

Abstract
The neurotrophin (NT) brain-derived neurotrophic factor (BDNF) plays an essential role in the formation of long-term potentiation (LTP). Here, we address whether this modulation by BDNF requires its continuous presence, or whether a local increase in BDNF is necessary during a specific time period of LTP initiation. Using electrical field stimulation of primary cultures of hippocampal neurons, we demonstrate that short high-frequency bursts of stimuli that induce LTP evoke also an instantaneous secretion of BDNF. In contrast, stimuli at low frequencies, inducing long-term depression, do not enhance BDNF secretion, suggesting that BDNF is specifically present, and thus required, at the time of LTP induction. The field-stimulation-mediated BDNF secretion depends on the formation of action potentials and is induced by IP3-mediated Ca2+ release from intracellular stores. Experiments, aimed at determining the sites of NT secretion that use NT6, showed similar patterns of surface labeling by field stimulation to those shown previously by high potassium.
Neurotrophins (NTs), in particular brain-derived neurotrophic factor (BDNF), play essential modulatory roles in activity-dependent neuronal plasticity (1–4). That this modulatory role is of physiological relevance evolves from observations in the developing visual cortex of various species. There, not only the local administration (5–7) or enhanced expression of NTs (8), but also blocking agents—specific antibodies or TrkB-IgG fusion proteins (9, 10)—interfere with the normal development of ocular dominance. Moreover, hippocampal long-term potentiation (LTP) is drastically reduced in both homozygous and heterozygous BDNF knockout mice (11, 12), and can be rescued by local administration (12) or viral re-expression (13) of BDNF. Furthermore, interference with endogenous BDNF function by anti-BDNF antibodies or TrkB-IgG fusion proteins (14–16) reduces LTP. Taken together, these observations indicate that the establishment of LTP depends on the immediate availability of critical levels of BDNF and that the impairment of LTP in BDNF knockout mice does not result from subtle cumulative developmental defects. Nonetheless, it is a matter of debate whether continuous, uniform concentrations of BDNF are sufficient to modulate LTP (15) or whether augmented quantities have to become available during a specific period of LTP induction and expression (16). Indeed, regulated secretion of NTs from brain slices, cultured neurons, and neuronal cell lines has previously been demonstrated using high potassium, Glu, carbachol, NTs, and cAMP (17–20). To evaluate the immediate availability of enlarged quantities of BDNF during LTP induction, we analyzed BDNF secretion in primary cultures of hippocampal neurons following electrical field stimulation. We demonstrate that BDNF secretion is triggered by brief bursts of action potentials (APs) at high frequencies that are known to induce LTP, whereas stimuli inducing long-term depression (LTD) failed to enhance BDNF secretion.
Materials and Methods
Tetrodotoxin (TTX), 2-aminoethoxydiphenyl borate (2-ABP), and thapsigargin were from Biotrend (Cologne, Germany). Picrotoxin, caffeine, and cytosine β-d-arabino-furanoside (AraC) were from Sigma.
Recombinant Adenovirus Constructs.
Construction of BDNFmyc adenovirus (AdVBDNF) and of NT6myc adenovirus (AdVNT6) is described in detail in refs. 19 and 21, respectively. Virus titers ranged from 1010-1011 plaque-forming units (pfu)/ml.
Neurons were transduced with adenovirus for 36–42 h (0.8 μl/500 μl). Transduction efficiency (40–80%) was controlled by immunocytochemistry or by using an EGFP-expressing adenoviral vector. Cultures with low infection rates (<30% neurons) or with more than 3% transduced astrocytes were discarded.
Cell Culture.
Hippocampi were dissected from E19 Wistar rat embryos, and neurons were dissociated by incubation (20 min, 37°C) in Trypsin-EDTA (GIBCO) and trituration in DMEM/10% horse serum (GIBCO). Cells were plated [for electron microscopy: 50,000 cells per cm2; for ELISA: 60,000–80,000 cells per cm2) on poly-dl-ornithine (0.5 mg/ml; Sigma) coated glass coverslips. After 3 h the medium was changed to defined medium (22). AraC (1.5 μM) was added at day 3. Neurons were used after 14 to 20 days.
Enzyme Immunoassay (ELISA).
BDNF concentrations were determined by two-site ELISA (19, 23).
Secretion of BDNF from Dissociated Hippocampal Neurons.
Neurons in 24-well plates were incubated for 30 min at 32°C in 300 μl of Hepes-buffered medium (in mM: NaCl 145, KCl 3, Hepes 10, MgCl2 2, CaCl2 3, glucose 8, BSA 0.25%) and washed three times. A first sample (300 μl) was collected after 1 or 5 min incubation in normal medium and served as control (basal secretion), and a second sample was taken after incubation (1 or 5 min) under the given experimental conditions. Mechanical turbulence from medium changes did not cause BDNF secretion. At the end of each experiment we added 10 μM Glu to check the responsiveness of neurons, otherwise they were discarded.
To obtain detectable levels of BDNF, BDNF was expressed by adenoviral vectors, which lead to an about 10–20-fold higher expression compared with endogenous BDNF levels (≈0.2 ng per 100,000 cells). Regulated secretion from AdVBDNF-transduced cells was 0.3–2% of the total BDNF, which is comparable to the calculated fractional secretion from endogenous stores (acute slices, 0.8%; dissociated neurons, 0.3–0.5%).
Viability of Neurons Under the Given Experimental Conditions.
After each experiment the viability of the neurons was evaluated by fluorescein diacetate (20 μg/ml) and propidium iodide (5 μg/ml) staining. Neuron viability was further evaluated by their response to Glu. Moreover, the membrane potential returned to the prestimulation level even after more intense stimulation (25 V, 200 Hz), as used in our experiments.
Statistical Analysis.
Despite standardized cell densities and transduction procedures, basal secretion varied from experiment to experiment (from about 90–200 pg/ml BDNF). Basal secretion results from spontaneous neuronal activity, because it is reduced by 1 μM TTX, and from constitutive secretion from astrocytes, because it is strongly reduced if neurons are cultured in presence of AraC. Therefore, we normalized values of evoked secretion to basal secretion from the same culture (% of basal). In addition, all comparative experiments were performed with neurons from the same preparation to exclude differences due to culture quality and transduction efficiency. All experiments were performed with at least three different preparations. The significance of secretion over basal levels was determined by t test (paired, two-tailed). Percentage values were averaged and differences between experiments evaluated by a nonpaired t test.
Secretion of NT6myc for Localization of Sites of Secretion by Electron Microscopy.
The experimental procedure is described in all detail in ref. 21. In accordance with the electrophysiological experiments, release experiments were performed at 32°C.
Electrophysiology.
Stimulation by field electrodes.
Neurons were stimulated with a homogenous electrical field, placing coverslips between two parallel platinum wires (7 mm long, distance of 7 mm, 1 mm distance from cells). Field electrodes are mounted in a plastic lid and are moved from well to well. Stimulus intensity was regulated by a stimulus isolator (Hivotronic) and stimulus patterns were controlled by a Master-8 (AMPI, Jerusalem, Israel) pulse generator. The field was homogenous because neuronal responses were independent of the location within the field. The following stimulation patterns were applied: continuous trains at different frequencies, burst patterns (each burst consisting of five stimuli at 50 or 100 Hz, interburst interval 500 ms), theta burst stimulation (TBS) (ten bursts, each of four pulses, at 100 Hz, with an interburst interval of 200 ms, TBS are 10 s apart), and tetani (three trains of 100 or 30 stimuli at 100 Hz, interburst interval 20 s).
Patch clamp experiments.
Neuronal responses to different stimuli and pharmacological treatments were analyzed by whole-cell patch clamp. Glass electrodes were filled with internal solution [in mM: 136.5 K-gluconat, 17.5 KCl, 9.0 NaCl, 1.0 MgCl2, 10 Hepes, 0.2 EGTA; for perforated patches 200 μg/ml Amphotericin B (ICN) were added, and for ruptured patches 2 mM ATP and 0.2 mM GTP were added]. Neurons were perfused (0.5 ml/min) at 32°C and patched in voltage clamp mode (Axoclamp 2B, Axon Instruments, Foster City, CA). Recordings were done in current clamp mode. Data were collected at 5 kHz and analyzed with a self-made labview program.
Results
The goal was to analyze the ability of various frequencies and patterns of electrical stimuli to trigger BDNF secretion from dissociated hippocampal neurons. To this end, we used field electrodes (Fig. 1A) to electrically stimulate a large number of neurons in a temporally defined manner, allowing us to subject the neurons to electrical activity reminiscent of that used in vitro to induce LTP or LTD.
Figure 1.

BDNF secretion in response to different stimulation frequencies. (A) Field stimulation setup: a culture of hippocampal neurons is placed between parallel oriented platinum wires, which are connected to a stimulus isolator and a pulse generator. (B) Current clamp recordings were performed in hippocampal pyramidal neurons in response to 50-Hz stimulus trains and 50-Hz burst patterns. Each stimulus (stimulus artifact, ▴) is followed by one AP (Δ). (C) Trains of 600 pulses at 50 Hz (n = 52) but not at 10 Hz (n = 37), elicited a significant BDNF secretion. (D) BDNF secretion in response to a train of 300 pulses at 50 Hz (n = 19) was comparable to secretion evoked by a 50-Hz burst pattern (n = 17). Stimuli (300 pulses) at 100 Hz (applied as bursts or trains, n = 36) did not further enhance BDNF secretion as compared with trains or bursts at 50 Hz (n = 36). The difference (in C and D) of secretion levels induced by 50 Hz results from the fact that experiments were performed independently with different cell preparations. Fractions were collected over 5 min. Values represent the mean ± SE. n.s., Not significant (P > 0.05); **, P < 0.01.
Determination of Stimulation Parameters.
Optimal stimulation parameters were determined by patch clamp recordings from individual neurons in parallel cultures of those used for the secretion experiments. Electrical pulses of 1 ms with 15 V reproducibly elicited APs. Neurons responded to a train of stimuli at a frequency of 10 or 50 Hz with one AP per stimulus (Fig. 1B). Some neurons did not react to each individual stimulus during prolonged trains (>5 s) at 50 Hz (data not shown). Therefore, we also stimulated with a 50-Hz burst pattern, to which neurons respond consistently with one AP per stimulus (Fig. 1B)
BDNF Secretion Is Frequency-Dependent.
We used hippocampal neurons expressing BDNF after transduction with an adenoviral vector carrying BDNF, because levels of endogenous BDNF are too low to detect BDNF secretion in our low-density cultures with the short stimulation periods used. Characteristics of BDNF secretion were not affected by adenoviral transduction of primary cultures of hippocampal neurons as compared with secretion from nontransduced hippocampal slices (19, 24, 25). Field-stimulation-induced BDNF secretion is specific to neurons, because we could not evoke BDNF secretion from AdVBDNF-transduced astrocyte cultures (see Fig. 7, which is published as supporting information on the PNAS web site, www.pnas.org).
To compare different frequencies or stimulation patterns, we kept the number of stimuli constant within given sets of experiments. There was a reliable increase in BDNF secretion in response to trains of 250–600 stimuli at a frequency of 50 Hz. A 50-Hz burst stimulation (300 stimuli; Fig. 1 B and D) was as effective as a continuous train of 300 stimuli at 50 Hz (Fig. 1D). Trains of 600 stimuli at a frequency of 10 Hz for 1 min (Fig. 1C) or of 5 Hz for 2 min (data not shown) did not evoke a significant increase in BDNF secretion (P > 0.5). In contrast, 600 stimuli at 50 Hz, applied either as bursts (1 min) or as a continuous train (12 s), evoked significant (P = 0.0002) BDNF secretion (Fig. 1C). In another set of experiments we compared BDNF secretion evoked by trains or bursts of stimuli at 50 Hz and 100 Hz, but did not detect further increase of BDNF secretion by stimuli at 100 Hz (Fig. 1D).
Neurotrophin Secretion Is Evoked by LTP- but not by LTD-Inducing Stimulus Patterns.
The difference in BDNF secretion by stimuli at either high or low frequencies is also reflected by a difference in LTP induction in hippocampal primary cultures (ref. 26; see Fig. 8A, which is published as supporting information on the PNAS web site), or slices (see Fig. 8B) where LTP was induced only by high frequencies (50 or 100 Hz), while low frequency stimulation (1–10 Hz) lead to LTD or no change in synaptic strength. Consequentially, we investigated whether “classical” LTP-inducing stimulus patterns such as TBS or stimulation with 100-Hz trains would also trigger BDNF secretion as compared with LTD-inducing (long trains of 1 Hz) stimuli. We compared BDNF secretion after three trains of 100 stimuli at 100 Hz with secretion resulting from 7× TBS, keeping the number of stimuli constant (300 stimuli). Substantial BDNF secretion was triggered by both stimulus patterns. In contrast, a train of 300 stimuli at 1 Hz did not result in enhanced BDNF secretion (P = 0.99; Fig. 2). Field stimulation using even milder LTP-induction protocols was also sufficient to evoke BDNF secretion. For instance, three trains of 30 stimuli at 100 Hz, or the “classical” TBS (only 3× TBS) triggered BDNF secretion to a significant extent (P < 0.001; Fig. 2). We could neither detect a significant difference between 7× TBS stimulation and 3 × 100-Hz trains (P = 0.79), nor between 3× TBS and 3 × 30 100-Hz trains (P = 0.31; Fig. 2).
Figure 2.

BDNF secretion and LTP are triggered by the same stimulation patterns, whereas LTD-inducing patterns do not induce BDNF secretion. BDNF secretion in response to different LTP- and LTD-inducing stimulation patterns was measured over a time period of 5 min. Numbers of stimuli (300) within the first three columns were identical: 3 × 100 (three trains of 100 stimuli at 100 Hz, interburst interval of 20 s; n = 38), 7× TBS (one TBS consists of ten bursts each of four pulses at 100 Hz, with an interburst interval of 200 ms, inter-TBS interval 10 s; n = 46), and 1 Hz (train of 300 pulses at 1 Hz; n = 18). Also, weaker LTP-inducing stimuli (3 × 30: three bursts each of 30 pulses at 100 Hz, interburst interval 20 s; 3× TBS) are sufficient to evoke a substantial BDNF secretion (n = 22). Values given represent the mean ± SE. n.s., Not significant (P > 0.05); **, P < 0.01.
Minimal Number of APs at 50 Hz and Time Course of Secretion.
In previous studies analyzing regulated NT secretion, very long stimuli (5–60 min) were generally used for technical reasons [dead volume of the perfusion system, bath application of stimulating agents (19, 24, 27)] or to reach detectable levels of secreted BDNF (18, 28). Electrical stimulation allowed us to determine the minimal number of stimuli necessary to reliably evoke NT secretion. We compared BDNF secretion elicited by short 50-Hz bursts of 1–5 s. A burst of 2 s was sufficient to evoke BDNF secretion above basal levels (P = 0.03), and BDNF secretion evoked by a 3-s burst was comparable to that by 5-s bursts (Fig. 3A). BDNF concentration was already at its maximum within the first minute of collection (Fig. 3B), whether the stimuli were distributed over the whole time period (TBS) or lasted only 5 s (50 Hz train). In subsequently collected fractions, BDNF concentration decayed rapidly.
Figure 3.

Short bursts of stimuli are sufficient to evoke an instantaneous BDNF secretion. (A) Hippocampal neurons were stimulated for 1–5 s (50–250 pulses) with trains of stimuli at 50 Hz. BDNF concentrations of supernatants taken in 1-min intervals were measured. A pulse of 2 s was sufficient to stimulate BDNF secretion. Values given represent the mean ± SE. n.s., Not significant (P > 0.05); **, P < 0.01; *, P < 0.05. (1–3 s, n = 11; 5 s, n = 28). (B) Hippocampal neurons were stimulated either with 100 Hz tetani (three trains of 100 stimuli at 100 Hz, interburst interval of 20 s; n = 11), 7 TBS (n = 13), or trains of 250 pulses at 50 Hz (n = 6). Supernatants were collected each minute. Values are normalized to the average of the first two basal values and are represented as the mean ± SE. The fat line represents the mean of all experiments (n = 30). All experiments show a similar time course: a constant baseline and immediate BDNF secretion in the fraction collected during the stimulus.
BDNF Secretion Depends on the Firing of APs.
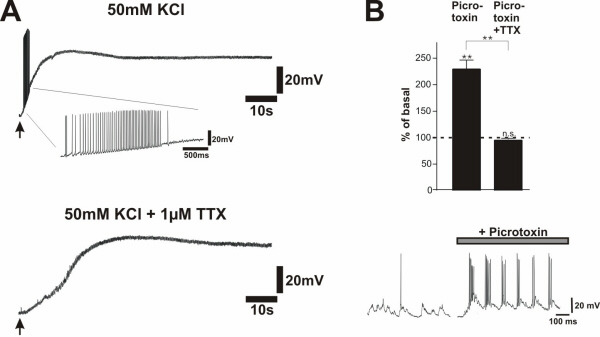
To evaluate which component of the neuronal response to the field stimulus is essential for NT secretion, voltage-dependent sodium channels were blocked by 1 μM TTX, preventing APs. Under these conditions, BDNF secretion in response to 50 Hz field stimulation was abolished (Fig. 4A). Thus, AP firing rather than depolarization of neurons (Fig. 4B) as such, is responsible for BDNF secretion. This interpretation is supported by the observation that KCl-evoked NT secretion was abolished in the presence of TTX (27), where neurons remained depolarized (−10 mV; see Fig. 9A, which is published as supporting information on the PNAS web site). In our cultures the inhibitory GABAergic system is sufficiently developed so that application of the GABAA-receptor antagonist picrotoxin resulted in short-lasting, high-frequency bursts that triggered BDNF secretion comparable to electrical stimulation, which was completely abolished with 1 μM TTX (see Fig. 9B).
Figure 4.

AP firing is required for regulated BDNF secretion. (A) BDNF secretion in response to trains of 250 pulses at 50 Hz in absence (n = 17) and presence (n = 22) of 1 μM TTX. BDNF secretion measured in supernatants collected over 5 min was completely abolished if APs were blocked. Values given represent the mean ± SE. n.s., Not significant (P > 0.05). (B) Patch clamp recordings in current clamp mode of hippocampal neurons under the same conditions as in A. TTX (1 μM) abolished the formation of APs in response to field stimulation. One stimulus in presence and in absence of TTX is enlarged.
Field-Stimulation-Induced BDNF Secretion Depends on Intact Intracellular Ca++ Stores.
In previous experiments it has been demonstrated that regulated NT secretion, in particular secretion initiated by activation of Trk- and Glu-receptors, is mediated by Ca2+ release from intracellular stores, rather than by Ca2+ influx via NMDA receptors or voltage-gated Ca2+ channels (19, 24, 25, 27). Here, we demonstrate that depletion of intracellular Ca2+ stores by caffeine and thapsigargin prevents BDNF secretion by 50 Hz field stimulation in the presence of normal Ca2+ concentration in the medium (Fig. 5). In previous experiments we have shown that both Trk- and Glu-receptor-mediated Ca2+ release from intracellular stores results from the activation of PLC and the subsequent formation of IP3 (25). Therefore, we performed additional experiments using the membrane-permeant 2-aminoethoxydiphenyl borate (2-ABP) compound, which selectively inhibits IP3-induced Ca2+ release (29). Incubation of hippocampal neurons with 75 μM 2-ABP blocked high-frequency-induced BDNF secretion in the presence of normal extracellular Ca2+ concentrations (Fig. 5).
Figure 5.

Electrical field stimulation-induced BDNF secretion depends on Ca2+ mobilization from intracellular stores. In the first set of experiments Ca2+ stores were depleted by preincubation with caffeine (10 mM) and thapsigargin (10 μM) for 20 min. This pretreatment abolished subsequent BDNF secretion by 50 Hz field stimulation (n = 33). In a second experiment hippocampal neurons were preincubated (2–20 min) with 75 μM 2-APB, an inhibitor of IP3 receptor function. 2-ABP blocked BDNF secretion induced by 50 Hz field stimulation (n = 42). Values given represent the mean ± SE. n.s., Not significant (P > 0.05); **, P < 0.01.
Site of Field-Stimulation-Induced Secretion.
We have previously demonstrated (21) by using immunogold cytochemistry that KCl (50 mM)-induced NT6myc secretion occurs all over the neuronal surface, but preferentially from neurites. NT6myc secretion induced by electrical field stimulation leads to a similar pattern of surface labeling (Fig. 6A). We identified NT6myc immunoreactivity along the surface of dendrites and axons without any preferences for specific structures such as synapses or spines. A train of 3,000 pulses at 50 Hz (1 min) resulted in a significant (P = 0.003) surface labeling in comparison to the same number of stimuli at 10 Hz, which did not result in a detectable NT6myc secretion. Moreover, 50-Hz-mediated surface labeling was prevented by 1 μM TTX, and stimulation with 3× TBS (Fig. 6A) induced a similar NT6myc secretion as 50 mM KCl and the 50-Hz train (Fig. 6B).
Figure 6.

Sites of field-stimulation-induced NT6myc secretion: (A) Representative examples of NT6myc surface distribution at the ultrastructural level after field stimulation with TBS or 50-Hz trains. Surface bound NT6myc was fixed immediately after secretion and detected via its myc tag, using preembedding immunogold cytochemistry (4 nm gold) at the neuronal surface. Secretion occurs from dendrites and axons and is not restricted to synaptic sites. Arrowheads point to secretion sites. a, axon; d, dendrite. (Scale bar, 100 nm.) (B) Quantitative evaluation of the NT6myc secretion at the electron microscopic level by stereological procedures. Pooled data from three independent experiments are presented. Values given represent the mean ± SE of three independent experiments. **, Statistically significant differences (gold particle per μm; P < 0,01) in comparison with stimulation with 10 Hz or 50 Hz in the presence of 1 μM TTX.
Discussion
The modulatory action of NTs on activity-dependent neuronal plasticity is determined by the spatial and temporal availability of individual NTs and their respective receptors. Both NTs and the corresponding Trk receptors show a characteristic distribution and are differentially regulated during development (30, 31), and their expression is influenced by epigenetic factors (1, 32).
The modulatory actions of BDNF have been analyzed extensively in the process of LTP in hippocampal slices (11–16). Nevertheless, it remained to be established whether a continuous, constant secretion of BDNF is sufficient to modulate LTP or whether augmented quantities of BDNF have to become available during a specific time period of the initiation of hippocampal LTP. This question has previously been approached by administration of BDNF (12, 14) and by blocking endogenous BDNF (14–16). However, the temporal resolution of these studies was limited by the slow diffusion of BDNF or BDNF-blocking agents into slices. Therefore, we addressed the question in a different way, overcoming these limitations by determining whether and how long BDNF secretion is evoked by LTP-inducing electrical stimulation.
To this end, we stimulated dissociated hippocampal neurons with field electrodes. Thus we could analyze BDNF secretion with a high time resolution that would not be possible in hippocampal slices, where BDNF had to diffuse over long distances, paved with TrkB-receptors, before reaching the incubation medium. Because LTP and LTD can be elicited by stimulation at high or low frequencies, respectively, in sufficiently differentiated primary cultures of hippocampal neurons (ref. 26; see Fig. 8A), this experimental system served our purposes favorably. To achieve a high time resolution and a reproducible BDNF secretion, we could not rely on endogenous BDNF because, as reported by Balkowiec and Katz in nodose-petrosal ganglion cells (28), very long stimulation periods of 30–60 min and high cell densities were required. Thus, we used AdVBDNF-transduced hippocampal neurons, which in previous experiments showed the same BDNF secretion characteristics as nontransduced hippocampal slices (19, 24, 25).
Field-stimulation-induced secretion of BDNF from cultivated hippocampal neurons depended on the stimulation frequency (Fig. 1C). Importantly, very short stimulation periods at high frequencies of 50–100 Hz were sufficient to induce an instantaneous and transient BDNF secretion (Fig. 2 and Fig. 3 A and B). The same or very similar stimuli as in our experiments were shown to induce LTP in dissociated hippocampal neurons (ref. 26; see Fig. 8A), as used for our secretion experiments and also in hippocampal slices. In contrast, low-frequency stimulation without detectable increase in BDNF secretion (Fig. 1C) did not result in LTP formation in cultured neurons (ref. 26; see Fig. 8A) or hippocampal slices (see Fig. 8B). Thus, our observations are compatible with a scenario in which BDNF secretion is initiated specifically by LTP-inducing stimuli. Hence, the highest BDNF concentrations are reached during and shortly after LTP induction, suggesting that BDNF modulates LTP at the initiation stage. This interpretation is supported by very recent experiments of Kossel et al. (35), who showed that the availability of endogenous BDNF during the LTP-inducing stimulus is important for full expression of LTP. These authors blocked endogenous BDNF function during a defined time period by pretreating hippocampal slices with caged BDNF antibodies, which were uncaged 2 min before and during the TBS stimulus used for LTP induction. This temporal blockade of endogenous BDNF function was sufficient to reduce LTP. The reduction was visible at the very first excitatory postsynaptic potentials measured after LTP induction, indicating that BDNF is necessary at the initiation of LTP and exerts its effect on synaptic potentiation within 1 min. This, however, does not exclude that BDNF also initiates long-term effects at the translational or transcriptional level (36, 37), which then affect synaptic transmission by another mechanism at later stages (38, 39).
Although under certain experimental conditions, such as “puff-application,” BDNF elicits bursts of APs (40) appropriate to initiate LTP, it is questionable whether under physiological conditions such a mechanism is relevant, because in most (11, 12, 14), but not all (41), experiments in hippocampal slices, the bath administration of BDNF was not sufficient to induce LTP. In view of our experiments suggesting that high concentrations of BDNF are only available in coincidence with activity, a modulatory rather than a causal action of BDNF is more likely. In accordance with that idea, Boulanger and Poo (42) demonstrated that BDNF potentiates synaptic efficacy more efficiently at active synapses, which also would serve as an additional mechanism to confine BDNF action to active synapses. In addition, our current and previous experiments demonstrate that regulated BDNF secretion occurs in conditions under which LTP is prevented—e.g., in the presence of NMDA receptor antagonists (data not shown) and in absence of extracellular Ca2+ (24). Thus, the coincidence of neuronal activity and BDNF availability, rather than a shared mechanism of LTP induction and BDNF secretion, must be important for the modulatory action of BDNF. This coincidence would be triggered by the shared stimulus for LTP and BDNF secretion.
Furthermore, our experiments demonstrate that stimulation patterns, which lead to LTD or do not influence synaptic strength, do not induce BDNF secretion, suggesting that BDNF selectively potentiates synaptic strength after secretion during LTP induction, but does not play a role in LTD formation. Indeed, in hippocampal slices, LTP induction is impaired in BDNF and TrkB-receptor knockout mice (11, 34) and by infusion of Trk-IgG fusion proteins (14–16). For the hippocampus, no information is available as to whether LTD induction is affected in BDNF- or TrkB-receptor-deficient mice. However, in the rat visual cortex LTD induction was not changed by infusion of Trk-IgG fusion proteins (43). Synaptic strength under conditions of LTD induction in the visual cortex was enhanced by addition of BDNF, thus preventing LTD formation (44, 45). These observations support the idea that BDNF is released during LTP, but not LTD, and thus locally enhances synaptic strength, but is not necessary for the reduction of synaptic strength—i.e., LTD.
Field-stimulation-induced BDNF secretion does not depend on depolarization as such, but on the formation of APs (Fig. 4A), because it is abolished by TTX, as is BDNF secretion by picrotoxin-elicited bursts of neuronal activity (see Fig. 9B). This is also true for high-potassium-mediated NGF secretion (27). The molecular mechanisms of BDNF secretion initiated by field stimulation seem to be very similar to those initiated by the activation of Trk- and non-NMDA Glu-receptors (25). The exact pathways leading to NT secretion initiated by these different stimuli are not yet known in full detail. However, they all converge on IP3-mediated Ca2+ release from endogenous stores. Accordingly, depletion of Ca2+ stores by caffeine/thapsigargin abolished field-stimulation-mediated BDNF secretion, even in the presence of extracellular Ca2+ (Fig. 5). Moreover, the specific IP3 receptor antagonist 2-APB blocked field-stimulation-induced BDNF secretion (Fig. 5). Because release of intracellular Ca2+ is crucial for NT secretion, the temporal summation of Ca2+ release from intracellular Ca2+ stores may be a mechanism responsible for the fact that stimulation at high, but not low, frequencies results in BDNF secretion (46). The responsible Ca2+ storage compartment, the smooth ER, is widely distributed throughout dendrites and axons including dendritic spines (47), which could explain the localization of NT6 secretion along axonal and dendritic processes (Fig. 6A). In a situation in which the neurons are locally, synaptically stimulated, NT secretion would be triggered by local Ca2+ increases only in the stimulated area, and would thus provide a mechanism for a locally confined, activity-dependent modulation of synaptic strength.
Supplementary Material
Acknowledgments
We thank M. Korte, V. Nägerl, and B. Berninger for comments on the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft.
Abbreviations
- BDNF
brain-derived neurotrophic factor
- LTP
long-term potentiation
- LTD
long-term depression
- AP
action potential
- NT
neurotrophin
- TBS
theta burst stimulation
- TTX
tetrodotoxin
References
- 1.Thoenen H. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 2.Cellerino A, Maffei L. Prog Neurobiol. 1996;49:53–63. doi: 10.1016/0301-0082(96)00008-1. [DOI] [PubMed] [Google Scholar]
- 3.McAllister A K, Katz L C, Lo D C. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 4.Schinder A F, Poo M. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 5.Maffei L, Berardi N, Domenici L, Parisi V, Pizzorusso T. J Neurosci. 1992;12:4651–4662. doi: 10.1523/JNEUROSCI.12-12-04651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domenici L, Berardi N, Carmignoto G, Vantini G, Maffei L. Proc Natl Acad Sci USA. 1991;88:8811–8815. doi: 10.1073/pnas.88.19.8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabelli R J, Hohn A, Shatz C J. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 8.Huang Z J, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear M F, Maffei L, Tonegawa S. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 9.Berardi N, Cellerino A, Domenici L, Fagiolini M, Pizzorusso T, Cattaneo A, Maffei L. Proc Natl Acad Sci USA. 1994;91:684–688. doi: 10.1073/pnas.91.2.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cabelli R J, Shelton D L, Segal R A, Shatz C J. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- 11.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patterson S L, Abel T, Deuel T A S, Martin K C, Rose J C, Kandel E R. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 13.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Figurov A, Pozzo-Miller L D, Olafsson P, Wang T, Lu B. Nature (London) 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 15.Kang H J, Welcher A A, Shelton D, Schuman E M. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Kolbeck R, Barde Y A, Bonhoeffer T, Kossel A. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blöchl A, Thoenen H. Mol Cell Neurosci. 1996;7:173–190. doi: 10.1006/mcne.1996.0014. [DOI] [PubMed] [Google Scholar]
- 18.Goodman L J, Valverde J, Lim F, Geschwind M D, Federoff H J, Geller A I, Hefti F. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 19.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Proc Natl Acad Sci USA. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krüttgen A, Möller J C, Heymach J V, Shooter E M. Proc Natl Acad Sci USA. 1998;95:15867–15867. doi: 10.1073/pnas.95.16.9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gärtner A, Shostak Y, Hackel N, Ethell I M, Thoenen H. Mol Cell Neurosci. 2000;15:215–234. doi: 10.1006/mcne.1999.0825. [DOI] [PubMed] [Google Scholar]
- 22.Brewer G J, Cotman C W. Brain Res. 1989;494:65–74. doi: 10.1016/0006-8993(89)90144-3. [DOI] [PubMed] [Google Scholar]
- 23.Kolbeck R, Bartke I, Eberle W, Barde Y A. J Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- 24.Griesbeck O, Canossa M, Campana G, Gärtner A, Hoener M C, Nawa H, Kolbeck R, Thoenen H. Microsc Res Tech. 1999;45:262–275. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<262::AID-JEMT10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 25.Canossa M, Gärtner A, Campana G, Inagaki N, Thoenen H. EMBO J. 2001;20:1640–1650. doi: 10.1093/emboj/20.7.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deisseroth K, Bito H, Tsien R W. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 27.Blöchl A, Thoenen H. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 28.Balkowiec A, Katz D M. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. J Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- 30.Maisonpierre P C, Belluscio L, Friedman B, Alderson R F, Wiegand S J, Furth M E, Lindsay R M, Yancopoulos G D. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 31.Ringstedt T, Lagercrantz H, Persson H. Dev Brain Res. 1993;72:119–131. doi: 10.1016/0165-3806(93)90165-7. [DOI] [PubMed] [Google Scholar]
- 32.Lindholm D, Castrén E, Berzaghi M, Blöchl A, Thoenen H. J Neurobiol. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- 33.Xu B, Gottschalk W, Chow A, Wilson R I, Schnell E, Zang K, Wang D, Nicoll R A, Lu B, Reichardt L F. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp H P, Bonhoeffer T, Klein R. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 35.Kossel A, Cambridge S B, Wagner U, Bonhoeffer T. Proc Natl Acad Sci USA. 2001;98:14702–14707. doi: 10.1073/pnas.251326998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang H J, Schuman E M. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 37.Korte M, Kang H, Bonhoeffer T, Schuman E. Neuropharmacology. 1998;37:553–559. doi: 10.1016/s0028-3908(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 38.Gottschalk W, Pozzo-Miller L D, Figurov A, Lu B. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pozzo-Miller L D, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng Z H, Lu B. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kafitz K W, Rose C R, Thoenen H, Konnerth A. Nature (London) 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- 41.Kang H, Schuman E M. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 42.Boulanger L, Poo M. Nat Neurosci. 1999;2:346–351. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- 43.Sermasi E, Margotti E, Cattaneo A, Domenici L. Eur J Neurosci. 2000;12:1411–1419. doi: 10.1046/j.1460-9568.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- 44.Kinoshita S, Yasuda H, Taniguchi N, Katohsemba R, Hatanaka H, Tsumoto T. J Neurosci. 1999;19:2122–2130. doi: 10.1523/JNEUROSCI.19-06-02122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huber K M, Sawtell N B, Bear M F. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 46.Jacobs J M, Meyer T. J Neurosci. 1997;17:4129–4135. doi: 10.1523/JNEUROSCI.17-11-04129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spacek J, Harris K M. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}