

Abstract
The molecular chaperone Hsp90 sequesters oncogenic mutants of the tumor suppressor p53 that have unstable core domains. It is not known whether p53 is bound in an unfolded, partly folded, or distorted structure, as is unknown for the structure of any bound substrate of Hsp90. It is a particularly difficult problem to analyze in detail the structures of large complexes in which one component is (partly) unfolded. We have shown by transverse relaxation-optimized NMR spectroscopy combined with cross-correlated relaxation-enhanced polarization transfer (CRINEPT-TROSY) that p53 core domain bound in an ≈200-kDa complex with Hsp90 was predominantly unfolded lacking helical or sheet secondary structure. This mode of binding might be a general feature of substrates of Hsp90.
Keywords: molecular chaperones, transcription factors, protein folding
The transcription factor p53 is the most frequently mutated gene in human tumors (1, 2). Most mutations map within its DNA binding core domain (p53core) (3, 4), and many mutations just destabilize the protein (5). Oncogenic mutants from one allele may inactivate wild-type protein from the other allele by formation of inactive tetramers (6, 7). A culprit in this process is the molecular chaperone Hsp90. It stabilizes unstable mutants such as p53-V143A, allowing subsequent hetero-oligomerization (8). A recent in vitro study suggests that Hsp90 might even bind to wild-type p53 (9, 10). Hsp90 became a drug target in cancer therapy when geldanamycin and similar compounds were found to block substrate release from Hsp90 (11–13), which prevents the formation of p53 hetero-oligomers (8). Hsp90 is a dimer, abundant in the cytosol. Although Hsp90 is a heat shock protein that binds to unfolded proteins in general, it is thought to be required for a specific set of kinases and transcription factors (14–16). It is unknown how the chaperone is specific for these proteins, which are unrelated in sequence and structure. It is also unclear in which shape Hsp90 holds its substrate; whether it is fully denatured or whether it is a late folding intermediate, as proposed for citrate synthase (15, 17). Because p53 is a natural substrate of Hsp90, the Hsp90–p53 complex is a suitable model to study these interactions.
p53 has a well defined domain structure, including the core and tetramerization domains that fold independently. The most unfortunate p53 mutants are those whose instability is restricted to the core domain. The isolated monomeric core domain fragment has proved to be a suitable system in which to study the destabilizing effects of mutations (5, 18). The residues affected by various mutations have been mapped by NMR spectroscopy (19). Conventional NMR spectroscopy is limited to protein complexes of less than 50 kDa and cannot be applied to the Hsp90–p53core complex, which is close to 200 kDa. Wüthrich and colleagues (20, 21) have recently pioneered novel methods of NMR that can be applied to such large complexes. We set out to analyze the structural requirements for p53core interaction with Hsp90 and to characterize the complex by transverse relaxation-optimized NMR spectroscopy combined with cross-correlated relaxation-enhanced polarization transfer (CRINEPT-TROSY) NMR spectroscopy.
Materials and Methods
Protein Purification.
The purification of human Hsp90 β was modified from published protocols (22) by using Rosetta cells (Novagen) and Q-Sepharose HiTrap, heparin-Sepharose and MonoQ media (Amersham Pharmacia). p53core was purified as described (19).
Fluorescence Spectroscopy.
Experiments were performed with a Perkin–Elmer LS50B fluorimeter equipped with a Neslab RTE-111 water circulator, controlled by laboratory software. Temperature dependence of fluorescence was measured by stepwise increases of temperature of 1°C and equilibrating the sample for at least 5 min before measurement. The temperature was monitored by a thermometer inside the cuvette. The samples were in P1 buffer (25 mM phosphate buffer (pH 7.2)/150 mM NaCl/150 mM KCl/5 mM DTT). An excitation wavelength of 270 nm was found to be optimal for the Hsp90–p53core system. Spectra were recorded between 300 and 400 nm.
Size Exclusion Chromatography.
The samples (50 μl) were separated in 10-drop fractions (≈300 μl) from a G75 size exclusion column (6 × 1.6 cm) in 25 mM phosphate buffer (pH 6.8), 150 mM NaCl, and 5 mM DTT. The samples were centrifuged for 5 min at 12,000 × g before loading on the column.
NMR Spectroscopy.
NMR spectra were acquired on a DRX800 (Bruker) with Z-gradients and a triple resonance probe utilizing pulse sequences published by Wüthrich and colleagues (21). Transfer periods of 3.6 ms provided optimal efficiency for Hsp90 and Hsp90–p53core. Typical acquisition times were in the order of 12 h for a matrix of 1,024 × 128 complex points. The samples were in P1 buffer containing 2% D2O. Addition of Complete protease inhibitor (Roche Molecular Biochemicals) stabilized the samples without affecting the spectra.
Results
Binding of Wild-Type and Mutant p53core to Hsp90 Depends on Their Stability.
To apply these new NMR methods, we first determined the conditions where Hsp90 binds to p53core wild-type and mutant proteins. We used temperature-dependent fluorescence to monitor the denaturation of p53core and its association with Hsp90 (Fig. 1A). The fluorescence of p53core strongly increased above 35°C, indicating unfolding (18). At above ≈41°C the fluorescence dropped because of light scattering caused by aggregation of the core domain and by thermoquenching. In contrast, the fluorescence of Hsp90 alone decreased monotonically until around 50°C, when Hsp90 unfolded. We calculated a curve out of the sum of the spectra of Hsp90 and p53core (Fig. 1A). Any deviation from that curve indicates an interaction between both proteins. There was not the strong increase in fluorescence around 35°C when p53core was heated together with Hsp90. Above the melting temperature of p53core, we observed only the slight decrease of fluorescence intensity due to thermoquenching but no drop due to light scattering. The sum of fluorescence for the separate samples of Hsp90 and p53core between 35°C and 41°C was considerably higher than for the combined sample. These data showed that Hsp90 bound p53core and prevented its aggregation.
Fig 1.
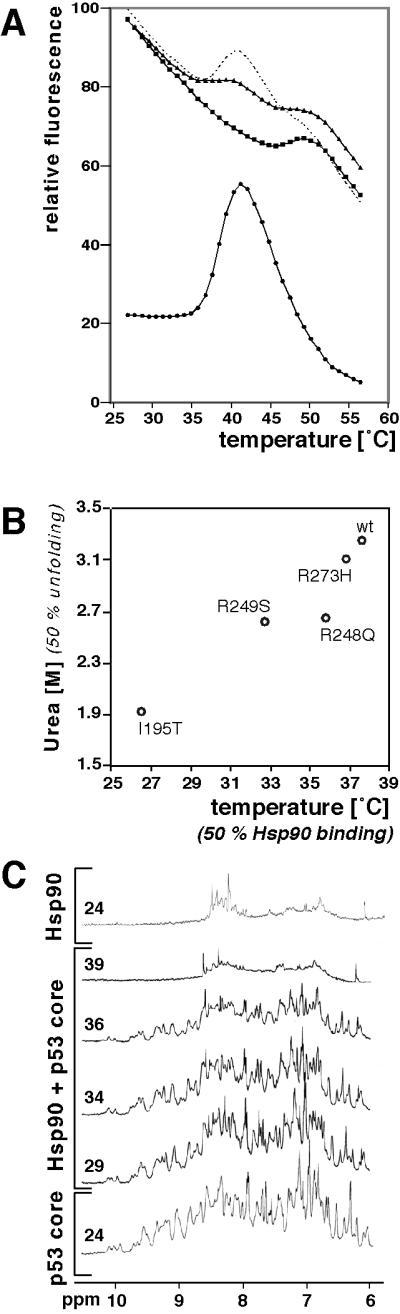
Hsp90 bound to p53core wild type and mutants after thermal denaturation. (A) Melting curves of wild type p53core (7.5 μM) in the presence (triangles) and absence (circles) of Hsp90 (10 μM dimer), and of Hsp90 in the absence of p53core (squares). Hsp90 unfolded at ≈50°C, whereas p53core unfolded at ≈37°C. In the absence of Hsp90 the fluorescence signal of p53core increased after unfolding because aggregates caused light scattering. In the presence of Hsp90 no aggregation was observed; therefore, the fluorescence values were significantly below the combined values of the isolated Hsp90 and p53core species. The summed curve of Hsp90 and p53core (dotted line; corrected for the inner filter effect) had a more prominent maximum than the curve of the combined sample. Fluorescence at 360 nm is shown at an excitation of 270 nm. (B) Hsp90 binding correlated with p53core unfolding. The urea concentrations used by Bullock et al. (23) to reach the 50% unfolding transition point of p53core species indicated were plotted versus the midpoints of the complex formation with Hsp90. (C) Complex formation between p53core (50 μM) and Hsp90 (60 μM dimer) was monitored by one-dimensional NMR. The amide region is shown of spectra acquired at temperature indicated in °C. At 39°C, the spectrum of the sample containing both p53core and Hsp90 was close to that of Hsp90 alone at 24°C, whereas up to 34°C the spectra were close to that of p53core alone at 24°C. At 36°C the signal intensity began already to decrease.
The thermal unfolding of mutants of different stability (R273H, R249S, R248Q, and I195T) was qualitatively similar to wild type except that both the melting points and the Hsp90 binding transitions were shifted to lower temperatures (not shown). We could calculate the transition temperatures for every p53core species in the presence of Hsp90. The midpoints for the urea denaturation curves of these mutants (23), which are measures of relative stability, were linearly correlated with the transition temperatures in the presence of Hsp90 (Fig. 1B). Thus, the binding of p53 mutants to Hsp90 depended on their being unfolded and was inversely proportional to their stability. Together, these data show that Hsp90 interaction with p53 was not restricted to particular mutants but depended solely on the stability of each mutant.
Hsp90 and p53core Do Not Interact at Lower Temperatures.
We monitored by one-dimensional NMR samples containing Hsp90 and p53core at increasing temperature (Fig. 1C) to see whether a complex was formed at lower temperatures. In general, the intensity of NMR signals decreases due to transverse relaxation with increasing size of complexes because the tumbling rate of larger particles is lower. The size of the complex of p53core bound to Hsp90 dimer is 191 kDa if the Hsp90 dimer binds one core domain, 216 kDa if it binds two. If the small 25-kDa core domain is bound stably by Hsp90, its signals should, therefore, be reduced significantly. The spectra of samples containing both, Hsp90 and p53core were, up to 34°C, similar to that of p53core alone (Fig. 1C), showing that no stable complex was formed below 34°C at the experimental concentration. At 39°C, however, the spectrum of the sample containing both proteins was very similar to that of Hsp90 at 24°C (Fig. 1C), indicating that no significant amount of free p53core was left in solution. There was some decrease in intensity at 36°C (Fig. 1C), consistent with the fluorescence data (Fig. 1A). Importantly, the solution remained mainly clear at 39°C in the presence of Hsp90, whereas there was strong aggregation in its absence. Further, SDS/PAGE confirmed that p53core remained in solution after heating in the presence but not the absence of Hsp90 (Fig. 2A). Analysis by size exclusion chromatography showed binding to Hsp90, too (Fig. 2B). These data indicated that p53core formed a stable complex with Hsp90 only after unfolding but not at lower temperatures. Two-dimensional NMR spectra using 15N-labeled p53core (100 μM) were identical in the presence and the absence of Hsp90 (70 μM dimer) at 24°C and 29°C (not shown). This excluded transient interaction below the transition point.
Fig 2.

Hsp90 prevents aggregation of p53core. (A) Hsp90 and p53core were slowly heated in the NMR up to 36°C. Fractions of total and supernatant were separated on SDS/PAGE. In this case, denaturation of p53core was not driven to completion; therefore, a small amount of p53core could be detected by SDS/PAGE (and also by NMR), even in the absence of Hsp90 (indicated by “90”). (B) p53core comigrates with Hsp90 on a size exclusion column. p53core migrates in the included volume but is shifted by Hsp90 into the void volume. p53core (89 μM) was either in the presence of Hsp90 (64-μM dimer) subjected to a mild heating protocol similar to that in Fig. 1C or left alone at 4°C (p53core heated in the absence of Hsp90 aggregated and could not be detected anymore). Fractions were analyzed by SDS/PAGE. Fraction numbers were indicated on top. Presence of Hsp90 is indicated at the bottom (“90”).
CRINEPT-TROSY NMR Is Suitable for 166-kDa Hsp90.
The strong reduction in signal intensity due to transverse relaxation makes it difficult to gain structural information about the Hsp90–p53core complex. Transverse relaxation is caused by proton spin–spin interactions, which could be overcome in two ways. First, perdeuteration of side chains reduced the number of potential spin–spin interactions. Second, transverse relaxation optimized spectroscopy, TROSY, was used (20). To establish NMR conditions, we initially acquired spectra of the 166-kDa 15N-labeled perdeuterated Hsp90 dimer. However, the TROSY spectrum of Hsp90 revealed only a few signals in the random coil region corresponding to flexible parts of Hsp90 (Fig. 3, red signals). We could not detect any signals appearing in regions normally associated with helical or sheet secondary structure.
Fig 3.

Hsp90 dimer is suitable for CRINEPT-TROSY NMR spectroscopy. The TROSY spectrum of 15N-labeled perdeuterated Hsp90 (158 μM dimer) showed signals in the random coil region only (red). The CRINEPT-TROSY spectrum of the same sample revealed a broad distribution of signals outside of this region (blue). The flexible signals visible already in the TROSY were doubled in the CRINEPT-TROSY. They were shifted by 90 Hz in both dimensions caused by the 1H-15N coupling, which is visualized by an additional overlay of the TROSY in yellow.
For further gain in sensitivity, we had to overcome relaxation during magnetization transfer periods, which becomes limiting for very large systems and which is not suppressed by TROSY. To optimize transfer efficiency, we combined TROSY with CRINEPT, cross-correlated relaxation enhanced polarization transfer (21). This should allow spectra of molecules even above 200 kDa to be obtained (24), but has yet to be applied to key biological questions. The CRINEPT-TROSY spectrum of 15N-labeled perdeuterated Hsp90 showed the wide-ranging distribution of signals over the 1H dimension, as expected for a folded protein (Fig. 3, blue). Most of these signals were not detected in the conventional TROSY spectrum. However, the signals that could be detected in the TROSY appeared with doublets separated by 90 Hz in both dimensions caused by 1H-15N coupling (indicated in yellow in Fig. 3). This is one potential disadvantage of CRINEPT-TROSY, that for flexible regions of a large molecule a second faster relaxing component is retained, causing doubling of the corresponding signals. The distinctly different relaxation behavior of these signals marked them as signals from flexible residues, all other signals corresponded, therefore, to rigid parts. A subset of signals appeared in the region typical for β-sheets, while the majority of signals was seen in the α-region. This spectrum indicated that it is possible to obtain structural information by NMR spectroscopy of proteins of nearly 200 kDa at close to physiological concentration.
Hsp90-Bound p53core Lacks Sheet and Helix Secondary Structure.
The CRINEPT-TROSY spectrum of the complex of 15N-labeled perdeuterated p53core bound to nonlabeled perdeuterated Hsp90 revealed signals in the region typical for random coil only (Fig. 4, blue). The selective labeling allowed detection of p53core only. We succeeded in forming a complex with Hsp90 by slowly increasing the temperature up to 36°C. This temperature is very close to the physiological value of 37°C. This emphasizes the biological significance of our approach. Completion of the formation of the complex was monitored by one-dimensional NMR spectroscopy. Afterward, the temperature was lowered to 29°C to stabilize the solution during acquisition of spectra. Free p53core aggregates even at a 20-fold lower concentration at this temperature over 12 h, whereas the NMR samples of p53core-Hsp90 complex were stable for up to 72 h, which was also checked by SDS/PAGE (Fig. 2A). Further, complexes were stable during gel filtration runs (Fig. 2B). We would not expect structural changes in the complex on decreasing the temperature of the complex after its formation.
Fig 4.

Hsp90-bound p53core is predominantly unfolded. The CRINEPT-TROSY spectrum of 15N-labeled perdeuterated p53core (100 μM) bound to perdeuterated Hsp90 (147 μM dimer) exhibited signals in the region typical for random coil (blue). It lacked significant contributions in the regions typical for sheets or helices as it can be found for folded p53core (green; ref. 19). The complex was formed by increasing the temperature slowly up to 36°C; the spectrum was taken at 29°C. The heteronuclear sequential quantum correlation (HSQC) spectrum of p53core unfolded in 3.5 M urea at 298 K was underlaid (orange). Signals occurred in the random coil region only, although differences to the spectrum of p53core bound to Hsp90 did exist. All three spectra could be matched on the common signals belonging to the residues of the flexible C terminus.
The characteristic distribution of signals of folded p53core disappeared (Fig. 4, green; ref. 19). A small subset of signals remained unchanged compared with the spectrum of folded p53core. These signals were also the only ones that could be found in the TROSY spectrum of the same sample (not shown). They could be assigned to the very C terminus of the core domain fragment (residues 289–312). This tail was found to be very flexible by NMR (B. S. DeDecker, S.M.V.F., and A.R.F., unpublished data) and was invisible in the crystal structure of the core domain (25). All other signals observed in the spectrum of the complex were not present in the complex of folded p53core. The appearance of these new signals is again positive evidence for the formation of the p53core–Hsp90 complex. Despite the sensitivity of the CRINEPT-TROSY approach as shown in Fig. 3, no signals were found in the regions typical for helical and sheet secondary structure, and all signals that appeared in the complex but which were absent in the spectrum of free, folded p53core were found in the random coil region (Fig. 4, blue). This suggests that p53core bound to Hsp90 was predominantly characterized by random coil secondary structure. Because all signals clustered in a small region of the spectrum, which was additionally crowded by the more intensive duplicated signals, it is not possible to estimate the number of residues for which signals can be observed. However, many of the signals appearing only in the CRINEPT-TROSY spectrum were not duplicated. Therefore, there must have been restricted flexibility of the core domain in the chaperone complex, although p53core was held by Hsp90 in random coil secondary structure.
The spectrum of p53core that was fully unfolded in urea was similar, although not identical, to that of the complex (Fig. 4, orange). Both spectra had signals in the random coil region only; in particular, the residues of the flexible C terminus matched with each other. It should be noted that the spectrum of p53core in urea shows sharp signals detectable by conventional HSQC NMR technique. Such signals would be expected of p53core if it was present in solution after thermal denaturation in a free form and not bound to Hsp90. This also confirms the existence of the complex of Hsp90 and p53core reported in Fig. 4.
Discussion
We have shown that wild-type p53core and mutants on denaturation bind to Hsp90, and that CRINEPT-TROSY NMR spectroscopy is suitable for obtaining structural information about the p53core–Hsp90 complex. The NMR spectrum of p53core bound to Hsp90 was predominantly characterized by signals typical for an unfolded protein.
The most likely interpretations of the spectral data are illustrated in Fig. 5. The possibility that p53core is bound in its native state to Hsp90 (Fig. 5A) can be excluded because p53core wild type and mutants bound Hsp90 only at temperatures at which they unfold (Fig. 1 A and B), and the NMR spectrum of Hsp90-bound p53core showed only signals belonging to unfolded but not to folded protein (Fig. 4). The possibility of there being a partially unfolded p53core that retained a significant part of its structure stabilized by Hsp90 in the native conformation (Fig. 5B) would be compatible with the fluorescence data in Fig. 1. But, such a complex would require that a subset of the signals of native p53core should appear in the NMR spectrum, which was not observed (Fig. 4). A third possibility is that p53core is bound by Hsp90 in an unfolded form that is in equilibrium with partially folded conformations or that the complex is present as ensemble of different conformations (Fig. 5C). This interpretation is consistent with the data presented here. But, the appearance of a partially native structure would require the Hsp90-dependent induction of an unusual stabilization of a part of the β-sandwich core of p53core. An interchange of several native-like forms seems unlikely because lack of conformational variability is a defining criterion of the native state in contrast to the unfolded state. The simplest interpretation is that Hsp90-bound p53core adopts an unfolded conformation (Fig. 5D). Unfolded states are flexible; therefore, this mode is not very different from that represented by Fig. 5C. Both 5 C and D lead to the conclusion that Hsp90-bound p53core is predominantly unfolded.
Fig 5.

Modes of binding of p53core to Hsp90. Schematic illustration of the binding of p53core (black) to the Hsp90 dimer (gray). Four complexes are shown that represent different modes of binding. (A) p53 core binds Hsp90 in the native state. This is incompatible with the data shown in Figs. 1 and 4. (B) p53core is partially stabilized in the native state. This is incompatible with the data shown in Fig. 4. (C) Bound p53core is in an equilibrium of unfolded and partially folded form, or present as a mixture of such states. This is compatible with the data, but requires a unusual stabilization of the partially folded β-sandwich of p53core. (D) Bound p53core is unfolded. This is compatible with the data. Variations of A–D are possible, but their main features should be found in one of the four modes. This figure does not imply a specific stoichiometry of the complex.
These interpretations assume that the complex is stable on the NMR time scale. The stability of the complex was shown by one-dimensional NMR (Fig. 1C) and by size-exclusion chromatography (Fig. 2B). Further, dissociation of unfolded p53core in the experiment shown in Fig. 4 would have resulted in its precipitation, which was not observed (Fig. 2A). Dissociation of native p53core would have resulted in strong signals of free, native p53core in the NMR spectrum of the p53core–Hsp90 complex, which were not observed (Fig. 4). Accordingly, native p53core did not dissociate from Hsp90 under our experimental conditions.
Oncogenic mutants of p53 that are destabilized melt at lower temperatures than wild type (5). The destabilized mutants will be preferentially targeted by Hsp90. Perhaps the oncogenic effects of these mutants depends on Hsp90 keeping them soluble. In the absence of Hsp90, the core domains of the destabilized mutants aggregate (5), so taking them out of solution. But, in the presence of Hsp90, they can be solubilized and their tetramerization domains can be accessible to forming mixed, inactive hybrids with wild-type p53.
The structural information obtained for p53 is the first example for a natural protein substrate bound to Hsp90 or, indeed, any other chaperone. It is dangerous to extrapolate from a database of just one example and so it cannot be ruled out that other substrates of Hsp90 exhibit different overall structural properties on binding to the chaperone. However, based on these data for the natural substrate p53 it seems unlikely that Hsp90 binding requires conserved three-dimensional properties that could be specific for late folding states or a defined native motif. Binding modes as illustrated in Fig. 5 C and D can explain why Hsp90 is able to recognize diverse substrates, varying from those with β-sheet cores to all α-helical proteins, which are only expected to have common structural features in their unfolded state. Nevertheless, GroEL, which is specific for denatured states of proteins, binds some features of denatured states, such as extended β-strands (26) and exposed hydrophobic faces of amphipathic helices (27) can be present in partly denatured states or even in native structures (28).
In terms of methodology, not only is CRINEPT-TROSY NMR spectroscopy (21) a demonstrably useful tool for studying such large complexes, it is the only method available for obtaining detailed structural information on large complexes in which one component is unfolded.
Acknowledgments
We thank M. Bycroft for valuable intellectual contributions, T. R. Rippin for providing samples of p53core, M. R. Proctor for advice for the purification of p53core, and T. Wiederkehr and B. Bukau for a plasmid encoding hsp90. S.R. was supported by an EMBO Long-Term Fellowship.
Abbreviations
CRINEPT-TROSY, transverse relaxation-optimized NMR spectroscopy combined with cross-correlated relaxation-enhanced polarization transfer
References
- 1.Hainaut P. (2002) Nat. Med. 8, 21-23. [DOI] [PubMed] [Google Scholar]
- 2.Hollstein M., Hergenhahn, M., Yang, Q., Bartsch, H., Wang, Z. Q. & Hainaut, P. (1999) Mutat. Res. 431, 199-209. [DOI] [PubMed] [Google Scholar]
- 3.Lane D. P. & Lain, S. (2002) Trends Mol. Med. 8, S38-S42. [DOI] [PubMed] [Google Scholar]
- 4.Hainaut P. & Hollstein, M. (2000) Adv. Cancer Res. 77, 81-137. [DOI] [PubMed] [Google Scholar]
- 5.Bullock A. N. & Fersht, A. R. (2001) Nat. Rev. Cancer 1, 68-76. [DOI] [PubMed] [Google Scholar]
- 6.Deb D., Chakraborti, A. S., Lanyi, A., Troyer, D. A. & Deb, S. (1999) Int. J. Oncol. 15, 413-422. [DOI] [PubMed] [Google Scholar]
- 7.McLure K. G. & Lee, P. W. (1998) EMBO J. 17, 3342-3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blagosklonny M. V., Toretsky, J., Bohen, S. & Neckers, L. (1996) Proc. Natl. Acad. Sci. USA 93, 8379-8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King F. W., Wawrzynow, A., Hohfeld, J. & Zylicz, M. (2001) EMBO J. 20, 6297-6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zylicz M., King, F. W. & Wawrzynow, A. (2001) EMBO J. 20, 4634-4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neckers L. (2002) Trends Mol. Med. 8, S55-S61. [DOI] [PubMed] [Google Scholar]
- 12.Kelland L. R., Sharp, S. Y., Rogers, P. M., Myers, T. G. & Workman, P. (1999) J. Natl. Cancer Inst. 91, 1940-1949. [DOI] [PubMed] [Google Scholar]
- 13.Young J. C. & Hartl, F. U. (2000) EMBO J. 19, 5930-5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter K. & Buchner, J. (2001) J. Cell. Physiol. 188, 281-290. [DOI] [PubMed] [Google Scholar]
- 15.Pearl L. H. & Prodromou, C. (2001) Adv. Protein Chem. 59, 157-186. [DOI] [PubMed] [Google Scholar]
- 16.Mayer M. P. & Bukau, B. (1999) Curr. Biol. 9, R322-R325. [DOI] [PubMed] [Google Scholar]
- 17.Jakob U., Lilie, H., Meyer, I. & Buchner, J. (1995) J. Biol. Chem. 270, 7288-7294. [DOI] [PubMed] [Google Scholar]
- 18.Bullock A. N., Henckel, J., DeDecker, B. S., Johnson, C. M., Nikolova, P. V., Proctor, M. R., Lane, D. P. & Fersht, A. R. (1997) Proc. Natl. Acad. Sci. USA 94, 14338-14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong K. B., DeDecker, B. S., Freund, S. M., Proctor, M. R., Bycroft, M. & Fersht, A. R. (1999) Proc. Natl. Acad. Sci. USA 96, 8438-8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pervushin K., Riek, R., Wider, G. & Wüthrich, K. (1997) Proc. Natl. Acad. Sci. USA 94, 12366-12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riek R., Wider, G., Pervushin, K. & Wüthrich, K. (1999) Proc. Natl. Acad. Sci. USA 96, 4918-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grenert J. P., Johnson, B. D. & Toft, D. O. (1999) J. Biol. Chem. 274, 17525-17533. [DOI] [PubMed] [Google Scholar]
- 23.Bullock A. N., Henckel, J. & Fersht, A. R. (2000) Oncogene 19, 1245-1256. [DOI] [PubMed] [Google Scholar]
- 24.Riek R., Pervushin, K. & Wüthrich, K. (2000) Trends Biochem. Sci. 25, 462-468. [DOI] [PubMed] [Google Scholar]
- 25.Cho Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P. (1994) Science 265, 346-355. [DOI] [PubMed] [Google Scholar]
- 26.Buckle A. M., Zahn, R. & Fersht, A. R. (1997) Proc. Natl. Acad. Sci. USA 94, 3571-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi N., Freund, S. M., Chatellier, J., Zahn, R. & Fersht, A. R. (1999) J. Mol. Biol. 292, 181-190. [DOI] [PubMed] [Google Scholar]
- 28.Chatellier J., Buckle, A. M. & Fersht, A. R. (1999) J. Mol. Biol. 292, 163-172. [DOI] [PubMed] [Google Scholar]