

Abstract
SKI-binding protein (SKIP) is a transcription cofactor present in all eukaryotes. Here we show that SKIP is a unique protein that is required for Caenorhabditis elegans viability and development. Expression of CeSKIP(skp-1) assayed by RT-PCR and by GFP fluorescence in transgenic lines starts in embryos and continues to adulthood. Loss of CeSKIP activity by RNA-mediated inhibition results in early embryonic arrest similar to that seen following inhibition of RNA polymerase II. RNA polymerase II phosphorylation appears normal early in CeSKIP RNA-mediated inhibition treated embryos although the expression of several embryonic GFP reporter genes is severely restricted or absent. Our data suggest that CeSKIP is an essential component of many RNA polymerase II transcription complexes and is indispensable for C. elegans development.
Keywords: transcription‖nuclear hormone receptors‖cofactors‖molting
The RNA polymerase II (RNA Pol II) transcription complex consists of the polymerase holoenzyme, general transcription factors, and gene-specific transcription factors. General factors include components such as TATA box-binding protein-associated factors (TAFs) and thyroid hormone receptor-associated protein (TRAP)/SRB- and MED-containing cofactor complex (SMCC)/mediator comprised of several proteins (1). The RNA Pol II complex is also regulated by gene-specific transcription factors that recognize cis-acting regulatory sequences in promoters resulting in activation or repression of the target gene.
One cofactor that has been identified in both stimulatory and inhibitory Pol II complexes is the SKI-binding protein (SKIP). SKIP was first identified in Drosophila melanogaster (2), and was subsequently identified in many other species (3–7). SKIP is an essential protein in yeast (3). Several papers (4–10) report interactions between SKIP and other proteins involved in the repression and/or activation of transcription. In addition, SKIP is known to be a cofactor in Notch, transforming growth factor (TGF)-β and vitamin D receptor (VDR) pathways where it acts as a coactivator. The ubiquitous presence of SKIP, from yeast to plants and animals, and its possible involvement in the regulation of transcription by diverse regulatory systems raises the possibility that this protein may be functioning as a fundamental cofactor in the RNA Pol II complex.
We have been studying the role of SKIP during Caenorhabditis elegans development. In this study, we demonstrate that C. elegans SKIP (CeSKIP) is essential for viability and is a critical component of multiple regulatory pathways during C. elegans development. Our results also show that despite phosphorylation of the RNA Pol II C-terminal domain (CTD), CeSKIP RNA-mediated inhibition (RNAi) embryos arrest with a phenotype similar to the loss of RNA Pol II activity and fail to express multiple embryonic green fluorescent protein (GFP) reporter genes. In C. elegans, CeSKIP is cotranscribed with bir-1, an inhibitor of apoptosis that is homologous to mammalian Survivin. We confirm earlier studies demonstrating that bir-1 inhibition results in early embryonic cell division defects and lethality. Loss of bir-1 activity postembryonically suggests additional roles for bir-1 including possible RNA Pol II regulation.
Materials and Methods
Sequence analysis was performed by using the Genetics Computer Group wisconsin (http://www.gcg.com), hmmer (http://hmmer.wustl.edu) software packages and the NCBI blast server (http://www.ncbi.nlm.nih.gov/blast/).
Strains.
C. elegans Bristol strain was used in this study wherever not specifically stated and was maintained as described (11). The following strains expressing GFP fusion proteins were used: JM 63 (elt-2∷gfp) integrated line (12, 13); PD7963 (hlh-1∷gfp) integrated line (14); PD8097 (hlh-2∷gfp) (15); egl-15∷gfp (16), and Nde∷gfp (17).
CeSKIP RNAi.
Approximately 1,500 bp of cDNA from the clone yk459d6 was amplified by PCR and cloned into L4440 vector (a kind gift from A. Fire, Carnegie Institute of Washington, Baltimore). For RNAi by microinjections and soaking the double stranded (ds) RNA was prepared by in vitro transcription reaction with T7 RNA polymerase (Promega) after linearization with XbaI and PstI. The annealing of complementary transcripts was done at 60°C for 10 min. The dsRNA was purified by phenol/chloroform extraction and ethanol precipitation with 1 μg of glycogen (GIBCO/BRL) per complete reaction volume.
The dsRNA was injected into syncytial gonad at a concentration of about 1 μg/μl using a Tritech microinjector attached to an Olympus IX70 microscope. Embryos of injected hermaphrodites were collected in a 10- to 12-h interval, incubated for another 12 h, and scored. For soaking experiments, young larvae and adult worms were incubated in 10 μl of dsRNA (1 μg/μl) at room temperature for 16 to 17 h. The incubations were performed in caps of inverted Eppendorf tubes that were sealed by parafilm. Up to 50 larvae L1 and L2 and 20 L3, L4, and adult worms were treated in a single drop. Treated animals were transferred in 12-h intervals to new plates. For feeding RNAi we used the methods as described by Timmons and Fire (18). The construct was transformed into HT115 E. coli and induced with isopropyl-beta -d-thiogalactoside (IPTG). Ampicillin was used for bacterial selection at concentration 100 μg/ml. For controls, HT115 E. coli were transformed with empty vector and induced with IPTG.
Transgenic Strain Generation.
The regions −3,330 and −1,850 bp from the CeSKIP ATG were amplified by PCR with the downstream primer covering the CeSKIP region for codons 3 and or 120 (9,360 bp). The amplified fragments were cloned in to pPD vectors (kind gifts from A. Fire) as follow: 4816: promoter A contains −1,850 to 360 bp region from CeSKIP gDNA in pPD 95.69 vector; 4912: promoter B contains −3,300 to 9 bp region from CeSKIP gDNA in pPD 95.69. The DNA prepared as Qiagen midipreparations was injected at 100 ng/μl to N2 worms together with a marker plasmid pRF4, rol-6 (su 1006), which leads to characteristic movement of transgenic worms (rollers).
Reverse Transcription (RT)-PCR.
The RT-PCR was performed from total and poly(A)+ RNA from synchronized stages of nematodes. The total DNA was treated with DNase (Promega). The poly(A)+ RNA was prepared from total RNA by using Invitrogen poly(A)+ kit. First-strand cDNA for each developmental stage was prepared from 1 μg of total RNA or 100 ng of poly(A)+ RNA. The reverse transcriptase (Superscript II; Life Technologies, Gaithersburg, MD) reaction was primed with random hexamers by using conditions recommended by the supplier. The cDNA reaction products were diluted 1:10 in water.
The PCR reactions were performed with 2 pairs of primers. One pair was specific for CeSKIP the other targeted an internal control gene, ama-1, which encodes the large subunit of RNA polymerase II (19). The primers have been designed so that any contamination of genomic DNA products (if present) would include several introns and be larger and less favored than the cDNA products. The number of PCR cycles was determined empirically after testing of each pair of primers separately; 30 cycles has been used. The cycling conditions were: 94°C for 3 min., followed by 30 cycles of 94°C for 30 s, 60°C for 40 s, and 72°C for 1 min. The reactions were incubated at 72°C for 10 min after cycling.
Electrophoresis of PCR products was performed on 2% agarose gels. The gels were stained 30 min with ethidium bromide and destained two times with water. The densitometric analysis of bands was performed by imaging with a Stratagene Eagle Eye digital camera followed by quantification of bands using IP LABS software.
The CeSKIP primers were designed: 4868: TGA TGG ACG TGG TCT CCA ACA GAC; 4869: TGA ACA GGT CCA GAT CCA CGA CTC. The ama-1 primers were designed as described by Johnstone and Barry (19).
Rapid Amplification of cDNA Ends and Sequencing.
Total and poly(A)+ RNA was prepared from mixed stage culture as described above. To obtain 5′end of the CeSKIP we used RACE kit (rapid amplification of cDNA ends) (Roche Molecular Biochemicals). Reverse transcription was performed by using the AMV (avian myeloblastosis virus) reverse transcriptase and random hexamer primer. The first strand of cDNA was purified with High Pure PCR Product Purification kit. The terminal transferase was used to add a homopolymeric A-tail to the 5′end of the cDNA. Tailed cDNA was amplified by PCR using a gene specific primer SP1 (4818: ATC ATC ATC CTC GTT CCA CGT CTT) and oligo dT-anchor primer. Second PCR was done with SL2 and SP2 (SL2 and SL1 primers were designed as described (20) and 4819: CCA TCA GTT CCA TAT TGC AAG GCG AG). The products were ligated by TA cloning strategy (Stratagene), transformed to Top 10 Escherichia coli competent cells (Stratagene). Several clones were sequenced for both genes by using ABI 473 automated sequencer.
Immunocytochemistry.
K76 mouse monoclonal IgG antibody (against P granules) was obtained from The Developmental Studis Hybridoma Bank at the University of Iowa. The embryos were frozen, cracked, fixed with methanol acetone, and air dried, and the primary antibody was used at dilution 1:50. Rhodamine conjugated goat anti-mouse IgG secondary antibody (Jackson Immunological) was used at a dilution of 1:400.
H5 antibody (mouse monoclonal IgM, a kind gift from Geraldine Seydoux) was used at dilution 1:10. The secondary antibody was FITC labeled goat anti-mouse IgM (Pears). In these experiments, DAPI (4′,6-diamidino-2-phenylindole) counterstaining was omitted to eliminate bleed through fluorescence of DAPI on the FITC channel.
Results
Characterization of CeSKIP.
GenBank contains 59 expressed sequence tags (ESTs) containing CeSKIP-related sequence. They can be all assembled into a single cDNA from the predicted T27F2.1 gene sequence; no splicing variant was identified. The EST clone yk459d6 was kindly provided by Yuji Kohara and used for this study because it appeared to be one of the longest and covered almost the entire coding region. We sequenced the entire cDNA clone and it was identical with the predicted coding sequence reported in GenBank. The yk459d6 cDNA sequence begins downstream of the predicted start codon at nucleotide position 10 and ends with a poly(A) tract beginning 76 nt downstream of the predicted stop codon (TAA). The predicted CeSKIP protein has 56% identity and 77% similarity to human SKIP.
The alignment of SKIP sequences from different species makes it possible to delineate distinct domains (Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). The low complexity segments (21) (observable best on the Dictyostelium sequence) split the protein into N-terminal, central and C-terminal domains. The central domain contains a highly conserved segment previously named the SNW domain caused by the presence of serine, asparagine, and tryptophan residues. Closer analysis shows, however, that this is just a central part of a much larger domain. This central domain has been shown recently to be responsible for the interaction with SKI (38). All three domains are unique to SKIP; their HMM profile (22) defines them fully and uniquely. None of the three domains can be attributed to a known domain family and none exists separately within another protein. The C-terminal domain, contrary to the reports in the literature and databases annotations, is not an SH2 domain according to SMART (23) and PFAM (24) HMM profiles. All proteins are predicted to be nonsecreted (25), and most of them contain a clear nuclear localization signal (26) as expected for nuclear proteins. Although the initial low-complexity segment preceding the N-terminal domain shows no overall conservation in sequence, it contains an absolutely conserved motive LPxP conserved in all reported SKIP proteins (Figs. 1A and 6). Searches in fully sequenced genomes revealed only one paralog per organism in eukaryota, and no paralogs in prokaryota. The human genome contains the SKIP gene on chromosome 14 (Homo sapiens working draft sequence segment) (contig NT_026437) (27); a 95% identical sequence can be found on chromosome 1 (contig NT_021928), but this is most likely an intronless pseudogene, the ORF of which is interrupted by several STOP codons.
Figure 1.

(A) Schematic representation of putative domains in SKIP. Boxes indicate the approximate size and position in the human orthologue, conservation >40% and >90% are indicated in gray and black, respectively. NL is a nuclear localization motive and LPxP is another conserved motive. SNW-box denotes the most conserved part of the protein. (B) Schematic representation of bir-1/CeSKIP operon obtained by analysis of the cosmid T27F2. The position of SL1 and SL2 trans-splicing is indicated.
Expression of CeSKIP.
Analysis of gene organization indicates that CeSKIP is likely part of a two-gene operon (Fig. 1B); about 25% of C. elegans genes are arranged in operons (28). The first gene of this operon is bir-1, which belongs to the class of genes known as inhibitors of apopotosis. bir-1 encodes a Survivin homologue and is necessary for spindle function during cell division (29, 30). RT-PCR with primers derived from bir-1 and CeSKIP in combination with SL1 and SL2 primers demonstrated that bir-1 was SL1 trans-spliced and CeSKIP was SL2 trans-spliced. These results are consistent with the prediction that CeSKIP is the second gene expressed in the operon. Trans-splicing of SL2 to the CeSKIP cDNA was confirmed by rapid amplification of cDNA ends with trans-splicing of SL2 occurring at position −17 relative to ATG.
The expression of CeSKIP during C. elegans development was assayed by RT-PCR using total RNA and poly(A)+ RNA prepared from synchronized cultures. CeSKIP transcripts were normalized against ama-1 (RNA Pol II large subunit; ref. 19). CeSKIP is expressed abundantly in embryos and continues to be expressed in all larval stages up to the adulthood (Fig. 2 A and B).
Figure 2.
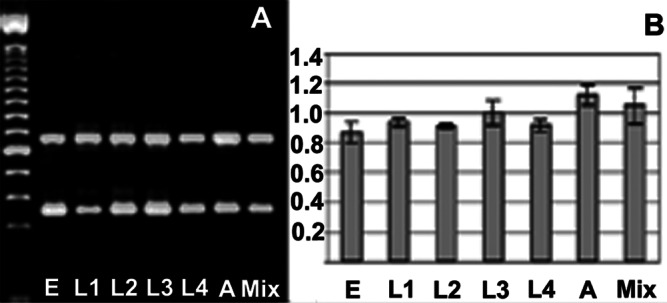
Expression of CeSKIP during C. elegans development. (A) Agarose gel electrophoresis of simultaneous PCR products for CeSKIP (upper band) and ama-1 (lower band). E, L1, L2, L3, L4, A, and Mix represent embryos, larvae in L1–L4 stage, adult, and mixed population of worms, respectively. (B) Densitometric quantification combined from two independent experiments and quantified on four PCRs. The values are expressed as relative units of a ratio CeSKIP/ama-1, the bars represent standard deviation.
To visualize the expression of CeSKIP we prepared strains expressing a GFP driven by a predicted promoter fragment consisting of 3,300 bp upstream of the CeSKIP ATG. This DNA fragment includes the entire coding region of bir-1 as well as 2,565 bp upstream of the bir-1 ATG. Embryos expressed CeSKIP∷gfp ubiquitously beginning at approximately the 30 cell stage. The expression of CeSKIP∷gfp continues in larval development and adult animals although the level of expression varies in different tissues (Fig. 3). The most pronounced expression is in pharyngeal cells, in head and neurons (Fig. 3 D and E), neurons of dorsal and ventral neuronal cord (Fig. 3 D, F, and G), in muscle cells (Fig. 3F), and epidermal cells mostly in head and in tail (Fig. 3 D, E, and G), and in some seam cells. In L4 larvae and adults, egg laying muscle cells, and anal muscle cells are also strongly labeled (Fig. 3G).
Figure 3.

Expression of CeSKIP∷gfp in transgenic strains. (A) Wide, probably ubiquitous expression of transgene in an embryo at about the 70-cell stage. (B) Dorsal view of an about 150-cell embryo expressing CeSKIP∷gfp in most cells. (C) Two fold embryo strongly expressing CeSKIP∷gfp in head, ventral, and tail regions. Each embryo is about 50 μm. (D) Larva L1 with pronounced transgene expression in neurons, epidermal cells, muscles, and gut. (E) Larva L4 showing expression of CeSKIP∷gfp in pharynx (arrowhead) and pharyngeal neurons. (F) Central part of body of larva L3. The arrow indicates ventral neuronal cord and arrowheads point to body wall muscles expressing CeSKIP∷gfp. (G) The expression of CeSKIP∷gfp in vulval muscles (arrowhead), ventral neuronal cord, and anal muscles (arrow) of a L4 larva. Epidermal cells and neurons have also strong expression of the transgene.
Interestingly, microinjections of a GFP fusion with the first 120 aa of CeSKIP had a strong embryonic and larval lethal effect in the progeny of injected animals and no lines were established. Unlike this translational fusion, there was no lethality associated with the transcriptional reporter gene fusion. This finding suggests that the presence of bir-1 on the transgene is not in itself toxic, in agreement with published data (29). However, the CeSKIP N-terminal domain may be acting as a dominant negative protein resulting in embryonic and larval lethality.
CeSKIP Loss of Function During Embryonic Development Leads to Complete Arrest Similar to RNA Polymerase II Inhibition.
dsRNA from the CeSKIP cDNA was injected into gonadal syncytia of adult hermaphrodites to assay CeSKIP function by RNAi. Progeny of injected animals showed 100% embryonic arrest between the 50 and 100 cell stage (n = 3,420; Fig. 4 A and B). The CeSKIP RNAi embryonic arrest appeared similar to that reported following inhibition of the large subunit of RNA Pol II encoded by the ama-1 gene (31). Like ama-1 RNAi, CeSKIP RNAi arrested embryos had defects in gastrulation, the gut precursors cells Ea and Ep remained at the embryonic surface (Fig. 4A), and the P lineage was arrested at the P4 stage (Fig. 4B).
Figure 4.
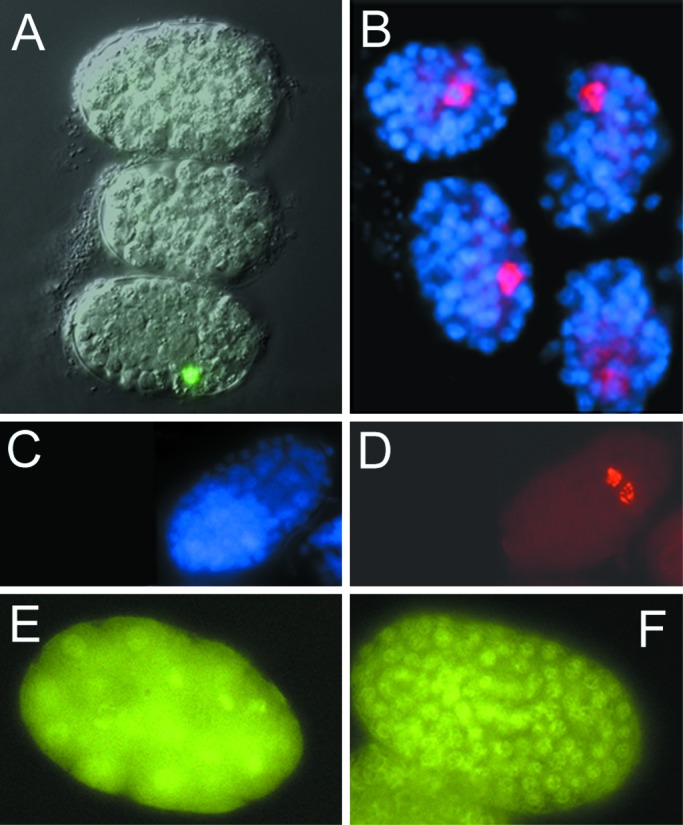
Embryonic arrest in CeSKIP RNAi. (A) Embryos from a line JM63 (expressing elt-2∷gfp) arrested at stage of about 70 cells. Note only one GFP-positive cell in one of the three embryos indicating the arrest of gastrulation. (B) SKIP RNAi treated embryos stained with K76 antibodies showing the single P4 germline precursor cell in these arrested embryos. All nuclei are contrastained with DAPI. (C and D) Control embryo stained with K 76 antibody showing number of cells (C) and the desendents of P4, Z2, and Z3 (D). (E) H5 antibody staining of phosphorylated ama-1 in CeSKIP RNAi-treated embryo. (F) Control embryo at the ≈150 cell stage phosphorylated ama-1 as detected by the H5 antibody. Each embryo is about 50 μm.
As a further assay of gene expression we used three GFP reporters that are expressed in early embryogenesis. hlh2∷gfp is ubiquitously expressed in early embryos beginning at the 30 cell stage (15). CeSKIP RNAi arrested embryos had decreased fluorescence in all embryos (n = 354), however, a few cells (one to seven per embryo) remained GFP positive. elt-2∷gfp is expressed in the gut (E) lineage starting at the 2E cell stage (12, 13, 32). The expression of elt-2∷gfp was almost completely inhibited in CeSKIP RNAi arrested embryos with only 1 or 2 cells expressing the transgene in ≈1% of the embryos examined (n = 366; Fig. 4A). The hlh-1∷gfp reporter is normally expressed in body wall muscles and their precursors; CeSKIP RNAi arrested embryos had no expression of the hlh1∷gfp (n = 194).
The lack of reporter gene expression following CeSKIP RNAi led us to assay the activation of RNA Pol II by using the antibody H5, which recognizes CTD phosphorylation (33–35). Embryos from parents microinjected with CeSKIP dsRNA had easily detectable phosphorylated RNA Pol II in early embryos up to arrested stage (Fig. 4E). The H5 staining disappeared over time in terminally arrested embryos (not shown).
CeSKIP RNAi Leads to Multiple Defects in Larval Development.
We sought to examine the role of CeSKIP during larval development by using an RNAi technique that would allow us to bypass the embryonic requirement for CeSKIP. This effect can be achieved by incubation of worms in solutions containing dsRNA prepared by in vitro transcription or by feeding worms bacteria producing specific dsRNA from a vector with inducible T7 promoters flanking the cDNA insert. Treatment of larval animals with CeSKIP RNAi by both methods resulted in a wide spectrum of postembryonic defects. The degree of defect depended on the time and dose of CeSKIP RNAi treatment. The most prominent defects included molting defects, larval arrest, defects in movement (increased amplitudes of movement tracks) and body shape irregularities in L4 and adults (Fig. 5). L1 larvae (n = 647) fed bacteria producing CeSKIP dsRNA had molting defects that appeared by the L2 and L3 stage followed by developmental arrest in 50–70% of animals. Animals that escaped developmental arrest had nearly 100% penetrant defects in the development of the uterus and vulva resulting in a protruding vulva (Pvul) phenotype (Fig. 5 C and D) and died as L4s with a dramatic lysis of the body. L2 larvae (n = 473) fed bacteria producing CeSKIP dsRNA had milder molting defects in L3 (50% of the treated animals) with the rest of the animals proceeding to adulthood with most animals having irregularities in germ line growth and a mild to medium egg laying (Egl) phenotype (Fig. 5 E and F). The Egl phenotype in these animals led us to assay the vulval and uterine muscles. Both sets of muscle were formed as assayed by egl-15∷gfp (16) and NdE∷gfp (17) reporter genes. L2 and L3 larvae (n = 175) fed bacteria producing CeSKIP dsRNA gave raise to nearly 100% arrested embryos, although embryos developed to later stage (about 150 cells) then seen after microinjection CeSKIP RNAi experiments.
Figure 5.
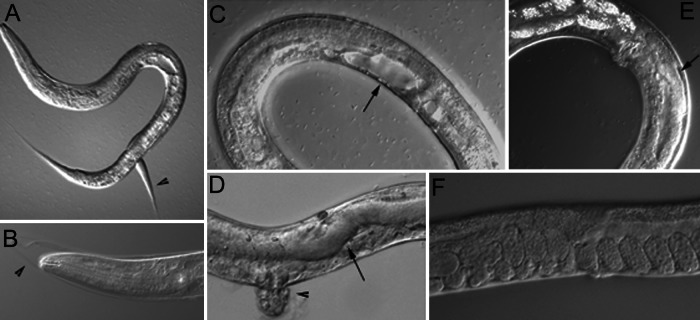
Larval defects resulting from CeSKIP RNAi. (A) Larva L3 with typical molting defect. Arrowhead indicates the attached cuticle from a previous molt. (B) Molting defect in L3 shown in the head area. The attached cuticle is indicated by the arrowhead. (C) Impaired development of the uterus and presence of a large vacuole (arrow). (D) Enlarged intestine (arrow) and protrusion of the vulva (arrowhead) in a L4 larva. (E) A young adult hermaphrodite with a defect in germline growth (arrow). (F) Adult worm showing retention of eggs (Egl phenotype).
bir-1 Has Functions Beyond Cell Division.
Because it is possible that operons link the expression of genes that are functionally related, we assayed bir-1 RNAi for phenotypes related to loss of SKIP activity in addition to the cell division functions that have been reported (29, 30). Surprisingly, larvae fed bacteria producing bir-1 dsRNA led to some phenotypes that were similar to that seen with CeSKIP loss of function (not shown). Approximately 10% of treated adult worms (n = 223) had changes in the gut with signs of vacuoles and lysis. About 15% of treated animals were Pvul–Egl as adults and they were also often short and fat (Dpy). We did not observe in bir-1 RNAi larvae the molting defects, movement defects, or larval arrest observed after CeSKIP RNAi.
Discussion
CeSKIP Is Essential for Development.
Our work shows that CeSKIP is an indispensable factor for multiple regulatory pathways during C. elegans development. CeSKIP loss of function during embryonic development leads to developmental arrest and lethality that is similar to RNA Pol II large subunit inhibition (31). RNA Pol II CTD phosphorylation appears normal in early CeSKIP RNAi embryos, but is eliminated in arrested embryos. The expression of several early embryonic GFP reporter genes is inhibited or eliminated in treated embryos.
Postembryonically, loss of CeSKIP activity results in numerous developmental defects including molting and vulvagenesis defects. These defects may reflect a role of CeSKIP in several specific developmental processes. The molting defect induced by CeSKIP RNAi is particularly interesting because of its similarity to the phenotype of CHR3 RNAi. CHR3 is a nuclear hormone receptor in C. elegans that is required for proper molting and development (36, 37). The molting defect induced by SKIP RNAi is phenotypically identical to that of CHR3 RNAi, though it is not as penetrant (≈60% vs. 100% respectively). In other systems SKIP is known to interact with nuclear hormone receptors, where it functions as a cofactor (5). This observation suggests that SKIP may be used for the regulation of development and tissue specific transcription in a way that has yet to be appreciated. The different levels of SKIP expression observed in mammalian tissues (5) may in fact reflect developmental programs that are differentially dependent on SKIP function.
SKIP Structure and Function.
The high sequence conservation of SKIP across phyla indicates an ancient and vital function. SKIP has been found to participate in a complex with many nuclear hormone receptors (5), CBF1 (7), and TGF-β (10), and to activate various viral promoters (38). It can be anticipated that it will be found in many additional transcription factor complexes.
The amino acid segment 201–333 (human sequence) was sufficient to interact with Smad-2 and Smad-3 transcription factors (10) and the segment 220–438 with VDR (5). The comparison of these data with the amino acid alignments suggests that the central domain may be the domain responsible for the interaction with transcription factors in general. Our observation of a possible dominant negative effect of transgenes containing the N-terminal region of CeSKIP, coupled with the high conservation of this domain across the phyla, suggests functional importance of this domain as well.
SKIP interacts with the protein SKI (6). This is an essential member of histone deacetylase (HDAC) (39), connecting mSin3 through its C terminus to NcoR or SMRT (40). Whereas NCoR and SMRT act as transcriptional corepressors, SKIP acts as a coactivator (7, 8, 10). NCoR and SMRT are well known general corepressors recruiting HDAC to transcription factors (39, 41). The disruption of their interaction with SKI leads to activation caused by the decoupling of HDAC and the transcription factor. This disruption has been observed on the action of the viral oncogene vSKI (42); it lacks the mSin3-binding C-terminal domain and acts therefore in a dominant negative fashion, not allowing the link between the transcription factor and HDAC. The resulting effect is the disruption of the link factor –NCoR–SKI–mSIN3–HDAC. Another viral oncogene, EBNA2, interacts with SKIP and disrupts the HDAC mediated repression (8). The intracellular signaling through NotchIC uses a similar mechanism (7).
Considering that the SKIP gene is present in all eukaryotic genomes as a single and well conserved paralog, we propose that SKIP plays an essential role in the transcription activation and repression in eukaryota in general and may even participate in the switching from the repression to the activation.
Supplementary Material
Acknowledgments
We thank Dr. Mike Krause for support, advice, and help on numerous experiments, and for critical reading of the manuscript. We thank Drs. Yuji Kohara for EST clones, Andy Fire and Lisa Timmons for the vector and host used in RNAi and transgenic line NdE∷gfp, Jim McGhee for JM63 strain, Michael Stern for egl-15∷gfp, and Geraldine Seydoux for H5 antibody. We thank Hana Prouzova and Robert Littlejohn for technical assistance. M.K. and Z.K. were partially supported by Grants 304-99-0682, NC 5261-3, and 11110000-3 from the Grant Agency of the Czech Republic (GACR), and the Ministry of Health and Ministry of Education of the Czech Republic, respectively.
Abbreviations
- RNAi
RNA-mediated interference
- SKIP
SKI-binding protein
- GFP
green fluorescent protein
- RNA Pol II
RNA polymerase II
- CTD
C-terminal domain
References
- 1.Ito M, Roeder R G. Trends Endocrinol Metab. 2001;12:127–134. doi: 10.1016/s1043-2760(00)00355-6. [DOI] [PubMed] [Google Scholar]
- 2.Saumweber H, Frasch M, Korge G. Chromosoma. 1990;99:52–60. doi: 10.1007/BF01737289. [DOI] [PubMed] [Google Scholar]
- 3.Harris S D, Cheng J, Pugh T A, Pringle J R. J Mol Biol. 1992;225:53–65. doi: 10.1016/0022-2836(92)91025-k. [DOI] [PubMed] [Google Scholar]
- 4.Folk P, Puta F, Krpejsova L, Blahuskova A, Markos A, Rabino M, Dottin R P. Gene. 1996;181:229–231. doi: 10.1016/s0378-1119(96)00483-0. [DOI] [PubMed] [Google Scholar]
- 5.Baudino T A, Kraichely D M, Jefcoat S C, Jr, Winchester S K, Partridge N C, MacDonald P N. J Biol Chem. 1998;273:16434–16441. doi: 10.1074/jbc.273.26.16434. [DOI] [PubMed] [Google Scholar]
- 6.Dahl R, Wani B, Hayman M J. Oncogene. 1998;16:1579–1586. doi: 10.1038/sj.onc.1201687. [DOI] [PubMed] [Google Scholar]
- 7.Zhou S, Fujimuro M, Hsieh J J, Chen L, Miyamoto A, Weinmaster G, Hayward S D. Mol Cell Biol. 2000;20:2400–2410. doi: 10.1128/mcb.20.7.2400-2410.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou S, Fujimuro M, Hsieh J J, Chen L, Hayward S D. J Virol. 2000;74:1939–1947. doi: 10.1128/jvi.74.4.1939-1947.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Chen H, Weinmaster G, Hayward S D. J Virol. 2001;75:2946–2956. doi: 10.1128/JVI.75.6.2946-2956.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leong G M, Subramaniam N, Figueroa J, Flanagan J L, Hayman M J, Eisman J A, Kouzmenko A P. J Biol Chem. 2001;276:18243–18248. doi: 10.1074/jbc.M010815200. [DOI] [PubMed] [Google Scholar]
- 11.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukushige T, Hawkins M G, McGhee J D. Dev Biol. 1998;198:286–302. [PubMed] [Google Scholar]
- 13.Fukushige T, Hendzel M J, Bazett-Jones D P, McGhee J D. Proc Natl Acad Sci USA. 1999;96:11883–11888. doi: 10.1073/pnas.96.21.11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krause M, Harrison S W, Xu S Q, Chen L, Fire A. Dev Biol. 1994;166:133–148. doi: 10.1006/dbio.1994.1302. [DOI] [PubMed] [Google Scholar]
- 15.Krause M, Park M, Zhang J M, Yuan J, Harfe B, Xu S Q, Greenwald I, Cole M, Paterson B, Fire A. Development (Cambridge, UK) 1997;124:2179–2189. doi: 10.1242/dev.124.11.2179. [DOI] [PubMed] [Google Scholar]
- 16.DeVore D L, Horvitz H R, Stern M J. Cell. 1995;83:611–620. doi: 10.1016/0092-8674(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 17.Harfe B D, Fire A. Development (Cambridge, UK) 1998;125:421–429. doi: 10.1242/dev.125.3.421. [DOI] [PubMed] [Google Scholar]
- 18.Timmons L, Fire A. Nature (London) 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 19.Johnstone I L, Barry J D. EMBO J. 1996;15:3633–3639. [PMC free article] [PubMed] [Google Scholar]
- 20.Krause M. In: Caenorhabditis elegans: Modern Biological Analysis of an Organism. Epstein H F, Shakes D C, editors. Vol. 48. San Diego: Academic; 1995. pp. 483–512. [Google Scholar]
- 21.Wootton J C, Federhen S. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- 22.Eddy S R. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 23.Schultz J, Copley R R, Doerks T, Ponting C P, Bork P. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bateman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 26.Reinhardt A, Hubbard T. Nucleic Acids Res. 1998;26:2230–2236. doi: 10.1093/nar/26.9.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lander E S, Linton L M, Birren B, Nusbaum C, Zody M C, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Nature (London) 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 28.Blumenthal T, Spieth J. Curr Opin Genet Dev. 1996;6:692–698. doi: 10.1016/s0959-437x(96)80022-0. [DOI] [PubMed] [Google Scholar]
- 29.Fraser A G, James C, Evan G I, Hengartner M O. Curr Biol. 1999;9:292–301. doi: 10.1016/s0960-9822(99)80137-7. [DOI] [PubMed] [Google Scholar]
- 30.Speliotes E K, Uren A, Vaux D, Horvitz H R. Mol Cell. 2000;6:211–223. doi: 10.1016/s1097-2765(00)00023-x. [DOI] [PubMed] [Google Scholar]
- 31.Powell-Coffman J A, Knight J, Wood W B. Dev Biol. 1996;178:472–483. doi: 10.1006/dbio.1996.0232. [DOI] [PubMed] [Google Scholar]
- 32.Zhu J, Fukushige T, McGhee J D, Rothman J H. Genes Dev. 1998;12:3809–3814. doi: 10.1101/gad.12.24.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warren S L, Landolfi A S, Curtis C, Morrow J S. J Cell Sci. 1992;103:381–388. doi: 10.1242/jcs.103.2.381. [DOI] [PubMed] [Google Scholar]
- 34.Bregman D B, Du L, van der Zee S, Warren S L. J Cell Biol. 1995;129:287–298. doi: 10.1083/jcb.129.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seydoux G, Dunn M A. Development (Cambridge, UK) 1997;124:2191–2201. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]
- 36.Kostrouchova M, Krause M, Kostrouch Z, Rall J E. Development (Cambridge, UK) 1998;125:1617–1626. doi: 10.1242/dev.125.9.1617. [DOI] [PubMed] [Google Scholar]
- 37.Kostrouchova M, Krause M, Kostrouch Z, Rall J E. Proc Natl Acad Sci USA. 2001;98:7360–7365. doi: 10.1073/pnas.131171898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prathapam T, Kuhne C, Hayman M, Banks L. Nucleic Acids Res. 2001;29:3469–3476. doi: 10.1093/nar/29.17.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinzel T, Lavinsky R M, Mullen T M, Soderstrom M, Laherty C D, Torchia J, Yang W M, Brard G, Ngo S D, Davie J R, et al. Nature (London) 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 40.Nomura T, Khan M M, Kaul S C, Dong H D, Wadhwa R, Colmenares C, Kohno I, Ishii S. Genes Dev. 1999;13:412–423. doi: 10.1101/gad.13.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horlein A J, Naar A M, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass C K, et al. Nature (London) 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 42.Hammond K L, Hanson I M, Brown A G, Lettice L A, Hill R E. Mech Dev. 1998;74:121–131. doi: 10.1016/s0925-4773(98)00071-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.