

Abstract
Intramyocardial injection of genes encoding angiogenic factors could provide a useful approach for the treatment of ischemic heart disease. However, uncontrolled expression of angiogenic factors in vivo may cause some unwanted side effects, such as hemangioma formation, retinopathy, and arthritis. It may also induce occult tumor growth and artherosclerotic plaque progression. Because hypoxia-inducible factor 1 is up-regulated in a variety of hypoxic conditions and it regulates gene expression by binding to a cis-acting hypoxia-responsive element (HRE), we propose to use HRE, found in the 3′ end of the erythropoietin gene to control gene expression in ischemic myocardium. A concatemer of nine copies of the consensus sequence of HRE isolated from the erythropoietin enhancer was used to mediate hypoxia induction. We constructed two adeno-associated viral vectors in which LacZ and vascular endothelial growth factor (VEGF) expressions were controlled by this HRE concatemer and a minimal simian virus 40 promoter. Both LacZ and VEGF expression were induced by hypoxia and/or anoxia in several cell lines transduced with these vectors. The functions of these vectors in ischemic myocardium were tested by injecting them into normal and ischemic mouse myocardium created by occlusion of the left anterior descending coronary artery. The expression of LacZ gene was induced eight times and of VEGF 20 times in ischemic myocardium compared with normal myocardium after the viral vector transduction. Hence, HRE is a good candidate for the control of angiogenic factor gene expression in ischemic myocardium.
Gene transfer of angiogenic factor genes to ischemic myocardium may provide a useful approach for treatment of coronary artery disease. We have shown that direct injection of an AAV vector carrying the vascular endothelial growth factor (VEGF) to ischemic myocardium can induce neovasculature formation (1). A potential problem associated with prolonged and high-level expression of VEGF is the formation of hemangioma that has been seen in the heart (2) and limb (3). Although we did not see such a complication in our previous experiments on mice, for clinical application it is important to safeguard against this complication. Examples of inducible promoters that have been used to control gene expression include the tetracycline operons, RU 486, edyasone, and other inducible systems (4–11). For the treatment of ischemic heart disease, an ideal control seems to be for angiogenic factor expression to respond to hypoxia.
Hypoxia-inducible factor-1 (HIF-1) is one of the key mammalian transcription factors that exhibit dramatic increases in both protein stability and intrinsic transcriptional potency during low-oxygen stress. It transactivates genes encoding several glucose transporters and glycolytic enzymes, as well as genes that increase tissue perfusion such as VEGF, inducible nitric oxide synthase, and erythropoietin (Epo) (12). HIF-1 is a heterodimeric basic helix–loop–helix protein (13, 14) that is composed of a labile α-subunit (HIF-1α) and a β-subunit. The β-subunit is constitutively and stably expressed in tissues in normal and hypoxic conditions. HIF-1α protein is rapidly degraded by ubiquitination and proteasomal digestion mediated by a 200-aa oxygen-dependent degradation domain (15, 16). This subunit is stabilized in hypoxic tissues and dimerizes with the β-subunit to form the functional HIF-1 protein (17), which in turn binds to hypoxic response elements (HRE) in the enhancers of several genes to up-regulate gene expression.
HRE has been reported in the 5′ or 3′ flanking regions of VEGF (18, 19) and Epo (20) and several other genes. The core consensus sequence is (A/G)CGT(G/C)C. HREs isolated from Epo and VEGF genes have been used to regulate several genes, such as suicide gene (21, 22) and apoptosis gene (23) expression in hypoxic tumors to enhance tumor killing.
In this study, we investigated the feasibility of using the HRE found in the Epo gene to control the expression of angiogenic factors in ischemic myocardium. We showed that HIF-1α expression was increased in ischemic myocardium and that nine copies of the Epo HRE could induce LacZ and VEGF expression under hypoxic and anoxic conditions in both cultured cells in vitro and ischemic mouse myocardium in vivo.
Materials and Methods
AAV Vector Construction and Production.
We inserted nine copies of the Epo HRE consensus sequence (CCGGGTAGCTGGCGTACGTGCTGCAG) (24, 25) to the pβgal-promoter plasmid between SmaI and HindIII sites (CLONTECH) upstream of the simian virus 40 (SV40) minimal promoter. We cloned the expression cassette (nine copies of Epo HRE, SV40 minimal promoter, LacZ gene, and SV40 polyadenylation signal) into an AAV vector between two inverted terminal repeats to generate the AAVH9LacZ vector (Fig. 1). AAVH9VEGF was generated by replacing LacZ gene in AAVH9LacZ with human VEGF165 cDNA (Fig. 1). AAV vectors were prepared by using the three-plasmid cotransfection system (26). AAV vector was cotransfected with two helper plasmids (provided by Avigen, Alameda, CA) to 293 cells by the calcium phosphate precipitate method. One helper plasmid, pLadeno5, has the adenoviral VA, E2A, and E4 regions that mediate AAV vector replication. The other, pHLP19, has AAV rep and cap genes. Cell lysates were produced by using three freeze-and-thaw cycles 3 days after the transfection. AAV vector was purified by CsCl2 centrifugation. Viral titers were determined by dot blot analysis of the DNA content.
Figure 1.
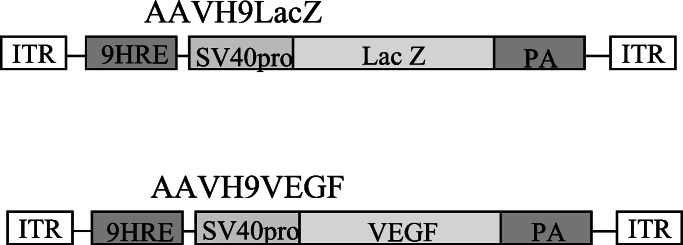
Structure of AAVH9LacZ and AAVH9VEGF. ITR, inverted terminal repeat.
Cell Culture, Infection, Hypoxic, and Anoxic Treatments.
HeLa, Hep 3B, and NIH 3T3 cells (American Type Culture Collection) were maintained in DMEM supplemented with 10% FBS. To infect cultured cells, AAV vectors were added to tissue culture dishes and incubated with the cells for 24 h at 37°C with 5% CO2. The infected cells were split into two groups 2 days afterward. One group was cultured under normoxic conditions (95% air, 5% CO2), and the other under anoxic (95% N2 and 5% CO2) or hypoxic (94% N2, 5% O2, and 1% CO2) conditions in serum-free medium for 16 h.
Chemiluminescent β-Galactosidase Assay.
β-Galactosidase activity was measured by using the Luminescent β-galactosidase Reporter System 3 (CLONTECH) according to the manufacturer's instructions. Cells were harvested 3 days after infection. The cells were washed with ice-cold PBS, and scraped off the plates by using a rubber policeman. After washing in ice-cold PBS, the cells were lysed in 100 mM potassium phosphate buffer (pH 7.8) containing 1 mM DTT by three freeze-and-thaw cycles. The protein concentration in each cell lysate was determined by Bio-Rad DC Protein Assay System (Bio-Rad) and used to normalize the LacZ activities for each sample. Fifty microliters of lysate from each sample was added to the reaction buffer mixture provided by the kit. Light emission was recorded by use of a Luminometer. Nontransduced cells of each cell line were treated and analyzed the same way as viral-vector-transduced cells and used as negative controls.
ELISA for VEGF in Tissue Culture Supernatant.
Cells were cultured in a complete serum-free medium for 16 h before collecting the supernatant. The amount of human VEGF in tissue culture supernatant was quantified by using an ELISA kit for human VEGF (Quantikine, R&D Systems). Samples were measured in duplicates.
Ischemic Heart Model and Intramyocardial Injection of AAV Vectors.
CD1 mice (Charles River Breeding Laboratories) were anesthetized with 15–16 μl of 2.5% Avertin per gram of body weight by i.p. injection. After the respiration of the animal had been controlled by a Small Animal Volume Controlled Ventilator (Harvard Rodent Ventilator, model 683, South Natick, MA), a thoracotomy incision was made in the fourth intercostal space. A surgical retractor was put in the incision to expose the heart. The anterior descending coronary artery was ligated permanently with a 6-0 nonabsorbable surgical suture. 1 × 1011 genomes of viral vectors in 50 μl of Hepes saline (pH 7.4) were injected directly to multiple sites of the myocardium on the left ventricle wall around the ischemic region (pale area) as described (1).
Histological Analysis.
Hearts were fixed in 10% formalin and embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Antibody against HIF-1α (Novus Biological, Littleton, CO) was used to stain the HIF-1α protein in ischemic myocardium. The immunohistochemical staining was done by the protocol of standard Elite Vectastain ABC Kit (Vector Laboratories).
Reverse Transcription (RT)-PCR Analysis.
TRIzoL RNA isolation system (GIBCO/BRL) was used to isolate RNA from hearts. cDNA syntheses were performed by using SuperScript II reverse transcriptase (GIBCO/BRL) with oligo(dT) (Roche Diagnostics) as primer. Total RNA (1.5 μg) was used for each sample. Five microliters of RT products were used for PCR amplification with LacZ and VEGF gene-specific primers. The primer sequences were 5′-GGCGTAAGTGAAGCGACCCG-3′ (sense) and 5′-GCGTGCAGCAGTGGCGATGG-3′ (antisense) for LacZ, and 5′-GGCAGAAGGAGGAGGGACAGAATC-3′ (sense) and 5′-CATTTACACGTCTGCGGATCTTGT-3′ (antisense) for VEGF. Semiquantitative RT-PCR was conducted by PCR amplification of 5 μl of RT product from each sample with limited cycles. The PCR products were electrophoresed in 1.2% agarose gel and transferred to N+ Hybond nylon membrane (Amersham Pharmacia Life Science). The amplified cDNA fragments were detected by hybridizing with 32P-labeled LacZ or VEGF probes and quantified by PhosphorImager analysis. Mouse hypoxanthine phosphoribosyltransferase (HPRT) was used as an internal control for both qualitative and semiquantitative PCR. The primer sequences for HPRT amplification were 5′-AAGGACCTCTCGAAGTGTTGGATA-3′ (sense) and 5′-CATTTAAAAGGAACTGTTGACAACG-3′ (antisense).
Western Blot Analysis.
After isolation of RNA by TRIzoL RNA isolation system (GIBCO/BRL), protein was collected from the same heart. The protein concentration was determined by the Black 10B method (27). In brief, a 200-μl protein sample was mixed with 600 μl of methanol, 200 μl of chloroform, and 500 μl of H2O. The upper phase was removed carefully and discarded. Methanol (800 μl) was added to the interphase and lower phase. After centrifugation, the protein pellet was dried in a Speed Vacuum for 5 min and dissolved in 25 μl of SDS-polyacrylamide gel loading buffer (0.06 M Tris⋅HCl, pH 6.8/1% SDS/1% 2-mercaptoethanol/10% glycerol/0.025% bromophenol blue) and boiled for 3 min. One microliter of sample was spotted on Hybond-C membrane (Amersham Pharmacia Life Science). One microliter of BSA solution in different concentrations (0.1–5 mg/ml) was spotted on the same membrane as standard. The membrane was dried at room temperature for 5 min, stained with Black 10B solution (0.1% wt/vol in 45% methanol and 5% acetic acid) for 10 min, washed in a solution containing 90% methanol and 2% acetic acid, and air dried. The density of the color of the protein dots was measured with alphaImager (Alpha Innotech, San Leandro, CA).
Equal amounts of proteins were separated on a SDS-7% polyacrylamide gel and transferred to a poly(vinylidene difluoride) membrane (Amersham Pharmacia Life Science) by using an electroblotting apparatus (Bio-Rad). The membrane was stained for total protein by using Ponceau-S (Sigma) and blocked with 5% skim milk powder, 0.1% Tween 20 in PBS, pH 7.4 overnight. Antibody against HIF-1α (Novus Biological) was used to stain the HIF-1α protein. ECL+ Western blotting detecting reagent (Amersham Pharmacia Life Science) was used to detect the signals. The signals were visualized by exposing the blot to x-ray films.
Results
Detection of Increased Level of HIF-1α in Ischemic Myocardium.
To test the feasibility of using HRE to induce gene expression in ischemic myocardium in vivo, we analyzed HIF-1α expression in ischemic hearts. Proteins were isolated from normal and ischemic mouse hearts 2 weeks after the occlusion of the anterior descending coronary artery. HIF-1α was detected by Western blot analysis in the ischemic hearts at a much higher level than the normal nonischemic hearts (Fig. 2). To define the site of expression of HIF-1α, immunohistochemical staining with an antibody against HIF-1α showed that HIF-1α was detectable in the region perfused by the anterior descending coronary artery 2 days after its occlusion (Fig. 3). Thus, the expression of HIF-1α in ischemic myocardium indicates that it would be feasible to use HRE to control angiogenic factor expression in ischemic hearts.
Figure 2.
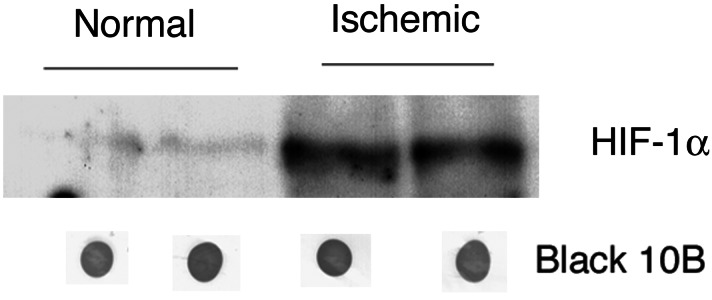
HIF-1α expression in ischemic myocardium by Western blot analysis. Two animals were studied in each of the normal and ischemic groups. The protein loaded for each sample is quantified by the Black 10B method.
Figure 3.

HIF-1α expression by ischemic myocardium. The arrows indicate ischemic myocardium [hematoxylin and eosin (H&E) stain, Left] that stains positively for HIF-1α (Right). No HIF-1α stain was seen on other parts of the myocardium or in normal nonischemic heart (data not show).
Anoxic Induction of LacZ Gene Expression in Cultured Cells.
An AAV vector, AAVH9LacZ, was constructed. Nine copies of HRE isolated from Epo enhancer and a minimal SV40 promoter were used to control the LacZ gene expression. The function of HRE was tested in HeLa, Hep 3B, and NIH 3T3 cells, which were known to express HIF-1 protein in hypoxic conditions. AAV-LacZ in which CMV promoter was used to control gene expression was also used to infect the same cell lines as control. Cells were infected with the AAV vectors at a multiplicity of infection of 106 genomes per cell. Nontransduced cells of each cell line were treated and analyzed in the same way as viral vector-transduced cells and used as negative control for the baseline light emission. The results demonstrated that the LacZ activities were increased 10 times in NIH 3T3 cells, 28 times in HeLa cells, and 30 times in Hep 3B cells after AAVH9LacZ transduction in anoxic condition (95%N2 and 5%CO2) (Table 1). However, the LacZ activities controlled by CMV promoter were not significantly different for the AAV-LacZ-transduced cells under anoxic and normoxic conditions (Table 1). Therefore, the HRE of Epo can induce the AAV delivered LacZ gene expression under anoxic condition in cultured cells.
Table 1.
Increased LacZ activity in AAVH9LacZ-transduced cells under anoxic condition
| Cells | Vectors/treatments
|
|||||
|---|---|---|---|---|---|---|
| AAV9H9LacZ
|
AAV-LacZ
|
|||||
| Anoxic* | Normoxic* | Ratio† | Anoxic* | Normoxic* | Ratio† | |
| NIH3T3 | 2,434 | 249 | 10 | 4,951 | 3,566 | 1.4 |
| HeLa | 21,370 | 763 | 28 | 21,448 | 27,766 | 0.8 |
| Hep 3B | 132 | 4.4 | 30 | 480 | 476 | 1 |
Values are relative light units (RLU) per microgram.
Ratio: anoxic/normoxic.
Hypoxic Induction of VEGF Expression in Hep 3B Cells.
To test whether Epo HRE can also induce AAV-delivered VEGF gene expression under hypoxic conditions, we constructed AAVH9VEGF vector by replacing the LacZ gene in AAVH9LacZ with human VEGF165 cDNA (Fig. 1). We infected Hep 3B cells with AAVH9VEGF. AAV-VEGF, in which CMV promoter was used to control the VEGF gene expression, was also used to infect the same cell line as control. The transduced cells were split into two groups 48 h after the infection. One group was cultured under normoxic conditions, and the other under anoxic conditions (95% N2 and 5% CO2) or hypoxic conditions (94% N2, 5%O2, and 1% CO2) in serum-free medium for 16 h. Nontransduced cells of each cell lines were also cultured and analyzed in the same way as the transduced cells as control to correct for endogenous VEGF gene expression. The number of cells in each well was counted to normalize the VEGF protein level. AAVH9VEGF-transduced Hep 3B cells expressed VEGF 119 times higher in hypoxic conditions, and 33 times higher in anoxic conditions than that in normoxic conditions. AAV-VEGF transduced Hep 3B cells also expressed 16.4 times higher VEGF in hypoxic condition and 14 times higher VEGF in anoxic conditions than that in normoxic conditions. The increased expression of VEGF by AAV-VEGF-transduced Hep 3B cell in hypoxic and anoxic conditions could be the result of induction of endogenous VEGF gene expression under these conditions, because the endogenous VEGF gene in Hep 3B cells has HRE in its enhancer and could respond to hypoxia induction. However, the induction levels were much higher with AAVH9VEGF-transduced cells than AAV-VEGF-transduced cells (Table 2). The lower expression of VEGF in anoxic conditions than in hypoxic conditions could be the result of the intolerance of Hep3B cells to the anoxic treatment, because the cells seemed unhealthy after the anoxic treatment.
Table 2.
Increased VEGF expression in AAVH9VEGF-transduced Hep 3B cells under hypoxic and anoxic conditions*
| AAVH9VEGF
|
AAV-VEGF
|
||||
|---|---|---|---|---|---|
| Hypoxic | Normoxic | Ratio | Hypoxic | Normoxic | Ratio |
| 1,682 | 14.1 | 119 | 2,825 | 173 | 16.4 |
| Anoxic | Normoxic | Ratio | Anoxic | Normoxic | Ratio |
| 620 | 19 | 33 | 1,507 | 107 | 14 |
Values stated are in picograms per 105 cells for 24 h.
Induction of LacZ and VEGF Expression by HRE in Ischemic Myocardium.
The function of the nine copies HRE in ischemic myocardium in vivo was studied by injecting 1 × 1011 genomes of AAVH9LacZ or AAVH9VEGF vectors to ischemic and normal mouse hearts. AAV-LacZ, in which CMV promoter was used to control LacZ gene expression, was injected to the same mouse model and used as control. LacZ and VEGF gene expression was analyzed by semiquantitative RT-PCR with the total RNA isolated from these hearts 2 weeks and 2 months after the injection, respectively. LacZ gene induction in AAVH9LacZ-transduced ischemic hearts (Fig. 4) was eight times higher than its induction in normal hearts (Fig. 5). The gene expression level is about the same in AAV-LacZ-transduced normal and ischemic hearts (Fig. 4). VEGF gene expression was also 20 times higher in ischemic heart than in normal heart after AAVH9VEGF transduction (Figs. 5 and 6). These results indicated that nine copies of Epo HRE could induce LacZ and VEGF expression in ischemic myocardium in vivo.
Figure 4.
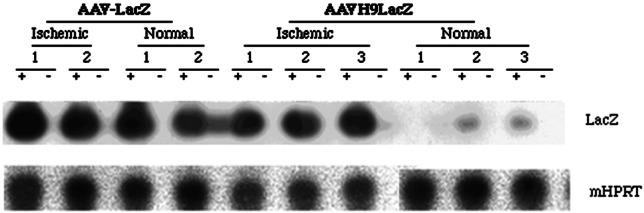
Semiquantitative RT-PCR analysis for LacZ expression with 35 PCR cycles. Mouse HPRT (mHPRT) was used as internal control and amplified with 15 PCR cycles. +, PCR with RT products; −, PCR with RNA without RT. The quantification was done by PhosphorImager analyses. The numbers indicate the individual animals in each group.
Figure 5.

Fold increase of LacZ and VEGF expression in ischemic hearts. Gray and black bars represent gene expression levels in ischemic and normal hearts, respectively. The expression levels of these two genes in normal hearts were arbitrarily assigned as one. The relative fold increases of gene expression in ischemic hearts versus in normal hearts were calculated according to the PhosphorImager analyses of semiquantitative RT-PCR data.
Figure 6.
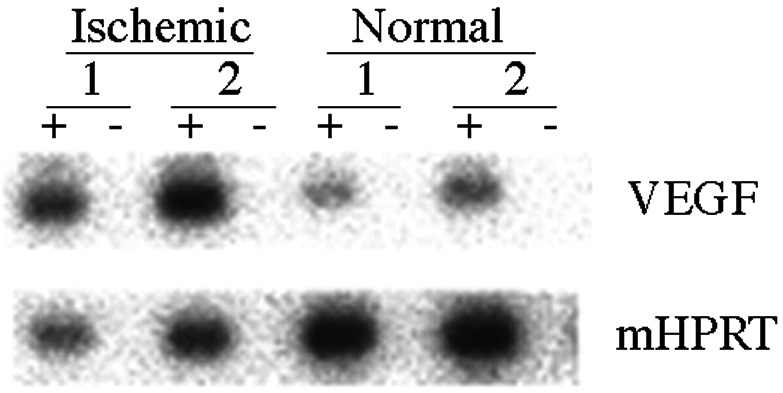
Semiquantitative RT-PCR analysis for VEGF expression in AAVH9VEGF vector-inoculated hearts. VEGF was amplified with 35 cycles. Mouse HPRT (mHPRT) was amplified 15 cycles and used as internal control. +, PCR products with RT; −, PCR without RT. The quantification was done by PhosphoImager analyses. The numbers indicate each individual animal.
Discussion
Despite angioplasty and bypass surgery, many patients have areas of myocardial ischemia that cannot be revascularized because of diffuse coronary artherosclerosis, occluded bypass grafts, or excess risk of surgery. The discovery of growth factors that stimulate blood vessel formation has opened up new approaches for the investigation of treatment of vascular insufficiencies (28–30). The growth factors that have been investigated for new blood vessel formations are VEGFs (31–33), fibroblast growth factors (34–36), and angiopoietins (37).
VEGF is a potent and highly specific mitogen for endothelial cells and induces angiogenesis (38–44). It is also synthesized and secreted by many differentiated cells in response to a variety of stimuli including hypoxia (19, 45–55). VEGF has been administered to ischemic hearts by various routes to improve coronary circulation. VEGF injected intravenously improved myocardial blood flow but also produced hypotension as a side effect (56). Alternatively, VEGF has been injected directly into the coronary arteries (32). The effect was transient because most of the angiogenic factors were not retained in the myocardium. The results in pig experiments with recombinant VEGF protein were revealing. Even a single intracoronary administration of VEGF was effective in increasing coronary blood flow and resulted in functional improvement despite the fact that only a small fraction of protein was localized to the ischemic area (57). However, brief exposure to rhVEGF165 in humans was insufficient to trigger and maintain a therapeutically meaningful angiogenic response in clinical trials, especially in the context of extensive atherosclerotic disease (30). Thus, long-lasting VEGF stimulation is needed for treatment of patients. Intramyocardial injection of the proteins has also been tried (36). Because the protein is likely to have limited lifespan, this mode of administration is not expected to have lasting effects. More prolonged effect could be achieved by injecting DNA encoding the genes for the angiogenic factors. Intramuscular injections of plasmid encoding for VEGF have been effective in animal experiments and in some patients with peripheral vascular disease (58–62). Injection of plasmid-carrying angiogenic factors for treatment of coronary artery disease is currently being tested (30, 31). The therapeutic usefulness of this approach has been limited by the low efficiency of cardiomyocyte transduction (0.1–1.0% of cardiomyocytes in the area of injection) (63). Moreover, Schwarz et al. (64) showed that injection of high doses of VEGF plasmid to ischemic heart caused the complication of angioma formation. This complication also occurred when VEGF was delivered by implantation of retroviral-vector-transduced myoblasts to ischemic rat leg (3) and ischemic rat heart (2). Moreover, increased systemic VEGF levels could cause some unwanted side effects such as inducing inappropriate angiogenesis at sites of vascular derangement or at sites where angiogenesis might have major adverse consequences; for example, in the retina, the synovium, and occult tumors (65). Hence, it is important to control VEGF expression when gene transduction is used.
Although other regulation systems are available, the ideal control system for expression of VEGF in ischemic heart is to let the gene expression respond to hypoxia, because evidence shows that different dosage and duration of VEGF stimulation may yield different results. For example, VEGF stimulates angiogenesis and vascular permeability at low doses but stimulates only angiogenesis at a high dose (66). Short-term administration of VEGF could not stimulate myocardial angiogenesis in initial clinical trials. In addition, transient VEGF expression after adenoviral gene transfer in the heart only induces a short-lived angiogenic response (67). Under inappropriate conditions, VEGF will not induce angiogenesis or only induce nonfunctional hypoperfused capillaries that will regress when VEGF levels drop (67). In these cases, no relief of ischemia occurs. For treatment of ischemic heart disease, the angiogenic therapy is aimed at stimulating functional and sustainable new blood vessels. VEGF expression should be turned off when the blood supply to the myocardium is adequate. Therefore, the HRE is an ideal regulatory system for controlling VEGF gene expression in the myocardium.
In these studies, we demonstrated an increased level of HIF-1α in ischemic myocardium as early as 2 days after coronary artery occlusion, and the increase was still present 2 weeks later. Thus, it is feasible to use the HIF-1/HRE system to control VEGF gene expression in the ischemic heart. We chose to use nine copies of Epo HRE in our studies because, in previous experiments with brain tumor cell lines, nine copies of HRE yielded the best induction of gene expression under hypoxic and anoxic conditions (23). Adeno-associated viral vector was used to deliver the transgenes because this vector can induce long-lasting transgene expression when injected into skeletal muscle (68–70), and we have shown that VEGF gene delivered by this viral vector can induce neovasculature formation in ischemic heart (1). Two AAV vectors were made, one carried the LacZ gene and the other the VEGF gene. Up-regulated gene expression in hypoxic and anoxic conditions was detected in several cell lines known to express HIF-1. The inductions were 10–30 times for LacZ in different cell lines (Table 1) under anoxic condition, and 119 and 33 times for VEGF in Hep3B cells in hypoxic and anoxic conditions, respectively (Table 2). Induction of gene expression was also observed in ischemic hearts that had been transduced with these two vectors, although the induction level is not as high as in in vitro experiment. We attribute this different induction level to the variable O2 tension in different regions of the ischemic heart; also some AAV vectors could have infected cardiac myocytes outside of the ischemic region.
In conclusion, we have demonstrated that nine copies of Epo HRE can induce LacZ and VEGF gene expression in cultured cell lines in vitro and ischemic mouse myocardium in vivo. These results indicate that HIF-1/HRE could provide a useful system for controlling angiogenic factor expression in the treatment of coronary artery disease. Its use can most likely be extended to the treatment of arterial insufficiency in the limb.
Acknowledgments
We thank Dr. Ronghua Lu for help in presurgery care of the mice and Dr. William Grossman for critical reading of the manuscript.
Abbreviations
- HRE
hypoxia-responsive element
- VEGF
vascular endothelial growth factor
- HIF-1
hypoxia-inducible factor-1
- Epo
erythropoietin
- HPRT
hypoxanthine phosphoribosyltransferase
- RT
reverse transcription
References
- 1.Su H, Lu R, Kan Y W. Proc Natl Acad Sci USA. 2000;97:13801–13806. doi: 10.1073/pnas.250488097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee R J, Springer M L, Blanco-Bose W E, Shaw R, Ursell P C, Blau H M. Circulation. 2000;102:898–901. doi: 10.1161/01.cir.102.8.898. [DOI] [PubMed] [Google Scholar]
- 3.Springer M L, Chen A S, Kraft P E, Bednarski M, Blau H M. Mol Cells. 1998;2:549–558. doi: 10.1016/s1097-2765(00)80154-9. [DOI] [PubMed] [Google Scholar]
- 4.Bohl D, Salvetti A, Moullier P, Heard J M. Blood. 1998;92:1512–1517. [PubMed] [Google Scholar]
- 5.Hofmann A, Nolan G P, Blau H M. Proc Natl Acad Sci USA. 1996;93:5185–5190. doi: 10.1073/pnas.93.11.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohl D, Naffakh N, Heard J M. Nat Med. 1997;3:299–305. doi: 10.1038/nm0397-299. [DOI] [PubMed] [Google Scholar]
- 7.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, O'Malley B W, Jr, Tsai S Y, O'Malley B W. Proc Natl Acad Sci USA. 1994;91:8180–8184. doi: 10.1073/pnas.91.17.8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blau H M, Rossi F M. Proc Natl Acad Sci USA. 1999;96:797–799. doi: 10.1073/pnas.96.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi F M, Guicherit O M, Spicher A, Kringstein A M, Fatyol K, Blakely B T, Blau H M. Nat Genet. 1998;20:389–393. doi: 10.1038/3871. [DOI] [PubMed] [Google Scholar]
- 11.Kringstein A M, Rossi F M, Hofmann A, Blau H M. Proc Natl Acad Sci USA. 1998;95:13670–13675. doi: 10.1073/pnas.95.23.13670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semenza G L. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 13.Wang G L, Jiang B H, Rue E A, Semenza G L. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G L, Semenza G L. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 15.Huang L E, Gu J, Schau M, Bunn H F. Proc Natl Acad Sci USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lando D, Peet D J, Whelan D A, Gorman J J, Whitelaw M L. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 17.Jiang B H, Rue E, Wang G L, Roe R, Semenza G L. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 18.Forsythe J A, Jiang B H, Iyer N V, Agani F, Leung S W, Koos R D, Semenza G L. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy A P, Levy N S, Wegner S, Goldberg M A. J Biol Chem. 1995;270:13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- 20.Gupta M, Mungai P T, Goldwasser E. Blood. 2000;96:491–497. [PubMed] [Google Scholar]
- 21.Koshikawa N, Takenaga K, Tagawa M, Sakiyama S. Cancer Res. 2000;60:2936–2941. [PubMed] [Google Scholar]
- 22.Dachs G U, Patterson A V, Firth J D, Ratcliffe P J, Townsend K M, Stratford I J, Harris A L. Nat Med. 1997;3:515–520. doi: 10.1038/nm0597-515. [DOI] [PubMed] [Google Scholar]
- 23.Ruan H, Su H, Hu L, Lamborn K R, Kan Y W, Deen D F. Neoplasia. 2001;3:255–263. doi: 10.1038/sj.neo.7900157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semenza G L, Wang G L. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang G L, Semenza G L. Proc Natl Acad Sci USA. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman G J, Iwaki Y, Colosi P. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 27.Chapdelaine P, Vignola K, Fortier M A. BioTechniques. 2001;31:478–482. doi: 10.2144/01313bm04. [DOI] [PubMed] [Google Scholar]
- 28.Folkman J. Circulation. 1998;97:628–629. doi: 10.1161/01.cir.97.7.628. [DOI] [PubMed] [Google Scholar]
- 29.Yla-Herttuala S, Martin J F. Lancet. 2000;355:213–222. doi: 10.1016/S0140-6736(99)04180-X. [DOI] [PubMed] [Google Scholar]
- 30.Ferrara N, Alitalo K. Nat Med. 1999;5:1359–1364. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- 31.Losordo D W, Vale P R, Symes J F, Dunnington C H, Esakof D D, Maysky M, Ashare A B, Lathi K, Isner J M. Circulation. 1998;98:2800–2804. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- 32.Pearlman J D, Hibberd M G, Chuang M L, Harada K, Lopez J J, Gladstone S R, Friedman M, Sellke F W, Simons M. Nat Med. 1995;1:1085–1089. doi: 10.1038/nm1095-1085. [DOI] [PubMed] [Google Scholar]
- 33.Takeshita S, Pu L Q, Stein L A, Sniderman A D, Bunting S, Ferrara N, Isner J M, Symes J F. Circulation. 1994;90:II228–234. [PubMed] [Google Scholar]
- 34.Giordano F J, Ping P, McKirnan M D, Nozaki S, DeMaria A N, Dillmann W H, Mathieu-Costello O, Hammond H K. Nat Med. 1996;2:534–559. doi: 10.1038/nm0596-534. [DOI] [PubMed] [Google Scholar]
- 35.Harada K, Grossman W, Friedman M, Edelman E R, Prasad P V, Keighley C S, Manning W J, Sellke F W, Simons M. J Clin Invest. 1994;94:623–630. doi: 10.1172/JCI117378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schumacher B, Pecher P, von Specht B U, Stegmann T. Circulation. 1998;97:645–650. doi: 10.1161/01.cir.97.7.645. [DOI] [PubMed] [Google Scholar]
- 37.Shyu K G, Manor O, Magner M, Yancopoulos G D, Isner J M. Circulation. 1998;98:2081–2087. doi: 10.1161/01.cir.98.19.2081. [DOI] [PubMed] [Google Scholar]
- 38.Muller Y A, Li B, Christinger H W, Wells J A, Cunningham B C, de Vos A M. Proc Natl Acad Sci USA. 1997;94:7192–7197. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrara N. Breast Cancer Res Treat. 1995;36:127–137. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 40.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 41.Dvorak H F, Brown L F, Detmar M, Dvorak A M. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 42.Neufeld G, Tessler S, Gitay-Goren H, Cohen T, Levi B Z. Prog Growth Factor Res. 1994;5:89–97. doi: 10.1016/0955-2235(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 43.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al. Nature (London) 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 44.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea K S, Powell-Braxton L, Hillan K J, Moore M W. Nature (London) 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 45.Ladoux A, Frelin C. Biochem Biophys Res Commun. 1993;195:1005–1010. doi: 10.1006/bbrc.1993.2144. [DOI] [PubMed] [Google Scholar]
- 46.Shweiki D, Itin A, Soffer D, Keshet E. Nature (London) 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 47.Finkenzeller G, Technau A, Marme D. Biochem Biophys Res Commun. 1995;208:432–439. doi: 10.1006/bbrc.1995.1356. [DOI] [PubMed] [Google Scholar]
- 48.Connolly D T, Heuvelman D M, Nelson R, Olander J V, Eppley B L, Delfino J J, Siegel N R, Leimgruber R M, Feder J. J Clin Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burger P C, Vogel F S, Green S B, Strike T A. Cancer. 1985;56:1106–1111. doi: 10.1002/1097-0142(19850901)56:5<1106::aid-cncr2820560525>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 50.Dvorak H F, Sioussat T M, Brown L F, Berse B, Nagy J A, Sotrel A, Manseau E J, Van de Water L, Senger D R. J Exp Med. 1991;174:1275–1278. doi: 10.1084/jem.174.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrara N, Houck K A, Jakeman L B, Winer J, Leung D W. J Cell Biochem. 1991;47:211–218. doi: 10.1002/jcb.240470305. [DOI] [PubMed] [Google Scholar]
- 52.Jakeman L B, Winer J, Bennett G L, Altar C A, Ferrara N. J Clin Invest. 1992;89:244–253. doi: 10.1172/JCI115568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Claffey K P, Shih S C, Mullen A, Dziennis S, Cusick J L, Abrams K R, Lee S W, Detmar M. Mol Biol Cell. 1998;9:469–481. doi: 10.1091/mbc.9.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Claffey K P, Wilkison W O, Spiegelman B M. J Biol Chem. 1992;267:16317–16322. [PubMed] [Google Scholar]
- 55.Detmar M, Brown L F, Berse B, Jackman R W, Elicker B M, Dvorak H F, Claffey K P. J Invest Dermatol. 1997;108:263–268. doi: 10.1111/1523-1747.ep12286453. [DOI] [PubMed] [Google Scholar]
- 56.Hariawala M D, Horowitz J R, Esakof D, Sheriff D D, Walter D H, Keyt B, Isner J M, Symes J F. J Surg Res. 1996;63:77–82. doi: 10.1006/jsre.1996.0226. [DOI] [PubMed] [Google Scholar]
- 57.Lopez J J, Laham R J, Stamler A, Pearlman J D, Bunting S, Kaplan A, Carrozza J P, Sellke F W, Simons M. Cardiovasc Res. 1998;40:272–281. doi: 10.1016/s0008-6363(98)00136-9. [DOI] [PubMed] [Google Scholar]
- 58.Takeshita S, Tsurumi Y, Couffinahl T, Asahara T, Bauters C, Symes J, Ferrara N, Isner J M. Lab Invest. 1996;75:487–501. [PubMed] [Google Scholar]
- 59.Tsurumi Y, Takeshita S, Chen D, Kearney M, Rossow S T, Passeri J, Horowitz J R, Symes J F, Isner J M. Circulation. 1996;94:3281–3290. doi: 10.1161/01.cir.94.12.3281. [DOI] [PubMed] [Google Scholar]
- 60.Bauters C, Asahara T, Zheng L P, Takeshita S, Bunting S, Ferrara N, Symes J F, Isner J M. Am J Physiol. 1994;267:H1263–H1271. doi: 10.1152/ajpheart.1994.267.4.H1263. [DOI] [PubMed] [Google Scholar]
- 61.Tsurumi Y, Kearney M, Chen D, Silver M, Takeshita S, Yang J, Symes J F, Isner J M. Circulation. 1997;96,Suppl. II:382–388. [PubMed] [Google Scholar]
- 62.Majesky M W. Circulation. 1996;94:3062–3064. doi: 10.1161/01.cir.94.12.3062. [DOI] [PubMed] [Google Scholar]
- 63.Fisher K J, Jooss K, Alston J, Yang Y, Haecker S E, High K, Pathak R, Raper S E, Wilson J M. Nat Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- 64.Schwarz E R, Speakman M T, Patterson M, Hale S S, Isner J M, Kedes L H, Kloner R A. J Am Coll Cardiol. 2000;35:1323–1330. doi: 10.1016/s0735-1097(00)00522-2. [DOI] [PubMed] [Google Scholar]
- 65.Folkman J, Shing Y. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 66.Carmeliet P. Nat Med. 2000;6:1102–1103. doi: 10.1038/80430. [DOI] [PubMed] [Google Scholar]
- 67.Pettersson A, Nagy J A, Brown L F, Sundberg C, Morgan E, Jungles S, Carter R, Krieger J E, Manseau E J, Harvey V S, et al. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- 68.Kessler P D, Podsakoff G M, Chen X, McQuiston S A, Colosi P C, Matelis L A, Kurtzman G J, Byrne B J. Proc Natl Acad Sci USA. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Herzog R W, Hagstrom J N, Kung S H, Tai S J, Wilson J M, Fisher K J, High K A. Proc Natl Acad Sci USA. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Snyder R O, Spratt S K, Lagarde C, Bohl D, Kaspar B, Sloan B, Cohen L K, Danos O. Hum Gene Ther. 1997;8:1891–1900. doi: 10.1089/hum.1997.8.16-1891. [DOI] [PubMed] [Google Scholar]