

Abstract
Monogenic lesions in pathways critical for effector functions responsible for immune surveillance, protection against autoinflammation, and appropriate responses to allergens and microorganisms underlie the pathophysiology of inborn errors of immunity (IEI). Variants in cytokine production, cytokine signaling, epithelial barrier function, antigen presentation, receptor signaling, and cellular processes and metabolism can drive autoimmunity, immunodeficiency, and/or allergic inflammation. Identification of these variants has improved our understanding of the role that many of these proteins play in skewing toward TH2-related allergic inflammation. Early-onset or atypical atopic disease, often in conjunction with immunodeficiency and/or autoimmunity, should raise suspicion for an IEI. This becomes a diagnostic dilemma if the initial clinical presentation is solely allergic inflammation, especially when the prevalence of allergic diseases is becoming more common. Genetic sequencing is necessary for IEI diagnosis and is helpful for early recognition and implementation of targeted treatment, if available. Although genetic evaluation is not feasible for all patients with atopy, identifying atopic patients with molecular immune abnormalities may be helpful for diagnostic, therapeutic, and prognostic purposes. In this review, we focus on IEI associated with TH2-driven allergic manifestations and classify them on the basis of the affected molecular pathways and predominant clinical manifestations.
Keywords: Inborn errors of immunity, atopy, allergic inflammation, dysregulation, atopic dermatitis, asthma, eosinophilia, mast cell dysregulation, primary immunodeficiency, primary atopic disorders, immunomodulation
Inborn errors of immunity (IEI) are diseases caused by variants in genes regulating immune system development and function. To date, there are more than 500 reported IEI, which may present with susceptibility to severe or recalcitrant infections, autoinflammation, autoimmune disease, lymphoproliferation, and allergic disease and other forms of type II inflammation.1
TH2 cells, which drive typical atopic diseases, are key elements of the type II immune response, which protects against helminth infection, stimulates tissue repair, neutralizes toxins and counteracts other forms of inflammation, and has other functions as well.2 TH2 cells can secrete IL-4, IL-5, IL-9, and IL-13, which recruit mast cells, eosinophils, and basophils and promote B-cell class switching to IgE.3–7 Failure to properly regulate TH2 cells and/or their downstream targets can lead to symptoms seen in clinical allergic and atopic diseases, such as IgE-mediated hypersensitivity and/or mast cell activation, allergic inflammation of the skin and mucosa, and interference with other forms of inflammation. Although, in general, immunologic markers of type II immunity such as elevated IgE, eosinophilia, and increased IL-4 and IL-13 are often associated with the clinical signs of atopy, such as allergic rhinitis, allergic asthma, and atopic dermatitis (AD), the clinical signs may not always correlate with laboratory abnormalities in IEI. Furthermore, there can be substantial variability in the type of type II immune-related phenotypes despite these abnormal laboratory values—such as AD without immediate hypersensitivity and vice versa.
Population data on the frequency of atopy in IEI are limited; however, estimates range from 10% to 28.8%, suggesting it may be more common than previously appreciated, with 5% to 25% of patients with IEI presenting initially with isolated complaints of atopy.8 Allergic diagnoses in the general population are becoming increasingly more common; nearly 1 in 3 adults and more than a quarter of children in the United States report having an allergic disease.8,9 Consequently, IEI may go unrecognized if allergic manifestations, such as AD, are the predominant initial symptoms and may result in delays in diagnosis.10 Conversely, atopic manifestation may not be addressed in the context of an IEI if severe infections or inflammations are the main clinical focus. The pathway of a patient with an IEI to clinical attention therefore significantly colors current conceptions of the relationship between IEI and atopic disease.
Although primary immunodeficiency refers to host defense defects presenting predominantly with infection, and primary immune regulation disorder refers to impaired tolerance and inflammation control presenting predominantly with increased autoimmunity and/or autoinflammation, primary atopic disorders is a term used to describe monogenic disorders with prominent atopic manifestations.11 Primary atopic disorders can include syndromes referred to previously as “hyper-IgE syndromes,” but also many other described disorders leading to this recent classification.11 This review will focus on allergic manifestations of IEI in general, with a focus on primary atopic disorders. We will highlight patterns of allergic disease linked to disturbances of specific molecular pathways and provide examples of precision treatment targeting these TH2-driven processes (Table I12–148; Fig 1).
TABLE I.
Selected IEI associated with TH2-related phenotypes
| Defect | Genes | Distinguishing phenotypic patterns | Insight/precision medicine/questions |
|---|---|---|---|
| Epithelial barrier disruption12–32 | DSG1, CDSN, DSP, SPINK5, FLG, COL7A1 | Severe dermatitis | Mechanism of positive IVIG effect for some? IL-4/IL-13R blockade improves skin immunity and inflammation |
| Attenuated antigen receptor signaling: actin cytoskeleton defects35–47 | WAS, WIPF1, ARPC1B, DOCK8, NCKAP1L, CARMIL2 | Viral skin infection Atopy and autoimmunity variable |
Dupilumab treatment improves viral skin infection Protection from mast cell–related symptoms in WAS |
| Attenuated antigen receptor signaling: CBM complex mutations48–50 | CARD11, CARD14, MALT1 | Viral skin infection/eczema herpeticum Atopy and autoimmunity variable |
NF-κB vs mTORC1 vs mTORC2 in driving TH2 phenotypes Lack of association of NF-κB–related IEI with TH2 phenotypes |
| Tolerance defects: enhanced PI3K/mTOR (APDS, IPEX)51–63,125 | PIK3CD, PIK3R1, FOXP3 | Mixed inflammation Variable phenotype |
PI3K/mTOR inhibition for TH2 phenotypes? |
| Omenn phenotype64–69 | Numerous SCID-related genes | Marked eosinophilia, erythroderma, IgE elevation Lack of antigen-specific allergy |
Leaky oligoclonal T-cell role in TH2-like phenotype? |
| Cytokine and cytokine signaling—diminished IL-6/STAT3 signaling70–83,126,127 | STAT3, IL6ST, IL6R ZNF341, IL6R, PGM3 | Elevated IgE, poor inflammatory response, relative protection from IgE-mediated severe allergic disease, fungal disease Rash at birth (STAT3 DN) |
Increased IL-4R expression–responsiveness to dupilumab AD but lack of significant hypersensitivity in STAT3 DN |
| Cytokine and cytokine signaling—increased JAK1/STAT5B/STAT684–102 | JAK1 GOF, STAT5B, STAT6 | Urticaria, failure to thrive, marked eosinophilia (JAK1/STAT5B) with eosinophilic GI disease, pan TH2-allergic/atopic phenotypes (STAT6) | Response of TH2-related phenotypes to JAK inhibition, dupilumab Extent of non-TH2 comorbidities in STAT5/STAT6 GOF? |
| Cytokine and cytokine signaling—TGF-β signaling100,101 | TGFβR1, TGFβR2, ERBIN | Eosinophilic gut disease, overlap with STAT3 DN pathway | Increased IL-4R expression–responsiveness to dupilumab |
| Cytokine and cytokine signaling—decreased IFN-γ production/signaling and counterregulation102–107,109,110,112–114 | IFNGR1, IFNGR2, IL12RB, IKZF1 GOF, TBX21 | Elevated IgE, eosinophilia | Use of biologics to improve TH1 skewing/IFN-γ signaling through IL-4 inhibition? |
| Mast cell degranulation142–148 | KIT, PLCG2, ADGRE2, KARS | Lifelong urticaria, increased anaphylaxis risk | Distinguish from mast cell extrinsic causes of allergic phenotypes |
ARPC1B, Actin-related protein 2/3 complex subunit 1B; CARD14, caspase recruitment domain family, member 14; CARMIL2, capping protein regulator and myosin 1 linker 2; CBM, CARD11-BCL10-MALT1; DSG1, desmoglein 1; DSP, desmoplakin; GI, gastrointestinal; IFNGR1/2, IFN-γ receptor 1/2; IVIG, intravenous immunoglobulin; NCKAP1L, NCK-associated protein 1 like; ZNF341, zinc finger protein 341.
FIG 1.
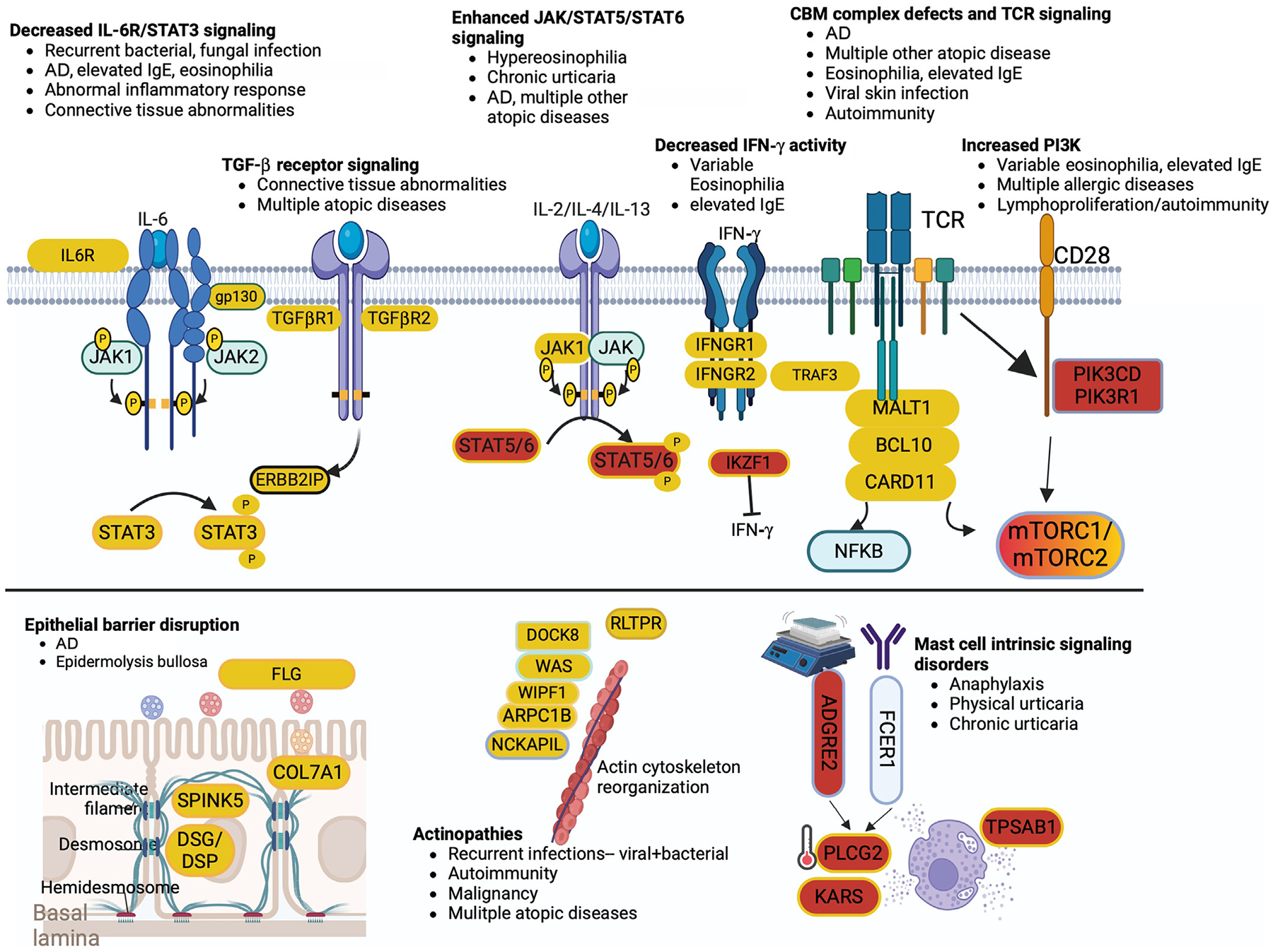
Outline of pathways implicated in genetic disorders of the immune system associated with atopic disease. Red backgrounds indicate genes/gene products with causal variants associated with increased function, and yellow backgrounds indicate decreased function. Mix of 2 suggests that both GOF and LOF can cause atopy. CBM, CARD11-BCL10-MALT1; IFNGR1/2, IFN-γ receptor 1/2; TCR, T-cell receptor.
ALLERGIC MANIFESTATIONS OF IEI AND UNIQUE PATHWAY-SPECIFIC PATTERNS OF DISEASE
Atopy in conjunction with other forms of inflammation, autoimmunity, or significant infection raises suspicion for underlying immune dysregulation. Elevated IgE in the context of recurrent pneumonia and staphylococcal infection and abscess was originally described in patients also referred to as having Job syndrome as the hyper-IgE syndrome. The initial description ultimately was found to be due to signal transducer and activator of transcription 3 (STAT3) dominant-negative (DN) variants; however, many other syndromes with a range of inheritance patterns associated with IgE elevation have been described with and without infection, with and without abscess formation, and with or without connective tissue or other immune and nonimmune comorbidities. Even the presence of IgE elevation itself can vary within any of these disorders. As such, it is important to classify IEI associated with TH2 and/or type II abnormalities by the pathways, genes, and presentations affected, instead of lumping them all together under a “hyper-IgE syndrome” umbrella.
Because of TH2 skewing, many conditions associated with excessive IgE production often have comorbid elevated eosinophilia. There are more than 30 IEI with associated eosinophilia as part of their clinical phenotype.119 Eosinophil counts higher than the upper limit of normal were present in almost 1 in 5 patients with IEI on the United States Immunodeficiency Network (USID-NET) registry,106 most notably in diseases such as STAT3 DN, Omenn syndrome, dedicator of cytokinesis 8 (DOCK8) deficiency, Wiskott-Aldrich syndrome (WAS), and immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX).120 Sustained and severe neonatal hypereosinophilia has been reported in somatic heterozygous gain-of-function (GOF) variants in STAT5B84,85,121 and Janus kinase 1 (JAK1) GOF.86 Eosinophilic gastrointestinal diseases (EGIDs) have also been reported in IEI, including Comel-Netherton syndrome, STAT3 DN, ERBB2-interacting protein (ERBIN) deficiency, Loeys-Dietz syndrome (LDS), IPEX, DOCK8 deficiency, CARD11 (caspase recruitment domain family, member 11) DN, STAT6 GOF, and others. Recent data from USIDNET reported that EGID was seen more frequently in patients with common variable immunodeficiency (CVID), chronic granulomatous disease (CGD), hyper-IgE syndrome (including STAT3 DN and phosphoglucomutase 3 [PGM3] deficiency), and autoimmune lymphoproliferative syndrome (ALPS),122 although this was a retrospective study dependent on centers’ reports and subject to ascertainment bias.
Monogenic autoinflammatory disorders, which are most commonly associated with IL-1, TNF, interferons, and/or IL-6 mediated inflammation, and thus not intuitively expected to have TH2 skewing, nonetheless display a range of TH2-related clinical phenotypes and TH2 cytokine production profiles. For instance, patients with adenosine deaminase 2 (ADA2) deficiency have autoinflammation and vasculitis associated with excessive TNF-α activity, and, as might be expected, serum IgE and eosinophils are significantly reduced compared with levels in the general population. However, cryopyrin-associated periodic syndromes due to nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain-containing protein 3 GOF variants leading to excessive IL-1β secretion are associated with marked eosinophilia and multiple clinical allergy diagnoses.123 The reasons for the diverging type II phenotypes in autoinflammatory diseases remain to be fully understood, but their study may ultimately offer insight into the interaction between autoinflammatory and atopic pathophysiology, even in contexts outside IEI.
Diseases associated with tolerance defects, the so-called regulatory T (Treg)-opathies, may include TH2-related symptoms as well. Food allergy can be an early or initial manifestation of IPEX syndrome or IPEX-like diseases. In a study,51 12% of patients with IPEX syndrome or IPEX-like diseases were initially diagnosed with food allergy, and only 15% of these patients were correctly diagnosed with IPEX at the time of presentation to medical care.
Patients with asthma are reported to have a higher prevalence of CVID compared with patients without asthma.124 Whether this signifies an expansion of the atopic phenotype or whether CVID is a predisposing factor for asthma is not clear, especially because asthma has both type II and non–type II endotypes. The association of asthma with CVID is interesting in light of CVID having the highest prevalence in the USIDNET report previously mentioned.106 Various monogenic etiologies encompassed under the diagnosis of CVID may provide opportunities to understand the underlying mechanism of atopy in these patients. For instance, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta (PI3KCD) GOF variants have been found as disease-causing in some patients originally carrying the CVID diagnosis and have been associated with atopic disease.125
It is noteworthy that there are IEI that are associated with relative protection from some forms of allergic disease. As examples, patients with STAT3 DN and WAS are relatively protected from severe IgE-mediated immediate hypersensitivity, partly because of defects in mast cell degranulation.126–131 In addition, GATA2 deficiency markedly impairs mast cell function and patients have lower rates of clinical allergy.132
PATHWAY-SPECIFIC IMPAIRMENTS ASSOCIATED WITH IEI
Enhanced numbers and activity of allergic effector cells and/or loss of genes that contribute to the regulation of these TH2 pathways result in pathologic allergic disease and inflammation.11,119,133 Severe combined immunodeficiencies (SCIDs) were some of the first known IEI presenting with atopic manifestations, such as eczematous dermatitis. Although complete SCID with absent T cells hinders the ability to mount an allergic response, hypomorphic variants allow for sufficient development of allergy effector cells despite otherwise compromised T-cell immunity against infection.134 The higher prevalence of severe clinical atopic disease in patients with IFN-γ receptor defects supports the notion that IFN-γsignaling may normally suppress TH2 responses via counterregulation of TH2 immunity pathways such as T-cell differentiation from naive T cells into TH2 cells or B-cell class switching to IgE.107 The following sections will highlight specific pathways whose derangements are associated with allergic inflammation (Fig 1).
Epithelial barrier disruption and risk for AD and skin infection
The integrity of the skin barrier is crucial to the prevention of allergic diseases. The link between atopy and an impaired skin barrier is not fully understood; however, activation of antigen-presenting cells by penetrating allergens, enhanced inflammation by resident microorganisms, allergic sensitization to Staphylococcus superantigens, and transepithelial water loss may all contribute to an environment more conducive to an allergic phenotype.11 Furthermore, TH2 inflammation appears to impair antimicrobial peptide production and activity, predisposing skin to infection and altered commensals.135,136 Monogenic disorders that primarily lead to disruption of the integrity of the skin barrier have been associated with inflammatory skin diseases, most notably atopic inflammation. Loss-of-function (LOF) variants in FLG, the gene encoding filaggrin, which is responsible for organization and crosslinking of keratin filaments, lead to severe eczematous dermatitis. Genome-wide association studies reveal a central role for rare and common FLG variants in the overall risk for AD.12 Although FLG is not expressed in the lower airways, LOF variants in FLG have been significantly associated with the development of asthma in children, which may be due to allergen sensitization in those children with AD.13
Desmosomes are junctional protein complexes that protect skin integrity by resisting mechanical forces. Pathogenic variants in genes that encode desmosomal junctional protein complex components corneodesmosin, desmoplakin, and desmoglein 1 are also associated with severe type II skin inflammation,14–16 and loss of corneodesmosome serine protease inhibitor of Kazal type 5 (SPINK5) leads to severe ichthyosis, erythroderma, bamboo hair, elevated IgE, and atopy.17 Although infants with AD are more likely to develop food allergies, data from infants with variants in SPINK5 and FLG have demonstrated a significant association with food allergy that is independent of AD status.18,19 DN and biallelic LOF variants in COL7A1, a collagen protein critical for dermal-epidermal basement membrane barrier function, typically lead to an epidermolysis bullosa phenotype characterized by skin blistering, but immune and inflammatory comorbidities including IgA nephropathy, significant itch, eosinophilic infiltrate, and TH2 cytokine production.20 There is some degree of phenotypic-genotypic correlation that has been observed in dystrophic epidermolysis bullosa whereby DN missense variants are generally associated with milder clinical disease. Dupilumab blockade of the shared receptor subunit for IL-4 and IL-13 (IL-4Rα) has proven effective for atopic manifestations associated with pathogenic variants in SPINK521–26 and COL7A1.20,27,28 Immunoglobulin replacement therapy is another treatment for skin lesions of Comel-Netherton syndrome (SPINK5), with some reports of significant improvement.29–31 The mechanism by which this improvement occurs is unknown; however, earlier literature highlighted improvement in AD symptoms in patients with severe disease.32
Abnormal antigen receptor signaling
Activation of T and B cells and propagation of signaling are dependent on a multitude of mediators for proper transcriptional regulation of effector function, migration, proliferation, and adhesion. Defects in genes involved in this process are highly associated with atopy and are detailed herein.
Impaired actin skeleton assembly.
T-cell migration and effector functions are dependent on actin polymerization and rearrangement of the cytoskeleton. LOF variants in the genes linked to this process have been associated with disorders of severe infections, autoimmunity, and neoplastic disease, as well as variably penetrant eczematous dermatitis and other forms of allergy. These include WAS due to LOF WAS protein, and LOF variants in WAS protein–interacting protein (encoded by WIP), actin-related protein 2/3 complex subunit 1B (encoded by ARPC1B), hematopoietic protein-1 (HEM-1) (encoded by NCKAP1L [NCK-associated protein 1 like]), and DOCK8.33–42 Abnormal actin dynamics and overlapping clinical phenotypes also feature prominently in patients with LOF in RLTPR encoding capping protein regulator and myosin 1 linker 2, which is important for CD28-mediated signaling.43–45 The drivers of allergic inflammation in these disorders may include Treg-cell dysfunction and loss of tolerance.11 The extensive viral skin infections frequently seen in DOCK8 deficiency are thought to be, in part, due to the inability of the DOCK8-deficient natural killer and T lymphocytes to migrate normally through the skin matrix, leading to lymphocyte destruction42,46 and subsequent environment conducive to allergic inflammation.47
CARD11-BCL10-MALT1-opathies.
The CARD11-BCL10-MALT1 (caspase recruitment domain family, member 11–B-cell chronic lymphocytic leukemia/lymphoma 10–mucosa-associated lymphoid tissue lymphoma translocation gene 1) complex is a key bridge between antigen receptor/coreceptor stimulation and activation of transcription factors necessary for regulatory and inflammatory responses, such as mediators involved in the nuclear factor-κB (NF-κB) pathway.48 Bacterial respiratory and viral skin infections as well as autoimmunity are prominent manifestations of DN variants in CARD11. These incompletely penetrant hypomorphic variants are associated with variable interference of NF-κB signaling, allowing some effector function that can be skewed to TH2 response with strong atopic phenotype.48–50 Although these rare variants that negatively affect CARD11-BCL10-MALT1 component function lead to rare IEI, common, noncoding, regulatory variants that associate with decreased expression of MALT1 and CARD11 are associated with increased allergy risk, highlighting the central importance of this pathway in regulating allergic disease.48
Activated PI3K delta syndrome.
Activated PI3K delta syndrome (APDS) is characterized by humoral immune deficiency, abnormal EBV immunity, autoimmunity, and susceptibility to lymphoproliferative phenotypes including lymphoma. It results from GOF variants in PIK3CD and PIK3R1, resulting in excessive signaling through the PI3K/AKT/mTORC1/mTORC2 pathways. Multiple atopic phenotypes including peripheral and symptomatic tissue eosinophilia (eg, EGID), allergy, eczema, and elevated IgE are also seen in APDS.52,53 Although impaired mTORC1 may be associated with the allergic disease seen in CARD11-associated atopy with dominant interference of NF-κB signaling, there is excessive mTORC1 activity in APDS, which can explain the autoimmune/TH1/TH17 phenotypes. How the TH2 phenotypes emerge may potentially be explained by excess mTORC2 activity, which has been shown to drive TH2 responses and impair Treg-cell function.54–56 Rapamycin, an mTORC1/mTORC2 inhibitor, has been used successfully in APDS and other primary immune regulation disorders by inhibiting effector T cells while improving the function of Treg cells.57–59 PI3K inhibition in the form of leniolisib, a small molecule inhibitor of PI3K, is highly effective in treating the clinical phenotypes of APDS,125 and PI3K inhibition has shown significant promise in several models of allergic inflammation.60–63 These observations raise the possibility of rapamycin and leniolisib as precision therapies for type II–associated inflammation.
Omenn syndrome.
Hypomorphic LOF variants in genes regulating T-cell receptor rearrangement or development, which lead to SCIDs—such as LIG4, DCLRE1C, RAG1 and RAG2, ADA, IL7RA, CDH7, and IL2RG—can result in Omenn syndrome with the onset of erythroderma and eczematous dermatitis, hypereosinophilia, increased IgE levels, histiocytic and lymphocytic infiltrate, as well as hepatosplenomegaly and lymphadenopathy.64–67 Hypomorphic, “leaky,” variants lead to oligoclonal expansion of T cells, which is not allergen-specific in neither clinical nor laboratory-based testing. Although not fully understood, leaky SCID phenotypes lead to a TH2-like phenotype potentially because of a combination of lymphopenia-driven proliferation, oligoclonal T-cell receptor repertoire, and thymic abnormalities that affect tolerance.68 This phenotype can manifest as early as the first few weeks of life.67 Microbial drivers of the inflammatory process tend to occur more often in infants with less impacted gene product levels and/or function.68 Frequently, management requires significant immune suppression before the curative hematopoietic stem cell transplantation.69 Immunomodulators such as cyclosporine and tacrolimus are often needed to manage immune dysregulation while waiting for transplant.
Disruption of cytokine and cytokine signaling
Effective immune regulation depends on intercellular signaling initiated by cytokines and chemokines. Disrupted cellular processes involving cytokine receptors and downstream signaling molecules have been found to be associated with allergic inflammation.
Impaired IL-6 receptor/IL-6 signal transducer/STAT3/zinc finger protein 341 signaling.
STAT3 is downstream of the IL-6 receptor (IL-6R) and the cytokine coreceptor GP130, and DN variants in STAT3 lead to an IEI characterized by eczematous dermatitis and markedly elevated IgE, as well as recurrent infections including reactivation of latent viruses (eg, varicella-zoster virus and EBV), connective tissue, dental, skeletal, and vascular abnormalities reflecting the ubiquitous expression of STAT3 in a multitude of tissues, and varied cytokines and chemokines that use this pathway.70 Additional disorders with a similar phenotype but varying nonimmunologic features along this pathway including LOF variants in IL-6 signal transducer (IL6ST), which encodes GP130, IL6R, and biallelic variants in STAT3’s promoter zinc finger protein 341.71–73 IL-6R deficiency is limited to atopic manifestations, reduced inflammatory responses, and recurrent skin and lung infections, whereas patients with IL6ST DN variants frequently develop severe destructive lung disease with pneumatoceles and bronchiectasis, frequently associated with Aspergillus infection or allergic bronchopulmonary aspergillosis.72–75 STAT3 DN leads to enhanced expression of IL-4R, likely explaining the success in treating atopic phenotypes with dupilumab in these patients.76
PGM3 deficiency due to biallelic hypomorphic variants in PGM3 can lead to profound allergic disease with severe AD, food allergy, asthma, EGIDs, allergic rhinitis, allergic bronchopulmonary aspergillosis, and food-protein–induced enteropathy syndrome.77–80 Infectious, inflammatory, and musculoskeletal and neurodevelopmental comorbidities are also present. PGM3 is critical for normal glycosylation, and the abnormal N-glycan pattern on the surface of lymphocytes can be detected via flow cytometry particularly in naive T cells.80 The process by which abnormal glycosylation contributes to allergic inflammation is yet to be elucidated; however, decreased N-glycosylation of GP130 in PGM3 deficiency has been observed, resulting in poor surface expression, which may explain some of the phenotypic overlap with IL6ST deficiency.81
Of note, although many clinical aspects of STAT3 GOF such as significant autoimmunity and lymphoproliferation are distinctly different than STAT3 DN, as expected, patients with STAT3 GOF have a predisposition to developing eczematous dermatitis,82 although they do not appear to have classical IgE-mediated disease. JAK inhibitors have been shown to be effective for the treatment of atopic manifestations in STAT3 GOF by blocking downstream cytokine-induced JAK activation.83
JAK1, STAT5, and STAT6 GOF.
Germline and/or somatic GOF variants in JAK1 or in STAT5B, which is directly downstream of JAK1,84–86 can lead to profound urticaria or eczematous dermatitis, autoimmunity, failure to thrive, and marked eosinophilia. Heterozygous GOF variants in STAT6, which is also downstream of JAK1 and propagates signal via IL-4 and IL-13 receptors,87 have recently been described in cohorts of patients with severe allergic disease.88–93 Patients with STAT6 GOF develop the full spectrum of atopic disorders including early-onset severe AD, EGIDs, asthma, eosinophilia, elevated IgE, IgE-mediated food allergies, and anaphylaxis.93 Functional studies revealed sustained STAT6 phosphorylation resulting in TH2 skewing. Clinical response to dupilumab has been seen, as expected given the role of STAT6 in IL-4 and IL-13 signaling.88
Improvements in AD, urticaria, autoimmunity and eosinophilia, and even food allergy, growth, and quality of life have been described with targeted inhibition of JAK1/2 in patients with JAK1 and STAT5B GOF.86,94,95 The food allergy response raises the possibility that JAK inhibition could be a potential therapy for IgE-mediated food allergy, and in fact JAK inhibitors are being trialed for the treatment of food allergy (NCT05069831). Oral and topical JAK inhibitors are now approved for the treatment of AD.96 For AD, JAK inhibitors have been demonstrated to be better than dupilumab in a network analysis of AD treatments97; however, the side effect profile must be considered when long-term therapy is planned. Insights from the use of these agents in IEI could help establish recommended therapies for specific defects (eg, dupilumab vs JAK inhibitors for JAK/STAT GOF83,88,95,98,99) to optimize risks and benefit considerations.
TGF-β pathway and atopy with connective tissue features.
An autosomal-dominant disorder called LDS secondary to TGF-β, TGFβR1, and TGFβR2 LOF variants predisposes to the development of AD, asthma, food allergy, and EGIDs. Although the exact mechanism by which this occurs is unknown, in vivo, these patients have increased TH2 cytokine–producing lymphocytes, leading to elevated IgE and eosinophils.100 Like STAT3 DN, LOF variants in TGF-β–related genes result in connective tissue abnormalities. A link between the 2 pathways involves the protein ERBIN (ERBB2IP), which complexes with STAT3 and limits nuclear localization of SMAD2/3. Disrupted STAT3 signaling or decreased lymphocytic ERBIN expression can lead to increased IL-4Rα expression.76 ERBIN deficiency shares features of elevated IgE, EGID, and connective tissue abnormalities with STAT3 DN and LDS. As expected, IL-4Rα blockade was effective in an otherwise treatment-resistant ERBIN patient with eosinophilic esophagitis.101
IFN-γ pathway.
Rare genetic variants in IFN-γ receptor pathway genes are enriched in patients with AD with the specific phenotype of having comorbid eczema herpeticum, associated with enhanced TH2 immunity, and suppressed production of antimicrobial peptides.102–105 This of course raises the question as to whether the susceptibility to viral skin infection is due to loss of IFN-γ–related host defense or rather enhanced TH2 cytokine production from loss of TH2 counterregulation from the TH1/IFN-γ pathway. Patients with rare and common LOF variants in IFN-γ receptor 1 are enriched for significant TH2 laboratory phenotypes, although they tend not to be enriched for overt allergic and atopic clinical phenotypes.102,104,106–111 Deficiency of T-bet (encoded by TBX21), the master transcription factor for TH1 cells, which is up-regulated by IFN-γ, has been reported in 1 patient with susceptibility to mycobacterial infection. The patient also had eosinophilia and TH2 cytokine–driven airway disease.112–114
IKZF1 is a transcriptional repressor of IFN-γ, and patients with IKZF1 GOF variants can develop impaired IFN-γ expression, significant allergic disease, eosinophilia, and increased IL-4 expression, in addition to autoimmunity, lymphoproliferation, and other immune dysregulations. This is in contrast to patients with IKZF1 LOF in whom atopy was not a feature and eosinopenia has been noted. It is tantalizing that immunomodulatory amide drugs such as lenalidomide cause activation of TH1 cells by inducing proteasomal degradation of IKAROS family proteins leading to increased IFN-γ production, and treatment of patient T cells with lenalidomide improves IFN-γ expression while diminishing excess IL-4 production.114
A good representation of the interplay between TH1/TH2 immunity is illustrated by the use of anti–IL-4 blockade. Dupilumab has shown efficacy in reducing the risk of eczema herpeticum in patients with severe AD because of increased type I immune response against herpes simplex virus type 1.115–118 Dupilumab treatment also has been reported to enhance TH1 host defense in a patient with refractory disseminated coccidioidomycosis,137 and various allergic fungal disorders have been successfully treated with dupilumab.138–140
Symptoms of mast cell dysregulation
Consideration of symptoms of mast cell degranulation in the context of IEI focuses on typical IgE-mediated activation as seen in many of the disorders described herein: autoimmune urticaria as comorbid immune dysregulation, such as autoimmune polyendocrinopathy and ectodermal dysplasia syndrome141; lesions that cause mast cell intrinsic abnormalities, such as cold urticaria seen in PLCG2-associated antibody deficiency and immune dysregulation (PLAID); or vibratory urticaria caused by LOF variants in adhesion G protein–coupled receptor E2 (ADGRE2).142
ADGRE2 encodes the receptor EGF-like module-containing mucin-like hormone receptor–like 2. Dissociation of a subunit of the receptor leading to mast cell degranulation occurs when physical forces such as vibration are applied.142 Subphysiologic temperatures can trigger spontaneous degranulation of mast cells in heterozygous genomic deletions in PLAID; cold urticaria, granulomatous skin disease, humoral deficiency, and autoimmunity are hallmarks of the disease.143 The mechanism of autoimmune urticaria in autoimmune polyendocrinopathy and ectodermal dysplasia syndrome remains not elucidated. However, some case reports showed positive FCεRI antibodies, which perhaps suggests autoantibody-induced mast cell degradation.141
The KIT receptor is necessary for appropriate mast cell differentiation, survival, and chemotaxis, and KIT signaling enhances mast cell degranulation. Somatic and rare germline GOF variants leading to constitutive activation of the tyrosine kinase KIT can cause episodic increase in mast cell mediator release, manifesting as symptoms of recurrent urticaria, flushing, wheezing, and severe anaphylaxis.144 Enhanced transcriptional activity of genes involved in anaphylaxis was also identified in an individual with a KARS missense variant who had severe anaphylaxis in response to paper wasp venom.145
Acute urticaria is common in childhood and triggered spontaneously or in response to food, viral infections, and medications, but it is largely self-limited.146 Recurrent urticaria beginning in infancy is potentially an early sign of IEI. Infants with somatic STAT5B GOF variants initially present with recurrent urticaria, but later develop AD, eosinophilia, and food allergy.84 Early-onset cold urticarias are present in cryopyrin-associated periodic syndrome and PLAID; they differ, however, in that evaporative cooling triggers lesions in PLAID. Urticaria in response to vibration would raise suspicion for familial vibratory urticaria associated with ADGRE2.147,148
Conclusions
TH2-driven allergic inflammation is a prominent feature of many IEI, and a broad spectrum of multiple atopic conditions may be present because of the dysregulation of host immunity. Allergic disease may be the initial presentation of several of these disorders, and with the rise in polygenic allergic disease, this may prove to be a diagnostic challenge for providers. Suspicion for IEI should occur when atopic conditions cooccur with autoimmunity and/or infection susceptibility. Timely identification of monogenic variants in IEI through genetic sequencing is crucial for optimal management, and targeted therapies for genetic lesions may also improve concurrent allergic inflammation. Future work that better characterizes allergic disease in the context of specific clinical or laboratory findings and development of algorithms that inform which patients with allergy should undergo genetic testing for IEI are needed.
DISCLOSURE STATEMENT
This work was supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
Disclosure of potential conflict of interest: J. D. Milner served on the scientific advisory board for Blueprint Medicine. The rest of the authors declare that they have no relevant conflicts of interest.
Abbreviations used
- AD
Atopic dermatitis
- ADGRE2
Adhesion G protein–coupled receptor E2
- APDS
Activated PI3K delta syndrome
- BCL10
B-cell chronic lymphocytic leukemia/lymphoma 10
- CARD11
Caspase recruitment domain family, member 11
- CVID
Common variable immunodeficiency
- DN
Dominant-negative
- DOCK8
Dedicator of cytokinesis 8
- EGID
Eosinophilic gastrointestinal disease
- ERBIN
ERBB2-interacting protein
- FLG
Filaggrin
- GOF
Gain of function
- IEI
Inborn errors of immunity
- IL-6R
IL-6 receptor
- IL6ST
IL-6 signal transducer
- IPEX
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked
- JAK
Janus kinase
- LDS
Loeys-Dietz syndrome
- LOF
Loss of function
- MALT1
Mucosa-associated lymphoid tissue lymphoma translocation gene 1
- NF-κB
Nuclear factor-κB
- PGM3
Phosphoglucomutase 3
- PI3K
Phosphoinositide 3-kinase
- PLAID
PLCG2-associated antibody deficiency and immune dysregulation
- SCID
Severe combined immunodeficiency
- SPINK5
Serine protease inhibitor of Kazal type 5
- STAT
Signal transducer and activator of transcription
- Treg
Regulatory T
- USIDNET
United States Immunodeficiency Network
- WAS
Wiskott-Aldrich syndrome
- WIP
WASP-interacting protein
REFERENCES
- 1.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022;42:1473–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris NL, Loke P. Recent advances in type-2-cell-mediated immunity: insights from helminth infection. Immunity 2018;48:396. [DOI] [PubMed] [Google Scholar]
- 3.Galli SJ, Gaudenzio N, Tsai M. Mast cells in inflammation and disease: recent progress and ongoing concerns. Annu Rev Immunol 2020;38:49–77. [DOI] [PubMed] [Google Scholar]
- 4.Lyons JJ, Yi T. Mast cell tryptases in allergic inflammation and immediate hyper-sensitivity. Curr Opin Immunol 2021;72:94–106. [DOI] [PubMed] [Google Scholar]
- 5.Siracusa MC, Kim BS, Spergel JM, Artis D. Basophils and allergic inflammation. J Allergy Clin Immunol 2013;132:789–801; quiz 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh ER, Stokes K, August A. The role of eosinophils in allergic airway inflammation. Discov Med 2010;9:357–62. [PubMed] [Google Scholar]
- 7.Iype J, Fux M. Basophils orchestrating eosinophils’ chemotaxis and function in allergic inflammation. Cells 2021;10:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng AE, Boersma P. Diagnosed allergic conditions in adults: United States, 2021. NCHS Data Brief 2023;460:1–8. [PubMed] [Google Scholar]
- 9.Zablotsky B, Black LI, Akinbami LJ. Diagnosed allergic conditions in children aged 0–17 years: United States, 2021. NCHS Data Brief 2023;459:1–8. [PubMed] [Google Scholar]
- 10.Vaseghi-Shanjani M, Smith KL, Sara RJ, Modi BP, Branch A, Sharma M, et al. Inborn errors of immunity manifesting as atopic disorders. J Allergy Clin Immunol 2021;148:1130–9. [DOI] [PubMed] [Google Scholar]
- 11.Milner JD. Primary atopic disorders. Annu Rev Immunol 2020;38:785–808. [DOI] [PubMed] [Google Scholar]
- 12.Bin L, Leung DY. Genetic and epigenetic studies of atopic dermatitis. Allergy Asthma Clin Immunol 2016;12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenn MW, Ellis AK. The clinical relevance of filaggrin mutations: effect on allergic disease. Ann Allergy Asthma Immunol 2016;117:483–9. [DOI] [PubMed] [Google Scholar]
- 14.Oji V, Eckl KM, Aufenvenne K, Nätebus M, Tarinski T, Ackermann K, et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet 2010;87:274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAleer MA, Pohler E, Smith FJ, Wilson NJ, Cole C, MacGowan S, et al. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N-terminal plakin domain of desmoplakin. J Allergy Clin Immunol 2015;136:1268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013;45:1244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000;25:141–2. [DOI] [PubMed] [Google Scholar]
- 18.Kalb B, Marenholz I, Jeanrenaud A, Meixner L, Arnau-Soler A, Rosillo-Salazar OD, et al. Filaggrin loss-of-function mutations are associated with persistence of egg and milk allergy. J Allergy Clin Immunol 2022;150:1125–34. [DOI] [PubMed] [Google Scholar]
- 19.Ashley SE, Tan HT, Vuillermin P, Dharmage SC, Tang MLK, Koplin J, et al. The skin barrier function gene SPINK5 is associated with challenge-proven IgE-mediated food allergy in infants. Allergy 2017;72:1356–64. [DOI] [PubMed] [Google Scholar]
- 20.Darbord D, Hickman G, Pironon N, Barbieux C, Bonnet-des-Claustres M, Titeux M, et al. Dystrophic epidermolysis bullosa pruriginosa: a new case series of a rare phenotype unveils skewed Th2 immunity. J Eur Acad Dermatol Venereol 2022; 36:133–43. [DOI] [PubMed] [Google Scholar]
- 21.Wu PC, Dai YX, Li CL, Chen CC, Chang YT, Ma SH. Dupilumab in the treatment of genodermatosis: a systematic review. J Dtsch Dermatol Ges 2023;21: 7–17. [DOI] [PubMed] [Google Scholar]
- 22.Yan S, Wu X, Jiang J, Yu S, Fang X, Yang H, et al. Dupilumab improves clinical symptoms in children with Netherton syndrome by suppressing Th2-mediated inflammation. Front Immunol 2022;13:1054422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Yu L, Zhang S, Wang C, Li Z, Li M, et al. Successful treatment of Netherton syndrome with dupilumab: a case report and review of the literature. J Dermatol 2022;49:165–7. [DOI] [PubMed] [Google Scholar]
- 24.Inaba Y, Kanazawa N, Muraoka K, Yariyama A, Kawaguchi A, Kunimoto K, et al. Dupilumab improves pruritus in Netherton syndrome: a case study. Children (Basel) 2022;9:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herz-Ruelas ME, Chavez-Alvarez S, Garza-Chapa JI, Ocampo-Candiani J, Cab-Morales VA, Kubelis-Lopez DE. Netherton syndrome: case report and review of the literature. Skin Appendage Disord 2021;7:346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andreasen TH, Karstensen HG, Duno M, Lei U, Zachariae C, Thyssen JP. Successful treatment with dupilumab of an adult with Netherton syndrome. Clin Exp Dermatol 2020;45:915–7. [DOI] [PubMed] [Google Scholar]
- 27.Zhao C, Cao S, Gao X, Xu X, Gu L. Identification of a novel COL7A1 variant associated with dystrophic epidermolysis bullosa pruriginosa responding effectively to dupilumab. Mol Genet Genomic Med 2023;11:e2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu XG, Yan S, Jiang JQ, Zhou TT, Fang X, Yang H, et al. Successful treatment of epidermolysis bullosa pruriginosa by dupilumab. J Dermatol 2023;50:837–42. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Pan C, Wei R, Li H, Yang Y, Chen J, et al. Netherton syndrome caused by compound heterozygous mutation, c.80A>G mutation in SPINK5 and large-sized genomic deletion mutation, and successful treatment of intravenous immunoglobulin. Mol Genet Genomic Med 2021;9:e1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zelieskova M, Banovcin P, Kozar M, Kozarova A, Nudzajova Z, Jesenak M. A novel SPINK5 mutation and successful subcutaneous immunoglobulin replacement therapy in a child with Netherton syndrome. Pediatr Dermatol 2020;37: 1202–4. [DOI] [PubMed] [Google Scholar]
- 31.Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G, et al. Com el-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol 2009;124:536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jolles S A review of high-dose intravenous immunoglobulin treatment for atopic dermatitis. Clin Exp Dermatol 2002;27:3–7. [DOI] [PubMed] [Google Scholar]
- 33.Cook SA, Comrie WA, Poli MC, Similuk M, Oler AJ, Faruqi AJ, et al. HEM1 deficiency disrupts mTORC2 and F-actin control in inherited immunodysregulatory disease. Science 2020;369:202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med 2012;209:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ochs HD. Mutations of the Wiskott-Aldrich syndrome protein affect protein expression and dictate the clinical phenotypes. Immunol Res 2009;44:84–8. [DOI] [PubMed] [Google Scholar]
- 36.Kuijpers TW, Tool ATJ, van der Bijl I, de Boer M, van Houdt M, de Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol 2017;140:273–7.e10. [DOI] [PubMed] [Google Scholar]
- 37.Papadatou I, Marinakis N, Botsa E, Tzanoudaki M, Kanariou M, Orfanou I, et al. Case report: a novel synonymous ARPC1B gene mutation causes a syndrome of combined immunodeficiency, asthma, and allergy with significant intrafamilial clinical heterogeneity. Front Immunol 2021;12:634313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol 2019;143:2296–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook S, Lenardo MJ, Freeman AF. HEM1 actin immunodysregulatory disorder: genotypes, phenotypes, and future directions. J Clin Immunol 2022;42:1583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, Ramesh N, et al. A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J Clin Invest 2016;126:3837–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janssen E, Wilkie H, Geha RS. Macabre T(H)2 skewing in DOCK8 deficiency. J Allergy Clin Immunol 2021;148:73–5. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med 2009;361:2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat Commun 2017;8:14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorte HS, Osnes LT, Fevang B, Aukrust P, Erichsen HC, Backe PH, et al. A potential founder variant in CARMIL2/RLTPR in three Norwegian families with warts, molluscum contagiosum, and T-cell dysfunction. Mol Genet Genomic Med 2016;4:604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med 2016;213:2413–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Q, Dove CG, Hor JL, Murdock HM, Strauss-Albee DM, Garcia JA, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med 2014;211:2549–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider C, Shen C, Gopal AA, Douglas T, Forestell B, Kauffman KD, et al. Migration-induced cell shattering due to DOCK8 deficiency causes a type 2-biased helper T cell response. Nat Immunol 2020;21:1528–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeVore SB, Khurana Hershey GK. The role of the CBM complex in allergic inflammation and disease. J Allergy Clin Immunol 2022;150:1011–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immunol 2018;141:1818–30.e2. [DOI] [PubMed] [Google Scholar]
- 50.Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet 2017; 49:1192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jamee M, Zaki-Dizaji M, Lo B, Abolhassani H, Aghamahdi F, Mosavian M, et al. Clinical, immunological, and genetic features in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like syndrome. J Allergy Clin Immunol Pract 2020;8:2747–60.e7. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen T, Lau A, Bier J, Cooke KC, Lenthall H, Ruiz-Diaz S, et al. Human PIK3R1 mutations disrupt lymphocyte differentiation to cause activated PI3K-delta syndrome 2. J Exp Med 2023;220:e20221020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bier J, Rao G, Payne K, Brigden H, French E, Pelham SJ, et al. Activating mutations in PIK3CD disrupt the differentiation and function of human and murine CD4(1) T cells. J Allergy Clin Immunol 2019;144:236–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009;30:832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 2011; 12:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai K, Zhang W, Li S, Zhang Z, Xie S, Xu M, et al. mTOR pathway regulates the differentiation of peripheral blood Th2/Treg cell subsets in patients with pemphigus vulgaris. Acta Biochim Biophys Sin (Shanghai) 2021;53:438–45. [DOI] [PubMed] [Google Scholar]
- 57.Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, et al. Treatment with rapamycin can restore regulatory T-cell function in IPEX patients. J Allergy Clin Immunol 2020;145:1262–71.e13. [DOI] [PubMed] [Google Scholar]
- 58.Rao VK. Approaches to managing autoimmune cytopenias in novel immunological disorders with genetic underpinnings like autoimmune lymphoproliferative syndrome. Front Pediatr 2015;3:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Redenbaugh V, Coulter T. Disorders related to PI3Kdelta hyperactivation: characterizing the clinical and immunological features of activated PI3-kinase delta syndromes. Front Pediatr 2021;9:702872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cutrina-Pons A, De Sa A, Fear DJ, Gould HJ, Ramadani F. Inhibition of PI3K p110delta activity reduces IgE production in IL-4 and anti-CD40 stimulated human B cell cultures. Immunology 2023;170:483–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim JS, Jeong JS, Kwon SH, Kim SR, Lee YC. Roles of PI3K pan-inhibitors and PI3K-delta inhibitors in allergic lung inflammation: a systematic review and meta-analysis. Sci Rep 2020;10:7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee HY, Lee GH, Kim HR, Lee YC, Chae HJ. Phosphatidylinositol 3-kinase-delta controls endoplasmic reticulum membrane fluidity and permeability in fungus-induced allergic inflammation in mice. Br J Pharmacol 2020;177:1556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamaki K, Tamura Y, Suzuki T, Uesaki Y, Dougan A, Koyama Y. PI3K/mTOR inhibitor dactolisib attenuates allergic response through inhibitions of the sensitization and mast cell activation. Pharmazie 2023;78:128–33. [DOI] [PubMed] [Google Scholar]
- 64.Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide AL, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet 2006;43:211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalman L, Lindegren ML, Kobrynski L, Vogt R, Hannon H, Howard JT, et al. Mutations in genes required for T-cell development: IL7R, CD45, IL2RG, JAK3, RAG1, RAG2, ARTEMIS, and ADA and severe combined immunodeficiency: HuGE review. Genet Med 2004;6:16–26. [DOI] [PubMed] [Google Scholar]
- 66.Kurosawa A, Adachi N. Functions and regulation of Artemis: a goddess in the maintenance of genome integrity. J Radiat Res 2010;51:503–9. [DOI] [PubMed] [Google Scholar]
- 67.Delmonte OM, Schuetz C, Notarangelo LD. RAG deficiency: two genes, many diseases. J Clin Immunol 2018;38:646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Somech R, Simon AJ, Lev A, Dalal I, Spirer Z, Goldstein I, et al. Reduced central tolerance in Omenn syndrome leads to immature self-reactive oligoclonal T cells. J Allergy Clin Immunol 2009;124:793–800. [DOI] [PubMed] [Google Scholar]
- 69.Mazzolari E, Moshous D, Forino C, De Martiis D, Offer C, Lanfranchi A, et al. Hematopoietic stem cell transplantation in Omenn syndrome: a single-center experience. Bone Marrow Transplant 2005;36:107–14. [DOI] [PubMed] [Google Scholar]
- 70.Hsu AP, Davis J, Puck JM, Holland SM, Freeman AF. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. STAT3 hyper IgE syndrome. Seattle: University of Washington; 1993. [PubMed] [Google Scholar]
- 71.Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol 2018;3:eaat4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med 2017;214:2547–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spencer S, Köstel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med 2019;216:1986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Béziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med 2020;217:e20191804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arlabosse T, Materna M, Riccio O, Schnider C, Angelini F, Perreau M, et al. New dominant-negative IL6ST variants expand the immunological and clinical spectrum of GP130-dependent hyper-IgE syndrome. J Clin Immunol 2023;43:1566–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med 2017;214:669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ben-Khemis L, Mekki N, Ben-Mustapha I, Rouault K, Mellouli F, Khemiri M, et al. A founder mutation underlies a severe form of phosphoglutamase 3 (PGM3) deficiency in Tunisian patients. Mol Immunol 2017;90:57–63. [DOI] [PubMed] [Google Scholar]
- 78.Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol 2014;133:1410–9, 1419.e1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol 2014;133:1400–9, 1409.e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bernth-Jensen JM, Holm M, Christiansen M. Neonatal-onset T(−)B(−)NK(+) severe combined immunodeficiency and neutropenia caused by mutated phosphoglucomutase 3. J Allergy Clin Immunol 2016;137:321–4. [DOI] [PubMed] [Google Scholar]
- 81.Ben-Ali M, Ben-Khemis L, Mekki N, Yaakoubi R, Ouni R, Benabdessalem C, et al. Defective glycosylation leads to defective gp130-dependent STAT3 signaling in PGM3-deficient patients. J Allergy Clin Immunol 2019;143:1638–40.e2. [DOI] [PubMed] [Google Scholar]
- 82.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015;125:591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol 2018;142:1665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma CA, Xi L, Cauff B, DeZure A, Freeman AF, Hambleton S, Kleiner G, et al. Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood 2017;129:650–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nadeau K,Hwa V, Rosenfeld RG. STAT5b deficiency:an unsuspected cause of growth failure, immunodeficiency, and severe pulmonary disease. J Pediatr 2011;158:701–8. [DOI] [PubMed] [Google Scholar]
- 86.Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol 2017;139:2016–20.e5. [DOI] [PubMed] [Google Scholar]
- 87.Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther 2021;6:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sharma M, Leung D, Momenilandi M, Jones LCW, Pacillo L, James AE, et al. Human germline heterozygous gain-of-function STAT6 variants cause severe allergic disease. J Exp Med 2023;220:e20221755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamashita M, Morio T. Germline STAT6 gain-of-function variants cause severe allergy. J Allergy Clin Immunol 2023;151:1252–4. [DOI] [PubMed] [Google Scholar]
- 90.Takeuchi I, Yanagi K, Takada S, Uchiyama T, Igarashi A, Motomura K, et al. STAT6 gain-of-function variant exacerbates multiple allergic symptoms. J Allergy Clin Immunol 2023;151:1402–9.e6. [DOI] [PubMed] [Google Scholar]
- 91.Suratannon N, Ittiwut C, Dik WA, Ittiwut R, Meesilpavikkai K, Israsena N, et al. A germline STAT6 gain-of-function variant is associated with early-onset allergies. J Allergy Clin Immunol 2023;151:565–71.e9. [DOI] [PubMed] [Google Scholar]
- 92.Minskaia E, Maimaris J, Jenkins P, Albuquerque AS, Hong Y, Eleftheriou D, et al. Autosomal dominant STAT6 gain of function causes severe atopy associated with lymphoma. J Clin Immunol 2023;43:1611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baris S, Benamar M, Chen Q, Catak MC, Martinez-Blanco M, Wang M, et al. Severe allergic dysregulation due to a gain of function mutation in the transcription factor STAT6. J Allergy Clin Immunol 2023;152:182–94.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fayand A, Hentgen V, Posseme C, Lacout C, Picard C, Moguelet P, et al. Successful treatment of JAK1-associated inflammatory disease. J Allergy Clin Immunol 2023;152:972–83. [DOI] [PubMed] [Google Scholar]
- 95.Eisenberg R, Gans MD, Leahy TR, Gothe F, Perry C, Raffeld M, et al. JAK inhibition in early-onset somatic, nonclonal STAT5B gain-of-function disease. J Allergy Clin Immunol Pract 2021;9:1008–10.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mikhaylov D, Ungar B, Renert-Yuval Y, Guttman-Yassky E. Oral Janus kinase inhibitors for atopic dermatitis. Ann Allergy Asthma Immunol 2023;130:577–92. [DOI] [PubMed] [Google Scholar]
- 97.Blauvelt A, Teixeira HD, Simpson EL, Costanzo A, De Bruin-Weller M, Barbarot S, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol 2021; 157:1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deyà-Martínez A, Rivière JG, Roxo-Junior P, Ramakers J, Bloomfield M, Guisado Hernandez P, et al. Impact of JAK inhibitors in pediatric patients with STAT1 gain of function (GOF) mutations—10 children and review of the literature. J Clin Immunol 2022;42:1071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hadjadj J, Frémond ML, Neven B. Emerging place of JAK inhibitors in the treatment of inborn errors of immunity. Front Immunol 2021;12:717388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, Halushka MK, et al. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med 2013;5:195ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Droghini HR, Abonia JP, Collins MH, Milner JD, Lyons JJ, Freeman AF, et al. Targeted IL-4Ralpha blockade ameliorates refractory allergic eosinophilic inflammation in a patient with dysregulated TGF-beta signaling due to ERBIN deficiency. J Allergy Clin Immunol Pract 2022;10:1903–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leung DY, Gao PS, Grigoryev DN, Rafaels NM, Streib JE, Howell MD, et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-gamma response. J Allergy Clin Immunol 2011;127:965–73.e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao PS, Leung DY, Rafaels NM, Boguniewicz M, Hand T, Gao L, et al. Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J Invest Dermatol 2012;132:650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao L, Bin L, Rafaels NM, Huang L, Potee J, Ruczinski I, et al. Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol 2015; 136:1591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Watcharanurak K, Nishikawa M, Takahashi Y, Kabashima K, Takahashi R, Takakura Y. Regulation of immunological balance by sustained interferon-gamma gene transfer for acute phase of atopic dermatitis in mice. Gene Ther 2013;20: 538–44. [DOI] [PubMed] [Google Scholar]
- 106.Smith KL, Dai D, Modi BP, Sara R, Garabedian E, Marsh RA, et al. Inborn errors of immunity associated with type 2 inflammation in the USIDNET registry. Front Immunol 2022;13:831279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wood PM, Fieschi C, Picard C, Ottenhoff TH, Casanova JL, Kumararatne DS. Inherited defects in the interferon-gamma receptor or interleukin-12 signalling pathways are not sufficient to cause allergic disease in children. Eur J Pediatr 2005; 164:741–7. [DOI] [PubMed] [Google Scholar]
- 108.Barnes KC. Genetic epidemiology of health disparities in allergy and clinical immunology. J Allergy Clin Immunol 2006;117:243, 54; quiz 55–6. [DOI] [PubMed] [Google Scholar]
- 109.Matsuda A, Ebihara N, Kumagai N, Fukuda K, Ebe K, Hirano K, et al. Genetic polymorphisms in the promoter of the interferon gamma receptor 1 gene are associated with atopic cataracts. Invest Ophthalmol Vis Sci 2007;48:583–9. [DOI] [PubMed] [Google Scholar]
- 110.Nakao F, Ihara K, Kusuhara K, Sasaki Y, Kinukawa N, Takabayashi A, et al. Association of IFN-gamma and IFN regulatory factor 1 polymorphisms with childhood atopic asthma. J Allergy Clin Immunol 2001;107:499–504. [DOI] [PubMed] [Google Scholar]
- 111.Traidl S, Roesner L, Zeitvogel J, Werfel T. Eczema herpeticum in atopic dermatitis. Allergy 2021;76:3017–27. [DOI] [PubMed] [Google Scholar]
- 112.Yang R, Mele F, Worley L, Langlais D, Rosain J, Benhsaien I, et al. Human T-bet governs innate and innate-like adaptive IFN-gamma immunity against mycobacteria. Cell 2020;183:1826–47.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang R, Weisshaar M, Mele F, Benhsaien I, Dorgham K, Han J, et al. High Th2 cytokine levels and upper airway inflammation in human inherited T-bet deficiency. J Exp Med 2021;218:e20202726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoshino A, Boutboul D, Zhang Y, Kuehn HS, Hadjadj J, Ozdemir N, et al. Gain-of-function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci Immunol 2022;7:eabi7160. [DOI] [PubMed] [Google Scholar]
- 115.Eichenfield LF, Bieber T, Beck LA, Simpson EL, Thaci D, de Bruin-Weller M, et al. Infections in dupilumab clinical trials in atopic dermatitis: a comprehensive pooled analysis. Am J Clin Dermatol 2019;20:443–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fleming P, Drucker AM. Risk of infection in patients with atopic dermatitis treated with dupilumab: a meta-analysis of randomized controlled trials. J Am Acad Dermatol 2018;78:62–9.e1. [DOI] [PubMed] [Google Scholar]
- 117.Traidl S, Harries L, Kienlin P, Begemann G, Roesner LM, Werfel T. Dupilumab strengthens herpes simplex virus type 1-specific immune responses in atopic dermatitis. J Allergy Clin Immunol 2023;152:1460–9.e5. [DOI] [PubMed] [Google Scholar]
- 118.Johar RA, Hasanain A, Khouqeer Y. Efficacy of dupilumab in treating atopic dermatitis with recurrent eczema herpeticum in a patient with DOCK8-deficiency hyper-IgE syndrome: a case report. Cureus 2023;15:e43360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vaseghi-Shanjani M, Snow AL, Margolis DJ, Latrous M, Milner JD, Turvey SE, et al. Atopy as immune dysregulation: offender genes and targets. J Allergy Clin Immunol Pract 2022;10:1737–56. [DOI] [PubMed] [Google Scholar]
- 120.Williams KW, Milner JD, Freeman AF. Eosinophilia associated with disorders of immune deficiency or immune dysregulation. Immunol Allergy Clin North Am 2015;35:523–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shahein AR, Gonzalez S, Carcao M, Abdelhaleem M, Hellmann J, McNamara PJ. A case of severe eosinophilia in a preterm infant. Pediatrics 2011;127:e767–71. [DOI] [PubMed] [Google Scholar]
- 122.Tran P, Gober L, Garabedian EK, Fuleihan RL, Puck JM, Sullivan KE, et al. Eosinophilic gastrointestinal disorders in patients with inborn errors of immunity: data from the USIDNET registry. Front Immunol 2022;13:987895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schwartz DM, Kitakule MM, Dizon BL, Gutierrez-Huerta C, Blackstone SA, Burma AM, et al. Systematic evaluation of nine monogenic autoinflammatory diseases reveals common and disease-specific correlations with allergy-associated features. Ann Rheum Dis 2021;80:788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Urm SH, Yun HD, Fenta YA, Yoo KH, Abraham RS, Hagan J, et al. Asthma and risk of selective IgA deficiency or common variable immunodeficiency: a population-based case-control study. Mayo Clin Proc 2013;88:813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rao VK, Webster S, Šedivá A, Plebani A, Schuetz C, Shcherbina A, et al. A randomized, placebo-controlled phase 3 trial of the PI3Kd inhibitor leniolisib for activated PI3Kδ syndrome. Blood 2023;141:971–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Siegel AM, Stone KD, Cruse G, Lawrence MG, Olivera A, Jung MY, et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J Allergy Clin Immunol 2013;132:1388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hox V, O’Connell MP, Lyons JJ, Sackstein P, Dimaggio T, Jones N, et al. Diminution of signal transducer and activator of transcription 3 signaling inhibits vascular permeability and anaphylaxis. J Allergy Clin Immunol 2016;138:187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boos AC, Hagl B, Schlesinger A, Halm BE, Ballenberger N, Pinarci M, et al. Atopic dermatitis, STAT3- and DOCK8-hyper-IgE syndromes differ in IgE-based sensitization pattern. Allergy 2014;69:943–53. [DOI] [PubMed] [Google Scholar]
- 129.Ihekweazu FD, Versalovic J. Development of the pediatric gut microbiome: impact on health and disease. Am J Med Sci 2018;356:413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pivniouk VI, Snapper SB, Kettner A, Alenius H, Laouini D, Falet H, et al. Impaired signaling via the high-affinity IgE receptor in Wiskott-Aldrich syndrome protein-deficient mast cells. Int Immunol 2003;15:1431–40. [DOI] [PubMed] [Google Scholar]
- 131.Lexmond WS, Goettel JA, Lyons JJ, Jacobse J, Deken MM, Lawrence MG, et al. FOXP31 Tregs require WASP to restrain Th2-mediated food allergy. J Clin Invest 2016;126:4030–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Desai A, Sowerwine K, Liu Y, Lawrence MG, Chovanec J, Hsu AP, et al. GATA-2-deficient mast cells limit IgE-mediated immediate hypersensitivity reactions in human subjects. J Allergy Clin Immunol 2019;144:613–7.e14. [DOI] [PubMed] [Google Scholar]
- 133.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone, I: definition according to profiles of lymphokine activities and secreted proteins. J Immunol 1986;136:2348–57. [PubMed] [Google Scholar]
- 134.Milner JD, Fasth A, Etzioni A. Autoimmunity in severe combined immunodeficiency (SCID): lessons from patients and experimental models. J Clin Immunol 2008;28:S29–33. [DOI] [PubMed] [Google Scholar]
- 135.Kopfnagel V, Harder J, Werfel T. Expression of antimicrobial peptides in atopic dermatitis and possible immunoregulatory functions. Curr Opin Allergy Clin Immunol 2013;13:531–6. [DOI] [PubMed] [Google Scholar]
- 136.Kolls JK, McCray PB Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol 2008;8:829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsai M, Thauland TJ, Huang AY, Bun C, Fitzwater S, Krogstad P, et al. Disseminated coccidioidomycosis treated with interferon-gamma and dupilumab. N Engl J Med 2020;382:2337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bernstein JS, Wechsler ME. Eosinophilic respiratory disorders and the impact of biologics. Curr Opin Pulm Med 2023;29:202–8. [DOI] [PubMed] [Google Scholar]
- 139.Bulkhi AA, Mirza AA, Aburiziza AJ, Marglani OA. Dupilumab: an emerging therapy in allergic fungal rhinosinusitis. World Allergy Organ J 2022;15:100638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lo RM, Liu AY, Valdez TA, Gernez Y. Dupilumab use in recalcitrant allergic fungal rhinosinusitis. Ann Allergy Asthma Immunol 2020;125:617–9. [DOI] [PubMed] [Google Scholar]
- 141.Huebner S, Nelson M, Martinez B. Chronic urticaria as possible manifestation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). J Allergy Clin Immunol 2012;129:AB216. [Google Scholar]
- 142.Boyden SE, Desai A, Cruse G, Young ML, Bolan HC, Scott LM, et al. Vibratory urticaria associated with a missense variant in ADGRE2. N Engl J Med 2016;374: 656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med 2012;366:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ke H, Kazi JU, Zhao H, Sun J. Germline mutations of KIT in gastrointestinal stromal tumor (GIST) and mastocytosis. Cell Biosci 2016;6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ribó P, Guo Y, Aranda J, Ainsua-Enrich E, Navinés-Ferrer A, Guerrero M, et al. Mutation in KARS: a novel mechanism for severe anaphylaxis. J Allergy Clin Immunol 2021;147:1855–64.e9. [DOI] [PubMed] [Google Scholar]
- 146.Ansotegui IJ, Bernstein JA, Canonica GW, Gonzalez-Diaz SN, Martin BL, Morais-Almeida M, et al. Insights into urticaria in pediatric and adult populations and its management with fexofenadine hydrochloride. Allergy Asthma Clin Immunol 2022;18:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McSweeney SM, Saklatvala J, Rispoli R, Ganier C, Woszczek G, Thomas L, et al. Genome-wide meta-analysis implicates variation affecting mast cell biology in urticaria. J Allergy Clin Immunol 2024;153:521–6.e11. [DOI] [PubMed] [Google Scholar]
- 148.Kristjansson RP, Oskarsson GR, Skuladottir A, Oddsson A, Rognvaldsson S, Sveinbjornsson G, et al. Sequence variant affects GCSAML splicing, mast cell specific proteins, and risk of urticaria. Commun Biol 2023;6:703. [DOI] [PMC free article] [PubMed] [Google Scholar]