 10.1016/j.chembiol.2017.05.003. [DOI] [PubMed] [Google Scholar]
- Skoulidis F., Li B. T., Dy G. K., Price T. J., Falchook G. S., Wolf J., Italiano A., Schuler M., Borghaei H., Barlesi F.. et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021;384(25):2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima E. C., Drezner N., Li X., Mishra-Kalyani P. S., Liu Y., Zhao H., Bi Y., Liu J., Rahman A., Wearne E.. et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer. Res. 2022;28(8):1482–1486. doi: 10.1158/1078-0432.CCR-21-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telliez J.-B., Dowty M. E., Wang L., Jussif J., Lin T., Li L., Moy E., Balbo P., Li W., Zhao Y.. et al. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem. Biol. 2016;11(12):3442–3451. doi: 10.1021/acschembio.6b00677. [DOI] [PubMed] [Google Scholar]
- Piliang M., Soung J., King B., Shapiro J., Rudnicka L., Farrant P., Magnolo N., Piraccini B. M., Luo X., Wolk R.. et al. Efficacy and safety of the oral Janus kinase 3/tyrosine kinase expressed in hepatocellular carcinoma family kinase inhibitor ritlecitinib over 24 months: integrated analysis of the ALLEGRO phase IIb/III and long-term phase III clinical studies in alopecia areata. Br. J. Dermatol. 2025;192(2):215–227. doi: 10.1093/bjd/ljae365. [DOI] [PubMed] [Google Scholar]
- Shawky A. M., Almalki F. A., Abdalla A. N., Abdelazeem A. H., Gouda A. M.. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics. 2022;14(5):1001. doi: 10.3390/pharmaceutics14051001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H., He L., Shao M., Yang Z., Ran Y., Li D., Zhou Y., Tang M., Wang T., Gong Y.. et al. Discovery of a highly selective JAK3 inhibitor for the treatment of rheumatoid arthritis. Sci. Rep. 2018;8(1):5273. doi: 10.1038/s41598-018-23569-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Jesson M. I., Seneviratne U. I., Lin T. H., Sharif M. N., Xue L., Nguyen C., Everley R. A., Trujillo J. I., Johnson D. S.. et al. PF-06651600, a Dual JAK3/TEC Family Kinase Inhibitor. ACS Chem. Biol. 2019;14(6):1235–1242. doi: 10.1021/acschembio.9b00188. [DOI] [PubMed] [Google Scholar]
- Blair H. A.. Ritlecitinib: First Approval. Drugs. 2023;83(14):1315–1321. doi: 10.1007/s40265-023-01928-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordinsky M., Hebert A. A., Gooderham M., Kwon O., Murashkin N., Fang H., Harada K., Law E., Wajsbrot D., Takiya L.. et al. Efficacy and safety of ritlecitinib in adolescents with alopecia areata: Results from the ALLEGRO phase 2b/3 randomized, double-blind, placebo-controlled trial. Pediatr. Dermatol. 2023;40(6):1003–1009. doi: 10.1111/pde.15378. [DOI] [PubMed] [Google Scholar]
- Clementi C., Carloni P., Maritan A.. Protein design is a key factor for subunit-subunit association. Proc. Natl. Acad. Sci. U. S. A. 1999;96(17):9616–9621. doi: 10.1073/pnas.96.17.9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brulet J. W., Borne A. L., Yuan K., Libby A. H., Hsu K.-L.. Liganding Functional Tyrosine Sites on Proteins Using Sulfur-Triazole Exchange Chemistry. J. Am. Chem. Soc. 2020;142(18):8270–8280. doi: 10.1021/jacs.0c00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker S. M., Backus K. M., Lazear M. R., Forli S., Correia B. E., Cravatt B. F.. Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 2017;9(12):1181–1190. doi: 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y., Zhang X., Chen Z., Yan D., Zhu L., Zhang Z., Wang X., Tian K., Huang Y., Yang X.. et al. Global profiling of functional histidines in live cells using small-molecule photosensitizer and chemical probe relay labelling. Nat. Chem. 2024;16(9):1546–1557. doi: 10.1038/s41557-024-01545-6. [DOI] [PubMed] [Google Scholar]
- Bach K., Beerkens B. L. H., Zanon P. R. A., Hacker S. M.. Light-Activatable, 2,5-Disubstituted Tetrazoles for the Proteome-wide Profiling of Aspartates and Glutamates in Living Bacteria. ACS Cent. Sci. 2020;6(4):546–554. doi: 10.1021/acscentsci.9b01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanon P. R. A., Yu F., Musacchio P. Z., Lewald L., Zollo M., Krauskopf K., Mrdović D., Raunft P., Maher T. E., Cigler M.. et al. Profiling the proteome-wide selectivity of diverse electrophiles. ChemRxiv. 2021 doi: 10.26434/chemrxiv-2021-w7rss-v2. [DOI] [Google Scholar]
- Nakamura T., Kawai Y., Kitamoto N., Osawa T., Kato Y.. Covalent Modification of Lysine Residues by Allyl Isothiocyanate in Physiological Conditions: Plausible Transformation of Isothiocyanate from Thiol to Amine. Chem. Res. Toxicol. 2009;22(3):536–542. doi: 10.1021/tx8003906. [DOI] [PubMed] [Google Scholar]
- Sun H., Xi M., Jin Q., Zhu Z., Zhang Y., Jia G., Zhu G., Sun M., Zhang H., Ren X.. et al. Chemo- and Site-Selective Lysine Modification of Peptides and Proteins under Native Conditions Using the Water-Soluble Zolinium. J. Med. Chem. 2022;65(17):11840–11853. doi: 10.1021/acs.jmedchem.2c00937. [DOI] [PubMed] [Google Scholar]
- Borne A. L., Brulet J. W., Yuan K., Hsu K.-L.. Development and biological applications of sulfur-triazole exchange (SuTEx) chemistry. RSC Chem. Biol. 2021;2(2):322–337. doi: 10.1039/D0CB00180E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer G. H., Garrod S., Woods V. L., Taylor S. S.. Catalytic Independent Functions of a Protein Kinase as Revealed by a Kinase-dead Mutant: Study of the Lys72His Mutant of cAMP-dependent Kinase. J. Mol. Biol. 2005;351(5):1110–1122. doi: 10.1016/j.jmb.2005.06.011. [DOI] [PubMed] [Google Scholar]
- De Jesus I. S., Vélez J. A. C., Pissinati E. F., Correia J. T. M., Rivera D. G., Paixao M. W.. Recent Advances in Photoinduced Modification of Amino Acids, Peptides, and Proteins. Chem. Rec. 2024;24(3):e202300322. doi: 10.1002/tcr.202300322. [DOI] [PubMed] [Google Scholar]
- Xiang F., Hu F., Weng Y., Gao M.. Photo-and Electrochemical Modification of Amino Acids, Peptides and Proteins through Cysteine: Recent Advances and Future Perspectives. Eur. J. Org. Chem. 2024;27(45):e202400772. doi: 10.1002/ejoc.202400772. [DOI] [Google Scholar]
- Debon A., Siirola E., Snajdrova R.. Enzymatic Bioconjugation: A Perspective from the Pharmaceutical Industry. JACS Au. 2023;3(5):1267–1283. doi: 10.1021/jacsau.2c00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malins L. R.. Peptide modification and cyclization via transition-metal catalysis. Curr. Opin. Chem. Biol. 2018;46:25–32. doi: 10.1016/j.cbpa.2018.03.019. [DOI] [PubMed] [Google Scholar]
- Wijesinghe A., Kumari S., Booth V.. Conjugates for use in peptide therapeutics: A systematic review and meta-analysis. PLoS One. 2022;17(3):e0255753. doi: 10.1371/journal.pone.0255753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons E., Roet S., Kim R. Q., Mulder M. P. C.. A Comprehensive Guide for Assessing Covalent Inhibition in Enzymatic Assays Illustrated with Kinetic Simulations. Curr. Protoc. 2022;2(6):e419. doi: 10.1002/cpz1.419. [DOI] [PubMed] [Google Scholar]
- Lu H., England K., am Ende C., Truglio J. J., Luckner S., Reddy B. G., Marlenee N. L., Knudson S. E., Knudson D. L., Bowen R. A.. et al. Slow-Onset Inhibition of the FabI Enoyl Reductase from Francisella tularensis: Residence Time and in Vivo Activity. ACS Chem. Biol. 2009;4(3):221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup G. K., You Z., Ross P. L., Allen E. K. H., Daryaee F., Hale M. R., O’Donnell J., Ehmann D. E., Schuck V. J. A., Buurman E. T.. et al. Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat. Chem. Biol. 2015;11(6):416–423. doi: 10.1038/nchembio.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee P., Botello-Smith W. M., Zhang H., Qian L., Alsamarah A., Kent D., Lacroix J. J., Baudry M., Luo Y.. Can Relative Binding Free Energy Predict Selectivity of Reversible Covalent Inhibitors? J. Am. Chem. Soc. 2017;139(49):17945–17952. doi: 10.1021/jacs.7b08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw J. M., McFarland J. M., Paavilainen V. O., Bisconte A., Tam D., Phan V. T., Romanov S., Finkle D., Shu J., Patel V.. et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015;11(7):525–531. doi: 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faridoon, Ng R., Zhang G., Li J. J.. An update on the discovery and development of reversible covalent inhibitors. Med. Chem. Res. 2023;32(6):1039–1062. doi: 10.1007/s00044-023-03065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorarensen A., Balbo P., Banker M. E., Czerwinski R. M., Kuhn M., Maurer T. S., Telliez J.-B., Vincent F., Wittwer A. J.. The advantages of describing covalent inhibitor in vitro potencies by IC50 at a fixed time point. IC50 determination of covalent inhibitors provides meaningful data to medicinal chemistry for SAR optimization. Bioorg. Med. Chem. 2021;29:115865. doi: 10.1016/j.bmc.2020.115865. [DOI] [PubMed] [Google Scholar]
- Strelow J. M.. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discovery. 2017;22(1):3–20. doi: 10.1177/1087057116671509. [DOI] [PubMed] [Google Scholar]
- Tonge P. J.. Quantifying the Interactions between Biomolecules: Guidelines for Assay Design and Data Analysis. ACS Infect. Dis. 2019;5(6):796–808. doi: 10.1021/acsinfecdis.9b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahisa I., Sameshima T., Hixon M. S.. Rapid Determination of the Specificity Constant of Irreversible Inhibitors (kinact/KI) by Means of an Endpoint Competition Assay. Angew. Chem., Int. Ed. 2015;54(47):14099–14102. doi: 10.1002/anie.201505800. [DOI] [PubMed] [Google Scholar]
- Shirley J. D., Gillingham J. R., Nauta K. M., Diwakar S., Carlson E. E.. kinact/KI Value Determination for Penicillin-Binding Proteins in Live Cells. ACS Infect. Dis. 2024;10(12):4137–4145. doi: 10.1021/acsinfecdis.4c00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling D., Barayeu U., Steimbach R. R., Talwar D., Miller A. K., Dick T. P.. Commonly Used Alkylating Agents Limit Persulfide Detection by Converting Protein Persulfides into Thioethers. Angew. Chem., Int. Ed. 2022;61(30):e202203684. doi: 10.1002/anie.202203684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill B. G., Reily C., Oh J.-Y., Johnson M. S., Landar A.. Methods for the determination and quantification of the reactive thiol proteome. Free Radic. Biol. Med. 2009;47(6):675–683. doi: 10.1016/j.freeradbiomed.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W. X., Tsou C. L.. Determination of the rate constant of enzyme modification by measuring the substrate reaction in the presence of the modifier. Biochemistry. 1982;21(5):1028–1032. doi: 10.1021/bi00534a031. [DOI] [PubMed] [Google Scholar]
- Zhang J., Lim S. M., Yu M. R., Chen C., Wang J., Wang W., Rui H., Lu J., Lu S., Mok T.. et al. D3S-001, a KRAS G12C Inhibitor with Rapid Target Engagement Kinetics, Overcomes Nucleotide Cycling, and Demonstrates Robust Preclinical and Clinical Activities. Cancer Discovery. 2024;14(9):1675–1698. doi: 10.1158/2159-8290.CD-24-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M., Peters U., Sos M. L., Wells J. A., Shokat K. M.. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J., Cravatt B. F.. Ligand discovery by activity-based protein profiling. Cell Chemical Biology. 2024;31(9):1636–1651. doi: 10.1016/j.chembiol.2024.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. G., Pratt M. R.. Click Chemistry in Proteomic Investigations. Cell. 2020;180(4):605–632. doi: 10.1016/j.cell.2020.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barglow K. T., Cravatt B. F.. Activity-based protein profiling for the functional annotation of enzymes. Nat. Methods. 2007;4(10):822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]
- Dreiman G. H. S., Bictash M., Fish P. V., Griffin L., Svensson F.. Changing the HTS Paradigm: AI-Driven Iterative Screening for Hit Finding. SLAS Discovery. 2021;26(2):257–262. doi: 10.1177/2472555220949495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R., Kumar Singh S., Ahmad N., Misra S.. Challenges and advancements in high-throughput screening strategies for cancer therapeutics. Global Translational Medicine. 2024;3(1):2448. doi: 10.36922/gtm.2448. [DOI] [Google Scholar]
- Chan W. C., Liu X., Magin R. S., Girardi N. M., Ficarro S. B., Hu W., Tarazona Guzman M. I., Starnbach C. A., Felix A., Adelmant G.. et al. Accelerating inhibitor discovery for deubiquitinating enzymes. Nat. Commun. 2023;14(1):686. doi: 10.1038/s41467-023-36246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach I., Bernard D., Nguyen K., Ho G., Morrison A., Stecula A., Rosnik A., O’Sullivan A. M., Davtyan A., Samudio B.. et al. AI is a viable alternative to high throughput screening: a 318-target study. Sci. Rep. 2024;14(1):7526. doi: 10.1038/s41598-024-54655-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarron R., Banks M. N., Bojanic D., Burns D. J., Cirovic D. A., Garyantes T., Green D. V. S., Hertzberg R. P., Janzen W. P., Paslay J. W.. et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discovery. 2011;10(3):188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Zheng Z., Zeng Y., Lai K., Liao B., Li P., Tan C. S. H.. Protein painting for structural and binding site analysis via intracellular lysine reactivity profiling with o-phthalaldehyde. Chem. Sci. 2024;15(16):6064–6075. doi: 10.1039/D4SC00032C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Y., Zheng R., Bayer F. P., Wong C., Chang Y.-C., Meng C., Zolg D. P., Reinecke M., Zecha J., Wiechmann S.. et al. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC-MS/MS. Nat. Commun. 2020;11(1):157. doi: 10.1038/s41467-019-13973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonović M., Bogyo M.. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev. Proteomics. 2008;5(5):721–730. doi: 10.1586/14789450.5.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.-E., Mann M.. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat. Protoc. 2006;1(6):2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- Li J., Van Vranken J. G., Pontano Vaites L., Schweppe D. K., Huttlin E. L., Etienne C., Nandhikonda P., Viner R., Robitaille A. M., Thompson A. H.. et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods. 2020;17(4):399–404. doi: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E., Speers A. E., Cravatt B. F.. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)a general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2007;2(6):1414–1425. doi: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- Eng J. K., McCormack A. L., Yates J. R.. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Eng J. K., Jahan T. A., Hoopmann M. R.. Comet: An open-source MS/MS sequence database search tool. PROTEOMICS. 2013;13(1):22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Bern M., Kil Y. J., Becker C.. Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinform. 2012;40(1):13.20.11–13.20.14. doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toroitich E. K., Ciancone A. M., Hahm H. S., Brodowski S. M., Libby A. H., Hsu K.-L.. Discovery of a Cell-Active SuTEx Ligand of Prostaglandin Reductase 2. ChemBioChem. 2021;22(12):2134–2139. doi: 10.1002/cbic.202000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin Grams R., Yuan K., Founds M. W., Ware M. L., Pilar M. G., Hsu K.-L.. Imidazoles are Tunable Nucleofuges for Developing Tyrosine-Reactive Electrophiles. ChemBioChem. 2024;25(16):e202400382. doi: 10.1002/cbic.202400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A., Jones L. H.. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015;6(5):2650–2659. doi: 10.1039/C5SC00408J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee H., Debreczeni J., Breed J., Tentarelli S., Aquila B., Dowling J. E., Whitty A., Grimster N. P.. A study of the reactivity of S(VI)-F containing warheads with nucleophilic amino-acid side chains under physiological conditions. Org. Biomol. Chem. 2017;15(45):9685–9695. doi: 10.1039/C7OB02028G. [DOI] [PubMed] [Google Scholar]
- Mortenson D. E., Brighty G. J., Plate L., Bare G., Chen W., Li S., Wang H., Cravatt B. F., Forli S., Powers E. T.. et al. “Inverse Drug Discovery” Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J. Am. Chem. Soc. 2018;140(1):200–210. doi: 10.1021/jacs.7b08366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighty G. J., Botham R. C., Li S., Nelson L., Mortenson D. E., Li G., Morisseau C., Wang H., Hammock B. D., Sharpless K. B.. et al. Using sulfuramidimidoyl fluorides that undergo sulfur(vi) fluoride exchange for inverse drug discovery. Nat. Chem. 2020;12(10):906–913. doi: 10.1038/s41557-020-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline G. M., Nugroho K., Kelly J. W.. Inverse Drug Discovery identifies weak electrophiles affording protein conjugates. Curr. Opin. Chem. Biol. 2022;67:102113. doi: 10.1016/j.cbpa.2021.102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bálint D., Póti Á. L., Alexa A., Sok P., Albert K., Torda L., Földesi-Nagy D., Csókás D., Turczel G., Imre T.. et al. Reversible covalent c-Jun N-terminal kinase inhibitors targeting a specific cysteine by precision-guided Michael-acceptor warheads. Nat. Commun. 2024;15(1):8606. doi: 10.1038/s41467-024-52573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Hatcher J. M., Teng M., Gray N. S., Kostic M.. Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation. Cell Chem. Biol. 2019;26(11):1486–1500. doi: 10.1016/j.chembiol.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker J. M., Bernardes G. J. L., Lin Y. A., Davis B. G.. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. Asian J. 2009;4(5):630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- Spicer C. D., Davis B. G.. Selective chemical protein modification. Nat. Commun. 2014;5(1):4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Maurais A. J., Weerapana E.. Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol. 2019;50:29–36. doi: 10.1016/j.cbpa.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. D. R., Qian B., Baker D., Kothary R.. Conservation, Variability and the Modeling of Active Protein Kinases. PLoS One. 2007;2(10):e982. doi: 10.1371/journal.pone.0000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. L., Rule S., Martin P., Goy A., Auer R., Kahl B. S., Jurczak W., Advani R. H., Romaguera J. E., Williams M. E.. et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2013;369(6):507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munir T., Brown J. R., O’Brien S., Barrientos J. C., Barr P. M., Reddy N. M., Coutre S., Tam C. S., Mulligan S. P., Jaeger U.. et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am. J. Hematol. 2019;94(12):1353–1363. doi: 10.1002/ajh.25638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg L. A., Smith A. M., Sirisawad M., Verner E., Loury D., Chang B., Li S., Pan Z., Thamm D. H., Miller R. A.. et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010;107(29):13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. C.-H., Schuler M. H., Yamamoto N., O’Byrne K. J., Hirsh V., Mok T., Geater S. L., Orlov S. V., Tsai C.-M., Boyer M. J.. et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J. Clin. Oncol. 2012;30(15_suppl):LBA7500–LBA7500. doi: 10.1200/jco.2012.30.15_suppl.lba7500. [DOI] [Google Scholar]
- Yang J. C. H., Sequist L. V., Geater S. L., Tsai C.-M., Mok T. S. K., Schuler M., Yamamoto N., Yu C.-J., Ou S.-H. I., Zhou C.. et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16(7):830–838. doi: 10.1016/S1470-2045(15)00026-1. [DOI] [PubMed] [Google Scholar]
- Khozin S., Weinstock C., Blumenthal G. M., Cheng J., He K., Zhuang L., Zhao H., Charlab R., Fan I., Keegan P.. et al. Osimertinib for the Treatment of Metastatic EGFR T790M Mutation-Positive Non-Small Cell Lung Cancer. Clin. Cancer. Res. 2017;23(9):2131–2135. doi: 10.1158/1078-0432.CCR-16-1773. [DOI] [PubMed] [Google Scholar]
- Finlay M. R. V., Anderton M., Ashton S., Ballard P., Bethel P. A., Box M. R., Bradbury R. H., Brown S. J., Butterworth S., Campbell A.. et al. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J. Med. Chem. 2014;57(20):8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Mons E., Kim R. Q., van Doodewaerd B. R., van Veelen P. A., Mulder M. P. C., Ovaa H.. Exploring the Versatility of the Covalent Thiol-Alkyne Reaction with Substituted Propargyl Warheads: A Deciding Role for the Cysteine Protease. J. Am. Chem. Soc. 2021;143(17):6423–6433. doi: 10.1021/jacs.0c10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus K. M., Correia B. E., Lum K. M., Forli S., Horning B. D., González-Páez G. E., Chatterjee S., Lanning B. R., Teijaro J. R., Olson A. J.. et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534(7608):570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi R. N., Resnick E., Rogel A., Rao B. V., Gabizon R., Goldenberg K., Gurwicz N., Zaidman D., Plotnikov A., Barr H.. et al. Tunable Methacrylamides for Covalent Ligand Directed Release Chemistry. J. Am. Chem. Soc. 2021;143(13):4979–4992. doi: 10.1021/jacs.0c10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo N., Fuchida H., Sato M., Watari K., Shibata T., Kuwata K., Miura C., Okamoto K., Hatsuyama Y., Tokunaga K.. et al. Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat. Chem. Biol. 2019;15(3):250–258. doi: 10.1038/s41589-018-0204-3. [DOI] [PubMed] [Google Scholar]
- PyMOL Molecular Graphics System, Version 3.1; Schrödinger, LLC, 2024. [Google Scholar]
- Isom D. G., Castañeda C. A., Cannon B. R., García-Moreno E B.. Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U. S. A. 2011;108(13):5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Yu N. J., Kleiner R. E.. Chemoproteomic Approaches to Studying RNA Modification-Associated Proteins. Acc. Chem. Res. 2023;56(19):2726–2739. doi: 10.1021/acs.accounts.3c00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel A. J., Brulet J. W., Wang X., Founds M. W., Libby A. H., Bai D. L., Lemke M. C., Leace D. M., Harris T. E., Hafner M.. et al. Chemoproteomic capture of RNA binding activity in living cells. Nat. Commun. 2023;14(1):6282. doi: 10.1038/s41467-023-41844-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-R., Byun D. P., Thakur K., Ritchie J., Xie Y., Holewinski R., Suazo K. F., Stevens M., Liechty H., Tagirasa R.. et al. Discovery of a Tunable Heterocyclic Electrophile 4-Chloro-pyrazolopyridine That Defines a Unique Subset of Ligandable Cysteines. ACS Chem. Biol. 2024;19(5):1082–1092. doi: 10.1021/acschembio.4c00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakemeyer M., Bogyo M.. Uncovering an overlooked consequence of phosphorylation: change in cysteine reactivity. Nat. Methods. 2022;19(3):281–283. doi: 10.1038/s41592-022-01414-5. [DOI] [PubMed] [Google Scholar]
- Xiao W., Chen Y., Wang C.. Quantitative Chemoproteomic Methods for Reactive Cysteinome Profiling. Isr. J. Chem. 2023;63(3–4):e202200100. doi: 10.1002/ijch.202200100. [DOI] [Google Scholar]
- Huang F., Han X., Xiao X., Zhou J.. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules. 2022;27(22):7728. doi: 10.3390/molecules27227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta N. V., Degani M. S.. The expanding repertoire of covalent warheads for drug discovery. Drug Discovery Today. 2023;28(12):103799. doi: 10.1016/j.drudis.2023.103799. [DOI] [PubMed] [Google Scholar]
- Krüger D. M., Neubacher S., Grossmann T. N.. Protein-RNA interactions: structural characteristics and hotspot amino acids. RNA. 2018;24(11):1457–1465. doi: 10.1261/rna.066464.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S., Daley D. T. A., Luscombe N. M., Berman H. M., Thornton J. M.. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29(4):943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. J., Broom M., Jones S.. Protein-RNA interactions: Structural analysis and functional classes. Proteins: Struct., Funct., Bioinf. 2007;66(4):903–911. doi: 10.1002/prot.21211. [DOI] [PubMed] [Google Scholar]
- Thurlkill R. L., Grimsley G. R., Scholtz J. M., Pace C. N.. pK values of the ionizable groups of proteins. Protein Sci. 2006;15(5):1214–1218. doi: 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M. J., Castañeda C. A., Schlessman J. L., Sue G. R., Isom D. G., Cannon B. R., García-Moreno E B.. The pKa Values of Acidic and Basic Residues Buried at the Same Internal Location in a Protein Are Governed by Different Factors. J. Mol. Biol. 2009;389(1):34–47. doi: 10.1016/j.jmb.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch C. A., Karp D. A., Lee K. K., Stites W. E., Lattman E. E., García-Moreno E. B.. Experimental pKa Values of Buried Residues: Analysis with Continuum Methods and Role of Water Penetration. Biophys. J. 2002;82(6):3289–3304. doi: 10.1016/S0006-3495(02)75670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broo K. S., Brive L., Sott R. S., Baltzer L.. Site-selective control of the reactivity of surface-exposed histidine residues in designed four-helix-bundle catalysts. Fold. Des. 1998;3(4):303–312. doi: 10.1016/S1359-0278(98)00041-8. [DOI] [PubMed] [Google Scholar]
- Liao S.-M., Du Q.-S., Meng J.-Z., Pang Z.-W., Huang R.-B.. The multiple roles of histidine in protein interactions. Chem. Cent. J. 2013;7(1):44. doi: 10.1186/1752-153X-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza J., Smallegan M. J., Denu J. M.. Site-Specific Reactivity of Nonenzymatic Lysine Acetylation. ACS Chem. Biol. 2015;10(1):122–128. doi: 10.1021/cb500848p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom D. G., Cannon B. R., Castañeda C. A., Robinson A., García-Moreno E B.. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc. Natl. Acad. Sci. U. S. A. 2008;105(46):17784–17788. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto R., Saloura V., Nakamura Y.. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer. 2015;15(2):110–124. doi: 10.1038/nrc3884. [DOI] [PubMed] [Google Scholar]
- Luo M.. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev. 2018;118(14):6656–6705. doi: 10.1021/acs.chemrev.8b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. A., Policastro R. A., Savant S. S., Sriramkumar S., Ding N., Lu X., Mohammad H. P., Cao S., Kalin J. H., Cole P. A.. et al. Lysine-Specific Demethylase 1 Mediates AKT Activity and Promotes Epithelial-to-Mesenchymal Transition in PIK3CA-Mutant Colorectal Cancer. Mol. Cancer Res. 2020;18(2):264–277. doi: 10.1158/1541-7786.MCR-19-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-J. C., Koutelou E., Dent S. Y. R.. Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell. 2022;82(4):716–727. doi: 10.1016/j.molcel.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M.. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40(D1):D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. A., Cole P. A.. The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem. Biol. 2020;27(8):953–969. doi: 10.1016/j.chembiol.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B., Chen K., Zhai L., Liu M., Liu B., Tan M.. Substrate and Functional Diversity of Protein Lysine Post-translational Modifications. Genom. Proteom. Bioinf. 2024;22(1):qzae019. doi: 10.1093/gpbjnl/qzae019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali I., Conrad R. J., Verdin E., Ott M.. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018;118(3):1216–1252. doi: 10.1021/acs.chemrev.7b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen D. G., Xie X., Basisty N., Byrnes J., McSweeney S., Schilling B., Wolfe A. J.. Post-translational Protein Acetylation: An Elegant Mechanism for Bacteria to Dynamically Regulate Metabolic Functions. Front. Microbiol. 2019;10:1604. doi: 10.3389/fmicb.2019.01604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Bardenys G., Peiró S.. Enzymatic lysine oxidation as a posttranslational modification. FEBS J. 2022;289(24):8020–8031. doi: 10.1111/febs.16205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney J., Moon H.-J., Ronnebaum T., Lantz M., Mure M.. Human copper-dependent amine oxidases. Arch. Biochem. Biophys. 2014;546:19–32. doi: 10.1016/j.abb.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucero H. A., Kagan H. M.. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006;63(19):2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar K.. Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 2001;70:1–32. doi: 10.1016/S0079-6603(01)70012-8. [DOI] [PubMed] [Google Scholar]
- Molnar J., Fong K. S. K., He Q. P., Hayashi K., Kim Y., Fong S. F. T., Fogelgren B., Molnarne Szauter K., Mink M., Csiszar K.. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta. 2003;1647(1):220–224. doi: 10.1016/S1570-9639(03)00053-0. [DOI] [PubMed] [Google Scholar]
- Grams R. J., Hsu K.-L.. Catch your breath. Nat. Chem. Biol. 2022;18(7):686–687. doi: 10.1038/s41589-022-01063-x. [DOI] [PubMed] [Google Scholar]
- King D. T., Zhu S., Hardie D. B., Serrano-Negrón J. E., Madden Z., Kolappan S., Vocadlo D. J.. Chemoproteomic identification of CO2-dependent lysine carboxylation in proteins. Nat. Chem. Biol. 2022;18(7):782–791. doi: 10.1038/s41589-022-01043-1. [DOI] [PubMed] [Google Scholar]
- Wang W. W., Chen L.-Y., Wozniak J. M., Jadhav A. M., Anderson H., Malone T. E., Parker C. G.. Targeted Protein Acetylation in Cells Using Heterobifunctional Molecules. J. Am. Chem. Soc. 2021;143(40):16700–16708. doi: 10.1021/jacs.1c07850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Wang X., Huang H., Xu M., Ma X., Miao F., Lu X., Zhang C.-J., Gao L., Zhang Z.-M.. et al. Small Molecule-Induced Post-Translational Acetylation of Catalytic Lysine of Kinases in Mammalian Cells. J. Am. Chem. Soc. 2024;146(34):23978–23988. doi: 10.1021/jacs.4c07181. [DOI] [PubMed] [Google Scholar]
- Hamajima W., Fujimura A., Fujiwara Y., Yamatsugu K., Kawashima S. A., Kanai M.. Site-Selective Synthetic Acylation of a Target Protein in Living Cells Promoted by a Chemical Catalyst/Donor System. ACS Chem. Biol. 2019;14(6):1102–1109. doi: 10.1021/acschembio.9b00102. [DOI] [PubMed] [Google Scholar]
- Wang Z. A., Kurra Y., Wang X., Zeng Y., Lee Y.-J., Sharma V., Lin H., Dai S. Y., Liu W. R.. A Versatile Approach for Site-Specific Lysine Acylation in Proteins. Angew. Chem., Int. Ed. 2017;56(6):1643–1647. doi: 10.1002/anie.201611415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering R. E., Cravatt B. F.. Functional Lysine Modification by an Intrinsically Reactive Primary Glycolytic Metabolite. Science. 2013;341(6145):549–553. doi: 10.1126/science.1238327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Du S., Liu Z., Liu M., Xu Z., Wang X., Kee J. X., Yi F., Sun H., Yao S. Q.. Chemical Biology Tools for Protein Lysine Acylation. Angew. Chem., Int. Ed. 2022;61(21):e202200303. doi: 10.1002/anie.202200303. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A., Gao J.. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016;34:110–116. doi: 10.1016/j.cbpa.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crugeiras J., Rios A., Riveiros E., Amyes T. L., Richard J. P.. Glycine Enolates: The Effect of Formation of Iminium Ions to Simple Ketones on α-Amino Carbon Acidity and a Comparison with Pyridoxal Iminium Ions. J. Am. Chem. Soc. 2008;130(6):2041–2050. doi: 10.1021/ja078006c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crugeiras J., Rios A., Riveiros E., Richard J. P.. Substituent Effects on the Thermodynamic Stability of Imines Formed from Glycine and Aromatic Aldehydes: Implications for the Catalytic Activity of Pyridoxal-5′-phosphate. J. Am. Chem. Soc. 2009;131(43):15815–15824. doi: 10.1021/ja906230n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque M., Forte N., Baker J. R.. Site-selective lysine conjugation methods and applications towards antibody-drug conjugates. Chem. Commun. 2021;57(82):10689–10702. doi: 10.1039/D1CC03976H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H., Mizuguchi H., Kagamiyama H.. The Imine-Pyridine Torsion of the Pyridoxal 5‘-Phosphate Schiff Base of Aspartate Aminotransferase Lowers Its pKa in the Unliganded Enzyme and Is Crucial for the Successive Increase in the pKa during Catalysis. Biochemistry. 1998;37(43):15076–15085. doi: 10.1021/bi981517e. [DOI] [PubMed] [Google Scholar]
- Parra M., Stahl S., Hellmann H.. Vitamin B6 and Its Role in Cell Metabolism and Physiology. Cells. 2018;7(7):84. doi: 10.3390/cells7070084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuchair Ebadi C. F. G.. Azhar Al-Sayegh. Drug-pyridoxal phosphate interactions. Drug Metab. Drug Interact. 1982;4:289–331. doi: 10.1515/DMDI.1982.4.4.289. [DOI] [PubMed] [Google Scholar]
- French T. C., Auld D. S., Bruice T. C.. Catalytic Reactions Involving Azomethines. V. Rates and Equilibria of Imine Formation with 3-Hydroxypyridine-4-aldehyde and Amino Acids*. Biochemistry. 1965;4(1):77–84. doi: 10.1021/bi00877a014. [DOI] [PubMed] [Google Scholar]
- Eliot A. C., Kirsch J. F.. Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu. Rev. Biochem. 2004;73(1):383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- Liang J., Han Q., Tan Y., Ding H., Li J.. Current Advances on Structure-Function Relationships of Pyridoxal 5′-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 2019;6:4. doi: 10.3389/fmolb.2019.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A., Yoshimura T., Mikami B., Esaki N.. Tyrosine 265 of Alanine Racemase Serves as a Base Abstracting α-Hydrogen from L-Alanine: The Counterpart Residue to Lysine 39 Specific to D-Alanine. J. Biochem. 1999;126(4):781–786. doi: 10.1093/oxfordjournals.jbchem.a022517. [DOI] [PubMed] [Google Scholar]
- Watanabe A., Yoshimura T., Mikami B., Hayashi H., Kagamiyama H., Esaki N.. Reaction Mechanism of Alanine Racemase from Bacillus stearothermophilus: X-RAY CRYSTALLOGRAPHIC STUDIES OF THE ENZYME BOUND WITH N-(5′-PHOSPHOPYRIDOXYL)ALANINE*. J. Biol. Chem. 2002;277(21):19166–19172. doi: 10.1074/jbc.M201615200. [DOI] [PubMed] [Google Scholar]
- Fonda M. L., Auerbach S. B.. The interaction of pyridoxal phosphate with aspartate apoaminotransferase. Biochim. Biophys. Acta, Enzymol. 1976;422(1):38–47. doi: 10.1016/0005-2744(76)90006-1. [DOI] [PubMed] [Google Scholar]
- Toney M. D.. Aspartate aminotransferase: An old dog teaches new tricks. Arch. Biochem. Biophys. 2014;544:119–127. doi: 10.1016/j.abb.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H., Tsukiyama F., Ishii S., Mizuguchi H., Kagamiyama H.. Acid-Base Chemistry of the Reaction of Aromatic l-Amino Acid Decarboxylase and Dopa Analyzed by Transient and Steady-State Kinetics: Preferential Binding of the Substrate with Its Amino Group Unprotonated. Biochemistry. 1999;38(47):15615–15622. doi: 10.1021/bi9909795. [DOI] [PubMed] [Google Scholar]
- Takano Y., Kondo H. X., Nakamura H.. Quantum chemical studies on hydrogen bonds in helical secondary structures. Biophys. Rev. 2022;14(6):1369–1378. doi: 10.1007/s12551-022-01034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt M. R., Ganguly H. K., Zondlo N. J.. Acyl Capping Group Identity Effects on α-Helicity: On the Importance of Amide·Water Hydrogen Bonds to α-Helix Stability. Biochemistry. 2024;63(9):1118–1130. doi: 10.1021/acs.biochem.3c00646. [DOI] [PubMed] [Google Scholar]
- Tramonti A., Ghatge M. S., Babor J. T., Musayev F. N., di Salvo M. L., Barile A., Colotti G., Giorgi A., Paredes S. D., Donkor A. K.. et al. Characterization of the Escherichia coli pyridoxal 5′-phosphate homeostasis protein (YggS): Role of lysine residues in PLP binding and protein stability. Protein Sci. 2022;31(11):e4471. doi: 10.1002/pro.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. P., Plecko B., Mills P. B., Clayton P. T.. Disorders affecting vitamin B6 metabolism. J. Inherit. Metab. Dis. 2019;42(4):629–646. doi: 10.1002/jimd.12060. [DOI] [PubMed] [Google Scholar]
- Wang P., Pradhan K., Zhong X.-b., Ma X.. Isoniazid metabolism and hepatotoxicity. Acta Pharm. Sin. B. 2016;6(5):384–392. doi: 10.1016/j.apsb.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilliers K., Labadarios D., Schaaf H., Willemse M., Maritz J., Werely C., Hussey G., Donald P.. Pyridoxal-5-phosphate plasma concentrations in children receiving tuberculosis chemotherapy including isoniazid. Acta Paediatr. 2010;99(5):705–710. doi: 10.1111/j.1651-2227.2010.01696.x. [DOI] [PubMed] [Google Scholar]
- Wason S., Lacouture P. G., Lovejoy F. H. Jr. Single High-Dose Pyridoxine Treatment for Isoniazid Overdose. JAMA. 1981;246(10):1102–1104. doi: 10.1001/jama.1981.03320100038026. [DOI] [PubMed] [Google Scholar]
- Li F., Miao Y., Zhang L., Neuenswander S. A., Douglas J. T., Ma X.. Metabolomic Analysis Reveals Novel Isoniazid Metabolites and Hydrazones in Human Urine. Drug Metab. Pharmacokinet. 2011;26(6):569–576. doi: 10.2133/dmpk.DMPK-11-RG-055. [DOI] [PubMed] [Google Scholar]
- Khan S. R., Morgan A. G. M., Michail K., Srivastava N., Whittal R. M., Aljuhani N., Siraki A. G.. Metabolism of isoniazid by neutrophil myeloperoxidase leads to isoniazid-NAD+ adduct formation: A comparison of the reactivity of isoniazid with its known human metabolites. Biochem. Pharmacol. 2016;106:46–55. doi: 10.1016/j.bcp.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Hall H. K. Jr. Correlation of the Base Strengths of Amines1. J. Am. Chem. Soc. 1957;79(20):5441–5444. doi: 10.1021/ja01577a030. [DOI] [Google Scholar]
- Echevarría-Gorostidi G. R., Basagoitia A., Pizarro E., Goldsmid R., Blanco J. G. S., Blanco F. G.. Kinetic Study of the Reaction of Pyridoxal 5′-Phosphate with Hydrazino Compounds of Pharmacological Activity. Helv. Chim. Acta. 1998;81(5–8):837–844. doi: 10.1002/hlca.19980810505. [DOI] [Google Scholar]
- Airoldi L., Watkins C. J., Wiggins J. F., Wurtman R. J.. Effect of pyridoxine on the depletion of tissue pyridoxal phosphate by carbidopa. Metabolism. 1978;27(7):771–779. doi: 10.1016/0026-0495(78)90211-1. [DOI] [PubMed] [Google Scholar]
- Wise A., Lemus H. N., Fields M., Swan M., Bressman S.. Refractory Seizures Secondary to Vitamin B6 Deficiency in Parkinson Disease: The Role of Carbidopa-Levodopa. Case Rep. Neurol. 2022;14(2):291–295. doi: 10.1159/000525234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleineke J., Peters H., Söling H. D.. Inhibition of hepatic gluconeogenesis by phenethylhydrazine (phenelzine) Biochem. Pharmacol. 1979;28(8):1379–1389. doi: 10.1016/0006-2952(79)90441-6. [DOI] [PubMed] [Google Scholar]
- Lowther J., Yard B. A., Johnson K. A., Carter L. G., Bhat V. T., Raman M. C. C., Clarke D. J., Ramakers B., McMahon S. A., Naismith J. H.. et al. Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: evidence for a novel decarboxylative mechanism of inactivation. Mol. BioSyst. 2010;6(9):1682–1693. doi: 10.1039/c003743e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R., Ogawa S., Takahashi Y., Fujishiro T.. Cycloserine enantiomers inhibit PLP-dependent cysteine desulfurase SufS via distinct mechanisms. FEBS J. 2022;289(19):5947–5970. doi: 10.1111/febs.16455. [DOI] [PubMed] [Google Scholar]
- Peisach D., Chipman D. M., Van Ophem P. W., Manning J. M., Ringe D.. d-Cycloserine Inactivation of d-Amino Acid Aminotransferase Leads to a Stable Noncovalent Protein Complex with an Aromatic Cycloserine-PLP Derivative. J. Am. Chem. Soc. 1998;120(10):2268–2274. doi: 10.1021/ja973353f. [DOI] [Google Scholar]
- Gao S., Liu H., de Crécy-Lagard V., Zhu W., Richards N. G. J., Naismith J. H.. PMP-diketopiperazine adducts form at the active site of a PLP dependent enzyme involved in formycin biosynthesis. Chem. Commun. 2019;55(96):14502–14505. doi: 10.1039/C9CC06975E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther J., Beattie A. E., Langridge-Smith P. R. R., Clarke D. J., Campopiano D. J.. l-Penicillamine is a mechanism-based inhibitor of serine palmitoyltransferase by forming a pyridoxal-5′-phosphate-thiazolidine adduct. Med. Chem. Commun. 2012;3(8):1003–1008. doi: 10.1039/c2md20020a. [DOI] [Google Scholar]
- Elezaby M. S., Moussa N. M., Elhilaly A. E., Farid S.. The Binding Affinity of Amphetamine to 4-Pyridinecarboxaldehyde and the Coenzymes Pyridoxal and Pyridoxal-5-phosphate. Chem. Pharm. Bull. 1977;25(3):401–412. doi: 10.1248/cpb.25.401. [DOI] [PubMed] [Google Scholar]
- Hoegl A., Nodwell M. B., Kirsch V. C., Bach N. C., Pfanzelt M., Stahl M., Schneider S., Sieber S. A.. Mining the cellular inventory of pyridoxal phosphate-dependent enzymes with functionalized cofactor mimics. Nat. Chem. 2018;10(12):1234–1245. doi: 10.1038/s41557-018-0144-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fux A., Pfanzelt M., Kirsch V. C., Hoegl A., Sieber S. A.. Customizing Functionalized Cofactor Mimics to Study the Human Pyridoxal 5′-Phosphate-Binding Proteome. Cell Chem. Biol. 2019;26(10):1461–1468. doi: 10.1016/j.chembiol.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanzelt M., Maher T. E., Absmeier R. M., Schwarz M., Sieber S. A.. Tailored Pyridoxal Probes Unravel Novel Cofactor-Dependent Targets and Antibiotic Hits in Critical Bacterial Pathogens. Angew. Chem., Int. Ed. 2022;61(24):e202117724. doi: 10.1002/anie.202117724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkers K., Shizukuishi S., Scudder S. L., Willis R., Takemura K., Longenecker J. B.. Biochemical evidence for a deficiency of vitamin B6 in subjects reacting to monosodium L-glutamate by the Chinese Restaurant Syndrome. Biochem. Biophys. Res. Commun. 1981;100(3):972–977. doi: 10.1016/0006-291X(81)91918-5. [DOI] [PubMed] [Google Scholar]
- Zhou T., Parillon L., Li F., Wang Y., Keats J., Lamore S., Xu Q., Shakespeare W., Dalgarno D., Zhu X.. Crystal Structure of the T315I Mutant of Abl Kinase. Chem. Biol. Drug Des. 2007;70(3):171–181. doi: 10.1111/j.1747-0285.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- Wilde J. A., Bolton P. H., Mazumder A., Manoharan M., Gerlt J. A.. Characterization of the equilibrating forms of the aldehydic abasic site in duplex DNA by oxygen-17 NMR. J. Am. Chem. Soc. 1989;111(5):1894–1896. doi: 10.1021/ja00187a062. [DOI] [Google Scholar]
- Melton D., Lewis C. D., Price N. E., Gates K. S.. Covalent Adduct Formation between the Antihypertensive Drug Hydralazine and Abasic Sites in Double- and Single-Stranded DNA. Chem. Res. Toxicol. 2014;27(12):2113–2118. doi: 10.1021/tx5003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M.. ADVANCED PROTEIN GLYCOSYLATION IN DIABETES AND AGING. Annu. Rev. Med. 1995;46(1):223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- Goldin A., Beckman J. A., Schmidt A. M., Creager M. A.. Advanced Glycation End Products. Circulation. 2006;114(6):597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- Rungratanawanich W., Qu Y., Wang X., Essa M. M., Song B.-J.. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021;53(2):168–188. doi: 10.1038/s12276-021-00561-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-y., Zhang J.-Q., Li L., Guo M.-m., He Y.-f., Dong Y.-m., Meng H., Yi F.. Advanced Glycation End Products in the Skin: Molecular Mechanisms, Methods of Measurement, and Inhibitory Pathways. Front. Med. 2022;9:837222. doi: 10.3389/fmed.2022.837222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twarda-Clapa A., Olczak A., Białkowska A. M., Koziołkiewicz M.. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells. 2022;11(8):1312. doi: 10.3390/cells11081312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibbersen C., Schou Oxvig A.-M., Bisgaard Olesen S., Nielsen C. B., Galligan J. J., Jørgensen K. A., Palmfeldt J., Johannsen M.. Profiling of Methylglyoxal Blood Metabolism and Advanced Glycation End-Product Proteome Using a Chemical Probe. ACS Chem. Biol. 2018;13(12):3294–3305. doi: 10.1021/acschembio.8b00732. [DOI] [PubMed] [Google Scholar]
- Chen X., Liu Y., Kong L., Wen Z., Wang W., Wang C.. Quantitative Chemoproteomic Profiling of Protein Cross-Links Induced by Methylglyoxal. ACS Chem. Biol. 2022;17(8):2010–2017. doi: 10.1021/acschembio.2c00017. [DOI] [PubMed] [Google Scholar]
- Reddy V. P., Aryal P., Darkwah E. K.. Advanced Glycation End Products in Health and Disease. Microorganisms. 2022;10(9):1848. doi: 10.3390/microorganisms10091848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C., Sabol J., Mitsuhashi T., Vlassara H.. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes. 1999;48(6):1308–1315. doi: 10.2337/diabetes.48.6.1308. [DOI] [PubMed] [Google Scholar]
- Mason M., Belvisi L., Pignataro L., Dal Corso A.. A Tight Contact: The Expanding Application of Salicylaldehydes in Lysine-Targeting Covalent Drugs. ChemBioChem. 2024;25:e202300743. doi: 10.1002/cbic.202300743. [DOI] [PubMed] [Google Scholar]
- Weaver J., Craven G. B., Tram L., Chen H., Taunton J.. Aminomethyl Salicylaldehydes Lock onto a Surface Lysine by Forming an Extended Intramolecular Hydrogen Bond Network. J. Am. Chem. Soc. 2024;146(35):24233–24237. doi: 10.1021/jacs.4c04314. [DOI] [PubMed] [Google Scholar]
- Gampe C., Verma V. A.. Curse or Cure? A Perspective on the Developability of Aldehydes as Active Pharmaceutical Ingredients. J. Med. Chem. 2020;63(23):14357–14381. doi: 10.1021/acs.jmedchem.0c01177. [DOI] [PubMed] [Google Scholar]
- Volkmann K., Lucas J. L., Vuga D., Wang X., Brumm D., Stiles C., Kriebel D., Der-Sarkissian A., Krishnan K., Schweitzer C.. et al. Potent and Selective Inhibitors of the Inositol-requiring Enzyme 1 Endoribonuclease. J. Biol. Chem. 2011;286(14):12743–12755. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Sun J., Zhu C., Tang G., Wang W., Xu M., Xiang M., Zhang C.-J., Zhang Z.-M., Gao L.. et al. Cell-Active, Reversible, and Irreversible Covalent Inhibitors That Selectively Target the Catalytic Lysine of BCR-ABL Kinase. Angew. Chem., Int. Ed. 2022;61(26):e202203878. doi: 10.1002/anie.202203878. [DOI] [PubMed] [Google Scholar]
- Quach D., Tang G., Anantharajan J., Baburajendran N., Poulsen A., Wee J. L. K., Retna P., Li R., Liu B., Tee D. H. Y.. et al. Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors. Angew. Chem., Int. Ed. 2021;60(31):17131–17137. doi: 10.1002/anie.202105383. [DOI] [PubMed] [Google Scholar]
- Ahmed Laskar A., Younus H.. Aldehyde toxicity and metabolism: the role of aldehyde dehydrogenases in detoxification, drug resistance and carcinogenesis. Drug Metab. Rev. 2019;51(1):42–64. doi: 10.1080/03602532.2018.1555587. [DOI] [PubMed] [Google Scholar]
- Zheng L., Li Y., Wu D., Xiao H., Zheng S., Wang G., Sun Q.. Development of covalent inhibitors: Principle, design, and application in cancer. MedComm - Oncol. 2023;2(4):e56. doi: 10.1002/mog2.56. [DOI] [Google Scholar]
- Liu R., Yue Z., Tsai C.-C., Shen J.. Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2019;141(16):6553–6560. doi: 10.1021/jacs.8b13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco K. R., Bruystens J., Varricchio C., McCurdy S., Wu J., Lopez-Ramirez M. A., Ginsberg M., Caffrey C. R., Brancale A., Gingras A. R.. et al. Targeted Reversible Covalent Modification of a Noncatalytic Lysine of the Krev Interaction Trapped 1 Protein Enables Site-Directed Screening for Protein-Protein Interaction Inhibitors. ACS Pharmacol. Transl. Sci. 2023;6(11):1651–1658. doi: 10.1021/acsptsci.3c00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Ramirez M. A., McCurdy S., Li W., Haynes M. K., Hale P., Francisco K., Oukoloff K., Bautista M., Choi C. H. J., Sun H.. et al. Inhibition of the HEG1-KRIT1 interaction increases KLF4 and KLF2 expression in endothelial cells. FASEB BioAdvances. 2021;3(5):334–355. doi: 10.1096/fba.2020-00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven G. B., Chu H., Sun J. D., Carelli J. D., Coyne B., Chen H., Chen Y., Ma X., Das S., Kong W.. et al. Mutant-selective AKT inhibition through lysine targeting and neo-zinc chelation. Nature. 2025;637:205. doi: 10.1038/s41586-024-08176-4. [DOI] [PubMed] [Google Scholar]
- Lapierre J.-M., Eathiraj S., Vensel D., Liu Y., Bull C. O., Cornell-Kennon S., Iimura S., Kelleher E. W., Kizer D. E., Koerner S.. et al. Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An Orally Bioavailable, Selective, and Potent Allosteric AKT Inhibitor. J. Med. Chem. 2016;59(13):6455–6469. doi: 10.1021/acs.jmedchem.6b00619. [DOI] [PubMed] [Google Scholar]
- Hyman D. M., Smyth L. M., Donoghue M. T. A., Westin S. N., Bedard P. L., Dean E. J., Bando H., El-Khoueiry A. B., Pérez-Fidalgo J. A., Mita A.. et al. AKT Inhibition in Solid Tumors With AKT1Mutations. J. Clin. Oncol. 2017;35(20):2251–2259. doi: 10.1200/JCO.2017.73.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth L. M., Tamura K., Oliveira M., Ciruelos E. M., Mayer I. A., Sablin M.-P., Biganzoli L., Ambrose H. J., Ashton J., Barnicle A.. et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1E17K-Mutant, ER-Positive Metastatic Breast Cancer. Clin. Cancer. Res. 2020;26(15):3947–3957. doi: 10.1158/1078-0432.CCR-19-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji U., Dean E. J., Pérez-Fidalgo J. A., Batist G., Bedard P. L., You B., Westin S. N., Kabos P., Garrett M. D., Tall M.. et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer. Res. 2018;24(9):2050–2059. doi: 10.1158/1078-0432.CCR-17-2260. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A., Gao J.. Iminoboronate Formation Leads to Fast and Reversible Conjugation Chemistry of α-Nucleophiles at Neutral pH. Chem. Eur. J. 2015;21(42):14748–14752. doi: 10.1002/chem.201502077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reja R. M., Wang W., Lyu Y., Haeffner F., Gao J.. Lysine-Targeting Reversible Covalent Inhibitors with Long Residence Time. J. Am. Chem. Soc. 2022;144(3):1152–1157. doi: 10.1021/jacs.1c12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel C., Kasper M.-A., Stieger C. E., Hackenberger C. P. R., Christmann M.. Protein Modification of Lysine with 2-(2-Styrylcyclopropyl)ethanal. Org. Lett. 2019;21(24):10043–10047. doi: 10.1021/acs.orglett.9b03982. [DOI] [PubMed] [Google Scholar]
- Chilamari M., Kalra N., Shukla S., Rai V.. Single-site labeling of lysine in proteins through a metal-free multicomponent approach. Chem. Commun. 2018;54(53):7302–7305. doi: 10.1039/C8CC03311K. [DOI] [PubMed] [Google Scholar]
- Yang T., Cuesta A., Wan X., Craven G. B., Hirakawa B., Khamphavong P., May J. R., Kath J. C., Lapek J. D., Niessen S.. et al. Reversible lysine-targeted probes reveal residence time-based kinase selectivity. Nat. Chem. Biol. 2022;18(9):934–941. doi: 10.1038/s41589-022-01019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Jia S., Chen H., You L.. Modulation of imine chemistry with intramolecular hydrogen bonding: Effects from ortho-OH to NH. Tetrahedron. 2020;76(17):131128. doi: 10.1016/j.tet.2020.131128. [DOI] [Google Scholar]
- Abbasov M. E., Kavanagh M. E., Ichu T.-A., Lazear M. R., Tao Y., Crowley V. M., am Ende C. W., Hacker S. M., Ho J., Dix M. M.. et al. A proteome-wide atlas of lysine-reactive chemistry. Nat. Chem. 2021;13:1081–1092. doi: 10.1038/s41557-021-00765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken A. K., Gekko C. E., Suss N. O., Lueders E. E., Cui Q., Fu Q., Lui A. C. W., Anderson E. T., Zhang S., Abbasov M. E.. Biomimetic Synthesis and Chemical Proteomics Reveal the Mechanism of Action and Functional Targets of Phloroglucinol Meroterpenoids. J. Am. Chem. Soc. 2024;146(4):2524–2548. doi: 10.1021/jacs.3c10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Tang G., Zhu C., Sun J., Wang X., Xiang M., Huang H., Wang W., Li L., Zhang Z.-M.. et al. 2-Ethynylbenzaldehyde-Based, Lysine-Targeting Irreversible Covalent Inhibitors for Protein Kinases and Nonkinases. J. Am. Chem. Soc. 2023;145(7):3844–3849. doi: 10.1021/jacs.2c11595. [DOI] [PubMed] [Google Scholar]
- Deng J.-R., Lai N. C.-H., Kung K. K.-Y., Yang B., Chung S.-F., Leung A. S.-L., Choi M.-C., Leung Y.-C., Wong M.-K.. N-Terminal selective modification of peptides and proteins using 2-ethynylbenzaldehydes. Commun. Chem. 2020;3(1):67. doi: 10.1038/s42004-020-0309-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Huang R., Li Z., Zhu W., Chen J., Zhan Y., Jiang B.. Selective lysine modification of native peptides via aza-Michael addition. Org. Biomol. Chem. 2017;15(35):7339–7345. doi: 10.1039/C7OB01866E. [DOI] [PubMed] [Google Scholar]
- Ihle N. T., Williams R., Chow S., Chew W., Berggren M. I., Paine-Murrieta G., Minion D. J., Halter R. J., Wipf P., Abraham R.. et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol. Cancer Ther. 2004;3(7):763–772. doi: 10.1158/1535-7163.763.3.7. [DOI] [PubMed] [Google Scholar]
- Koul D., Shen R., Kim Y.-W., Kondo Y., Lu Y., Bankson J., Ronen S. M., Kirkpatrick D. L., Powis G., Yung W. K. A.. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro. Oncol. 2010;12(6):559–569. doi: 10.1093/neuonc/nop058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardcastle I. R., Arris C. E., Bentley J., Boyle F. T., Chen Y., Curtin N. J., Endicott J. A., Gibson A. E., Golding B. T., Griffin R. J.. et al. N2-Substituted O6-Cyclohexylmethylguanine Derivatives: Potent Inhibitors of Cyclin-Dependent Kinases 1 and 2. J. Med. Chem. 2004;47(15):3710–3722. doi: 10.1021/jm0311442. [DOI] [PubMed] [Google Scholar]
- Anscombe E., Meschini E., Mora-Vidal R., Martin M. P., Staunton D., Geitmann M., Danielson U. H., Stanley W. A., Wang L. Z., Reuillon T.. et al. Identification and Characterization of an Irreversible Inhibitor of CDK2. Chem. Biol. 2015;22(9):1159–1164. doi: 10.1016/j.chembiol.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prucha G. R., Henry S., Hollander K., Carter Z. J., Spasov K. A., Jorgensen W. L., Anderson K. S.. Covalent and noncovalent strategies for targeting Lys102 in HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2023;262:115894. doi: 10.1016/j.ejmech.2023.115894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A. H., Lee W.-G., Spasov K. A., Cisneros J. A., Kudalkar S. N., Petrova Z. O., Buckingham A. B., Anderson K. S., Jorgensen W. L.. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proc. Natl. Acad. Sci. U. S. A. 2017;114(36):9725–9730. doi: 10.1073/pnas.1711463114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal U. P., Gilbert A. M., Obach R. S., Flanagan M. E., Chen J. M., Garcia-Irizarry C., Starr J. T., Schuff B., Uccello D. P., Young J. A.. Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design: N-α-acetyl-l-lysine as an amine nucleophile. MedChemComm. 2016;7(5):864–872. doi: 10.1039/C6MD00017G. [DOI] [Google Scholar]
- LoPachin R. M., Gavin T., DeCaprio A., Barber D. S.. Application of the Hard and Soft, Acids and Bases (HSAB) Theory to Toxicant-Target Interactions. Chem. Res. Toxicol. 2012;25(2):239–251. doi: 10.1021/tx2003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Hamon C.. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003;75(8):1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Pickens C. J., Johnson S. N., Pressnall M. M., Leon M. A., Berkland C. J.. Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide-Alkyne Cycloaddition. Bioconjugate Chem. 2018;29(3):686–701. doi: 10.1021/acs.bioconjchem.7b00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecha J., Satpathy S., Kanashova T., Avanessian S. C., Kane M. H., Clauser K. R., Mertins P., Carr S. A., Kuster B.. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol. Cell. Proteomics. 2019;18(7):1468–1478. doi: 10.1074/mcp.TIR119.001385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R., You P., Weng K., Li L., Song Y.. TMT Labeling under Acidic pH Overcomes Detrimental Overlabeling and Improves Peptide Identification Rates. Anal. Chem. 2023;95(28):10595–10602. doi: 10.1021/acs.analchem.3c00525. [DOI] [PubMed] [Google Scholar]
- Frey K. M., Bollini M., Mislak A. C., Cisneros J. A., Gallardo-Macias R., Jorgensen W. L., Anderson K. S.. Crystal Structures of HIV-1 Reverse Transcriptase with Picomolar Inhibitors Reveal Key Interactions for Drug Design. J. Am. Chem. Soc. 2012;134(48):19501–19503. doi: 10.1021/ja3092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito J. A., Niu H., Bertoletti N., Carter Z. J., Jin S., Spasov K. A., Cisneros J. A., Valhondo M., Cutrona K. J., Anderson K. S.. et al. Covalent Inhibition of Wild-Type HIV-1 Reverse Transcriptase Using a Fluorosulfate Warhead. ACS. Med. Chem. Lett. 2021;12(2):249–255. doi: 10.1021/acsmedchemlett.0c00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C., Yang D., An Y., Kong L., Zhou Z., Tang L., Zhang Z., Dai Y., Wang R.. Tunable Activated Esters Enable Lysine-Selective Protein Labeling and Profiling. Anal. Chem. 2024;96(46):18377–18383. doi: 10.1021/acs.analchem.4c02215. [DOI] [PubMed] [Google Scholar]
- Down K., Amour A., Baldwin I. R., Cooper A. W. J., Deakin A. M., Felton L. M., Guntrip S. B., Hardy C., Harrison Z. A., Jones K. L.. et al. Optimization of Novel Indazoles as Highly Potent and Selective Inhibitors of Phosphoinositide 3-Kinase δ for the Treatment of Respiratory Disease. J. Med. Chem. 2015;58(18):7381–7399. doi: 10.1021/acs.jmedchem.5b00767. [DOI] [PubMed] [Google Scholar]
- Dalton S. E., Dittus L., Thomas D. A., Convery M. A., Nunes J., Bush J. T., Evans J. P., Werner T., Bantscheff M., Murphy J. A.. et al. Selectively Targeting the Kinome-Conserved Lysine of PI3Kδ as a General Approach to Covalent Kinase Inhibition. J. Am. Chem. Soc. 2018;140(3):932–939. doi: 10.1021/jacs.7b08979. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P., Szardenings A. K., Liyanage M., Nomanbhoy T. K., Wu M., Weissig H., Aban A., Chun D., Tanner S., Kozarich J. W.. Functional Interrogation of the Kinome Using Nucleotide Acyl Phosphates. Biochemistry. 2007;46(2):350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- Nordin B. E., Liu Y., Aban A., Brown H. E., Wu J., Hainley A. K., Rosenblum J. S., Nomanbhoy T. K., Kozarich J. W.. ATP Acyl Phosphate Reactivity Reveals Native Conformations of Hsp90 Paralogs and Inhibitor Target Engagement. Biochemistry. 2015;54(19):3024–3036. doi: 10.1021/acs.biochem.5b00148. [DOI] [PubMed] [Google Scholar]
- Franks C. E., Hsu K.-L.. Activity-Based Kinome Profiling Using Chemical Proteomics and ATP Acyl Phosphates. Cur. Protoc. Chem. Biol. 2019;11(3):e72. doi: 10.1002/cpch.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerberg E. S., Dagbay K. B., Green J. L., Soni I., Aban A., Nomanbhoy T. K., Savinov S. N., Hardy J. A., Kozarich J. W.. Chemoproteomics Using Nucleotide Acyl Phosphates Reveals an ATP Binding Site at the Dimer Interface of Procaspase-6. Biochemistry. 2019;58(52):5320–5328. doi: 10.1021/acs.biochem.9b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Shin M., Ware T. B., Donvito G., Muchhala K. H., Mischel R., Mustafa M. A., Serbulea V., Upchurch C. M., Leitinger N.. et al. Endocannabinoid biosynthetic enzymes regulate pain response via LKB1-AMPK signaling. Proc. Natl. Acad. Sci. U. S. A. 2023;120(52):e2304900120. doi: 10.1073/pnas.2304900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M., Franks C. E., Hsu K.-L.. Isoform-selective activity-based profiling of ERK signaling. Chem. Sci. 2018;9(9):2419–2431. doi: 10.1039/C8SC00043C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. N., Noble M. E. M., Owen D. J.. Active and Inactive Protein Kinases: Structural Basis for Regulation. Cell. 1996;85(2):149–158. doi: 10.1016/S0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Nolen B., Taylor S., Ghosh G.. Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Mol. Cell. 2004;15(5):661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Xiao Y., Wang Y.. Global discovery of protein kinases and other nucleotide-binding proteins by mass spectrometry. Mass Spectrom. Rev. 2016;35(5):601–619. doi: 10.1002/mas.21447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenner G. W., McDermott J. R., Sheppard R. C.. The safety catch principle in solid phase peptide synthesis. Journal of the Chemical Society D: Chem. Commun. 1971;(12):636–637. doi: 10.1039/c29710000636. [DOI] [Google Scholar]
- Backes B. J., Ellman J. A.. Carbon-Carbon Bond-Forming Methods on Solid Support. Utilization of Kenner’s ″Safety-Catch″ Linker. J. Am. Chem. Soc. 1994;116(24):11171–11172. doi: 10.1021/ja00103a048. [DOI] [Google Scholar]
- Noki S., de la Torre B. G., Albericio F.. Safety-Catch Linkers for Solid-Phase Peptide Synthesis. Molecules. 2024;29(7):1429. doi: 10.3390/molecules29071429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T., Song Z., Amaike K., Lee S., Yin S., Kiyonaka S., Hamachi I.. Affinity-Guided Oxime Chemistry for Selective Protein Acylation in Live Tissue Systems. J. Am. Chem. Soc. 2017;139(40):14181–14191. doi: 10.1021/jacs.7b07339. [DOI] [PubMed] [Google Scholar]
- Tamura T., Ueda T., Goto T., Tsukidate T., Shapira Y., Nishikawa Y., Fujisawa A., Hamachi I.. Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat. Commun. 2018;9(1):1870. doi: 10.1038/s41467-018-04343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimaradka V., Hoon Oh J., Heroven C., Radu Aricescu A., Yuzaki M., Tamura T., Hamachi I.. Site-specific covalent labeling of His-tag fused proteins with N-acyl-N-alkyl sulfonamide reagent. Bioorg. Med. Chem. 2021;30:115947. doi: 10.1016/j.bmc.2020.115947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M., Murakawa S., Higashiguchi K., Matsuda K., Tamura T., Hamachi I.. Lysine-Reactive N-Acyl-N-aryl Sulfonamide Warheads: Improved Reaction Properties and Application in the Covalent Inhibition of an Ibrutinib-Resistant BTK Mutant. J. Am. Chem. Soc. 2023;145(48):26202–26212. doi: 10.1021/jacs.3c08740. [DOI] [PubMed] [Google Scholar]
- Zheng J., Trafny E. A., Knighton D. R., Xuong N., Taylor S. S., Ten Eyck L. F., Sowadski J. M.. 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. 1993;D49:362–365. doi: 10.1107/S0907444993000423. [DOI] [PubMed] [Google Scholar]
- Ueda T., Tamura T., Kawano M., Shiono K., Hobor F., Wilson A. J., Hamachi I.. Enhanced Suppression of a Protein-Protein Interaction in Cells Using Small-Molecule Covalent Inhibitors Based on an N-Acyl-N-alkyl Sulfonamide Warhead. J. Am. Chem. Soc. 2021;143(12):4766–4774. doi: 10.1021/jacs.1c00703. [DOI] [PubMed] [Google Scholar]
- Tivon Y., Falcone G., Deiters A.. Protein Labeling and Crosslinking by Covalent Aptamers. Angew. Chem., Int. Ed. 2021;60(29):15899–15904. doi: 10.1002/anie.202101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan E. M., Norinskiy M. A., Bracken A. K., Lueders E. E., Chen X., Fu Q., Anderson E. T., Zhang S., Abbasov M. E.. Activity-Based Acylome Profiling with N-(Cyanomethyl)-N-(phenylsulfonyl)amides for Targeted Lysine Acylation and Post-Translational Control of Protein Function in Cells. J. Am. Chem. Soc. 2024;146(40):27622–27643. doi: 10.1021/jacs.4c09073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M., Jiang J., Ficarro S. B., Seo H.-S., Bae J. H., Donovan K. A., Fischer E. S., Zhang T., Dhe-Paganon S., Marto J. A.. et al. Exploring Ligand-Directed N-Acyl-N-alkylsulfonamide-Based Acylation Chemistry for Potential Targeted Degrader Development. ACS. Med. Chem. Lett. 2021;12(8):1302–1307. doi: 10.1021/acsmedchemlett.1c00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnew-Francis K. A., Williams C. M.. Squaramides as Bioisosteres in Contemporary Drug Design. Chem. Rev. 2020;120(20):11616–11650. doi: 10.1021/acs.chemrev.0c00416. [DOI] [PubMed] [Google Scholar]
- Marchetti L. A., Kumawat L. K., Mao N., Stephens J. C., Elmes R. B. P.. The Versatility of Squaramides: From Supramolecular Chemistry to Chemical Biology. Chem. 2019;5(6):1398–1485. doi: 10.1016/j.chempr.2019.02.027. [DOI] [Google Scholar]
- Seio K., Miyashita T., Sato K., Sekine M.. Synthesis and Properties of New Nucleotide Analogues Possessing Squaramide Moieties as New Phosphate Isosters. Eur. J. Org. Chem. 2005;2005(24):5163–5170. doi: 10.1002/ejoc.200500520. [DOI] [Google Scholar]
- Lu M., Lu Q.-B., Honek J. F.. Squarate-based carbocyclic nucleosides: Syntheses, computational analyses and anticancer/antiviral evaluation. Bioorg. Med. Chem. Lett. 2017;27(2):282–287. doi: 10.1016/j.bmcl.2016.11.058. [DOI] [PubMed] [Google Scholar]
- Dürr E.-M., Doherty W., Lee S. Y., El-Sagheer A. H., Shivalingam A., McHugh P. J., Brown T., McGouran J. F.. Squaramide-Based 5′-Phosphate Replacements Bind to the DNA Repair Exonuclease SNM1A. ChemistrySelect. 2018;3(45):12824–12829. doi: 10.1002/slct.201803375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T., Shinada T., Ohfune Y.. Synthesis of novel amino squaric acids via addition of dianion enolates derived from N-Boc amino acid esters. Tetrahedron Lett. 2005;46(2):311–314. doi: 10.1016/j.tetlet.2004.11.044. [DOI] [Google Scholar]
- Shinada T., Ishida T., Hayashi K.-i., Yoshida Y., Shigeri Y., Ohfune Y.. Synthesis of leucine-enkephalin analogs containing α-amino squaric acid. Tetrahedron Lett. 2007;48(43):7614–7617. doi: 10.1016/j.tetlet.2007.08.103. [DOI] [Google Scholar]
- Maeda K., Kiniwa Y.-i., Ohfune Y., Ishiguro S., Suzuki K., Murata K., Matsuda H., Shinada T.. Solid phase synthesis of α-amino squaric acid-containing peptides. RSC Adv. 2014;4(92):50639–50643. doi: 10.1039/C4RA10442K. [DOI] [Google Scholar]
- Martínez L., Sampedro A., Sanna E., Costa A., Rotger C.. Synthesis and conformational studies of peptido-squaramide foldable modules: a new class of turn-mimetic compounds. Org. Biomol. Chem. 2012;10(9):1914–1921. doi: 10.1039/c2ob06715c. [DOI] [PubMed] [Google Scholar]
- Martínez-Crespo L., Escudero-Adán E. C., Costa A., Rotger C.. The Role of N-Methyl Squaramides in a Hydrogen-Bonding Strategy to Fold Peptidomimetic Compounds. Chem. Eur. J. 2018;24(67):17802–17813. doi: 10.1002/chem.201803930. [DOI] [PubMed] [Google Scholar]
- Martínez L., Martorell G., Sampedro Á., Ballester P., Costa A., Rotger C.. Hydrogen Bonded Squaramide-Based Foldable Module Induces Both β- and α-Turns in Hairpin Structures of α-Peptides in Water. Org. Lett. 2015;17(12):2980–2983. doi: 10.1021/acs.orglett.5b01268. [DOI] [PubMed] [Google Scholar]
- Noteborn W. E. M., Saez Talens V., Kieltyka R. E.. Reversible Loading of Nanoscale Elements on a Multicomponent Supramolecular Polymer System by Using DNA Strand Displacement. ChemBioChem. 2017;18(20):1995–1999. doi: 10.1002/cbic.201700441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotger C., Piña M. N., Vega M., Ballester P., Deyà P. M., Costa A.. Efficient Macrocyclization of Preorganized Palindromic Oligosquaramides. Angew. Chem., Int. Ed. 2006;45(41):6844–6848. doi: 10.1002/anie.200602790. [DOI] [PubMed] [Google Scholar]
- Narasimhan S. K., Sejwal P., Zhu S., Luk Y.-Y.. Enhanced cell adhesion and mature intracellular structure promoted by squaramide-based RGD mimics on bioinert surfaces. Bioorg. Med. Chem. 2013;21(8):2210–2216. doi: 10.1016/j.bmc.2013.02.032. [DOI] [PubMed] [Google Scholar]
- Sampedro A., Villalonga-Planells R., Vega M., Ramis G., Fernández de Mattos S., Villalonga P., Costa A., Rotger C.. Cell Uptake and Localization Studies of Squaramide Based Fluorescent Probes. Bioconjugate Chem. 2014;25(8):1537–1546. doi: 10.1021/bc500258b. [DOI] [PubMed] [Google Scholar]
- Fernández-Moreira V., Alegre-Requena J. V., Herrera R. P., Marzo I., Gimeno M. C.. Synthesis of luminescent squaramide monoesters: cytotoxicity and cell imaging studies in HeLa cells. RSC Adv. 2016;6(17):14171–14177. doi: 10.1039/C5RA24521D. [DOI] [Google Scholar]
- Taylor K. I., Ho J. S., Trial H. O., Carter A. W., Kiessling L. L.. Assessing Squarates as Amine-Reactive Probes. J. Am. Chem. Soc. 2023;145:25056. doi: 10.1021/jacs.2c05691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara K., Takeda Y., Daikoku S., Hachisu M., Seko A., Ito Y.. Profiling Aglycon-Recognizing Sites of UDP-glucose:glycoprotein Glucosyltransferase by Means of Squarate-Mediated Labeling. Biochemistry. 2015;54(31):4909–4917. doi: 10.1021/acs.biochem.5b00785. [DOI] [PubMed] [Google Scholar]
- Fonvielle M., Sakkas N., Iannazzo L., Le Fournis C., Patin D., Mengin-Lecreulx D., El-Sagheer A., Braud E., Cardon S., Brown T.. et al. Electrophilic RNA for Peptidyl-RNA Synthesis and Site-Specific Cross-Linking with tRNA-Binding Enzymes. Angew. Chem., Int. Ed. 2016;55(43):13553–13557. doi: 10.1002/anie.201606843. [DOI] [PubMed] [Google Scholar]
- Ivancová I., Pohl R., Hubálek M., Hocek M.. Squaramate-Modified Nucleotides and DNA for Specific Cross-Linking with Lysine-Containing Peptides and Proteins. Angew. Chem., Int. Ed. 2019;58(38):13345–13348. doi: 10.1002/anie.201906737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Krasnova L., Finn M. G., Sharpless K. B.. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem., Int. Ed. 2014;53(36):9430–9448. doi: 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- Baker B. R.. Irreversible enzyme inhibitors. CXLIX. Tissue-specific irreversible inhibitors of dihydrofolic reductase. Acc. Chem. Res. 1969;2(5):129–136. doi: 10.1021/ar50017a001. [DOI] [Google Scholar]
- Cuesta A., Taunton J.. Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu. Rev. Biochem. 2019;88:365–381. doi: 10.1146/annurev-biochem-061516-044805. [DOI] [PubMed] [Google Scholar]
- Cruite J. T., Dann G. P., Che J., Donovan K. A., Ferrao S., Ficarro S. B., Fischer E. S., Gray N. S., Huerta F., Kong N. R.. et al. Cereblon covalent modulation through structure-based design of histidine targeting chemical probes. RSC Chem. Biol. 2022;3(9):1105–1110. doi: 10.1039/D2CB00078D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. E., Vuorinen A., Aatkar A., Pogány P., Pettinger J., Grant E. K., Kirkpatrick J. M., Rittinger K., House D., Burley G. A.. et al. Profiling Sulfur(VI) Fluorides as Reactive Functionalities for Chemical Biology Tools and Expansion of the Ligandable Proteome. ACS Chem. Biol. 2023;18(2):285–295. doi: 10.1021/acschembio.2c00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aatkar A., Vuorinen A., Longfield O. E., Gilbert K., Peltier-Heap R., Wagner C. D., Zappacosta F., Rittinger K., Chung C.-w., House D.. et al. Efficient Ligand Discovery Using Sulfur(VI) Fluoride Reactive Fragments. ACS Chem. Biol. 2023;18(9):1926–1937. doi: 10.1021/acschembio.3c00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid S., Turk S., Volkamer A., Rippmann F., Fulle S.. KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics. 2017;18(1):16. doi: 10.1186/s12859-016-1433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Ouyang X., Wan X., Gajiwala K. S., Kath J. C., Jones L. H., Burlingame A. L., Taunton J.. Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc. 2017;139(2):680–685. doi: 10.1021/jacs.6b08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C. O. N., Yang Y., Leung R. W. H., So K. K. H., Guo H. J., Lei M. M. L., Muliawan G. K., Gao Y., Yu Q. Q., Yun J. P.. et al. Broad-spectrum kinome profiling identifies CDK6 upregulation as a driver of lenvatinib resistance in hepatocellular carcinoma. Nat. Commun. 2023;14(1):6699. doi: 10.1038/s41467-023-42360-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlenghi F., Scalvini L., Vacondio F., Castelli R., Bozza N., Marseglia G., Rivara S., Lodola A., La Monica S., Minari R.. et al. A sulfonyl fluoride derivative inhibits EGFRL858R/T790M/C797S by covalent modification of the catalytic lysine. Eur. J. Med. Chem. 2021;225:113786. doi: 10.1016/j.ejmech.2021.113786. [DOI] [PubMed] [Google Scholar]
- Rüegger J., Gagestein B., Janssen A. P. A., Valeanu A., Mori A. L., van der Peet M., Boutkan M. S., Florea B. I., Henneman A. A., Hochstrasser R.. et al. CellEKT: A robust chemical proteomics workflow to profile cellular target engagement of kinase inhibitors. Molecular Cellular Proteomics. 2025:100961. doi: 10.1016/j.mcpro.2025.100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J., Le Bihan Y.-V., Widya M., van Montfort R. L. M., Jones K., Cheeseman M. D.. An Irreversible Inhibitor of HSP72 that Unexpectedly Targets Lysine-56. Angew. Chem., Int. Ed. 2017;56(13):3536–3540. doi: 10.1002/anie.201611907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J., Carter M., Jones K., Cheeseman M. D.. Kinetic Optimization of Lysine-Targeting Covalent Inhibitors of HSP72. J. Med. Chem. 2019;62(24):11383–11398. doi: 10.1021/acs.jmedchem.9b01709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zhao X., Cappiello J. R., Yang Y., Cheng Y., Liu G., Fang W., Luo Y., Zhang Y., Dong J.. et al. Identification of simple arylfluorosulfates as potent agents against resistant bacteria. Proc. Natl. Acad. Sci. U. S. A. 2021;118(28):e2103513118. doi: 10.1073/pnas.2103513118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Li J., Li S., Li G., Sharpless K. B., Wu P.. SuFEx Click Chemistry Enabled Late-Stage Drug Functionalization. J. Am. Chem. Soc. 2018;140(8):2919–2925. doi: 10.1021/jacs.7b12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillebrand L., Liang X. J., Serafim R. A. M., Gehringer M.. Emerging and Re-emerging Warheads for Targeted Covalent Inhibitors: An Update. J. Med. Chem. 2024;67(10):7668–7758. doi: 10.1021/acs.jmedchem.3c01825. [DOI] [PubMed] [Google Scholar]
- Tang G., Wang W., Wang X., Ding K., Ngan S. C., Chen J.-Y., Sze S. K., Gao L., Yuan P., Lu X.. et al. Cell-active, irreversible covalent inhibitors that selectively target the catalytic lysine of EGFR by using fluorosulfate-based SuFEx chemistry. Eur. J. Med. Chem. 2023;259:115671. doi: 10.1016/j.ejmech.2023.115671. [DOI] [PubMed] [Google Scholar]
- Tang G., Wang W., Zhu C., Huang H., Chen P., Wang X., Xu M., Sun J., Zhang C.-J., Xiao Q.. et al. Global Reactivity Profiling of the Catalytic Lysine in Human Kinome for Covalent Inhibitor Development. Angew. Chem., Int. Ed. 2024;63(12):e202316394. doi: 10.1002/anie.202316394. [DOI] [PubMed] [Google Scholar]
- Brulet J. W., Ciancone A. M., Yuan K., Hsu K.-L.. Advances in Activity-Based Protein Profiling of Functional Tyrosines in Proteomes. Isr. J. Chem. 2023;63(3–4):e202300001. doi: 10.1002/ijch.202300001. [DOI] [Google Scholar]
- Huang T., Hosseinibarkooie S., Borne A. L., Granade M. E., Brulet J. W., Harris T. E., Ferris H. A., Hsu K.-L.. Chemoproteomic profiling of kinases in live cells using electrophilic sulfonyl triazole probes. Chem. Sci. 2021;12(9):3295–3307. doi: 10.1039/D0SC06623K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm H. S., Toroitich E. K., Borne A. L., Brulet J. W., Libby A. H., Yuan K., Ware T. B., McCloud R. L., Ciancone A. M., Hsu K.-L.. Global targeting of functional tyrosines using sulfur-triazole exchange chemistry. Nat. Chem. Biol. 2020;16(2):150–159. doi: 10.1038/s41589-019-0404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloud R. L., Yuan K., Mahoney K. E., Bai D. L., Shabanowitz J., Ross M. M., Hunt D. F., Hsu K.-L.. Direct Target Site Identification of a Sulfonyl-Triazole Covalent Kinase Probe by LC-MS Chemical Proteomics. Anal. Chem. 2021;93(35):11946–11955. doi: 10.1021/acs.analchem.1c01591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancone A. M., Seo K. W., Chen M., Borne A. L., Libby A. H., Bai D. L., Kleiner R. E., Hsu K.-L.. Global Discovery of Covalent Modulators of Ribonucleoprotein Granules. J. Am. Chem. Soc. 2023;145(20):11056–11066. doi: 10.1021/jacs.3c00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff R., Schlüter H.. Amino acids: Chemistry, functionality and selected non-enzymatic post-translational modifications. J. Proteomics. 2012;75(8):2275–2296. doi: 10.1016/j.jprot.2012.01.041. [DOI] [PubMed] [Google Scholar]
- Koide S., Sidhu S. S.. The Importance of Being Tyrosine: Lessons in Molecular Recognition from Minimalist Synthetic Binding Proteins. ACS Chem. Biol. 2009;4(5):325–334. doi: 10.1021/cb800314v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira I. S., Fernandes P. A., Ramos M. J.. Hot spotsA review of the protein-protein interface determinant amino-acid residues. Proteins: Struct., Funct., Bioinf. 2007;68(4):803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- Mian I. S., Bradwell A. R., Olson A. J.. Structure, function and properties of antibody binding sites. J. Mol. Biol. 1991;217(1):133–151. doi: 10.1016/0022-2836(91)90617-F. [DOI] [PubMed] [Google Scholar]
- Nada H., Choi Y., Kim S., Jeong K. S., Meanwell N. A., Lee K.. New insights into protein-protein interaction modulators in drug discovery and therapeutic advance. Signal Transduct. Target. Ther. 2024;9(1):341. doi: 10.1038/s41392-024-02036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. H., Narayanan A., Hett E. C.. Understanding and applying tyrosine biochemical diversity. Mol. Biosyst. 2014;10(5):952–969. doi: 10.1039/c4mb00018h. [DOI] [PubMed] [Google Scholar]
- Grossmann A., Benlasfer N., Birth P., Hegele A., Wachsmuth F., Apelt L., Stelzl U.. Phospho-tyrosine dependent protein-protein interaction network. Mol. Syst. Biol. 2015;11(3):794. doi: 10.15252/msb.20145968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe J. W., Bertozzi C. R.. Tyrosine sulfation: a modulator of extracellular protein-protein interactions. Chem. Biol. 2000;7(3):R57–R61. doi: 10.1016/S1074-5521(00)00093-4. [DOI] [PubMed] [Google Scholar]
- Ande S. R., Xu Y. X. Z., Mishra S.. Prohibitin: a potential therapeutic target in tyrosine kinase signaling. Signal Transduct. Target. Ther. 2017;2(1):17059. doi: 10.1038/sigtrans.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.-H., Zhang Z.-Y.. Regulatory Mechanisms and Novel Therapeutic Targeting Strategies for Protein Tyrosine Phosphatases. Chem. Rev. 2018;118(3):1069–1091. doi: 10.1021/acs.chemrev.7b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner Y., Downey G. P.. The Importance of Tyrosine Phosphorylation Control of Cellular Signaling Pathways in Respiratory Disease: pY and pY Not. Am. J. Respir. Cell Mol. Biol. 2018;59(5):535–547. doi: 10.1165/rcmb.2018-0049TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Dorta D., Deniaud D., Mével M., Gouin S. G.. Tyrosine Conjugation Methods for Protein Labelling. Chem. Eur. J. 2020;26(63):14257–14269. doi: 10.1002/chem.202001992. [DOI] [PubMed] [Google Scholar]
- Zeng W., Xue J., Geng H., Liu X., Yang J., Shen W., Yuan Y., Qiang Y., Zhu Q.. Research progress on chemical modifications of tyrosine residues in peptides and proteins. Biotechnol. Bioeng. 2024;121(3):799–822. doi: 10.1002/bit.28622. [DOI] [PubMed] [Google Scholar]
- Szijj P. A., Kostadinova K. A., Spears R. J., Chudasama V.. Tyrosine bioconjugation - an emergent alternative. Org. Biomol. Chem. 2020;18(44):9018–9028. doi: 10.1039/D0OB01912G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N. S., Whitaker L. R., Francis M. B.. A Three-Component Mannich-Type Reaction for Selective Tyrosine Bioconjugation. J. Am. Chem. Soc. 2004;126(49):15942–15943. doi: 10.1021/ja0439017. [DOI] [PubMed] [Google Scholar]
- McFarland J. M., Joshi N. S., Francis M. B.. Characterization of a Three-Component Coupling Reaction on Proteins by Isotopic Labeling and Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2008;130(24):7639–7644. doi: 10.1021/ja710927q. [DOI] [PubMed] [Google Scholar]
- Romanini D. W., Francis M. B.. Attachment of Peptide Building Blocks to Proteins Through Tyrosine Bioconjugation. Bioconjugate Chem. 2008;19(1):153–157. doi: 10.1021/bc700231v. [DOI] [PubMed] [Google Scholar]
- Fraenkel-Conrat H., Olcott H. S.. REACTION OF FORMALDEHYDE WITH PROTEINS: VI. CROSS-LINKING OF AMINO GROUPS WITH PHENOL, IMIDAZOLE, OR INDOLE GROUPS. J. Biol. Chem. 1948;174(3):827–843. doi: 10.1016/S0021-9258(18)57292-6. [DOI] [PubMed] [Google Scholar]
- Minakawa M., Guo H.-M., Tanaka F.. Imines that React with Phenols in Water over a Wide pH Range. J. Org. Chem. 2008;73(21):8669–8672. doi: 10.1021/jo8017389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.-M., Minakawa M., Ueno L., Tanaka F.. Synthesis and evaluation of a cyclic imine derivative conjugated to a fluorescent molecule for labeling of proteins. Bioorg. Med. Chem. Lett. 2009;19(4):1210–1213. doi: 10.1016/j.bmcl.2008.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckey J. I., Dickson B. M., Cheng N., Liu Y., Norris J. L., Cholensky S. H., Tempel W., Qin S., Huber K. G., Sagum C.. et al. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol. 2016;12(3):180–187. doi: 10.1038/nchembio.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Hadisurya M., Tao W. A., Dykhuizen E., Krusemark C.. Covalent Protein Inhibitors via Tyrosine Conjugation with Cyclic Imine Mannich Electrophiles. ChemRxiv. 2022 doi: 10.26434/chemrxiv-2022-tvgn1. [DOI] [Google Scholar]
- Ban H., Gavrilyuk J., Barbas C. F. III. Tyrosine Bioconjugation through Aqueous Ene-Type Reactions: A Click-Like Reaction for Tyrosine. J. Am. Chem. Soc. 2010;132(5):1523–1525. doi: 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- Ban H., Nagano M., Gavrilyuk J., Hakamata W., Inokuma T., Barbas C. F. III. Facile and Stabile Linkages through Tyrosine: Bioconjugation Strategies with the Tyrosine-Click Reaction. Bioconjugate Chem. 2013;24(4):520–532. doi: 10.1021/bc300665t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dorta D., Thobie-Gautier C., Croyal M., Bouzelha M., Mével M., Deniaud D., Boujtita M., Gouin S. G.. Electrochemically Promoted Tyrosine-Click-Chemistry for Protein Labeling. J. Am. Chem. Soc. 2018;140(49):17120–17126. doi: 10.1021/jacs.8b09372. [DOI] [PubMed] [Google Scholar]
- Bauer D. M., Ahmed I., Vigovskaya A., Fruk L.. Clickable Tyrosine Binding Bifunctional Linkers for Preparation of DNA-Protein Conjugates. Bioconjugate Chem. 2013;24(6):1094–1101. doi: 10.1021/bc4001799. [DOI] [PubMed] [Google Scholar]
- Cui L., Ma Y., Li M., Wei Z., Huan Y., Li H., Fei Q., Zheng L.. Tyrosine-Reactive Cross-Linker for Probing Protein Three-Dimensional Structures. Anal. Chem. 2021;93(10):4434–4440. doi: 10.1021/acs.analchem.0c04337. [DOI] [PubMed] [Google Scholar]
- Denijs E., Unal K., Bevernaege K., Kasmi S., De Geest B. G., Winne J. M.. Thermally Triggered Triazolinedione-Tyrosine Bioconjugation with Improved Chemo- and Site-Selectivity. J. Am. Chem. Soc. 2024;146(18):12672–12680. doi: 10.1021/jacs.4c02173. [DOI] [PubMed] [Google Scholar]
- Hu Q.-Y., Allan M., Adamo R., Quinn D., Zhai H., Wu G., Clark K., Zhou J., Ortiz S., Wang B.. et al. Synthesis of a well-defined glycoconjugate vaccine by a tyrosine-selective conjugation strategy. Chem. Sci. 2013;4(10):3827–3832. doi: 10.1039/c3sc51694f. [DOI] [Google Scholar]
- Nilo A., Allan M., Brogioni B., Proietti D., Cattaneo V., Crotti S., Sokup S., Zhai H., Margarit I., Berti F.. et al. Tyrosine-Directed Conjugation of Large Glycans to Proteins via Copper-Free Click Chemistry. Bioconjugate Chem. 2014;25(12):2105–2111. doi: 10.1021/bc500438h. [DOI] [PubMed] [Google Scholar]
- Madl C. M., Heilshorn S. C.. Tyrosine-Selective Functionalization for Bio-Orthogonal Cross-Linking of Engineered Protein Hydrogels. Bioconjugate Chem. 2017;28(3):724–730. doi: 10.1021/acs.bioconjchem.6b00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decoene K. W., Unal K., Staes A., Zwaenepoel O., Gettemans J., Gevaert K., Winne J. M., Madder A.. Triazolinedione protein modification: from an overlooked off-target effect to a tryptophan-based bioconjugation strategy. Chem. Sci. 2022;13(18):5390–5397. doi: 10.1039/D1SC06942J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moinpour M., Barker N. K., Guzman L. E., Jewett J. C., Langlais P. R., Schwartz J. C.. Discriminating changes in protein structure using tyrosine conjugation. Protein Sci. 2020;29(8):1784–1793. doi: 10.1002/pro.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammells P. J., Baker S. P., Belardinelli L., Olsson R. A.. Substituted 1,3-Dipropylxanthines as Irreversible Antagonists of A1 Adenosine Receptors. J. Med. Chem. 1994;37(17):2704–2712. doi: 10.1021/jm00043a010. [DOI] [PubMed] [Google Scholar]
- Beauglehole A. R., Baker S. P., Scammells P. J.. Fluorosulfonyl-Substituted Xanthines as Selective Irreversible Antagonists for the A1-Adenosine Receptor. J. Med. Chem. 2000;43(26):4973–4980. doi: 10.1021/jm000181f. [DOI] [PubMed] [Google Scholar]
- Glukhova A., Thal D. M., Nguyen A. T., Vecchio E. A., Jörg M., Scammells P. J., May L. T., Sexton P. M., Christopoulos A.. Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell. 2017;168(5):867–877. doi: 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- Yang X., Dilweg M. A., Osemwengie D., Burggraaff L., van der Es D., Heitman L. H., Ijzerman A. P.. Design and pharmacological profile of a novel covalent partial agonist for the adenosine A1 receptor. Biochem. Pharmacol. 2020;180:114144. doi: 10.1016/j.bcp.2020.114144. [DOI] [PubMed] [Google Scholar]
- Beerkens B. L. H., Koç Ç., Liu R., Florea B. I., Le Dévédec S. E., Heitman L. H., Ijzerman A. P., van der Es D.. A Chemical Biological Approach to Study G Protein-Coupled Receptors: Labeling the Adenosine A1 Receptor Using an Electrophilic Covalent Probe. ACS Chem. Biol. 2022;17(11):3131–3139. doi: 10.1021/acschembio.2c00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., van Veldhoven J. P. D., Offringa J., Kuiper B. J., Lenselink E. B., Heitman L. H., van der Es D., Ijzerman A. P.. Development of Covalent Ligands for G Protein-Coupled Receptors: A Case for the Human Adenosine A3 Receptor. J. Med. Chem. 2019;62(7):3539–3552. doi: 10.1021/acs.jmedchem.8b02026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G., Hou X., Byun W. S., Kim G., Jarhad D. B., Lee G., Hyun Y. E., Yu J., Lee C. S., Qu S.. et al. Structure-Activity Relationship of Truncated 2,8-Disubstituted-Adenosine Derivatives as Dual A2A/A3 Adenosine Receptor Antagonists and Their Cancer Immunotherapeutic Activity. J. Med. Chem. 2023;66(17):12249–12265. doi: 10.1021/acs.jmedchem.3c00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G., Jarhad D. B., Lee G., Kim G., Hou X., Yu J., Lee C. S., Warnick E., Gao Z.-G., Ahn S. Y.. et al. Structural Modification and Biological Evaluation of 2,8-Disubstituted Adenine and Its Nucleosides as A2A Adenosine Receptor Antagonists: Exploring the Roles of Ribose at Adenosine Receptors. J. Med. Chem. 2024;67(12):10490–10507. doi: 10.1021/acs.jmedchem.4c01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A., Merighi S., Varani K., Borea P. A., Baraldi S., Aghazadeh Tabrizi M., Romagnoli R., Baraldi P. G., Ciancetta A., Tosh D. K.. et al. A3 Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018;38(4):1031–1072. doi: 10.1002/med.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. L., Fallot L. B., Wan T. C., Keyes R. F., Suresh R. R., Rothwell A. C., Gao Z.-G., McCorvy J. D., Smith B. C., Jacobson K. A.. et al. Characterization of Dual-Acting A3 Adenosine Receptor Positive Allosteric Modulators That Preferentially Enhance Adenosine-Induced Gαi3 and GαoA Isoprotein Activation. ACS Pharmacol. Transl. Sci. 2022;5(8):625–641. doi: 10.1021/acsptsci.2c00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerkens B. L. H., Snijders I. M., Snoeck J., Liu R., Tool A. T. J., Le Dévédec S. E., Jespers W., Kuijpers T. W., van Westen G. J. P., Heitman L. H.. et al. Development of an Affinity-Based Probe to Profile Endogenous Human Adenosine A3 Receptor Expression. J. Med. Chem. 2023;66(16):11399–11413. doi: 10.1021/acs.jmedchem.3c00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hett E. C., Xu H., Geoghegan K. F., Gopalsamy A., Kyne R. E. Jr, Menard C. A., Narayanan A., Parikh M. D., Liu S., Roberts L.. et al. Rational Targeting of Active-Site Tyrosine Residues Using Sulfonyl Fluoride Probes. ACS Chem. Biol. 2015;10(4):1094–1098. doi: 10.1021/cb5009475. [DOI] [PubMed] [Google Scholar]
- Fadeyi O. O., Hoth L. R., Choi C., Feng X., Gopalsamy A., Hett E. C., Kyne R. E. Jr, Robinson R. P., Jones L. H.. Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue. ACS Chem. Biol. 2017;12(8):2015–2020. doi: 10.1021/acschembio.7b00403. [DOI] [PubMed] [Google Scholar]
- Chen W., Dong J., Plate L., Mortenson D. E., Brighty G. J., Li S., Liu Y., Galmozzi A., Lee P. S., Hulce J. J.. et al. Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s) through Chemoselective SuFEx Reaction with a Binding Site Tyr Residue. J. Am. Chem. Soc. 2016;138(23):7353–7364. doi: 10.1021/jacs.6b02960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolding J. E., Martín-Gago P., Rajabi N., Gamon L. F., Hansen T. N., Bartling C. R. O., Strømgaard K., Davies M. J., Olsen C. A.. Aryl Fluorosulfate Based Inhibitors That Covalently Target the SIRT5 Lysine Deacylase. Angew. Chem., Int. Ed. 2022;61(47):e202204565. doi: 10.1002/anie.202204565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi N., Auth M., Troelsen K. R., Pannek M., Bhatt D. P., Fontenas M., Hirschey M. D., Steegborn C., Madsen A. S., Olsen C. A.. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic Insight. Angew. Chem., Int. Ed. 2017;56(47):14836–14841. doi: 10.1002/anie.201709050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D., Franzini A., Pomicter A. D., Halverson B. J., Antelope O., Mason C. C., Ahmann J. M., Senina A. V., Vellore N. A., Jones C. L.. et al. SIRT5 Is a Druggable Metabolic Vulnerability in Acute Myeloid Leukemia. Blood Cancer Discovery. 2021;2(3):266–287. doi: 10.1158/2643-3230.BCD-20-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi N., Nielsen A. L., Olsen C. A.. Dethioacylation by Sirtuins 1–3: Considerations for Drug Design Using Mechanism-Based Sirtuin Inhibition. ACS. Med. Chem. Lett. 2020;11(10):1886–1892. doi: 10.1021/acsmedchemlett.9b00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi N., Hansen T. N., Nielsen A. L., Nguyen H. T., Bæk M., Bolding J. E., Bahlke O. Ø., Petersen S. E. G., Bartling C. R. O., Strømgaard K.. et al. Investigation of Carboxylic Acid Isosteres and Prodrugs for Inhibition of the Human SIRT5 Lysine Deacylase Enzyme. Angew. Chem., Int. Ed. 2022;61(22):e202115805. doi: 10.1002/anie.202115805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancone A. M., Hosseinibarkooie S., Bai D. L., Borne A. L., Ferris H. A., Hsu K.-L.. Global profiling identifies a stress-responsive tyrosine site on EDC3 regulating biomolecular condensate formation. Cell Chem. Biol. 2022;29(12):1709–1720. doi: 10.1016/j.chembiol.2022.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R., Shaikh M., Lemke M. C., Yuan K., Libby A. H., Bai D. L., Ross M. M., Harris T. E., Hsu K.-L.. Predicting small molecule binding pockets on diacylglycerol kinases using chemoproteomics and AlphaFold. RSC Chem. Biol. 2023;4(6):422–430. doi: 10.1039/D3CB00057E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri I. P., Colbon P. J. J.. The Growing Importance of Chirality in 3D Chemical Space Exploration and Modern Drug Discovery Approaches for Hit-ID. ACS Med. Chem. Lett. 2021;12(8):1220–1229. doi: 10.1021/acsmedchemlett.1c00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z.. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015;58(6):2584–2608. doi: 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Bunally S. B., Luscombe C. N., Young R. J.. Using Physicochemical Measurements to Influence Better Compound Design. SLAS Discovery. 2019;24(8):791–801. doi: 10.1177/2472555219859845. [DOI] [PubMed] [Google Scholar]
- Sivakumar K., Venkadasamy V. L., Amudhan G., Ann K. J., Goud G. K., Nayani K., Gogoi J., Kuncha Santosh K., Mainkar Prathama S., Kruparani Shobha P.. et al. Design principles and functional basis of enantioselectivity of alanyl-tRNA synthetase and a chiral proofreader during protein biosynthesis. Nucleic Acids Res. 2023;51(7):3327–3340. doi: 10.1093/nar/gkad205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger K.-E., Reetz M. T.. Directed evolution of enantioselective enzymes for organic chemistry. Curr. Opin. Chem. Biol. 2000;4(1):68–73. doi: 10.1016/S1367-5931(99)00054-X. [DOI] [PubMed] [Google Scholar]
- Li Z.-L., Pei S., Chen Z., Huang T.-Y., Wang X.-D., Shen L., Chen X., Wang Q.-Q., Wang D.-X., Ao Y.-F.. Machine learning-assisted amidase-catalytic enantioselectivity prediction and rational design of variants for improving enantioselectivity. Nat. Commun. 2024;15(1):8778. doi: 10.1038/s41467-024-53048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann C. M., Robustini L., Paradisi F.. Influence of Reaction Conditions on Enzymatic Enantiopreference: the Curious Case of HEwT in the Synthesis of THF-Amine. ChemBioChem. 2022;23(15):e202200335. doi: 10.1002/cbic.202200335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Remsberg J. R., Li H., Njomen E., DeMeester K. E., Tao Y., Xia G., Hayward R. E., Yoo M., Nguyen T.. et al. Proteomic Ligandability Maps of Spirocycle Acrylamide Stereoprobes Identify Covalent ERCC3 Degraders. J. Am. Chem. Soc. 2024;146(15):10393–10406. doi: 10.1021/jacs.3c13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Felber J. G., Zou Z., Njomen E., Remsberg J. R., Ogasawara D., Ye C., Melillo B., Schreiber S. L., He C.. et al. Chemical Proteomic Discovery of Isotype-Selective Covalent Inhibitors of the RNA Methyltransferase NSUN2. Angew. Chem., Int. Ed. 2023;62(51):e202311924. doi: 10.1002/anie.202311924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Remillard D., Vinogradova E. V., Yokoyama M., Banchenko S., Schwefel D., Melillo B., Schreiber S. L., Zhang X., Cravatt B. F.. Targeted Protein Degradation by Electrophilic PROTACs that Stereoselectively and Site-Specifically Engage DCAF1. J. Am. Chem. Soc. 2022;144(40):18688–18699. doi: 10.1021/jacs.2c08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman H. C., Merlini E., Guijas C., DeMeester K. E., Njomen E., Kozina E. M., Yokoyama M., Vinogradova E., Reardon H. T., Melillo B.. et al. Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc. Natl. Acad. Sci. U. S. A. 2022;119(35):e2208457119. doi: 10.1073/pnas.2208457119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Dix M. M., Bianco G., Remsberg J. R., Lee H.-Y., Kalocsay M., Gygi S. P., Forli S., Vite G., Lawrence R. M.. et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 2019;11(12):1113–1123. doi: 10.1038/s41557-019-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara D., Konrad D. B., Tan Z. Y., Carey K. L., Luo J., Won S. J., Li H., Carter T. R., DeMeester K. E., Njomen E.. et al. Chemical tools to expand the ligandable proteome: Diversity-oriented synthesis-based photoreactive stereoprobes. Cell Chem. Biol. 2024;31(12):2138–2155. doi: 10.1016/j.chembiol.2024.10.005. e2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njomen E., Hayward R. E., DeMeester K. E., Ogasawara D., Dix M. M., Nguyen T., Ashby P., Simon G. M., Schreiber S. L., Melillo B.. et al. Multi-tiered chemical proteomic maps of tryptoline acrylamide-protein interactions in cancer cells. Nat. Chem. 2024;16(10):1592–1604. doi: 10.1038/s41557-024-01601-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Craven G. B., Kamber R. A., Cuesta A., Zhersh S., Moroz Y. S., Bassik M. C., Taunton J.. Direct mapping of ligandable tyrosines and lysines in cells with chiral sulfonyl fluoride probes. Nat. Chem. 2023;15(11):1616–1625. doi: 10.1038/s41557-023-01281-3. [DOI] [PubMed] [Google Scholar]
- Xie X., Yu T., Li X., Zhang N., Foster L. J., Peng C., Huang W., He G.. Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials. Signal Transduct. Target. Ther. 2023;8(1):335. doi: 10.1038/s41392-023-01589-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin J. N., Zhang E., Nomura D. K.. Reimagining Druggability Using Chemoproteomic Platforms. Acc. Chem. Res. 2021;54(7):1801–1813. doi: 10.1021/acs.accounts.1c00065. [DOI] [PubMed] [Google Scholar]
- Duffy M. J., Crown J.. Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer. 2021;148(1):8–17. doi: 10.1002/ijc.33197. [DOI] [PubMed] [Google Scholar]
- Llombart V., Mansour M. R.. Therapeutic targeting of “undruggable” MYC. eBioMedicine. 2022;75:103756. doi: 10.1016/j.ebiom.2021.103756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Crown J., Duffy M. J.. Degradation of MYC by the mutant p53 reactivator drug, COTI-2 in breast cancer cells. Investigational New Drugs. 2023;41(4):541–550. doi: 10.1007/s10637-023-01368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S., Patterson J. T., Gaj T., Barbas C. F. Iii. Site-Selective Labeling of a Lysine Residue in Human Serum Albumin. Angew. Chem., Int. Ed. 2014;53(44):11783–11786. doi: 10.1002/anie.201405924. [DOI] [PubMed] [Google Scholar]
- Liu Z., Chen X.. Simple Bioconjugate Chem. serves great clinical advances: albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016;45(5):1432–1456. doi: 10.1039/C5CS00158G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson J. T., Wilson H. D., Asano S., Nilchan N., Fuller R. P., Roush W. R., Rader C., Barbas C. F. III. Human Serum Albumin Domain I Fusion Protein for Antibody Conjugation. Bioconjugate Chem. 2016;27(10):2271–2275. doi: 10.1021/acs.bioconjchem.6b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos M. J., Oliveira B. L., Martínez-Sáez N., Guerreiro A., Cal P. M. S. D., Bertoldo J., Maneiro M., Perkins E., Howard J., Deery M. J.. et al. Chemo- and Regioselective Lysine Modification on Native Proteins. J. Am. Chem. Soc. 2018;140(11):4004–4017. doi: 10.1021/jacs.7b12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adusumalli S. R., Rawale D. G., Thakur K., Purushottam L., Reddy N. C., Kalra N., Shukla S., Rai V.. Chemoselective and Site-Selective Lysine-Directed Lysine Modification Enables Single-Site Labeling of Native Proteins. Angew. Chem., Int. Ed. 2020;59(26):10332–10336. doi: 10.1002/anie.202000062. [DOI] [PubMed] [Google Scholar]
- Mons E., Jansen I. D. C., Loboda J., van Doodewaerd B. R., Hermans J., Verdoes M., van Boeckel C. A. A., van Veelen P. A., Turk B., Turk D.. et al. The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin K. J. Am. Chem. Soc. 2019;141(8):3507–3514. doi: 10.1021/jacs.8b11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Gago P., Olsen C. A.. Arylfluorosulfate-Based Electrophiles for Covalent Protein Labeling: A New Addition to the Arsenal. Angew. Chem., Int. Ed. 2019;58(4):957–966. doi: 10.1002/anie.201806037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Zhang X., Zhang H., Tang Y., Pan H., Wang H., Ji T., Guo Y., Gao Q., Song T.. et al. Discovery of a Fluorogenic Probe for In Situ Pyruvate Kinase M2 Isoform (PKM2) Labeling through Chemoselective SNAr with a Binding Site Lysine Residue. Anal. Chem. 2021;93(28):9669–9676. doi: 10.1021/acs.analchem.1c00208. [DOI] [PubMed] [Google Scholar]