

Abstract
Protein folding and binding are kindred processes. Many proteins in the cell are unfolded, so folding and function are coupled. This paper investigates how binding kinetics is influenced by the folding of a protein. We find that a relatively unstructured protein molecule can have a greater capture radius for a specific binding site than the folded state with its restricted conformational freedom. In this scenario of binding, the unfolded state binds weakly at a relatively large distance followed by folding as the protein approaches the binding site: the “fly-casting mechanism.” We illustrate this scenario with the hypothetical kinetics of binding a single repressor molecule to a DNA site and find that the binding rate can be significantly enhanced over the rate of binding of a fully folded protein.
We often glibly state that a protein must be folded to function. The reasoning underlying this statement is that organizing complex networks of chemical reactions in the cell requires these reactions to be highly specific. This specificity is achieved ultimately by having a high degree of geometrical precision in molecular binding—the famous “lock and key.” Geometric precision accompanies the increased rigidity of a biomolecule once it has folded; thus, apparently, folding is required for specific function. It does come as a surprise, then, that many proteins in the cell appear to be unfolded most of the time (see ref. 1 and refs. therein). Several ideas about potential biological advantages of being unfolded have been proposed. For example, the rapid turnover of unfolded proteins because of proteolytic degradation may be required for cell-cycle regulation (2). Thermodynamic arguments have also been made suggesting that coupling folding and binding may allow greater equilibrium distinctions for binding to different sites (3). In this note, we investigate whether too much rigidity may conflict with the need for biomolecules to move during their function and is therefore a kinetic disadvantage. There is much evidence for residual flexibility in the folded state, which must be thought of as having an ensemble of conformations (4). Still, the range of motions allowed in the folded state, as measured by Debye–Waller factors (5), is more restricted than those allowed in an unfolded molecule, thus slowing the exploration of configuration space. Here, we illustrate, by means of a specific example of operator binding to DNA, how the speed of molecular recognition can be enhanced by having folding (necessary for the required specificity) occur during binding rather than before.
Folding and binding are kindred phenomena. The similarity of binding and folding is clear at the thermodynamic level, where both processes involve accurately locating molecular fragments with respect to each other, reducing the configurational entropy, and simultaneously lowering the free energy by the exclusion of solvent and formation of hydrogen bonds and salt bridges (6, 7). At the structural level, the similarity between packing patterns between protein subunits and in protein interiors has been investigated (8, 9). The question of induced fit in binding (10) is clearly parallel to questions of specificity in the folding problem (11–14). Our goal is to unite folding and binding in a dynamic perspective. The dynamics of searching for specific conformations in folding has been treated by exploiting statistical mechanical theories of energy landscapes (15–18). These theories picture folding as diffusion through an ensemble of partially folded states characterized by reaction coordinates or order parameters. For this reduced description, the energy landscape approach accounts for the trends in average energy as the protein becomes more ordered and utilizes the statistics of the variations in energy, the “ruggedness” of the landscape that allows the possibility of kinetic traps, to understand the speed of the search. Rapid search through the landscape aimed towards a specific native structure requires the landscape to have an overall gradient that is large compared to the local ruggedness. Globally, the energy landscape resembles a funnel (19–22). The effect of the ruggedness on a funneled landscape is to provide a slowing of the diffusion through the configuration space. The rate of finding the folded state depends on the configurational diffusion rate as well as the free energy barriers arising from the tradeoff between energy and entropy captured by the funnel description. The free energy of each individual configuration of the chain falls as the ground state is approached, thus providing a driving force in that direction. Countering this trend, there are many more configurations, which are disorganized, at the top of a funnel landscape. The way in which energy and entropy compensate each other determines how big the thermodynamic barrier to folding is. The thermodynamic barriers for real proteins seem to be well described by perfectly funnel-like landscapes (18), so-called Gō models (23). These models account for the energetics of forming a native-like contact, but nonnative interactions are dismissed as thermodynamically insignificant. Such models finesse the issue of specificity by taking it as a phenomenological fact. Dynamics on perfectly funnel-like landscapes can be addressed by using several different mathematical approaches, including direct molecular dynamics or Monte Carlo simulation (24–28). An alternative approach develops a free energy functional that describes energy entropy compensation as different parts of the protein order. Such free energy functional methods make explicit the interplay of energetics and chain topology. Unlike detailed atomistic simulations, these methods use as input chain statistics directly accessible to experimental determination such as chain stiffness, etc. The free energy functional is described through local order parameters such as the fluctuation of each residue about the native structure (29) or the probability of forming specific contacts (30–32). Such free energy functionals can often be even further simplified by assuming perfect ordering of contiguous segments. These simplified theories account well for trends in folding kinetics (33–35).
Free Energy Profiles for Folding and Binding
To describe the combined processes of folding and binding, we first introduce a free energy functional that depends on the fraction of specific contacts made in the protein itself, {qijp} (30–32). Extending this contact free energy functional to describe binding requires additional order parameters: the fractional occupancy of contacts between the protein and its target-binding site (surface contacts), {qijs}, and geometrical parameters describing the approach of the protein molecule as a whole towards its target, e.g., a vector displacement of the center of mass of the protein, Rcm and a set of Euler angles, Ω, specifying the molecular orientation with respect to target fixed axes.
Although the explicit form of the free energy is outlined in the Appendix, it is useful here to briefly comment on the newer physical considerations incorporated in this functional. We consider the free energy functional
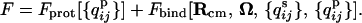 |
1 |
The first term, representing the free energy functional for protein contacts, is taken from previous publications (30–32). The second term accounts for the free energy of binding and couples local structure formation and the binding interactions through the entropy cost of forming surface contacts at a fixed distance and orientation from the binding site. The entropy of localizing surface residues near the binding site depends on the accessible conformations available to those residues. We describe the ensemble of accessible conformations of a partially ordered protein by the distribution of monomer positions about the native structure, {ρi(xi, ri)}. These densities depend on Rcm and Ω through the fiducial native positions {ri} (see Fig. 1). The monomer density depends implicitly on the structural characterization of the ensemble. Unstructured regions of the protein will have larger fluctuations and hence a lower entropy cost of binding compared to more localized residues at the same separation distance from the binding site.
Figure 1.

The position of a protein surface residue as the protein is displaced from its operator-binding site as used in the free energy functional model. The native crystal structure defines a constant fiducial position of a surface residue (r′i) from the protein center of mass (Rcm). The fluctuations of residue i are centered about the mean position ri = 〈xi〉 (relative to the binding location, riB). If the protein is translated only without rotation (Ω = 0), then riB equals r′i so that the probability density of residue i can be described by P(xi − ri).
Analogous to refs. 30–32, the contact probabilities are determined self consistently by minimizing the free energy with respect to {qijp} and {qijs} now at a fixed location from the binding site. Other global structural reaction coordinates such as the total fraction of internal protein contacts, Qp, and the fraction of contacts with the target, Qs, are constrained through Lagrange multipliers. If the internal chain dynamics is fast, this procedure leads to free energy profiles as a function of the approach geometry.
Reduced free energy profiles parameterized by Rcm and Ω are directly observable through single-molecule experiments by using atomic force microscope techniques (36–38). Even without detailed calculations, we can easily understand how such free energy profiles differ in the limiting cases in which the molecule is completely folded or completely unstructured. If the protein were completely folded at all times (Qp = 1), then binding could occur only when the protein approaches the binding site within a distance of the order of the folded state rms displacement (rmsd), typically on the order of 1 Å, and with an appropriate orientation so that binding contacts can be made across the interface. This gives rise to a very short-range anisotropic attraction because of the small rmsd in a completely folded protein. Effectively there is a large entropic barrier to binding. Conversely, when a protein is completely unfolded, binding can begin as soon as the protein approaches the binding site within a radius of gyration of the unfolded protein (a fraction of the end-to-end distance of a random coil of N bonds of length a, Rg ∼ 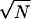 a). When unfolded, the attraction has a much longer range but will be quite weak because the binding contacts will not be strongly ordered until they cooperate. By coupling folding with binding, a weak attraction at large distances grows on approach, allowing further folding of the molecule to take place cooperatively, becoming a short-range specific attraction. These considerations suggest that the binding speed is enhanced relative to that of the fully folded protein.
a). When unfolded, the attraction has a much longer range but will be quite weak because the binding contacts will not be strongly ordered until they cooperate. By coupling folding with binding, a weak attraction at large distances grows on approach, allowing further folding of the molecule to take place cooperatively, becoming a short-range specific attraction. These considerations suggest that the binding speed is enhanced relative to that of the fully folded protein.
Assuming a concentration of one molecule per Å3, the binding flux for species α can be approximated by the simple formula
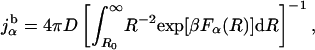 |
2 |
where D is the diffusion constant, Fα(R) is the free energy of species α at the location indicated by R, and R0 is the contact distance. The binding rate measured in the laboratory comes from each species, so ktot = juMu + jfMf, where Mα = exp[−βFα(∞)]/(exp[−βFu(∞)] + exp[−βFf(∞)]) represents the fraction of each species. This bimolecular rate is in units of inverse molar per second. As a point of reference, we also compute rates for binding of the completely folded (Q = 1) species as kQ=1 = jQ=1 MQ=1, where the subscript denoting the protein order parameter Qp has been suppressed for clarity.
Eq. 2 assumes three-dimensional isotropic diffusion and averages completely over orientations. Accounting for anisotropy is straightforward and would lead to a larger relative speed up. Here we calculate the free energy only as a function of distance from the binding site with the orientation fixed as in the folded complex. Somewhere along the approach to the binding site, the folding commences because the free energy curves cross. Precisely where this occurs depends on whether folding can be nucleated from the binding site outwards. This “nonadiabatic” effect, caused by diffusion through the protein conformation space, requires us to examine a two-dimensional free energy plot F(Qp, R).
An Illustrative Example of the Fly-Casting Mechanism
Speed will be important for binding when concentrations are low, as in gene regulation, where only few copies of a gene and its regulators are sometimes present. Unfolded gene regulators are often found (39–41). The protein synthesis parts of the genetic switch mechanism may well be slower than this search, in which event binding speed per se may not have a significant evolutionary advantage, whereas kinetic discrimination might (41). Despite large amounts of beautiful work on these kinetics over the years, there remain open questions of mechanism (42–46). Our study should therefore be considered as merely illustrative of the scenario. For concreteness, we consider a caricature of binding of arc repressor. When bound, arc repressor is dimeric. Early evidence suggested that preformed and folded dimers bind as a bimolecular event (47). Our model calculation used to illustrate the mechanism considers a different scenario where one monomer of the pair is already bound, and the final unfolded monomer approaches. The reality may well be somewhere in between, requiring the consideration of a true termolecular process in which the two arc repressor molecules move up and down the DNA chain, binding only when they both encounter the target DNA sequence. Other systems that might better conform to the present scenario may include bZIP proteins (41) and other proteins involved in protein–protein signaling (48). Through a nonspecific largely electrostatic attraction, the repressors are localized in the vicinity of the nucleic acid chain (44). Again, for purposes of illustration, we neglect this aspect, assuming for simplicity that one repressor has already found its site, has folded, and is waiting for the next unfolded repressor to arrive. The folding–binding coupling should have an even more dramatic effect on a true (simultaneous) termolecular process.
The free energy curves as a function of separation distance are plotted in Fig. 2. The two curves that correspond to the folded and unfolded minima remain distinct throughout much of the binding. Also shown on the same plot are the profiles for the limiting cases of a completely folded state that has no disorder (Qp = 1) and a completely unfolded state that has no order (Qp = 0). These limiting cases behave as expected: for the Qp = 1 species, the free energy switches rapidly from no interaction with the binding site to the full stabilization of binding within a short distance of approach to the well; in contrast, for the Qp = 0 species, a weak interaction is formed further away but remains weak at closest approach. We see that if the protein enters in its unfolded but partially ordered state, a long-range attraction is formed that is quite weak but that this surface crosses to the folded state at some point. If folding dynamics is sufficiently fast, then the system switches from the unfolded curve to the lower free energy folded curve, giving, ultimately, the strong binding of the folded state to the DNA molecule.
Figure 2.

Free energy of binding for specific ensembles of arc repressor. The F(ΔR) curves are shown for the folded (red) and unfolded (green) minima as well as the fully ordered [(Q = 1 (black)] and disordered [Q = 0 (blue)] states at Tf. ΔR = R − R0 is the separation distance relative to that of the bound complex (R0). The effective capture radius is expanded by 8 Å for the unfolded state over the folded (which is 16 Å) and by 14 Å over that for the completely folded Q = 1 state. The orange curve is the free energy of the steepest descent path on the F(ΔR, Qp) surface shown in Fig. 3. Note the broken scales of R used to delineate the folding events, which occur in a narrow range of approach distance. The radius of the square well potential is b = R0 + 6.5 Å. The Debye–Waller factor for the folded residues is Δf = 1 Å, and for the unfolded chain Δu = 17 Å, which is the end-to-end distance of a random coil with 20 bond segments (the number of residues in the binding site).
The two-dimensional plots of F(R, Qp) shown in Fig. 3 give a more complete picture. The transition from the unfolded to folded states does not occur precisely at the equilibrium thermodynamic point where the one-dimensional free energy profiles cross, but instead would most probably follow one of the steepest descent trajectories. Considering the variation of target contact probability along the steepest descent trajectory, shown in red in Fig. 3 and in orange in Fig. 2, we see that some weak binding contacts are made at a large distance in the unfolded state because of the large fluctuations of the unfolded molecule. These contacts, however, ease the formation of structure within the molecule, allowing the free energy to lower as the target is approached. One can picturesquely imagine the process as one of a randomly gesticulating unfolded molecule casting out pieces of polymer chain, waiting for these to bind to the target.
Figure 3.

Contour plots of the two-dimensional free energy surface (in units of kBTf) as a function of approach distance and contact ordering fraction Qp. ΔR = R − R0 is the separation distance relative to that of the bound complex (R0). The steepest descent paths are shown for the unfolded (red) and folded (green) states. At Tf, far from the binding site, the folded and unfolded structures are equally favorable. At ΔR = 20 Å, the unfolded state already feels the interaction falling to a lower contour while the folded state remains unaffected by the binding interaction. Qp for the unfolded ensemble hardly increases until ΔR = 8 Å, whereupon the free energy falls dramatically, while Qp increases by about 0.2. Closer in, the unfolded trajectory completes folding rapidly with binding. Parameters and broken scale in R are same as in Fig. 2.
Once the target has been engaged, the folding free energy comes into play. The whole molecule folds and reels in the target, as in fly fishing. The target has a smaller diffusion constant than the protein; so, in fact, the protein gets reeled into its DNA sequence target rather than the other way around. We call the present sequence of events the “fly-casting mechanism.” The main components of this mechanism are illustrated in Fig. 4. How effective the fly-casting mechanism is in speeding up rates depends on the speed of folding itself and on the concentrations of target protein molecule pairs, which determine the overall folding vs. binding rates. Our simplifications probably lead to an underestimate of the effect.
Figure 4.

A cartoon of how fly casting increases folding speed. At an approach distance Rcm, the partially folded ensemble is already able to form a few initial contacts to the binding site, while the folded structure remains out of range because of the smaller fluctuations in the folded state. Although these initial contacts are weak, they allow the protein to “reel” itself into the operator, completing folding and binding simultaneously. The increased capture radius allows the unfolded protein to find its specific binding site faster.
The free energy calculated as a function of separation distance allows us to compute the binding rate using Eq. 2. In Fig. 5, the ratio of rates ktot/kQ=1 calculated from the free energy along the steepest descents paths shown in Fig. 3, is plotted for a range of unfolded concentrations, Mu. This ratio indicates the effective speedup of the fly-casting mechanism over a range of temperatures and binding stabilities. When allowed to have the flexibility to latch onto the binding surface at a greater distance, the partially folded protein binds up to 1.6 times the rate, if only the completely folded (Qp = 1) protein could bind.
Figure 5.

The fly-casting speedup ratio, ktot/kQ=1, is plotted vs. the unfolded protein concentration assuming a binding stability corresponding to experiment. For Kbind = Kexp, fly casting speeds up binding 1.6 times the rate of the fully formed (Qp = 1) protein through a range of reasonable protein concentrations. Parameters for the free energy functional are as in Fig. 2, and the contact radius is taken to be R0 = 3 Å.
The fly-casting mechanism leads to characteristic and potentially testable dependences of measured rates on stability and temperature. The Arrhenius plot for ktot shown in Fig. 6 indicates that binding is an activated process (if only mildly) at the experimental binding stability, Keqb = 5.2 × 10−14 M (47). This arises because partial unfolding caused by temperature increase helps the mechanism come into play. This behavior is found to persist throughout a wide range of values of the equilibrium constant. For comparison, the binding rate for the completely folded (Qp = 1) state is also plotted in Fig. 6. For this reference situation, binding is virtually unactivated. The temperature dependence given by the model assumes the binding energies themselves are temperature independent. Of course, this assumption is not valid because of the entropic contributions to the hydrophobic interactions. Thus, the laboratory temperature dependence may well be nonmonotonic, as in the phenomenon of cold denaturation. A real test of the mechanism thus needs careful measurements of the equilibrium interactions as a function of temperature.
Figure 6.

Arrhenius plots for the total binding flux ktot (solid) and completely folded binding flux (dashed). Notice energetic interaction terms are constant here. Parameters are same as in Fig. 5.
In the process of binding, as in folding a single monomer, explicitly cooperative (i.e., many-body) forces caused by hydrophobic residue burial and side-chain ordering play a role. The free energy functional model implicitly takes these into account for binding in a number of ways through coarse-graining contacts. We have also considered the effects of introducing an explicit local cooperativity between surface contacts, Fbcoop = −∑ijqijsqijs. This cooperativity diminishes the role of fly casting in both the folded and unfolded minima. Significantly increased local cooperativity effectively turns off the ability of the folded state to fly cast, while still allowing the unfolded state to maintain its expanded capture radius (although with a smaller free energetic difference to the Qp = 1 state).
Because folding and binding are coupled, one consequence of the fly-casting mechanism is that even residues away from the binding site in the folded complex can influence the reaction. Indeed, preliminary calculations for this model suggest that changing the stability of contacts outside the binding site gives binding rates that vary with stability. By using an approach similar to the φ-value analysis developed to probe the transition state ensemble in protein folding (49), it should be possible to test this prediction by measuring the binding rates of site specific mutants that alter the relative stability of the folded and unfolded states, but that do not change contacts.
Conclusion
The fly-casting mechanism has some parallels to well-known dimensionality reduction mechanisms that speed the search for specific targets. The possibility of such schemes was pointed out by Adam and Delbrück for sensory receptor binding and by Eigen and Richter for DNA recognition (50, 51), who noticed that for DNA recognition to be fast, it was important to have a weak longer-range nonspecific binding to the DNA before a target sequence was encountered. Here, by exploiting the available folding free energy, an increased capture radius arises for the final recognition event, even though the configuration space of the proteins is high dimensional. The high-dimensional search is facile because the configurational diffusion is guided by the folding funnel. Our calculation, based on a purely funnel-like surface, has not discussed the role of nonspecific binding, but fly casting should also help avoid metastable nonspecific bound complexes arising from the ruggedness of the DNA-protein interaction landscape.
Acknowledgments
We thank José Onuchic and Don Crothers for critically reading the manuscript. This work was supported by the National Institutes of Health (Grant 1R01 GM44557) and the Petroleum Research Fund administered by the American Chemical Society (Grant PRF 30353-AC6).
Appendix
The free energy functional given by Eq. 1 has several parts, many of which we have described before. We refer the reader to ref. 32 for details about the protein free energy, Fprot. Although complicated in appearance, Fprot consists of easily interpretable contributions specifying the stabilization energy of making specific contacts, the entropy cost of making those same contacts, and cooperative interactions between contacts. These cooperative interactions arise from two sources. One source is the intrinsic chain statistics that make it easier to form contacts once some have already been made. Another source of cooperativity comes from degrees of freedom (such as the exclusion of solvent or the ordering of side chains) not explicitly taken into account in the reduced description of the protein. This cooperativity is represented by explicit many-body forces in the contact free energy. The values for the protein contact probabilities, {qp} are found by minimizing Fprot with respect to {qp}, keeping the global contact probability Qp = ∑ij qijp fixed, as described in ref. 32. These minimization equations resemble “titration” equations for individual contacts. Varying the constraint Qp and substituting {qp} into Fprot gives a free energy profile parameterized by Qp.
The binding part of the functional consists of energetic and entropic parts Fbind = Ebind [{qs}] − TSbind [ri, Ω, {qs}, {qp}], where {qs} are contact probabilities between the protein and the surface to which it binds. We take the energetic part within a potential well to be described by the same energy function as postulated for those contacts involved in folding, whereas the binding energy is
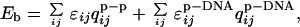 |
3 |
where qijp−p is the contact probability between protein monomers, and qijp−DNA is the contact probability between the protein and operator site. The protein monomer interactions, ɛij, are the same as those used for the intradomain contacts reported in ref. 32. In this work, {ɛijp−DNA} are taken to be uniform and scaled to match the experimental equilibrium constant reported for the binding of the arc repressor dimer to the dimer/operator complex A2O + A2 ⇌ A4O, Kexp = 5.2 × 10−14 M (47). The main new physics of the problem is contained in the entropic cost of forming a contact between a residue and its binding site partner. This cost depends on the location of the protein molecule as a whole but also on the partial ordering within the molecule.
The ensemble of configurations of the protein can be described by the distribution of the monomer positions, {ρi(xi, ri)}, about the fiducial native positions, {ri}. These densities depend on the center of mass position Rcm and orientation Ω through {ri}. For the folded and unfolded ensembles, the probability distributions are approximately Gaussian, ρiα(xi, ri) = (3/2πΔα2)3/2 exp[−3(xi − ri)2/2Δα2], where the mean square fluctuations, Δα2, are related to the Debye–Waller factor of the ith residue in the folded (α = f) and unfolded (α = u) states. This probability density is evaluated at the distance between the fiducial location of the residue and the location it will have in the bound state (see Fig. 1). The densities of monomer positions depend on the protein ordering through the Debye-Waller factors. Because the accessible excursions decrease as the protein orders, Δu2 > Δf2, where the subscripts u and f denote the completely unfolded (Qp = 0) and folded (Qp = 1) ensembles, respectively. Because even a single well-formed protein contact can greatly reduce the monomer fluctuations, the monomer density depends on the probability of not forming even a single contact φnc = ∏prot (1 − qijp). The protein structure-dependent density is then
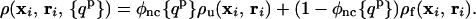 |
4 |
Taking the binding potential to be an attractive square well of strength ɛ near the binding site, the probability for the ith protein monomer to be within the well is
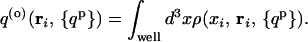 |
5 |
This probability depends on the local protein structure at the binding surface through the monomer density, accounting for the sensitive manner in which the local protein structure restricts motion as it forms. After forming only a small number of local contacts, the contribution of the unfolded distribution to the structure-dependent probability becomes negligible. In calculating q(o), we approximate the integral of each of the densities by neglecting orientation; for an isotropic spherical well of radius b, we have
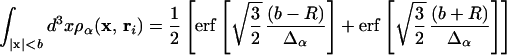 |
6 |
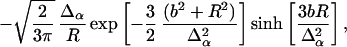 |
where R = |ri| is the distance from the binding site.
To construct the entropy of binding, we consider the number of ways of distributing the N surface contacts between the Nqs bound and N(1 − qs) unbound states. Denoting the degeneracies of a bound and unbound contact by gB and gU, respectively, we have
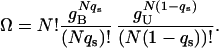 |
7 |
Because gB = Nq(o) (R) and gU = N(1 − q(o) (R)), the entropy of binding per residue is
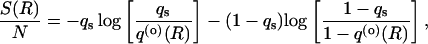 |
8 |
where R is the mean separation distance from the binding site. This is the form of the binding entropy for a particular contact; the total binding entropy Sbind is obtained by summing over the set of surface contacts {qijs}.
Minimizing the functional with respect to the individual surface contact probabilities, qijs, gives the expected Boltzmann distribution of surface contacts
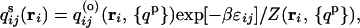 |
9 |
where Z(ri, {qp}) = exp[−βɛij]qij(o) (ri, {qp}) + (1 − qij(o) (ri, {qp})) is the restricted partition function for being bound or unbound. The mean field protein contact probabilities are obtained by minimizing the free energy with respect to {qp}, after eliminating {qs} through Eq. 9. The resulting contact probabilities are substituted into Eq. 1 to generate the reduced free energy profiles.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160259697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160259697
References
- 1.Wright P E, Dyson H J. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 2.Kriwacki R W, Hengst L, Tennant L, Reed S I, Wright P E. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ptashne M. A Genetic Switch: Phage λ and Higher Organisms. 2nd Ed. Cambridge, MA: Blackwell Scientific; 1992. [Google Scholar]
- 4.Frauenfelder H, Sligar S G, Wolynes P G. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 5.Frauenfelder H, Petsko G A, Tsernoglou D. Nature (London) 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 6.Ha J H, Spolar R S, Record M T., Jr J Mol Biol. 1989;209:801–816. doi: 10.1016/0022-2836(89)90608-6. [DOI] [PubMed] [Google Scholar]
- 7.Spolar R S, Record M T., Jr Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 8.Miller S. Protein Eng. 1989;3:77–83. doi: 10.1093/protein/3.2.77. [DOI] [PubMed] [Google Scholar]
- 9.Janin J, Chothia C. J Biol Chem. 1990;265:16027–16030. [PubMed] [Google Scholar]
- 10.Yankeelov J A, Jr, Koshland D E., Jr J Biol Chem. 1965;240:1593–1602. [PubMed] [Google Scholar]
- 11.Frankel A D, Kim P S. Cell. 1991;65:717–719. doi: 10.1016/0092-8674(91)90378-c. [DOI] [PubMed] [Google Scholar]
- 12.Alber T. Curr Biol. 1993;3:182–184. doi: 10.1016/0960-9822(93)90268-s. [DOI] [PubMed] [Google Scholar]
- 13.Holmbeck S M A, Dyson H J, Wright P E. J Mol Biol. 1998;284:533–539. doi: 10.1006/jmbi.1998.2207. [DOI] [PubMed] [Google Scholar]
- 14.Love J J. Ph.D. thesis. San Diego: Univ. of California; 1999. [Google Scholar]
- 15.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 16.Bryngelson J D, Onuchic N J, Socci N D, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 17.Abkevich V I, Gutin A M, Shakhnovich E I. Folding Des. 1996;1:221–230. doi: 10.1016/S1359-0278(96)00033-8. [DOI] [PubMed] [Google Scholar]
- 18.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 19.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onuchic J N, Wolynes P G, Luthey-Schulten Z A, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolynes P G, Onuchic J N, Thirumalai D. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 22.Chan H S, Dill K A. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 23.Gō N. Annu Rev Biophys Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 24.Honeycutt J D, Thirumalai D. Biopolymers. 1992;32:695–709. doi: 10.1002/bip.360320610. [DOI] [PubMed] [Google Scholar]
- 25.Socci N D, Onuchic J N. J Chem Phys. 1994;101:1519–1528. [Google Scholar]
- 26.Boczko E M, Brooks C L. Proc Natl Acad Sci USA. 1997;94:10161–10166. doi: 10.1073/pnas.94.19.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardin C, Luthey-Schulten Z, Wolynes P G. Proteins Struct Funct Genet. 1999;34:281–294. [PubMed] [Google Scholar]
- 28.Takada S, Luthey-Schulten Z, Wolynes P G. J Chem Phys. 1999;110:11616–11629. [Google Scholar]
- 29.Portman J J, Takada S, Wolynes P G. Phys Rev Lett. 1998;81:5237–5240. [Google Scholar]
- 30.Shoemaker B A, Wang J, Wolynes P G. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoemaker B A, Wolynes P G. J Mol Biol. 1999;287:657–674. doi: 10.1006/jmbi.1999.2612. [DOI] [PubMed] [Google Scholar]
- 32.Shoemaker B A, Wang J, Wolynes P G. J Mol Biol. 1999;287:675–694. doi: 10.1006/jmbi.1999.2613. [DOI] [PubMed] [Google Scholar]
- 33.Galzitskaya O V, Finkelstein A V. Proc Natl Acad Sci USA. 1999;96:11299–11304. doi: 10.1073/pnas.96.20.11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alm E, Baker D. Proc Natl Acad Sci USA. 1999;96:11305–11310. doi: 10.1073/pnas.96.20.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muñoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tskhovrebova L, Trinick J, Sleep J A, Simmons R M. Nature (London) 1997;387:308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- 37.Rief M, Gautel M, Oesterhelt F, Fernandez J M, Gaub H E. Science. 1997;276:1109–1111. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 38.Kellermayer M S Z, Smith S B, Granzier H L, Bustamante C. Science. 1997;276:1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 39.Weiss M A, Ellenberger T, Wobbe C R, Lee J P, Harrison S C, Struhl K. Nature (London) 1990;347:575–578. doi: 10.1038/347575a0. [DOI] [PubMed] [Google Scholar]
- 40.Rentzeperis D, Jonsson T, Sauer R T. Nat Struct Biol. 1999;6:569–573. doi: 10.1038/9353. [DOI] [PubMed] [Google Scholar]
- 41.Kohler J J, Metallo S J, Schneider T L, Schepartz A. Proc Natl Acad Sci USA. 1999;96:11735–11739. doi: 10.1073/pnas.96.21.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riggs A D, Bourgeois S, Cohn M. J Mol Biol. 1970;53:401–417. doi: 10.1016/0022-2836(70)90074-4. [DOI] [PubMed] [Google Scholar]
- 43.von Hippel P H, Revzin A, Gross C A, Wang A C. Interaction of lac Repressor with Non-Specific DNA Binding Sites. Berlin: Walter de Gruyter; 1975. pp. 270–288. [Google Scholar]
- 44.von Hippel P H, Berg O G. J Biol Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 45.Milla M E, Sauer R T. Biochemistry. 1994;33:1125–1133. doi: 10.1021/bi00171a011. [DOI] [PubMed] [Google Scholar]
- 46.Brown B M, Sauer R T. Proc Natl Acad Sci USA. 1999;96:1983–1988. doi: 10.1073/pnas.96.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown B M, Sauer R T. Biochemistry. 1993;32:1354–1363. doi: 10.1021/bi00056a022. [DOI] [PubMed] [Google Scholar]
- 48.Radhakrishnan I, Pérez-Alvarado G C, Parker D, Dyson H J, Montminy M R, Wright P E. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 49.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 50.Adam G, Delbrück M. In: Structural Chemistry and Molecular Biology. Rich A, Davidson N, editors. New York: Freeman; 1968. pp. 198–215. [Google Scholar]
- 51.Richter P H, Eigen M. Biophys Chem. 1974;2:255–263. doi: 10.1016/0301-4622(74)80050-5. [DOI] [PubMed] [Google Scholar]