

Abstract
Eph kinases are receptor tyrosine kinases whose ligands, the ephrins, are also expressed on the surface of cells. Interactions between Eph kinases and ephrins on adjacent cells play a central role in neuronal patterning and vasculogenesis. Here we examine the expression of ephrins and Eph kinases on human blood platelets and explore their role in the formation of the hemostatic plug. The results show that human platelets express EphA4 and EphB1, and the ligand, ephrinB1. Forced clustering of EphA4 or ephrinB1 led to cytoskeletal reorganization, adhesion to fibrinogen, and α-granule secretion. Clustering of ephrinB1 also caused activation of the Ras family member, Rap1B. In platelets that had been activated by ADP and allowed to aggregate, EphA4 formed complexes with two tyrosine kinases, Fyn and Lyn, and the cell adhesion molecule, L1. Blockade of Eph/ephrin interactions prevented the formation of these complexes and caused platelet aggregation at low ADP concentrations to become more readily reversible. We propose that when sustained contacts between platelets have occurred in response to agonists such as collagen, ADP, and thrombin, the binding of ephrins to Eph kinases on adjacent platelets provides a mechanism to perpetuate signaling and promote stable platelet aggregation.
Platelet activation at sites of vascular injury begins with the arrest of circulating platelets on collagen and von Willebrand factor. It continues as additional platelets are recruited into the growing hemostatic plug in response to thrombin, thromboxane A2, and ADP. Contacts between platelets are mediated to a large extent by fibrinogen and von Willebrand factor bound to the integrin αIIbβ3. As a result, many of the early events of platelet activation are directed toward placing αIIbβ3 in the activated conformation. Once this placement has occurred, platelets develop increasingly stable contacts with each other that are able to withstand the shear forces produced by arterial blood flow. Most of the events that recruit platelets into a growing plug and place their integrins in the activated state are initiated by soluble agonists that bind to G protein-coupled receptors on the platelet surface. Signaling initiated in this manner can be rapid and potent, but it is relatively limited in duration, because the receptors become desensitized and activated G proteins return to the GDP-bound resting state. Therefore, it is reasonable to propose that additional signaling mechanisms exist to perpetuate platelet activation once aggregation has begun and platelets have come into more than transient contact with each other. Outside–in signaling through integrins provides one such mechanism (1). Activation of Axl family tyrosine kinases by secreted Gas6 may provide another (2).
In the present studies, we have considered whether Eph kinases and ephrins might provide an additional means of signaling in the late phases of platelet aggregation. Eph kinases are a large family of receptor tyrosine kinases with an extracellular ligand-binding domain and an intracellular kinase domain. In contrast to most receptors, the ligands for Eph kinases (known as ephrins) are themselves held to the cell surface by either a glycosylphosphatidylinositol anchor (the ephrin A family) or a transmembrane domain (the ephrin B family), limiting signaling to contexts in which sustained cell-to-cell contacts can occur. Eph kinases and ephrins play a critical role in neuronal patterning and axonal guidance during development (3, 4), and they modulate adhesive interactions (5–8). Interactions between Eph kinases and ephrins occur in trans with ligands on one cell binding to receptors on another. The binding of ephrins to Eph kinases causes the clustering of both the ligands and the receptors, which triggers signaling and responses in both the receptor-expressing cells and the ligand-expressing cells (9–15). These events can depend on the phosphorylation of the Eph and ephrin, but phosphorylation-independent interactions have been described as well (10–12, 16–18).
Although the role of Eph kinases and ephrins has been studied extensively in the developing central nervous system and vasculature, little is known about their expression on cells that are not normally adherent, such as blood cells. In the present studies we show that human platelets express two Eph kinases, EphA4 and EphB1, and at least one ligand, ephrinB1. We also show that these molecules are exposed on the platelet surface and that forced clustering of either the Eph kinases or the ligand causes cytoskeletal reorganization, adhesion, α-granule secretion, and activation of Rap1B, a Ras family member that has been implicated in integrin activation (19). Furthermore, in platelets that have been activated by ADP and allowed to aggregate, EphA4 forms signaling complexes with two nonreceptor tyrosine kinases, Fyn and Lyn, and the cell adhesion molecule, L1, whereas blockade of Eph/ephrin interactions during platelet aggregation prevents the formation of these complexes and inhibits platelet aggregation at low ADP concentrations. Based on these observations, we now propose that once platelet activation begins at sites of vascular injury, an increase in contact time between platelets allows Eph/ephrin clustering and signaling to occur, thereby providing a mechanism to perpetuate platelet aggregation and help stabilize the hemostatic plug.
Materials and Methods
Reagents.
Antibodies used to detect EphB1 on immunoblots and to precipitate EphA4 were produced in chickens immunized with peptides corresponding to residues C522–R536 of human EphB1 and residues M976–V986 of human EphA4. Additional antibodies to EphA4, ephrinB1, ephrinA3, Src, Fyn, and Lyn were purchased from Santa Cruz Biotechnology. The monoclonal antibody directed against L1 was purchased from Transduction Laboratories (Lexington, KY). FITC-conjugated P-selectin antibody was from BioSource International (Camarillo, CA). Alexa-Fluor 594-conjugated fibrinogen was from Molecular Probes. Glutathione S-transferase (GST)-conjugated RalGDS-RBD was prepared from a cDNA provided by J. Meinkoth (University of Pennsylvania) and used to detect Rap-1 activation as described (20).
Aggregation, Secretion, and Fibrinogen Binding.
Blood was obtained from healthy volunteer donors and anticoagulated with ACD (65 mM trisodium citrate/70 mM citric acid/100 mM dextrose, pH 4.4) at a final ratio of one part to five parts blood. Platelets were resuspended in modified Tyrode's buffer (137 mM NaCl/20 mM Hepes/5.6 mM glucose/1 g/liter BSA/1 mM MgCl2/2.7 mM KCl/3.3 mM NaH2PO4, pH 7.4) at 1 × 109 platelets per milliliter. Gel-filtered platelets were isolated by passage through Sepharose 2B (Amersham Pharmacia). In aggregation studies, the platelets were preincubated for 5 min at 37°C with 300 μg/ml of fibrinogen and 1 mM CaCl2 before the addition of an agonist. To measure fibrinogen binding and α-granule secretion, 300 μl of gel-filtered platelets was incubated with 100 μg/ml of Alexa-Fluor 594-labeled fibrinogen and 10 μl of FITC-labeled anti P-selectin for 30 min in the dark at room temperature in presence of the appropriate agonist. Platelets were then fixed for 10 min at room temperature by adding an equal volume of 2% formaldehyde–Tyrode's buffer (+1 mM CaCl2), sedimented at 1,310 × g for 20 sec, resuspended in 0.5 ml of PBS and analyzed by flow cytometry.
Fluorescence Microscopy.
Actin staining of adherent platelets was performed as described (21). For staining with FITC-His-EphA4/B1 or His-ephrinA4/B2, platelets were incubated with 5 μg/ml of the protein for 20 min. Nonadherent platelets were removed by gentle washing. Attached platelets were fixed in 3.7% formaldehyde/PBS for 10 min followed by a blocking step for 30 min in 10% goat serum. Slides were then mounted in Citifluor.
Platelet Adhesion Essay.
Ninety-six-well plates were coated with fibrinogen (2 mg/ml) or BSA (4 mg/ml) and then blocked with BSA (5 mg/ml). Fifty microliters of platelets (2 × 107 platelets per ml) were added to each well. After 5 min at 37°C, agonists were added in 25 μl of Tyrode's buffer with 3 mM CaCl2. After incubation, the plates were washed four times in PBS. Adherent platelets were lysed with Triton X-100, and p-nitrophenyl phosphate was added as a substrate for released acid phosphatase (150 μl of 5 mM p-nitrophenyl phosphate in 0.1 M citrate/0.1% Triton X-100). The reaction was stopped after 1 hr by adding 100 μl of 2 N NaOH, and OD405 was measured.
Immunoblotting.
Platelets were lysed in RIPA buffer (1% Triton X-100/1% deoxycholate/0.1% SDS/158 mM NaCl/10 mM Tris, pH 7.2/5 mM NaEDTA) in the presence of protease inhibitors. The lysate was boiled in sample buffer (2% SDS/1% β-mercaptoethanol/0.008% bromophenol blue/80 mM Tris, pH 6.8/1 mM EDTA) before SDS/PAGE analysis. After transfer, the membranes were blocked in 5% milk or 5% BSA.
Immunoprecipitation.
Chicken anti-EphA4 was conjugated to CNBr-activated Sepharose 4B (Amersham Pharmacia). For each sample 5 × 108 washed platelets were lysed (1% Triton X-100/10 mM Tris base/50 mM NaCl/30 mM Na pyrophosphate/50 mM NaF, pH 7.3) in the presence of protease inhibitors and 1 mM NaVO4. The insoluble fraction was removed by centrifugation at 16,000 × g for 15 min at 4°C and the supernatant incubated overnight at 4°C with chicken anti-EphA4 conjugated to Sepharose 4 beads. After three washes in PBS, the beads were boiled in sample buffer for 10 min and released proteins analyzed by SDS/PAGE and Western blotting. Membranes that were to be reprobed were stripped at 63°C for 10 min in 62.5 mM Tris⋅HCl, pH 6.8, with 2% SDS and 100 mM β-mercaptoethanol.
Reverse Transcription–PCR.
Total RNA was isolated by using the Promega RNA extraction kit from platelets that had been passed through a leukocyte removal filter (Pall). The RNA was then pretreated with 0.2 unit of RQ1 DNase (Promega) for 30 min. After inactivation of the DNase for 10 min at 90°C, the RNA was incubated with to 100 units of reverse transcriptase (GIBCO/BRL) with 20 pmol of random primer, 200 nM DTT, 4 nM dNTP, 20 units of RNase inhibitor (Promega) for 30 min at 37°C. The reverse transcriptase was inactivated for 10 min at 90°C. PCR was performed with 0.1 unit of TAQ polymerase (Promega), 4 nM dNTP, and 20 pmol of each primer. Specific primer pairs were designed for each Eph kinase and ephrin, and all amplification products were sequenced to confirm their identities.
GST Fusion Proteins.
All GST-fusion proteins were made by subcloning part of each receptor ectodomain in the pGEX1λT vector (Amersham Pharmacia) (for primer sequences, see Table 1, which is published as supporting information on the PNAS web site, www.pnas.org). GST-EphB1 includes EphB1 residues V15 to G244. The truncated variant, GST-ΔEphB1, lacks residues K157 to S215. GST-EphrinA4 includes ephrinA4 residues L11 to I200. The resulting constructs were transformed into the E. colistrain BL21 DE3. Recombinant GST fusion proteins were purified using the Pharmacia GST purification kit.
His-6 Fusion Proteins.
His-6 fusion proteins were generated by subcloning fragments of each receptor's ectodomain into the pET29(+)b vector (Novagen) (see Table 1 for primer sequences). His-6-EphA4 includes residues T21 to V551. His-6-ΔEpHA4 lacks residues D130 to F142. His-6-EphB1 includes residues to D529. His-6-ΔEphB1 lacks D118 to I134. His-6-EphrinA4 includes residues L20 to L188. His-6-ΔephrinA4 lacks residues L20 to Y61. His-6-EphrinB2 includes residues R4 to E224. His-6-ΔephrinB2 lacks residues R4 to V64. The resulting constructs were transformed into the E. coli strain BL21 DE3. Recombinant His6 fusion proteins were purified by using the Qiagen QIA expressionist purification kit.
FITC Labeling of the His-6 Fusion Proteins.
One milliliter of each protein (2 mg/ml in PBS) was incubated with 1 mg FITC type I (Molecular Probes) for 1 h at room temperature. The reaction was stopped by adding 0.1 ml of freshly prepared 1.5 M hydroxylamine, pH 8.5. The conjugate was separated from residual labeling reagent by column chromatography with Sephadex G-25.
Results
RNA was isolated from human platelets that had been filtered to minimize leukocyte contamination, reverse transcribed, and then amplified by using primers specific for most of the known Eph kinases and ephrins. Products of the expected size were seen for two of the kinases (EphA4 and EphB1) and two of the ligands (ephrinA3 and ephrinB1) (Fig. 1A). Western analysis further confirmed the expression of EphA4, EphB1, and ephrinB1 (Fig. 1B), but ephrinA3 could not be detected in platelet lysates with any of the available antibodies.
Figure 1.

Evidence for Eph kinases and ephrins on the surface of human platelets. (A) Reverse transcription and amplification (RT-PCR) of platelet RNA using primers specific for EphB1, EphA4, ephrinA3, and ephrinB1. Platelet factor 4 (PF4) is specific for platelets, whereas CD18 was used as a marker for leukocyte contamination. Sequencing of the amplified products confirmed their identities. (B) Immunoblots of total platelet lysates with antibodies that recognize EphA4, EphB1, and ephrinB1. (C) Platelets were incubated with 100 nM phorbol 12-myristate 13-acetate, allowed to adhere and spread on a fibrinogen-coated surface, and then stained with FITC-conjugated, His-6-tagged recombinant proteins corresponding to the exodomains of EphA4, EphB1, and ephrinA4. Control proteins lacked residues required for Eph/ephrin interactions are denoted with a “Δ.”
The presence of the Eph kinases and ephrins on the platelet surface was demonstrated by fluorescence microscopy (Fig. 1C) and flow cytometry (data not shown) by using FITC-conjugated, His-tagged recombinant proteins that corresponded to the exodomains of EphA4, EphB1 or ephrinA4. In general, A ephrins bind to A kinases and B ephrins bind to B kinases. An exception is EphA4, which can bind to ephrin B forms as well as ephrin A forms (22). Therefore, between them, the FITC-conjugated EphA4, EphB1, and ephrinA4 exodomains would be expected to recognize at least ephrinB1 and EphA4. Fluorescence microscopy showed each of these recombinant proteins bound to the surface of adherent platelets with a homogeneous distribution, whereas control proteins (denoted in the figure with a “Δ”) lacking residues needed for Eph/ephrin interactions did not. Similar experiments were performed with FITC-conjugated, GST-tagged exodomains. However, in this case the distribution of the FITC-conjugated proteins on the platelet surface was heterogeneous, reflecting the clustering that would be expected to occur when GST-tagged proteins spontaneously oligomerize (N.P., T.T., M. Tognolini, W. Jian, R. Fortua, and L.F.B., manuscript in preparation).
To determine whether forced clustering of EphA4 and ephrinB1 can evoke physiologic responses, we incubated platelets with self-oligomerizing GST-fusion proteins containing the exodomains of EphB1 or ephrinA4. Oligomerization of the fusion proteins was confirmed by nondenaturing gel electrophoresis. Addition of GST-EphB1 (a binding partner for ephrinB1) caused human platelets to adhere and spread on immobilized fibrinogen. The appearance of the adherent platelets, including reorganization of the actin cytoskeleton visualized with rhodamine-phalloidin to illuminate actin, was identical with that seen after the addition of ADP (Fig. 2A). A similar result was obtained with GST-EphrinA4 (data not shown), but no adhesion was seen after the addition of (i) unconjugated GST, (ii) GST-ΔEphB1, or (iii) monomeric His-tagged EphB1, which can bind but does not cause clustering (Fig. 2A). As noted by previous investigators (23), we found that human platelets will eventually spontaneously adhere and spread on immobilized fibrinogen in the absence of agonist, but during the 20- to 30-min time frame used in the present studies, background adhesion was minimal. GST-ephrinA4 caused an 2.5 ± 0.3-fold increase in adhesion over background, compared with the 3.5 ± 0.6-fold increase caused by ADP. Adhesion was abolished by Arg–Gly–Asp–Ser or an anti-αIIbβ3 antibody that blocks fibrinogen binding (24), confirming the integrin dependence. Notably, neither GST-EphB1 nor GST-ephrinA4 caused platelet aggregation or the binding of soluble Alexa-Fluor 594-labeled fibrinogen to platelets, suggesting that they are unable to activate αIIbβ3 to its high-affinity state (data not shown).
Figure 2.

Clustering of Eph kinases and ephrins activates platelets. (A) Platelets were incubated on a fibrinogen-coated surface for 20 min either without an agonist or with ADP (20 μM), GST-EphB1 (2 μg/ml), or GST-ΔEphB1 (2 μg/ml), and then stained with rhodamine-phalloidin to visualize actin. (B) Secretion of platelet α-granules was detected by flow cytometry by using a FITC-labeled antibody that recognizes P-selectin. The results shown compare resting with stimulated platelets and are representative of five such experiments.
GST-EphB1 and GST-ephrinA4 also caused the secretion of platelet α-granules, which was detected by measuring the appearance of the α-granule membrane protein, P-selectin, on the platelet surface. Both GST-EphB1 and GST-ephrinA4 caused P-selectin expression, although neither was as robust as ADP. GST-EphB1 lacking the ephrin binding domain had little effect (Fig. 2B). Although dense granule secretion was not specifically measured, it is unlikely that secreted ADP is responsible for the adhesion that occurs in response to GST-EphB1 because addition of apyrase to metabolize secreted ADP had little if any effect under conditions in which the response to ADP was abolished (N.P. and L.F.B., unpublished observation).
Finally, GST-EphB1 also caused activation of Rap1B in platelets. Rap1B is a member of the Ras superfamily that is prominently expressed in platelets (25–27) and has been implicated in integrin regulation (19, 28). Although activation of Rap1B in platelets is usually ascribed to Gq-mediated Ca2+-dependent signaling (25, 27), Ca2+-independent mechanisms exist as well (29). In Fig. 3, platelets were incubated with ADP, GST-EphB1, or GST-ΔEphB1. Rap1-GTP was readily detectable in platelets activated with ADP or GST-EphB1, but not in untreated platelets or in platelets incubated with GST-ΔEphB1. This activation occurred despite the fact that GST-EphB1 does not cause an increase in cytosolic Ca2+ in platelets under these conditions (data not shown).
Figure 3.
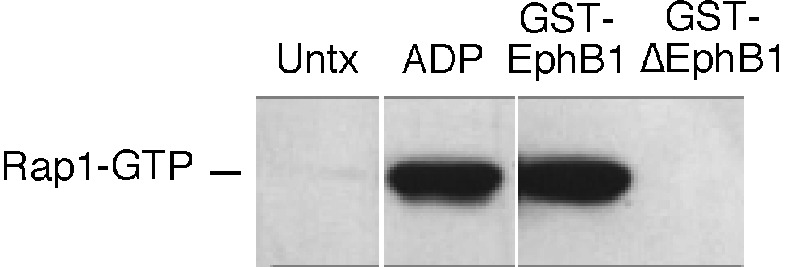
Activation of Rap1B. Platelets were incubated for 5 min with ADP (20 μM), GST-EphB1 (10 μg/ml), or GST-ΔEphB1(10 μg/ml), after which Rap1B in the GTP-bound state was precipitated with GST-RBD and detected with an anti-Rap1 antibody. The results shown are representative of three such experiments.
Association of Src Family Members and Cell Adhesion Molecules with EphA4 During Platelet Aggregation.
Because interactions between ephrins and Eph kinases on the surface of platelets would not normally be expected to occur until after platelet aggregation had begun, we next asked whether platelet aggregation initiated by ADP would result in the formation of signaling complexes involving Eph kinases and, if so, whether blockade of Eph/ephrin interactions would prevent the formation of those complexes. Known partners for Eph kinases and ephrins in other cells include Src family kinases and cell adhesion molecules (30–32). In Fig. 4, EphA4 was immunoprecipitated from resting and ADP-stimulated platelets that had been allowed to aggregate. Western blots showed two Src family members, Lyn and Fyn, had coprecipitated with EphA4, although Src itself did not. This association between EphA4, Lyn, and Fyn was not seen in resting platelets, and blockade of Eph/ephrin interactions with monomeric EphA4 and EphB1 exodomains abolished the association with Fyn and greatly reduced its association with Lyn.
Figure 4.
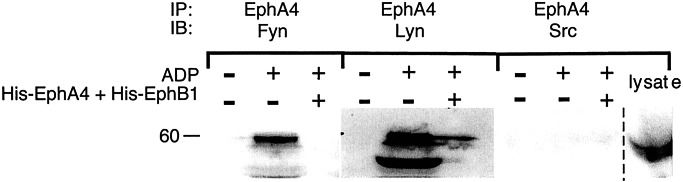
Platelet aggregation results in the formation of Fyn and Lyn complexes with EphA4. Platelets were incubated with ADP (20 μM) under conditions that allowed aggregation to occur. After 5 min, the cells were lysed and EphA4 was immunoprecipitated. The precipitates were then subjected to electrophoresis and probed with antibodies to Fyn, Lyn, or Src. Where indicated, Eph/ephrin interactions were blocked with a combination of His-EphA4 plus His-EphB1 at a final concentration of 10 μg/ml of each, a concentration found to be sufficient to block ephrin/Eph kinase interactions (58, 59). The lane farthest to the right shows total platelet lysates probed with a Src-specific antibody to document that Src is present. IP, the immunoprecipitating antibody; IB, the antibody used in the Western immunoblot.
Also present in complexes with EphA4 was the cell adhesion molecule, L1 (or Ng-CAM). L1 is of particular interest because of recent reports showing that it can bind to the αIIbβ3 and αvβ3 integrins, both of which are expressed on human platelets (33, 34). L1 is known to be expressed by neurons and endothelial cells (35, 36), but has not been shown to be expressed by human platelets. In total platelet lysates L1, which is highly glycosylated, was detectable as a single band (Fig. 5A). An antibody directed toward L1 detected it on the platelet surface (Fig. 5B). Like Fyn and Lyn, L1 coprecipitated with EphA4 in lysates from ADP-stimulated platelets, but not from resting platelets. Addition of monomeric His-EphA4 plus His-EphB1 prevented that association (Fig. 5C).
Figure 5.

L1/Ng-CAM is expressed in human platelets and forms complexes with EphA4 during platelet aggregation. (A) Western blot of total cell lysates from human brain, endothelial cells (HUVEC) and platelets showing the presence of L1/Ng-CAM. (B) Fluorescence-activated cell sorter analysis of human platelets with an antibody directed against the L1 exodomain. (C) Precipitation of L1 with EphA4 by using an anti-EphA4 antibody. The lysates were prepared from platelets incubated with 20 μM ADP under aggregating conditions in the presence or absence of His-EphA4 plus His-EphB1 (10 μg/ml of each). IP, the immunoprecipitating antibody; IB, the antibody used in the Western immunoblot.
Blockade of Eph/Ephrin Interactions Causes Platelets to Disaggregate.
To see if Eph/ephrin interactions help to promote the growth of platelet aggregates, we incubated platelets with ADP in the presence of either His-EphA4 plus His-EphB1 or His-ephrinA4 plus His-ephrinB2 with each combination added at a concentration separately established to be sufficient to block the binding of labeled Eph and ephrin exodomains. Typical results are shown in Fig. 6. At low concentrations of ADP, platelet aggregation was inhibited by the intact Eph and ephrin exodomains, but not by the binding domain-truncated “Δ” exodomains. In each case, platelet shape change (detected as an initial downward deflection in the light transmission tracing) occurred normally and aggregation began without a noticeable delay. However, full aggregation was not achieved before the platelets disaggregated (detected as a drop in the light transmission tracing toward baseline). Inhibition was most evident at low agonist concentrations. Increasing the ADP concentration to 20 μM overcame the effects of the His-tagged exodomains, restoring aggregation to control levels (data not shown) suggesting that Eph/ephrin interactions support or perpetuate platelet aggregation, particularly at submaximal agonist concentrations. Increasing the concentration of the interfering exodomains had no additional effect.
Figure 6.

Blockade of Eph/ephrin interactions inhibits platelet aggregation. Washed platelets supplemented with Ca2+ and fibrinogen and stirred at 37°C were activated with ADP in the presence or absence of the His-tagged fusion proteins that are indicated in the figure (10 μg/ml of each). The results shown are representative of seven such experiments.
Blockade of Eph/Ephrin Interactions Does Not Inhibit Fibrinogen Binding to Platelets.
Finally, to see whether the inhibition of aggregation at low ADP concentrations was caused by a failure to expose (or reversal of) high-affinity fibrinogen binding sites, we measured labeled fibrinogen binding to platelets in the presence and absence of His-EphA4 plus His-EphB1. A greater amount of fibrinogen bound to the platelets at 20 μM ADP than at 2 μM ADP, but in neither case was inhibition by the Eph exodomains detected (Fig. 7). Therefore, the disaggregation seen in Fig. 6 does not seem to be due to the down-regulation of high-affinity binding sites for fibrinogen on αIIbβ3.
Figure 7.

Blockade of Eph/ephrin interactions does not inhibit fibrinogen binding to αIIbβ3. The binding of labeled fibrinogen to platelets activated with ADP was measured by flow cytometry in the presence or absence of His-EphA4 plus His-EphB1 (10 μg/ml of each). The results shown compare resting with stimulated platelets and are representative of five such experiments.
Discussion
Formation of the hemostatic plug at sites of vascular injury can be thought of as occurring in several overlapping steps: an initiation phase in which platelets adhere to exposed collagen and von Willebrand factor or are activated by locally generated thrombin; a recruitment phase in which thrombin and agonists released from activated platelets help to incorporate additional platelets into the growing platelet plug; and a perpetuation phase in which the platelet plug is stabilized. Many of the early events in this process are initiated by agonists that bind to G protein-coupled receptors. In the present studies, we have considered whether the comparatively slow signals generated by Eph kinases and ephrins help to support and perpetuate platelet plug formation at a point when sustained contacts between platelets would allow ephrins expressed on the surface of one platelet to engage their cognate receptors on an adjacent platelet. If present, Eph kinases and ephrins would be well suited for this role because of their ability to generate signals in both the kinase and the ligand-expressing cells (37).
The results show that human platelets express at least one ephrin (B1) and two Eph kinases (A4 and B1). Although, in general, A kinases bind to A ephrins and B kinases bind to B ephrins, EphA4 can bind to both (22). Thus, platelets express at least one ligand for each of the Eph kinases. By fluorescence microscopy these molecules exist homogeneously on the platelet surface with their binding domains exposed. Forced clustering of either the kinases or the ephrin with soluble multimeric forms of these molecules causes reorganization of the platelet cytoskeleton, adhesion to fibrinogen, activation of Rap1B, and modest α-granule secretion. On the other hand, clustering did not cause an increase in cytosolic Ca2+, nor did it by itself cause exposure of high-affinity fibrinogen binding sites on αIIbβ3. These responses were not observed after the addition of fusion proteins lacking residues necessary for Eph/ephrin interactions or after the addition of nonclustering exodomain monomers. Therefore, binding is necessary, but not sufficient. Clustering must occur as well. Platelet activation by ADP caused the formation of signaling complexes that included EphA4, Fyn, Lyn, and the cell adhesion molecule L1, and quite possibly other molecules as well. These complexes were not present in resting platelets, and their formation in ADP-stimulated platelets was suppressed by blocking Eph/ephrin interactions with soluble exodomain monomers. The monomers also caused the platelet aggregates formed in response to ADP to degrade without affecting the exposure of fibrinogen-binding sites on αIIbβ3. The inhibition was most apparent at low ADP concentrations, where addition of saturating concentrations of the monomers resulted in reversible aggregation.
By becoming engaged after platelets come into contact with each other, Eph kinases and ephrins potentially provide a mechanism for sustained intracellular signaling within the platelet plug (illustrated in Fig. 8). As noted in the introduction, other examples of signaling mechanisms that may be operable during the late phases of platelet activation include outside-in signaling through αIIbβ3 (1) and activation of the Mer, Sky, and/or Axl kinases by secreted Gas6 (2). Each of these examples involves tyrosine kinases rather than the G protein-coupled receptors. Like Eph/ephrin signaling, outside-in signaling through αIIbβ3 would be expected to begin only after platelet aggregation has occurred. Signaling by Gas6/Axl would occur only after secretion of Gas6. It was recently shown that platelets from mice in which the cytoplasmic tyrosine residues of the β3 integrin subunit were mutated to phenylalanine tend to disaggregate, leaving the mice with a pronounced tendency to rebleed (38). Similarly, deletion of the gene encoding Gas-6 in mice produces a phenotype in which mouse platelets begin to aggregate, but the aggregates fall apart, resulting in aggregation tracings much like those observed here when Eph/ephrin interactions are blocked (2).
Figure 8.

A model for Eph/ephrin signaling in platelets. On the basis of the results of the present studies, it is proposed that when contacts between platelets become more than transient, the binding of ephrins on the surface of one platelet to cognate Eph kinases on adjacent platelets causes receptor clustering and promotes actin reorganization and α-granule secretion. Clustering of EphA4 in particular recruits Lyn, Fyn, and the cell adhesion molecule L1 (directly or through a potential intermediary protein X) into signaling complexes. In turn, that might affect the binding of L1, either with itself in a homophilic manner or with the β3 integrins, αIIbβ3, or αvβ3, although that has not yet been established.
The biology of Eph kinases and ephrins has been explored to the greatest extent in the developing nervous system where loss of the genes encoding selected Eph kinases and ephrins in mice produces phenotypes in which neural development is disrupted (39–44). In platelets, Eph/ephrin signaling by itself seems to be sufficient to reorganize the actin cytoskeleton and to cause the exocytosis of α-granules, but it does not cause an increase in cytosolic Ca2+, perhaps explaining its inability to promote αIIbβ3 activation. It also affects the adhesive state of the αIIbβ3 integrin without having the ability to shift the integrin into the high-affinity state needed to bind soluble fibrinogen. An increase in avidity might be insufficient to trigger platelet aggregation, but could promote the stability of the platelet plug. Notably, one consequence of ephrinB1 clustering in platelets is the activation of Rap1B, which has recently been implicated in pathways leading to integrin activation in lymphocytes and platelets (19, 28). The fact that ephrin B1 can activate Rap1B without causing the binding of soluble fibrinogen to the platelet surface would suggest that Rap1B activation alone is not sufficient to place αIIbβ3 into its high-affinity state. One of the molecules that becomes associated with EphA4 in ADP-activated platelets is L1/Ng-CAM, a cell surface molecule that binds to αIIbβ3 (33) as well as to itself. If the association of L1 with EphA4 increases the ability of L1 to bind to proteins on nearby cells, then this association provides a second mechanism by which Eph/ephrin interactions can promote the stability of the platelet plug (Fig. 8).
How might clustering of Eph kinases and ephrins produce platelet responses? Many studies have shown that Eph kinases and ephrins can associate with other signaling molecules, potentially allowing them to form dense signaling complexes. Some of these interactions are thought to lead to an association with adaptor proteins (Grb2, Grb4, Grb10, and Crk), second messenger-generating molecules (phospholipase Cγ and phosphatidylinositol 3-kinase), tyrosine kinases in the Src family, and protein tyrosine phosphatases (15, 30, 31, 45–48). Proteins that can bind to Eph kinases and ephrins regulate Ras family members (e.g., RasGAP, SHEP1) (9, 13, 49) and limit signaling through heterotrimeric G proteins by accelerating GTP hydrolysis (e.g., PDZ-RGS3) (50). Although not examined here, in cells other than platelets, clustering of Eph kinases and ephrins has been shown to affect the activation state of Rho and Rac, both of which have a role in the regulation of actin polymerization (51, 52), and the guanine nucleotide exchange factor, Ephexin, associates with EphA4 (52). Which of these might prove to be relevant in platelets remains to be fully explored.
In conclusion, the results obtained in this study suggest that once sustained contacts between platelets have occurred in responses to agonists such as ADP, interactions between Eph kinases and ephrins can provide a signaling mechanism that could help to promote the growth and stability of the growing hemostatic plug. Finally, Eph kinases and ephrins are expressed on endothelial cells in both the developing and the mature vasculature (53–57). Because our preliminary observations suggest that Eph kinases and ephrins are also expressed on leukocytes, signaling through these molecules may also play a larger role in heterotypic interactions involving platelets, endothelial cells, and inflammatory cells that remains to be explored.
Supplementary Material
Acknowledgments
We thank Peter O'Brien, Jing Yang, and Florence Martin for helpful discussions. These studies were supported by National Institutes of Health Grant HL-40387 and a postdoctoral fellowship from the American Heart Association (to D.W.). Peptide synthesis was supported by core grants of the Diabetes and Cancer Centers (DK-19525 and CA-16520).
Abbreviations
- GST
glutathione S-transferase
- CAM
cell adhesion molecule
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Shattil S J. Thromb Haemostasis. 1999;82:318–325. [PubMed] [Google Scholar]
- 2.Angelillo-Scherrer A, De Frutos P G, Aparicio C, Melis E, Savi P, Lupu F, Arnout J, Dewerchin M, Hoylaerts M F, Herbert M, et al. Nat Med. 2001;7:215–221. doi: 10.1038/84667. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson D G. Int Rev Cytol. 2000;196:177–244. doi: 10.1016/s0074-7696(00)96005-4. [DOI] [PubMed] [Google Scholar]
- 4.Dodelet V C, Pasquale E B. Oncogene. 2000;19:5614–5619. doi: 10.1038/sj.onc.1203856. [DOI] [PubMed] [Google Scholar]
- 5.Huynh-Do U, Stein E, Lane A A, Liu H, Cerretti D P, Daniel T O. EMBO J. 1999;18:2165–2173. doi: 10.1093/emboj/18.8.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou J X, Wang B, Kalo M S, Zisch A H, Pasquale E B, Ruoslahti E. Proc Natl Acad Sci USA. 1999;96:13813–13818. doi: 10.1073/pnas.96.24.13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker E, Huynh-Do U, Holland S, Dawson T, Daniel T O, Skolnik E Y. Mol Cell Biol. 2000;20:1537–1545. doi: 10.1128/mcb.20.5.1537-1545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huai J, Drescher U. J Biol Chem. 2001;276:6689–6694. doi: 10.1074/jbc.M008127200. [DOI] [PubMed] [Google Scholar]
- 9.Holland S J, Gale N W, Gish G D, Roth R A, Zhou S Y, Cantley L C, Henkemeyer M, Yancopoulos G D, Pawson T. EMBO J. 1997;16:3877–3888. doi: 10.1093/emboj/16.13.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hock B, Bohme B, Karn T, Yamamoto T, Kaibuchi K, Holtrich U, Holland S, Pawson T, Rubsamen-Waigmann H, Strebhardt K. Proc Natl Acad Sci USA. 1998;95:9779–9784. doi: 10.1073/pnas.95.17.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres R, Firestein B L, Dong H L, Staudinger J, Olson E N, Huganir R L, Bredt D S, Gale N W, Yancopoulos G D. Neuron. 1998;21:1453–1463. doi: 10.1016/s0896-6273(00)80663-7. [DOI] [PubMed] [Google Scholar]
- 12.Lin D, Gish G D, Songyang Z, Pawson T. J Biol Chem. 1999;274:3726–3733. doi: 10.1074/jbc.274.6.3726. [DOI] [PubMed] [Google Scholar]
- 13.Dodelet V C, Pazzagli C, Zisch A H, Hauser C A, Pasquale E B. J Biol Chem. 1999;274:31941–31943. doi: 10.1074/jbc.274.45.31941. [DOI] [PubMed] [Google Scholar]
- 14.Pandey A, Duan H, Dixit V M. J Biol Chem. 1995;270:19201–19204. doi: 10.1074/jbc.270.33.19201. [DOI] [PubMed] [Google Scholar]
- 15.Cowan C A, Henkemeyer M. Nature (London) 2001;413:174–179. doi: 10.1038/35093123. [DOI] [PubMed] [Google Scholar]
- 16.Birgbauer E, Cowan C A, Sretavan D W, Henkemeyer M. Development (Cambridge, UK) 2000;127:1231–1241. doi: 10.1242/dev.127.6.1231. [DOI] [PubMed] [Google Scholar]
- 17.Buchert M, Schneider S, Meskenaite V, Adams M T, Canaani E, Baechi T, Moelling K, Hovens C M. J Cell Biol. 1999;144:361–371. doi: 10.1083/jcb.144.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruckner K, Pablo Labrador J, Scheiffele P, Herb A, Seeburg P H, Klein R. Neuron. 1999;22:511–524. doi: 10.1016/s0896-6273(00)80706-0. [DOI] [PubMed] [Google Scholar]
- 19.Reedquist K A, Ross E, Koop E A, Wolthuis R M F, Zwartkruis F J T, Van Kooyk Y, Salmon M, Buckley C D, Bos J L. J Cell Biol. 2000;148:1151–1158. doi: 10.1083/jcb.148.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolthuis R M F, Franke B, Van Triest M, Bauer B, Cool R H, Camonis J H, Akkerman J W N, Bos J L. Mol Cell Biol. 1998;18:2486–2491. doi: 10.1128/mcb.18.5.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leng L J, Kashiwagi H, Ren X D, Shattil S J. Blood. 1998;91:4206–4215. [PubMed] [Google Scholar]
- 22.Gale N W, Holland S J, Valenzuela D M, Flenniken A, Pan L, Ryan T E, Henkemeyer M, Strebhardt K, Hirai H, Wilkinson D G, et al. Neuron. 1996;17:9–19. doi: 10.1016/s0896-6273(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 23.Haimovich B, Lipfert L, Brugge J S, Shattil S J. J Biol Chem. 1993;268:15868–15877. [PubMed] [Google Scholar]
- 24.Bennett J S, Hoxie J A, Leitman S S, Vilaire G, Cines D B. Proc Natl Acad Sci USA. 1983;80:2417–2421. doi: 10.1073/pnas.80.9.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franke B, Akkerman J W N, Bos J L. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer T H, Gatling M N, Lacal J-C, White G C, II. J Biol Chem. 1990;265:19405–19408. [PubMed] [Google Scholar]
- 27.Franke B, Van Triest M, De Bruijn K M T, Van Willligen G, Nieuwenhuis H K, Negrier C, Akkerman J W N, Bos J L. Mol Cell Biol. 2000;20:779–785. doi: 10.1128/mcb.20.3.779-785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bertoni, A., Tadokoro, S., Eto, K., Pampori, N., Parisi, L. V., White, G. C. & Shattil, S. J. (2002) J. Biol. Chem., in press. [DOI] [PubMed]
- 29. Woulfe, D., Jiang, H., Mortensen, R., Yang, J. & Brass, L. F. (2002) J. Biol. Chem., in press. [DOI] [PubMed]
- 30.Ellis C, Kasmi G, Ganju P, Walls E, Panayotou G, Reith A D. Oncogene. 1996;12:1727–1736. [PubMed] [Google Scholar]
- 31.Zisch A H, Kalos M S, Chong L D, Pasquale E B. Oncogene. 1998;16:2657–2670. doi: 10.1038/sj.onc.1201823. [DOI] [PubMed] [Google Scholar]
- 32.Zisch A H, Stallcup W B, Chong L D, Dahlin-Huppe K, Voshol J, Schachner M, Pasquale E B. J Neurosci Res. 1997;47:655–665. doi: 10.1002/(sici)1097-4547(19970315)47:6<655::aid-jnr12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 33.Blaess S, Kammerer R A, Hall H. J Neurochem. 1998;71:2615–2625. doi: 10.1046/j.1471-4159.1998.71062615.x. [DOI] [PubMed] [Google Scholar]
- 34.Felding-Habermann B, Silletti S, Mei F, Siu C H, Yip P M, Brooks P C, Cheresh D A, O'Toole T E, Ginsberg M H, Montgomery A M P. J Cell Biol. 1997;139:1567–1581. doi: 10.1083/jcb.139.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burden-Gulley S M, Pendergrast M, Lemmon V. Cell Tissue Res. 1997;290:415–422. doi: 10.1007/s004410050948. [DOI] [PubMed] [Google Scholar]
- 36.Grumet M. J Neurosci Res. 1992;31:1–13. doi: 10.1002/jnr.490310102. [DOI] [PubMed] [Google Scholar]
- 37.Holland S J, Gale N W, Mbamalu G, Vancopoulos G D, Henkemeyer M, Pawson T. Nature (London) 1996;383:722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- 38.Law D A, DeGuzman F R, Heiser P, Ministri-Madrid K, Killeen N, Phillips D R. Nature (London) 1999;401:808–811. doi: 10.1038/44599. [DOI] [PubMed] [Google Scholar]
- 39.Park S, Frisen J, Barbacid M. EMBO J. 1997;16:3106–3114. doi: 10.1093/emboj/16.11.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frisen J, Yates P A, McLaughlin T, Friedman G C, O'Leary D D, Barbacid M. Neuron. 1998;20:235–243. doi: 10.1016/s0896-6273(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 41.Dottori M, Hartley L, Galea M, Paxinos G, Polizzotto M, Kilpatrick T, Bartlett P F, Murphy M, Kontgen F, Boyd A W. Proc Natl Acad Sci USA. 2001;95:13248–13253. doi: 10.1073/pnas.95.22.13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cowan C A, Yokoyama N, Bianchi L M, Henkemeyer M, Fritzsch B. Neuron. 2000;2:417–430. doi: 10.1016/s0896-6273(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 43.Helmbacher F, Schneider-Maunoury S, Topilko P, Tiret L, Charney P. Development (Cambridge, UK) 2000;127:3313–3124. doi: 10.1242/dev.127.15.3313. [DOI] [PubMed] [Google Scholar]
- 44.Gerety S S, Wang H U, Chen Z-F, Anderson D J. Mol Cell. 1999;4:403–414. doi: 10.1016/s1097-2765(00)80342-1. [DOI] [PubMed] [Google Scholar]
- 45.Davy A, Robbins S M. EMBO J. 2000;19:5396–5405. doi: 10.1093/emboj/19.20.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davy A, Gale N W, Murray E W, Klinghoffer R A, Soriano P, Feuerstein C, Robbins S M. Genes Dev. 1999;13:3125–3135. doi: 10.1101/gad.13.23.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandey A, Lazart D F, Saltiel A R, Dixit V M. J Biol Chem. 1994;269:30154–30157. [PubMed] [Google Scholar]
- 48.Stein E, Cerretti D P, Daniel T O. J Biol Chem. 1996;271:23588–23593. doi: 10.1074/jbc.271.38.23588. [DOI] [PubMed] [Google Scholar]
- 49.Hock B, Bohme B, Karn T, Feller S, Rubsamen-Waigmann H, Strebhardt K. Oncogene. 1998;17:255–260. doi: 10.1038/sj.onc.1201907. [DOI] [PubMed] [Google Scholar]
- 50.Lu Q, Sun E E, Klein R S, Flanagan J G. Cell. 2001;105:69–79. doi: 10.1016/s0092-8674(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 51.Wahl S, Barth H, Ciossek T, Aktories K, Mueller B K. J Cell Biol. 2000;149:263–270. doi: 10.1083/jcb.149.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shamah S M, Lin M Z, Goldberg J L, Estrach S, Sahin M, Hu L, Bazalakova M, Neve R L, Corfas G, Debant A, Greenberg M E. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- 53.Sarma V, Wolf F W, Marks R M, Shows T B, Dixit V M. J Immunol. 1992;148:3302–3312. [PubMed] [Google Scholar]
- 54.Pandey A, Shao H, Marks R M, Polverini P J, Dixit V M. Science. 1995;268:567–569. doi: 10.1126/science.7536959. [DOI] [PubMed] [Google Scholar]
- 55.Daniel T O, Stein E, Cerretti D P, St. John P L, Robert B, Abrahamson D R. Kidney Int. 1996;50, Suppl. 57:S73–S81. [PubMed] [Google Scholar]
- 56.Wang H U, Chen Z F, Anderson D J. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 57.Adams R H, Wilkinson G A, Weiss C, Diella F, Gale N W, Deutsch U, Riasau W, Klein R. Genes Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.