

Abstract
Actin-myosin (actomyosin) generates mechanical force by consuming ATP molecules. We apply the energy landscape perspective to address a controversial issue as to whether the myosin head moves with multiple steps after a single ATP hydrolysis or only a single mechanical event of the lever-arm swinging follows a single ATP hydrolysis. Here we propose a theoretical model in which the refolding of the partially unfolded actomyosin complex and the movement of the myosin head along the actin filament are coupled. A single ATP hydrolysis is followed by the formation of a high free-energy partially unfolded actomyosin complex, which then gradually refolds with a concomitant multiple stepping movement on the way to the lowest free-energy rigor state. The model quantitatively explains the single-molecular observation of the multiple stepping movement and is consistent with structural observations of the disorder in the actomyosin-binding process. The model also explains the observed variety in dwell time before each step, which is not accounted for by previous models, such as the lever-arm or ratchet models.
Actin and myosin constitute thin and thick filaments, respectively, in muscle fibers, and their relative sliding movement generates mechanical force during muscle contraction. The elementary process of force generation has been a subject of intensive research for many years, and a commonly accepted hypothesis at present is the lever-arm model, which is consistent with various items of structural evidence (1–4). In this model, the neck domain of the myosin head rotates around the rest of the myosin head settled on the actin filament and thus acts as a lever arm. As ATP hydrolysis occurs while the myosin head is detached from the actin filament, and force is exerted only when the myosin head is attached to the actin filament, the lever-arm model intrinsically implies one-to-one correspondence, i.e., tight coupling between ATP hydrolysis and the mechanical event. However, using single-molecular measurements, the Yanagida group has reported dynamical behaviors that contradict the lever-arm model (5–8). In particular, Kitamura et al. (8) observed in an in vitro motility assay that a subfragment-1 of the myosin head moves along the actin filament in a stepwise manner for a successive two to five steps (11–30 nm) without detachment from the actin filament, with an average step size of 5.3 nm (approximately the size of actin subunit diameter) (8). The observation of Kitamura et al. (8) showed the one-to-many correspondence, i.e., loose coupling between ATP hydrolysis and mechanical events. Such movement of the myosin head is a biased Brownian movement along the actin filament (9, 10). The biased movement has been described by preceding models of mechanochemical coupling (11–13). Those models, however, dealt with the stochastic behavior of an ensemble of motor proteins at constant chemical nonequilibrium, i.e., constant consumption of ATP molecules, and were not meant to explain the loose coupling in a single ATP hydrolysis cycle. In this paper, we propose a theoretical model by using the energy landscape concept developed in protein folding studies (14, 15) and show that the model reasonably describes the multiple stepping movements that cannot be explained by the lever-arm or other preceding models.
Clues for constructing the model are the following experimental observations on the actomyosin-binding process. When the actomyosin complex is formed after ATP hydrolysis, it undergoes a transition from the weakly bound to the strongly bound state (16–21), which ends up with the structurally more restricted rigor state (1–4). This transition was spectroscopically shown to be a disorder-to-order transition (16–21). The transition is accompanied by structural changes, as evidenced by electron microscopy, which had been attributed to the cleft closure in the myosin head (22), and possibly by the folding of locally unfolded structures such as the melted helix (23). Multiple intermediate states were observed in the transition process (18). These observations of the disorder-to-order transition motivated us to use concepts and methods of protein folding study to describe the process.
Comparison in energy and time scale should further justify the use of analogy between actomyosin binding and protein folding. Free energy change accompanied with ATP hydrolysis, ∼15 kcal/mol (24), is within a typical range of that of protein folding from 10 to 20 kcal/mol (14). The time scale of both the transition from the weakly bound to the strongly bound state (18) and the stepping movement of the myosin head along the actin filament (8) coincides with that of protein folding from the millisecond to second order (14).
A Model Energy Landscape of Actomyosin Binding
We start with the postulate that the sliding movement of the myosin head is coupled to the gradual refolding of the partially unfolded actomyosin complex in the binding process of the myosin head to the actin filament. To embody this coupling, we represent the energy landscape of the actomyosin complex as a function of two parameters, x and ρ, as shown in Fig. 1A. Here, x denotes a position of the myosin head along the actin filament, and ρ is the order parameter onto which the conformational degree of freedom of the actomyosin complex is projected. ρ is defined as the ratio of the number of residues with the same configuration as in the free energy minimum (rigor) conformation to the number of residues concerned with the gradual refolding (see Appendix). Multiple basins arranged in line along the direction of ρ correspond to the multiple intermediate states or kinetic traps in the binding process (18). The free energy of these intermediate states has a global trend of decreasing free energy with increasing ρ, reflecting the formation of more contacts. Because identical actin subunits are repeatedly arranged along the actin filament, these intermediates are positioned periodically along the direction of x with a period of 5.5 nm (actin subunit diameter).
Figure 1.

A free energy landscape for the coupled refolding–stepping processes. (A) A contour plot of the free energy landscape as a function of x, the position of the myosin head along the actin filament, and ρ, the order parameter of refolding. The multiple intermediate states at ρ = 0.1, 0.3, 0.5, 0.7, and the rigor state at ρ = 0.9 are positioned periodically along the actin filament with a period of 5.5 nm. There is a global bias of decreasing free energy with increasing ρ, and kinetic connectivity between the intermediate states is in favor of stepping movement. (B) Kinetic representation of actomyosin binding as a sequential process through multiple intermediate states, I(x,ρ), toward rigor states, R(x,ρ).
The energy landscape of Fig. 1A was constructed by summing two parts, the position-independent gradual refolding free energy and the position-dependent binding free energy. As a model of the position-independent gradual refolding, we use the random energy model for protein folding by Bryngelson and Wolynes (25, 26). The main assumptions are that the free energy of conformations as a function of the order parameter is lower when the conformation is closer to the free-energy minimum conformation and that, as many conformations correspond to a given value of the order parameter, the free-energy distribution of the myosin conformations with given ρ can be approximated by a Gaussian function (see Appendix).
The position-dependent binding free energy is modeled in a similar manner as in Shoemaker et al. (27) to have a Gaussian distribution (see Appendix). We assume that the binding energy for the intermediate states at ρ1 = 0.1, ρ2 = 0.3, ρ3 = 0.5, ρ4 = 0.7, and the rigor state at ρ5 = 0.9 is large at x = na (a = 5.5 nm), and that the free-energy barrier between the intermediate states at (x,ρ) = (na, ρi) and ((n + 1)a, ρi+1) is relatively low, and thus they are kinetically well connected as shown in Fig. 1B, which couples conformational ordering and stepping movement. Such asymmetry with respect to x is a natural consequence of the asymmetry in the actomyosin-binding interaction with respect to the relative positional arrangement of actin and myosin. Interaction parameters were chosen so that the resultant free-energy difference between the intermediate states around ρ = 0.1 and the rigor state of ρ = 0.9 is about 100 pN⋅nm = 25 kT = 15 kcal/mol, which is the same order of free-energy difference in protein conformational changes or ATP hydrolysis (24).
Monte Carlo (MC) Simulation
We simulate movement of the myosin head along the actin filament by the MC simulation of random walk on the energy landscape shown in Fig. 1A, regarding MC steps as the time proceeding. Position of the myosin head along the actin filament is discretized as the one-dimensional lattice. The lattice spacing is δx = 0.275 nm, which is 1/20 of the size of the actin subunit, and one MC step corresponds to δt = 0.7 ns, which makes D = δx2/2δt = 0.054 nm2/ns, which is estimated by regarding the myosin head as an ellipsoid moving sidewise with semimajor axes of 8 nm and semiminor axes of 2.5 nm in water with viscosity η = 0.89 pN⋅ns⋅nm at 298 K (28).
Starting from random x and ρ chosen with probability proportional to a statistical weight Wconf(ρ) (see Appendix), which has a strong peak at about 0.2 < ρ < 0.4, the myosin head travels through the x–ρ space as a thermal process. Each MC step is composed of the following procedures: (i) The trial position and trial conformation are chosen. x is displaced by δx back or forth with equal probabilities. At the same time, the number of ordered amino acid residues, Nρ, is changed by one or kept constant. That is, order parameter ρ is changed by 1/N or not changed, with probability proportional to the number of conformations at each ρ. Change in x emulates the Brownian motion due to the thermal fluctuation, and change in ρ emulates conformational change induced within the time scale of nanosecond. (ii) Energy of the actomyosin complex is chosen randomly from the Gaussian distribution, which corresponds to a selection of single conformation out of multiple conformations with a given set of x and ρ. (iii) Whether the trial is accepted is determined by the energy difference between the trial and the preceding MC step according to the standard Metropolis criteria with kT = 298 K = 4.1 pN⋅nm. If the trial is not accepted, x and ρ are kept as in the preceding step, but time is elapsed by δt. These procedures are repeated until ρ reaches 0.9, the rigor state.
Results and Discussion
The results of the MC simulation described above are shown in Fig. 2. A typical trajectory in Fig. 2A shows that multiple stepwise movement occurred successively in a single binding event. The corresponding trajectory on the (x,ρ)-plane in Fig. 2B shows an example of variety in the direction of diffusion between the intermediate states. At first, the refolding progresses without movement along the actin filament. Then both the refolding and the movement along the actin filament occur at once, and movement along the actin filament occurs without the progress of the refolding. At last, they occur simultaneously again to reach ρ = 0.9. As a definite trend found in many trajectories, both occur mostly at once because of the existence of low-energy paths across the saddle point connecting the two intermediate states (basins) located diagonally. On the basis of the energy landscape, we can draw a kinetic scheme showing how the multiple stepping movement occurs with the progress of refolding as shown in Fig. 1B. The number of steps per single binding event is generally more than one and has a broad distribution, as shown in Fig. 2C. Thus, our model successfully reproduces the multiplicity and stochastic distribution of the stepping movement observed by Kitamura et al. (8), which cannot be explained by the tight coupling of ATP hydrolysis and the mechanical event in the lever-arm model. Free energy is “stored” for a long time due to the long lifetime of high free-energy conformations of proteins and is then “released,” not all at once but part by part, on the way to finding the free-energy minima of the strongly bound state.
Figure 2.

Results of MC simulation of the movement on the free-energy landscape shown in Fig. 1A. (A) A typical multiple stepping movement in position of the myosin head as a function of time, plotted every 7 μs. (B) Position vs. order parameter as in Fig. 1A, plotted every 7 μs. (C) Distribution of the number of steps per single binding process summed up for 1,000 trajectories.
The mechanism of energy storage and release in the myosin movement has also been discussed with ratchet models (29–31). In these models, myosin is modeled as a particle that is subjected to Brownian fluctuation in a thermal environment and moves on a periodic asymmetric potential energy surface, i.e., a ratchet. As expected from the second law of thermodynamics, no net motion of the particle occurs when the temperature of the system is spatially homogeneous (32). Although Vale and Oosawa proposed that the heterogeneous temperature distribution might be the origin of the unidirectional movement (29), spatial heterogeneity in temperature over the nanometer scale should disappear with a time scale of less than microseconds and cannot be sustained for the observed millisecond-to-second time scale of the myosin movement. In isothermal ratchet models, on the other hand, the temperature was assumed to be spatially homogeneous, but extra nonthermal fluctuations were added to the particle, which keeps the system out of equilibrium (30, 31). The nonthermal coherent fluctuation can resonate with the stochastic diffusive movement to bring about the unidirectional rectified movement of the particle. No explanation, however, has been given for the structural or physical origin of this hypothesized nonthermal fluctuation, which remains operative over more than milliseconds after ATP hydrolysis.
These models, as well as the lever-arm model (1–4), all assume that every step of the movement is equivalent. In contrast, our model postulates that the progress of multiple steps is correlated to the progress of refolding, and thus steps are not equivalent to each other (see Fig. 3). In our calculation, although the dwell-time distribution accumulated without discrimination of the order of steps is apparently fitted to a single exponential function, the dwell-time distributions before the first step, between the first and second steps, and between the second and third steps all show exponential decrease with different average dwell times (3.1, 4.0, and 6.7 ms, respectively). This is due to the heterogeneity of the energy surface around each intermediate state in their manner of refolding: the barrier height for the step is relatively low at small ρ, whereas it is relatively high at large ρ, which results in a longer dwell time as refolding proceeds. This variety in the dwell time before each step is observed in the single-molecule experiment. Although the dwell-time distribution accumulated without discrimination of the order of the steps is apparently fitted by a single exponential function (8) as in our result, the average dwell time between the first and second steps is shorter than that between the second and third steps by a factor of more than two (K. Kitamura and T. Yanagida, personal communication). Thus, our model sufficiently explains this experimental finding, which critically discriminates our model from the preceding models of the lever arm (1–4), Vale–Oosawa (29), and isothermal ratchet models (30, 31). The validity of the present model should further be probed experimentally by correlating the stepping movement with conformational ordering or by the use of the mechanical, chemical, or protein engineering modifications of the energy landscape.
Figure 3.

A change of the dwell-time distribution with the progress of stepping movement. The distribution of the dwell time before the first step, between the first and second steps, and between the second and third steps is shown (only successive forward steps are taken into account here). The dwell-time distributions before each step are well fitted by the single exponential curves. Even though the accumulated histogram is well fitted by a single exponential curve with an average dwell time of 4.2 ms, the average dwell time for each step elongates as the stepping movement proceeds (3.1, 4.0, and 6.7 ms, respectively).
Conclusion
In the present paper, we have provided a theoretical model that reproduces quantitative features of the experimentally observed multiple stepping movement of the myosin head along the actin filament (8). In our model, when the actomyosin complex is formed after ATP hydrolysis, it is in a high free-energy disordered state. Then, the global bias of the energy landscape of the conformational change leads to the coupled binding and sliding movement in the energy-descendent direction, which is the source of force generation. The kinetic connectivity between the intermediate states in the evolutionarily designed energy landscape produces a unidirectional multiple stepping movement in the fluctuating thermal environment. Our model should be experimentally distinguished from preceding models by correlating the progress of successive steps with the change in dwell time observed in single-molecule behavior.
There are a variety of biological molecular motors that work differently from the conventional myosin: In a class of myosin (myosin VI), the myosin head moves on the actin filament in a reverse direction (33). In the case of the ATP synthase (34, 35), the high efficiency of free energy transduction suggests tighter coupling between ATP hydrolysis and mechanical events than in the actomyosin case discussed here. Such diversity in the direction of movement or in the manner of coupling in free energy transduction can be attributed to the diversity of the shape of the energy landscape, such as the number of intermediate states or how they are connected. The difference in the energy landscape resulting from evolution should be the origin of the difference in the dynamical behavior in molecular motor systems.
Acknowledgments
We acknowledge Kazuo Kitamura and Toshio Yanagida for discussions on their experimental observations; Mitsunori Takano for critical reading of the manuscript; and Mio Nagai and Masataka Nagaoka for fruitful discussions at an early stage of this work. This work was supported by Research and Development Applying Advanced Computational Science and Technology of Japan Science and Technology Corporation and by grants from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Abbreviation
- MC
Monte Carlo
Appendix
In our model, the actomyosin complex takes disordered conformations after ATP hydrolysis and becomes more ordered with decreasing free energy on binding. This conformational ordering process should be analogous to the refolding process of small proteins and, if such analogy holds, at least N ≈ 101 − 102 residues should be involved in the process, and we assume N = 30. In a coarse-grained picture, each residue takes one of ν + 1 possible configurations: one is the same as that in the free energy minimum (rigor) conformation and ν other configurations with ν = 3. The conformation of the actomyosin complex is designated by a set of configurations at N residues.
The order parameter ρ is defined as the ratio of the number of residues that have the same configuration as in the rigor conformation to the number of residues concerned with gradual refolding N. Then, the number of conformations of the molecule with the order parameter ρ is
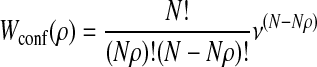 |
1 |
and therefore
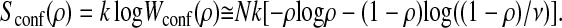 |
2 |
As a single value of ρ corresponds to multiple conformations, the energy of conformations with the given ρ is distributed with a certain width. We approximate that the energy of conformations with the order parameter ρ is an independent random variable and its probability between Econf and Econf + dE is
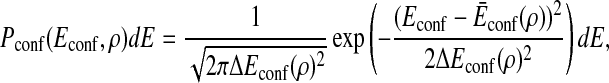 |
3 |
where the average and SD of conformation energy are
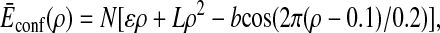 |
4 |
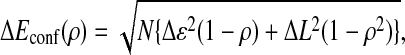 |
5 |
with ɛ = −2 pN⋅nm, L = −7 pN⋅nm, b = 0.7 pN⋅nm, Δɛ2 = 0.5 (pN⋅nm)2, and ΔL2 = 0.5 (pN⋅nm)2.
The relatively large negative values of ɛ and L in Ēconf(ρ) represent the global bias of energy surface toward the rigor conformation: the molecule becomes more stable when the conformation is more similar to rigor conformation. The model is an extension of the random energy model of protein folding developed by Bryngelson and Wolynes (25, 26). The last term of Eq. 4, which is not present in the formulation in ref. 25, is introduced here to model the existence of multiple intermediates, and b represents the barrier height between intermediates.
With a method similar to that used by Shoemaker et al. (27), the binding between the myosin head and the actin filament is modeled as the formation of Nb = 50 contacts with interaction energy per contact, ɛb = −5 pN⋅nm. We assume that the binding site of the myosin head spreads around x with the probability density of
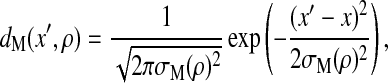 |
6 |
and that the binding position along the actin filament around x̄n(ρ) = na + 5aρ, with a = 5.5 nm and integer n, has spatial distribution
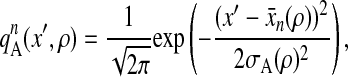 |
7 |
which is normalized to give the size of binding site σA when integrated with respect to position. Then, the probability that the myosin head is within the binding site on the actin filament is
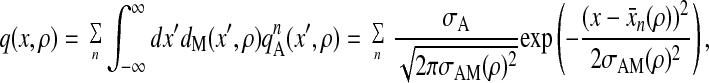 |
8 |
where σA = 1.0 nm, and
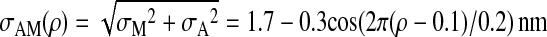 |
9 |
assures the large binding energy for the intermediate states at ρ = 0.1, 0.3, 0.5, 0.7, and the rigor state at ρ = 0.9. The ρ dependence of x̄n(ρ) models the low free-energy barrier between intermediate states (na, ρi) and ((n + 1)a, ρi+1) and brings about the kinetic connectivity between these states, which couples the gradual refolding and the stepping motion.
The Boltzmann distribution of contacts gives the contact formation probability
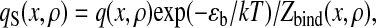 |
10 |
with the partition function for the formation of a contact
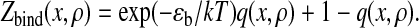 |
11 |
The number density of states with a given set of x and ρ is given by
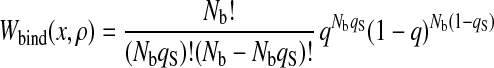 |
12 |
and therefore
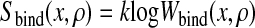 |
13 |
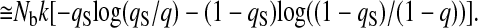 |
With an assumption similar to Eqs. 3–5, the probability that the total binding energy is between Ebind and Ebind + dE can be approximated by
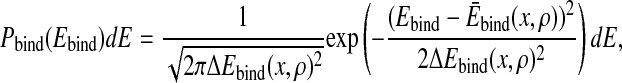 |
14 |
where the mean and SD are
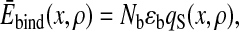 |
15 |
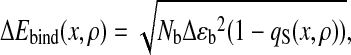 |
16 |
with the fluctuation in stabilization energy per unbound contact, Δɛb = 0.5 (pN⋅nm)2.
The total effective energy used for Metropolis judgment is
E(x,ρ) = Econf(ρ) +
Ebind(x,ρ) −
TSbind(x,ρ), which has a Gaussian
distribution with the mean Ē(x,ρ) =
Ēconf(ρ) +
Ēbind(x,ρ) −
TSbind(x,ρ) and SD ΔE(x,ρ)
=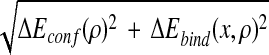 .
By multiplying the Boltzmann factor exp(−E/kT) to
this Gaussian distribution, the most probable value of energies,
conformational entropy, and free energy at temperature T is
found to be Ē*(x,ρ) = Ē(x,ρ) −
ΔE(x,ρ)2/kT,
S*conf(ρ) =
Sconf(ρ) −
ΔE(x,ρ)2/2(kT)2, and
F̄*(x,ρ) = Ē(x,ρ) −
TSconf(ρ) −
ΔE(x,ρ)2/2kT, respectively.
F̄*(x,ρ) thus obtained is shown in Fig.
1A. The interaction parameters defined above were
chosen so that the free energy difference between the intermediate
state around ρ = 0.1 and the rigor state of ρ = 0.9 is
about 100 pN⋅nm = 25 kT = 15 kcal/mol,
which is the same order of the free energy difference in protein
conformational changes or ATP hydrolysis.
.
By multiplying the Boltzmann factor exp(−E/kT) to
this Gaussian distribution, the most probable value of energies,
conformational entropy, and free energy at temperature T is
found to be Ē*(x,ρ) = Ē(x,ρ) −
ΔE(x,ρ)2/kT,
S*conf(ρ) =
Sconf(ρ) −
ΔE(x,ρ)2/2(kT)2, and
F̄*(x,ρ) = Ē(x,ρ) −
TSconf(ρ) −
ΔE(x,ρ)2/2kT, respectively.
F̄*(x,ρ) thus obtained is shown in Fig.
1A. The interaction parameters defined above were
chosen so that the free energy difference between the intermediate
state around ρ = 0.1 and the rigor state of ρ = 0.9 is
about 100 pN⋅nm = 25 kT = 15 kcal/mol,
which is the same order of the free energy difference in protein
conformational changes or ATP hydrolysis.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cooke R. Physiol Rev. 1997;77:671–697. doi: 10.1152/physrev.1997.77.3.671. [DOI] [PubMed] [Google Scholar]
- 2.Holmes K C. Curr Biol. 1997;7:R112–R118. doi: 10.1016/s0960-9822(06)00051-0. [DOI] [PubMed] [Google Scholar]
- 3.Highsmith S. Biochemistry. 1999;38:9791–9797. doi: 10.1021/bi9907633. [DOI] [PubMed] [Google Scholar]
- 4.Geeves M A, Holmes K C. Annu Rev Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- 5.Yanagida T, Arata T, Oosawa F. Nature (London) 1985;316:366–369. doi: 10.1038/316366a0. [DOI] [PubMed] [Google Scholar]
- 6.Saito K, Aoki T, Aoki T, Yanagida T. Biophys J. 1994;66:769–777. doi: 10.1016/s0006-3495(94)80853-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishijima A, Kojima H, Funatsu T, Tokunaga M, Higuchi H, Tanaka H, Yanagida T. Cell. 1998;92:161–171. doi: 10.1016/s0092-8674(00)80911-3. [DOI] [PubMed] [Google Scholar]
- 8.Kitamura K, Tokunaga M, Iwane A H, Yanagida T. Nature (London) 1999;397:129–134. doi: 10.1038/16403. [DOI] [PubMed] [Google Scholar]
- 9.Yanagida T, Iwane A H. Proc Natl Acad Sci USA. 2000;97:9357–9359. doi: 10.1073/pnas.97.17.9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geeves M A. Nature (London) 2002;415:129–131. doi: 10.1038/415129a. [DOI] [PubMed] [Google Scholar]
- 11.Qian H. Biophys Chem. 1997;67:263–267. doi: 10.1016/s0301-4622(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 12.Fisher M E, Kolomeisky A B. Proc Natl Acad Sci USA. 1999;96:6597–6602. doi: 10.1073/pnas.96.12.6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller D, Bustamante C. Biophys J. 2000;78:541–556. doi: 10.1016/S0006-3495(00)76615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 15.Dobson C M, Šali A, Karplus M. Angew Chem Int Ed Engl. 1998;37:868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 16.Baker J E, Brust-Mascher I, Ramachandran S, LaConte L E W, Thomas D D. Proc Natl Acad Sci USA. 1998;95:2944–2949. doi: 10.1073/pnas.95.6.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roopnarine O, Szent-Györgyi A G, Thomas D D. Biochemistry. 1998;37:14428–14436. doi: 10.1021/bi9808363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker M, Zhang X-Z, Jiang W, Trinick J, White H D. Proc Natl Acad Sci USA. 1999;96:465–470. doi: 10.1073/pnas.96.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warshaw D M, Hayes E, Gaffney D, Lauzon A M, Wu J, Kennedy G, Trybus K, Lowey S, Berger C. Proc Natl Acad Sci USA. 1998;95:8034–8039. doi: 10.1073/pnas.95.14.8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, Root D D. Biophys J. 2000;79:1498–1510. doi: 10.1016/S0006-3495(00)76401-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volkmann N, Hanein D. Curr Opin Cell Biol. 2000;12:26–34. doi: 10.1016/s0955-0674(99)00053-8. [DOI] [PubMed] [Google Scholar]
- 22.Volkmann N, Hanein D, Ouyang G, Trybus K M, DeRosier D J, Lowey S. Nat Struct Biol. 2000;7:1147–1155. doi: 10.1038/82008. [DOI] [PubMed] [Google Scholar]
- 23.Houdusse A, Kalabokis V N, Himmel D, Szent-Györgyi A G, Cohen C. Cell. 1999;97:459–470. doi: 10.1016/s0092-8674(00)80756-4. [DOI] [PubMed] [Google Scholar]
- 24.Kodama T. Physiol Rev. 1985;65:467–551. doi: 10.1152/physrev.1985.65.2.467. [DOI] [PubMed] [Google Scholar]
- 25.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 27.Shoemaker B A, Portman J J, Wolynes P G. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berg H C. Random Walks in Biology. Princeton: Princeton Univ. Press; 1983. p. 57. [Google Scholar]
- 29.Vale R D, Oosawa F. Adv Biophys. 1990;26:97–134. doi: 10.1016/0065-227x(90)90009-i. [DOI] [PubMed] [Google Scholar]
- 30.Astumian R D. Science. 1997;276:917–922. doi: 10.1126/science.276.5314.917. [DOI] [PubMed] [Google Scholar]
- 31.Jülicher F, Ajdari A, Prost J. Rev Mod Phys. 1997;69:1269–1281. [Google Scholar]
- 32.Feynman R P, Leighton R B, Sands M. The Feynman Lectures on Physics. Vol. 1. Reading, MA: Addison–Wesley; 1963. [Google Scholar]
- 33.Wells A L, Lin A W, Chen L Q, Safer D, Cain S M, Hasson T, Carragher B O, Milligan R A, Sweeney H L. Nature (London) 1999;401:505–508. doi: 10.1038/46835. [DOI] [PubMed] [Google Scholar]
- 34.Yasuda R, Noji H, Kinosita K, Jr, Yoshida M. Cell. 1998;93:1117–1124. doi: 10.1016/s0092-8674(00)81456-7. [DOI] [PubMed] [Google Scholar]
- 35.Yasuda R, Noji H, Yoshida M, Kinosita K, Jr, Itoh H. Nature (London) 2001;410:898–904. doi: 10.1038/35073513. [DOI] [PubMed] [Google Scholar]