

Abstract
The amyloid/tau/neurodegeneration (ATN) biomarker framework has greatly progressed the diagnosis and staging of Alzheimer's disease (AD). However, recent research highlights neuroinflammation as an equally critical factor in AD pathology across humans, rodents, and non‐human primates (NHPs). This review evaluates the combined use of ATN and neuroinflammatory biomarkers—such as glial fibrillary acidic protein (GFAP) (astrocytic marker) and triggering receptor expressed on myeloid cells 2 (TREM2)/ ionized calcium‐binding adapter molecule 1 (IBA‐1) (microglial markers)—in elucidating AD mechanisms, promoting early diagnosis, and shaping therapeutic strategies. It also summarizes the key features and translational potential of NHP models that closely mimic human AD pathology, highlighting the promising prospects of integrating these models with the ATN(X) biomarker system. These insights strengthen the link between biomarkers, NHP research, and clinical practice, opening new avenues for the early detection and treatment strategies of AD.
Highlights
Neuroinflammation biomarkers, including glial fibrillary acidic protein (GFAP), triggering receptor expressed on myeloid cells 2 (TREM2)/sTREM2, and YKL‐40, show strong clinical potential in Alzheimer's disease (AD).
Incorporating neuroinflammation biomarkers into the ATN(X) framework may enhance diagnostic precision.
Advanced non‐human primate (NHP) models closely replicate human brain pathology, addressing key limitations of mouse models.
Measuring ATN(X) biomarkers in NHPs may improve clinical translation and support early diagnosis of AD.
Optimizing NHP models—including ApoE4 status, injection protocols, and gene‐editing approaches—is crucial for reproducibility and efficiency.
Keywords: Alzheimer's disease, biomarkers, early diagnosis, models, neuroinflammation, non‐human primates, therapy
1. INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disease that aggravates over time, representing the most common cause of dementia. 1 , 2 It has stood out as a major public concern for its high mortality rate, 3 large patient population, 4 and substantial economic burden. 5 Due to increasing life expectancy and an aging global population, the number of global dementia cases is estimated to triple by 2050 compared to 50 million in 2018, most of which are composed of patients over 65 years old. 6 , 7
Aging is the primary risk factor for sporadic AD, with the risk doubling every 5 years after age 65. 8 Apolipoprotein E4 (ApoE4) remains the strongest genetic risk factor for sporadic AD, 9 present in over 50% of cases. 10 Familial AD is defined by mutations in amyloid precursor protein (APP) and PSEN1 and PSEN2 genes, while additional risk factors include high cholesterol diets, 11 alcohol consumption, 12 and stress. 13 Clinically, AD manifests as deficits in memory, language, learning, and thinking, which typically emerge during the mild cognitive impairment (MCI) stage caused by synaptic and neuronal damage. As the disease advances, dementia develops, characterized by severe memory deficits, reduced independence, and eventual complete dependency.
This review represents a pioneering effort to integrate neuroinflammation‐related biomarkers into the ATN(X) diagnostic framework while comprehensively categorizing recent advancements in AD NHP models based on pathological and biomarker outcomes. It adopts a novel perspective, exploring how these models contribute to understanding AD pathogenesis and critically summarizing their strengths and limitations. Through an in‐depth exploration of changes in key biomarkers across different disease stages, this review emphasizes the integration of biomarkers, especially neuroinflammation markers, into NHP models, thereby bridging the gap between preclinical research and clinical applications and advancing the development of more effective diagnostic and therapeutic strategies for AD.
2. HUMAN AD PATHOGENESIS AND BIOMARKER TYPES ACROSS DISEASE STAGES
2.1. Tracing amyloid aggregation: From toxic oligomers, fibrils, to senile plaques
APP is a transmembrane glycoprotein that plays a major role in amyloid pathology. 14 In the physiological state, α‐secretase cleaves APP that relates to normal synaptic transmission. 15 Pathologically, APP is sequentially cleaved by β‐secretase and γ‐secretase, releasing the amyloid‐beta (Aβ) peptide. 16 The presenilin genes are responsible for encoding the active component of the γ‐secretase enzyme, which performs intramembrane cleavage. 17 Mutations in the presenilin 1 and 2 genes perturb the enzymatic activity, increasing Aβ42 production and a higher Aβ42/Aβ40 ratio, 18 , 19 promoting amyloid plaque deposition. Variations within the APP gene have been identified to facilitate the production and self‐assembly of pathological Aβ peptides. 20 Collectively, these factors synergize to trigger the early onset of AD.
RESEARCH IN CONTEXT
Systematic review: The authors conducted a rigorous literature search using stringent criteria to identify studies pertaining to the pathological mechanisms and biomarkers associated with Alzheimer's disease (AD). Subsequently, a comprehensive search was conducted to identify studies focusing on the diagnostic and therapeutic assessment of biomarkers in the context of the disease. Additionally, a systematic search and screening process was applied to the literature on non‐human primate (NHP) models related to AD over the past three decades.
Interpretation: The study underscores neuroinflammation as a crucial component in the pathology of AD, supported by experimental and clinical evidence. Integrating neuroinflammatory biomarkers, particularly ATN(X) framework (TREM2), ionized calcium‐binding adapter molecule 1 (Iba1), glial fibrillary acidic protein (GFAP), and YKL‐40, into the amyloid/tau/neurodegeneration (ATN) framework holds significant potential for early diagnosis of AD. NHP models exhibit a high degree of similarity to humans. This review systematically summarizes the characteristics of various models (categorized into biomarker‐based outcomes and other results), their strengths and limitations, and proposes key questions for the building and improvement of these models.
Future directions: Once the feasibility of combining the proposed neuroinflammatory biomarkers into the ATN framework is confirmed, this will significantly enhance the accuracy and likelihood of early clinical diagnosis. Moreover, incorporating biomarker monitoring in NHP models and moving beyond a simple binary classification (positive or negative) will be more conducive to exploring the links between biomarkers and brain tissue pathology through quantitative analysis in positive models. Given the substantial advantages offered by NHP models, further research is needed to optimize these models, improve their reproducibility, shorten the modeling period, and ensure comprehensive expression of AD‐related pathological products.
Aβ40 and Aβ42 are two major components in Aβ deposits; notably, the structure of Aβ42 with two extra amino acids confers a greater tendency for aggregation and higher toxicity compared to Aβ40. 21 Aβ peptides are critical, as they can exist as monomers or aggregate into different structures, such as oligomers, protofibrils, and amyloid fibrils. 22 Oligomers are diminutive, soluble, and diffusible protein assemblies that subsequently transform into entities characterized by extensive β‐structures. 23 They are key contributors to the pathology of AD, as they demonstrate greater toxicity than other Aβ conformations. 22 As the oligomers develop into larger and more stable aggregates, the toxicity diminishes. 24 Aβ oligomers disrupt membrane’ integrity and organelle functions, spreading via cell‐to‐cell transmission. 25 Oligomeric Aβ disrupts the N‐methyl‐D‐aspartate receptor (NMDAR) signaling, leading to reactive oxygen species (ROS) activation, triggering calcium dysregulation, mitochondrial impairment, and synaptic dysfunction. 26 , 27 , 28 Notably, Aβ oligomers can exist without forming fibrils and exhibit significant neurotoxicity. Aβ oligomers and fibrils share β‐structures. 23 These β‐structured forms nucleate fibril formation, a repetitive substructure composed of β‐strands that run perpendicular to the fibril axis, forming cross‐β sheets aligned parallel to the fibril axis, 29 , 30 which further aggregate and deposit into plaques visible by light microscopy. According to later studies, Aβ1‐42 is the predominant component of amyloid‐β deposits found in the central cores of neuritic plaques as well as within the vascular tissues of the brain. 31 As Aβ1‐42 aggregates into plaques at an accelerated rate, the plasma and cerebral spinal fluid (CSF) Aβ1‐42/Aβ1‐40 ratios decline in individuals at early stages of subthreshold Aβ accumulation until the onset of dementia. 32 , 33 , 34 , 35 , 36 Since plaque build‐up is a complicated process that requires a high kinetic barrier and needs to undergo two phases of nucleation, the duration of developing such plaque is quite long, at an estimation of 15–20 years 37 , 38 (Figure 1).
FIGURE 1.

A schematic depiction of amyloid pathology and its progressive transformations in AD. The figure displays the sequential formation and accumulation of various Aβ species, initiated by the cleavage of APP via β‐ and γ‐secretases, resulting in the production of monomeric Aβ peptides. These monomers are categorized into on‐pathway and off‐pathway processes, with on‐pathway species displaying the capacity to aggregate into soluble oligomers, which are considered pivotal contributors to synaptic dysfunction. Off‐pathway processes involve certain Aβ monomers deviating from the conventional aggregation pathway, resulting in the failure to form typical oligomers or fibrils. Instead, these monomers may remain in their monomeric state or form alternative structures. Additional aggregation leads to the formation of protofibrils and eventually insoluble amyloid fibrils that deposit as amyloid plaques within the brain parenchyma, a defining feature of Alzheimer's pathology. The dynamic interconversions among these species, their potential reversibility, and their respective roles in neuronal toxicity are illustrated to emphasize the multifaceted nature of amyloid pathogenesis. Aβ, amyloid‐beta; AD, Alzheimer's disease; APP, amyloid precursor protein
According to the amyloid cascade hypothesis, the imbalance between the production and clearance of Aβ is a fundamental driver of Aβ aggregation, neurodegeneration, and cognitive impairment. 38 Glia, particularly microglia and astrocytes, are crucial in Aβ clearance, demonstrated by one study that degradation of Aβ by a synergistic interplay between microglia and astrocytes via cell‐to‐cell communication, 39 with microglia being more effective. While this crosstalk is beneficial for Aβ clearance, it may also pose a risk of aggravating Aβ pathology. 40 In the brain, Aβ spreads in a prion‐like manner, with Aβ deposits initially exhibiting robust metabolic activity in regions of the cortex, including the precuneus, medial orbitofrontal cortex, and posterior cingulate cortex. 41 Subsequently, these deposits extend throughout the neocortex, infiltrating the brainstem and subcortical nuclei. 42
With a deeper understanding of the disease, the Aβ cascade is now involved not only in its own pathogenesis but also in its interplay with tau pathology, neuroinflammation, and neurodegeneration. 43 , 44 , 45 Their intricate relationship between Aβ and tau has been extensively studied recently. It is renowned that Aβ functions as an upstream mechanism in tau pathology, 46 in the human brain, tau production positively correlates with amyloid plaque number. 47 Rodent studies with APP/PSEN1 mutations have confirmed tau as a critical factor, as knocking out the tau gene reduced neurotoxicity and memory impairment. 48 , 49 Additionally, a decrease in endogenous tau saved hAPP mice from cognitive deficits and plaque burden in the hippocampal area. 50 Furthermore, a study by Hurtado compared mice with mutations in frontotemporal dementia with Parkinsonism (PS19 mice), familial early‐onset AD (PDAPP mice), or their hybrids, showing that combined mutations accelerated tangle formation, while amyloid plaques remained unaffected. 48 , 51 Aβ initiates Aβ‐driven tau pathology by stimulating abnormal tau generation through phosphorylation, activated by kinases including GSK‐3β and CDK‐5. 52 , 53 Experimental studies in P301 mouse models have demonstrated that Aβ42 fibrils specifically accelerate tangle formation. 54 Notably, Aβ accumulation impacts tau pathology and triggers neurodegeneration through either functional loss or tau‐mediated toxicity, which is validated in cross‐sectional tau and amyloid positron emission tomography (PET) studies. 55 Collectively, these findings indicate an Aβ‐induced, tau‐dependent mechanism for pathological tau production. The loss of neurons and synapses forms the basis for tau‐driven cognitive impairment (Figure 2). After that, the joint accumulation of Aβ and tau triggers a cascade of deleterious events from synaptic loss and neuronal death to brain atrophy, which is thought to underlie the cognitive deficits that characterize the disease. 56
FIGURE 2.
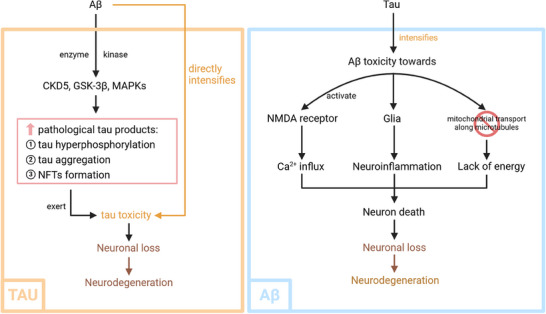
Interaction between Aβ pathology and tau pathology in AD. Aβ aggregation triggers a cascade that promotes tau hyperphosphorylation, aggregation, and the formation of NFTs, thereby accelerating neurodegeneration and cognitive decline. In turn, tau pathology establishes a feedback mechanism that exacerbates Aβ toxicity through its effects on NMDA receptor activity, glial cell activation, and mitochondrial dysfunction, forming a synergistic cycle of neurodegeneration. Aβ, amyloid‐beta; AD, Alzheimer's disease; NFT, neurofibrillary tangle; NMDA, N‐methyl‐D‐aspartate
2.2. Mapping tau dynamics: Biomarkers from fibrils to tangles
Tau was primarily found in neuronal axons as a microtubule‐associated protein. 57 In humans, the tau protein is encoded by the microtubule‐associated protein tau (MAPT) gene, which produces tau isoforms 0N, 1N, and 2N, as well as 3R and 4R, through alternative splicing. 58 In physiological conditions, tau is involved in microtubule assembly, transportation, and stabilization of neuronal axons. 52 During the disease process, tau proteins undergo hyperphosphorylation, a post‐translational modification that occurs at common phosphorylation sites such as threonine and serine in AD. 59 In human CSF, phosphorylated tau (p‐tau) rises first as p‐tau217, then p‐tau181 and p‐tau205, which are crucial for disease differentiation. 60 Along with total tau (t‐tau) and other tau isoforms, they collectively reflect the severity of tau pathology in AD. 60
Structural modifications in tau protein cause a transition from its native unfolded state to the formation of paired helical filaments or larger aggregates. Alternations detach tau from microtubules, often followed by aberrant aggregation. 61 Synaptic dysfunction therefore arises from loss of tau's microtubule stabilization, obstruction from tau aggregates, or downstream toxicity. 62 , 63 As AD progresses, tau proteins aggregate into insoluble neurofibrillary tangles (NFTs), which is a critical diagnostic hallmark. Tau deposition follows a stereotypical pattern, beginning in the entorhinal cortex and spreading to limbic areas and eventually the neocortex. 64 The spread of tau thus damages neurons and causes neurodegeneration. 65 , 66 In AD, neurodegeneration is widely detected using tau‐PET, which is highly sensitive for early and differential diagnosis of AD and provides valuable prognostic information. 1 Moreover, an analysis involving 982 participants in 2021 found that tau pathology interacts with vascular changes related to neurodegeneration, explaining the potential mechanism by which tau contributes to cognitive impairment in AD. 67 Tau‐PET is often used alongside the Braak staging system, a widely respected method that classifies AD by the spatial progression of pathological tau. This approach distinguishes different phases of AD and shows strong correlations with clinical, fluid, and imaging biomarkers. 68 , 69
2.3. Charting neurodegeneration: From synaptic dysfunction to neuron death
In AD, neurodegeneration plays a central role throughout the disease course, acting as a central driver of pathological progression and clinical symptoms. In the early stage, changes begin with Aβ plaques and early tau pathology, as tau pathology spreads, it leads to synaptic loss, brain atrophy, disrupted neural networks, and cognitive decline. 66 , 70 Longitudinal tau‐PET studies in human AD cases have been employed to quantify tau accumulation, showing a significant increase over time, ranging from 3.0% to 8.0% annually, among Aβ‐positive clinically diagnosed AD patients. 55 In addition to the respective effects of Aβ and tau, their combined interactions further accelerate neurodegenerative changes. 71 A recent clinical study involving elderly subjects with a family history of sporadic AD but no cognitive impairment revealed that tau deposition following Aβ accumulation triggered a swift transition in neuronal activity from hyperactivity to hypoactivity, 56 which was closely linked to cognitive decline, demonstrating the combined impacts of Aβ and tau on neurodegeneration.
NMDA receptors are critical for normal synaptic function and neuronal signal integration. In AD, pathological Aβ and tau proteins interact via fyn kinase, 72 with tau promoting fyn‐mediated NMDA receptor activity, 73 causing excessive Ca2⁺ influx, disrupted cellular signaling, mitochondrial dysfunction, 74 , 75 increased ROS production, ultimately resulting in neuronal damage and cell death. 51 Dysregulated Ca2⁺ not only directly impacts tau and Aβ but also creates a vicious cycle that exacerbates pathology. 76 Furthermore, two in‐depth clinical studies by van der Kant et al and Harrison revealed that Aβ accelerates tau progression, and together they worsen cognitive impairment and brain atrophy. 77 , 78
Neurofilament light (NfL) is a non‐specific biomarker indicative of axonal injury in neurodegenerative diseases, which was initially detected in CSF by enzyme‐linked immunosorbent assay (ELISA) and now is measurable in blood. 79 NfL levels rise in the early stage of AD and can forecast disease severity through CSF and serum. 80 , 81 However, given its broad nature, NfL should be complemented with other specific pathological markers for an accurate diagnosis of AD. 18F‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scanning is vital for monitoring AD progression, as it reflects glucose metabolism in regions such as the temporoparietal association cortex, anterior cingulate cortex, and posterior cingulate cortex. 82 This technique not only differentiates AD from other neurodegenerative diseases but also confirms clinical subtypes, evaluates disease severity, and predicts cognitive decline. 82 Magnetic resonance imaging (MRI) detects early brain changes, like hippocampal and cortical atrophy, before clinical symptoms emerge, 83 supporting differentiation from other dementias, 84 and enhancing diagnostic accuracy and individualized treatment guidance.
2.4. Neuroinflammatory signals: A key driver of neurodegeneration
2.4.1. Pathophysiological mechanisms of neuroinflammation in AD
Neuroinflammation refers to the inflammatory response within the central nervous system (CNS) and is integral to AD progression. 85 Astrocytes and microglia, the CNS's primary glial cells, form a critical line of defense 86 , 87 and are essential for clearing insoluble Aβ and tau deposits. 88 , 89 However, in AD, the overactivation of glial cells triggers uncontrolled inflammatory responses, which can be neurotoxic, furthering the disease progression. 90 They contribute by enhancing Aβ aggregation, 91 reducing neurotrophic support, causing inflammation‐induced neuronal damage, releasing abnormal phagocytic signals, 92 producing oxidative stress from ROS, and producing excitotoxicity from neurotoxic factors.
During AD, neuroinflammatory microglia induce a reactive astrocyte subtype termed A1 through cytokines like interleukin (IL) ‐1α, tumor necrosis factor (TNF), and C1q, which leads to neuronal death. 93 Communication between microglia and astrocytes may generate a positive feedback loop, resulting in a dysregulated and self‐sustaining inflammatory response. 94 Proinflammatory cytokines, notably plasma IL‐6 and TNF‐α, are elevated in Alzheimer's patients, driving neuroinflammation and neurotoxicity, providing compelling scientific evidence that supports the involvement of the inflammatory response in AD. 95 , 96 GFAP is an astrocyte‐specific and well‐established biomarker of neuroinflammation, with greater plasma increases in AD than in CSF. 97 Besides, YKL‐40, a glycoprotein produced by active astrocytes in AD and reflecting neuroinflammation, is also a promising biomarker for diagnosis and monitoring. 98 , 99 Although important in AD, there is still a lack of fluid markers truly specific to microglia, as proteins (such as TREM2) related to AD lack brain specificity. 100
2.4.2. Crosstalk between neuroinflammation and other AD pathologies
In AD pathogenesis, the activation of glial cells is also influenced by Aβ and tau. 101 , 102 The post mortem human brain study by Carrano et al observed activated microglia expressing NOX‐2 around Aβ‐burdened brain capillaries, suggesting a potential connection between microglia and aggregation of Aβ. 103 Research has shown that Aβ not only proliferates but also activates microglia through a process known as “priming,” which sensitizes microglia to secondary inflammatory stimuli, especially in the elderly, potentially triggering an exaggerated inflammatory response. 104 Activated microglia produce toxic ROS and reactive nitrogen species (RNS) through NADPH oxidase, contributing to amyloid formation and decreasing amyloid clearance. 105
Both Aβ and tau disrupt microglia homeostasis, 106 leading to distinct microglial subtypes, as confirmed in animal studies. 107 In AD, microglia dynamically transform from homeostatic to disease‐associated microglia (DAM) via sequential stages: early interferon‐responsiveness, mid‐stage DAM activation (Aβ clearance), and late major histocompatibility complex (MHC) ‐II hi proliferating states driving chronic inflammation and synaptic loss, highlighting the need for collaborative efforts to more precisely understand microglial identities and states. 108 , 109 , 110 Furthermore, tau pathology also intensifies glial activity, prompting innate immune cell activation and the release of both pro‐ and anti‐inflammatory molecules 111 that create a self‐amplifying neuroinflammation cycle. In addition to responding to Aβ and tau, microglia and astrocytes also promote Aβ and tau pathology, further accelerating AD progression. While microglia and astrocytes help Aβ clearance, they might increase pathological spread within and between interconnected networks, acting as a double‐edged sword. 39 Additionally, astrocytes lead to Aβ‐dependent tau pathology, as shown in a biomarker study on cognitively unimpaired AD patients, 112 indicating Aβ was linked to elevated levels of plasma p‐tau only in individuals with astrocyte reactivity (Ast+).
The blood–brain barrier (BBB) is a critical defense mechanism in the brain, maintaining homeostasis for neuronal signaling by regulating substance exchange and clearing pathological products. 113 In AD, BBB dysfunction is strongly linked to iron accumulation, which is observed from a young age and interferes with normal brain maturation. 114 Impaired BBB integrity allows excess iron influx, resulting in iron‐dependent programmed cell death, reinforcing oxidative stress, and intensifying chronic inflammation in the CNS, 115 , 116 which damages neurons and worsens BBB integrity. 117 , 118 A deficiency in BBB clearance promotes the aggregation of pathological products. This vicious cycle is exacerbated by genetic risk factors such as ApoE ε4, 119 which increases BBB permeability and iron overload, leading to accelerated cognitive decline. 120 , 121 , 122 , 123 Furthermore, Yuto Uchida's team also introduced advanced imaging methods, including quantitative susceptibility mapping (QSM) and dynamic contrast‐enhanced MRI (DCE‐MRI), 117 , 121 which now allow for non‐invasive detection and monitoring of BBB breakdown and iron deposition, serving as potential early biomarkers for monitoring AD.
Notably, abnormal neuroinflammation should not be regarded solely as a result of the disease; it is also a significant triggering factor. Biomarkers appearing according to disease stages have significantly advanced our understanding of the pathogenesis, diagnosis, and monitoring of AD. In the future, integrating multidimensional biomarkers within the ATN framework could enhance the accuracy of early diagnosis and treatment monitoring.
3. CURRENT USES OF BIOMARKERS IN HUMAN AD
3.1. Biomarkers in AD diagnosis: Integrating the ATN framework
Earlier diagnostic criteria for AD relied on clinical symptoms, such as the Clinical Dementia Rating (CDR), and PET imaging to detect NFTs and senile Aβ plaques, as approved by the U.S. Food and Drug Administration (FDA). 124 However, pathological changes may occur as early as 15 to 25 years before the onset of memory symptoms, often delaying diagnosis until the disease is advanced and treatment options are suboptimal, as they are unable to stop disease progression. 1 Therefore, early diagnosis of AD is desperately needed for earlier interventions. 125
Biomarker refers to specific, measurable indicators within a biological system that reflect normal physiological functions, pathological alterations, or pharmacological responses to therapeutic interventions. It is important that diagnostic AD biomarker tests introduced be accurate (a sensitivity and specificity of >85%, and a positive predictive value of >80%) and suitable for general medical practice in various routine clinical settings. 126 The National Institute on Aging‐Alzheimer's Association's (NIA‐AA's) A(amyloid)T(tau)N (neurodegeneration) framework, which is revised in 2024, provides a biomarker‐based approach for diagnosing AD, 127 , 128 with tau and Aβ recognized as two core pathological markers reflecting the key features of the disease.
Currently, the ATN framework has proven effective in identifying individuals with a higher risk of AD in clinical practice, with studies demonstrating that biomarker levels correlate with worse cognitive trajectories in individuals with MCI and those who are cognitively normal, irrespective of whether they report subjective memory complaints. 129 , 130 , 131 , 132 Furthermore, the framework can also predict the onset of dementia, potentially up to 5 years from MCI or cognitively normal states. 133 , 134 However, further in‐depth and extensive research is necessary before the ATN framework can be widely applied in clinical practice. Integrating multi‐omics biomarkers and advanced neuroimaging may provide the potential to refine the ATX(N) framework, thereby enhancing the accuracy of future classification algorithms across the spatial and temporal dimensions of AD. 135
3.1.1. Amyloid‐β (A): Marker of early pathological plaque deposition
Amyloid‐β serves as a primary component of senile plaques in the brain. Aside from amyloid‐PET, Aβ1‐42 (rather than Aβ1‐40 or total Aβ alone) in CSF correlates more strongly with a diagnosis of AD. 99 Blennow and Hampel's study included 13 independent studies, encompassing approximately 600 AD patients and 450 healthy controls. With a specificity of 90%, the average sensitivity of CSF Aβ1‐42 for distinguishing AD from normal aging is 86%. 136 Previous studies have demonstrated that Aβ42 is more likely to form plaques compared to Aβ40, which explains the strong correlation between the reduced Aβ42/Aβ40 ratio in plasma and CSF and plaque deposition. 137 This ratio is also closely associated with amyloid PET imaging results, 138 which surpass standalone CSF Aβ measurements in predicting amyloid positivity, highlighting its potential as a diagnostic marker for AD.
3.1.2. Tau (T): Indicator of tangle pathology and Braak staging
CSF p‐tau detection remains a standard in clinical practice. For the diagnosis of AD, tau phosphorylation at Thr181, Ser199, Thr231, Ser205, Thr217, and Ser404 have been identified as key isoforms, and they each offer unique pathological and diagnostic implications. 139 Among these, p‐tau231, p‐tau181, and p‐tau217 have shown their potential in distinguishing early AD in individuals with Aβ positivity. 140 p‐Tau217, in particular, increases during the early stages of amyloid plaque formation, whereas p‐tau205 demonstrates greater specificity and sensitivity in detecting the onset of neuronal dysfunction. 60 Furthermore, p‐tau217 is particularly noteworthy due to its strong correlation with longitudinal tau‐PET measurements. 141 , 142 In addition, CSF p‐tau262 and p‐tau356 enable sensitive detection of NFTs, allowing AD to be distinguished from other tauopathies and providing both diagnostic and therapeutic value. 143
Blood biomarkers are also highly valued for their non‐invasive nature, with comparable accuracy to CSF tests, lower cost, and greater acceptance among patients in clinical settings. Plasma p‐tau181, p‐tau217, and p‐tau231 prove strong performances in diagnosing AD, with p‐tau217 standing out for its superior accuracy in both early detection and differentiation 144 , 145 of AD compared to other established markers. 146 , 147 , 148 , 149 p‐Tau181 also effectively identifies Aβ‐PET positivity and predicts future AD in cognitively unimpaired and MCI individuals. 146 Recently, the plasma microtubule‐binding region (MTBR) containing residue 243 (eMTBR‐tau243) performed with high accuracy in identifying tau tangle pathology and late‐stage AD, with levels rising from the MCI stage and continuing to increase until dementia. 150
Tau‐PET, a non‐invasive method for visualizing tau protein accumulation in the brain, stands out as a priority biomarker for differential diagnosis with its high accuracy and specificity of over 90%. 151 , 152 By incorporating the advanced tau tracers (such as [18F]‐AV‐1451, MK‐6240, and PI‐2620), tau‐PET enables even more precise localization and quantification of tau deposits in specific brain regions, thereby supporting pathological staging based on the Braak staging scale. A longitudinal neuroimaging study on patients in the early stages of AD showed that initial tau‐PET measurements correlated with subsequent MRI‐detected neurodegeneration and forecasted cortical atrophy and cognitive performance over the following year. 153 , 154 Notably, Vogel's clinical study demonstrated tau‐PET in identifying both the typical pattern of tau pathology progression in AD (from the entorhinal cortex to the neocortex) and atypical tau distribution patterns associated with non‐typical clinical presentations, with reproducibility of these findings across different samples. 155 These discoveries highlight the diversity of AD, challenge the widely recognized model of tau propagation, and underscore the critical role of tau‐PET in individualized assessments.
3.1.3. Neurodegeneration (N): Clinical relevance with neuronal damage and functional decline
In the context of fluid biomarkers, elevated soluble t‐tau in CSF may result from the passive release of tau protein triggered by neurodegeneration, which is linked to Aβ‐related pathology. 101 Increased NfL levels in blood and CSF indicate neuroaxonal damage. 156 Mattsson's study using the ATN framework found that plasma NfL concentrations were higher in patients with AD dementia compared to those with MCI, and these levels correlated with disease severity. Furthermore, changes in NfL were also associated with the degree of alignment with the ATN criteria, suggesting the potential of plasma NfL as a marker for tracking disease progression and assessing pathophysiological processes in AD. 139 Brain‐derived neurotrophic factor (BDNF), which supports synaptic plasticity associated with learning and memory, is typically decreased in AD. 157 However, conflicting findings regarding plasma BDNF levels highlight the need for further investigation before its clinical application can be considered. 158 , 159
MRI demonstrates brain atrophy in AD patients, especially in the hippocampus and medial temporal lobe (MTL), providing a reliable basis for early diagnosis. Similarly, fluorodeoxyglucose 18FDG‐PET scans measure cerebral glucose metabolic function, and reduced metabolic activity in the bilateral temporoparietal regions is a diagnostic marker for AD, 160 helping to distinguish it from other types of dementia when combined with additional biomarkers.
3.1.4. Neuroinflammation (X): Emerging role in Alzheimer's pathology
GFAP serves as an indicator of astrocyte activation, with an increase in its levels detectable in both CSF and plasma. 161 Plasma GFAP levels are markedly elevated in preclinical AD, even in cognitively normal, Aβ‐positive individuals, and continue to increase as the disease progresses. 162 , 163 , 164 This elevation closely correlates with Aβ pathology, evidenced by decreased levels of Aβ1‐42 and a strong correlation with Aβ neuroimaging biomarkers. 165 , 166 However, since GFAP also fluctuates in various neurodegenerative diseases and its diagnostic accuracy is lower than that of Aβ and tau‐related biomarkers, 99 its reliability as a standalone biomarker is limited. In contrast, plasma GFAP changes are minimal in Parkinson's disease across cognitive stages 167 and in pure dementia with Lewy bodies without AD co‐pathology, 168 , 169 indicating a distinct AD‐specific, Aβ‐driven astrocyte activation pattern. GFAP levels are also significantly higher in AD compared to frontotemporal dementia, providing moderate effectiveness for differential diagnosis (area under the curve [AUC] 0.818). 161 In Alexander disease, GFAP elevation is primarily due to core GFAP pathology, whereas in AD, it is secondary to underlying disease processes, resulting in lower levels and slower GFAP increases. 170 , 171 Furthermore, GFAP levels may also rise after acute brain injury or systemic inflammation, complicating clinical interpretation. Notably, given the potential of GFAP in identifying early AD and elucidating Aβ changes and neuroinflammation, 172 its integration into biomarker panels alongside other core AD markers is promising to improve early and differential diagnosis. 173 YKL‐40 stands for the activation of microglia and astrocytes. 174 , 175 In AD, its levels in plasma and CSF show a moderate to large effect size (1.28–1.47). 99 While not directly reflecting core AD pathology, YKL‐40 can predict neuroinflammation and is involved in Aβ‐induced tau pathology and tau‐related neuronal injury. 173 It holds potential as a biomarker for neurodegeneration and glial activation and holds the potential of integration into neuroinflammation biomarkers in clinical assessments. 176
Microglia activation markers, including ionized calcium‐binding adapter molecule 1 (IBA‐1) and triggering receptors expressed on myeloid cells 2 (TREM2), typically increase during the AD continuum. 177 TREM2 is a microglial receptor that detects damage and regulates immune and metabolic responses, existing both as a membrane protein and as soluble sTREM2 (sTREM2) detectable in CSF and plasma. 178 However, sTREM2 is not supported as a stand‐alone biomarker due to its variability and limited disease specificity. 179 , 180 , 181 Additionally, CSF sTREM2 levels peak during the MCI stage and then plateau or decline during dementia, forming an inverted “U” curve. 178 Given that TREM2 and sTREM2 are also influenced by various factors such as NLRP3 182 and microglia subtypes, 183 their independent use remains controversial. However, it is undeniable that the TREM2 molecule is solely expressed by myeloid cells in the brain and serves as a direct marker of their activity. 184 , 185 Recent clinical studies also demonstrated its significant potential in understanding myeloid cell responses, predicting cognitive impairment, and potential as therapeutic targets. 186 , 187 , 188 , 189 , 190 , 191 Integrating TREM2/sTREM2 with other markers within the ATN(X) framework may improve diagnostic accuracy, enhance differential diagnosis, and support clinical monitoring, especially with advanced nanosensing platforms. Furthermore, proinflammatory cytokines such as IL‐6 and TNF‐α within the innate immune system are released by activated glial cells and have been shown to correlate with disease progression. 192 However, the high variability and low disease specificity of these biomarkers limit their clinical usefulness, as levels are easily affected by various systemic conditions. 193 Therefore, we excluded them from our clinical framework in favor of more specific markers.
Based on current pathological mechanisms and clinical research findings, we propose integrating neuroinflammation biomarkers as the “X” component within the ATN(X) framework, with the potential to enhance diagnostic accuracy and sensitivity for AD. Further investigations into neuroinflammation are crucial to better understand its role in AD and to refine the utility of these biomarkers for broader clinical application, which may offer a promising avenue for early diagnosis in AD. Biomarkers hold great potential in the screening, diagnosis, staging, and progression prediction of AD (Table 1). Although research on biomarkers has been rapidly advancing in recent years, current definitions based solely on biomarkers are not yet fully applicable to clinical practice and require further validation. For more accurate and personalized diagnosis in clinical settings, especially in cognitively unimpaired individuals, biomarkers should be considered closely alongside clinical phenotypic presentations. 194
TABLE 1.
Summary of molecular and imaging biomarkers associated with Alzheimer's disease
| Characteristic | Origin | Marker | Significance |
|---|---|---|---|
| Aβ | |||
| CSF, plasma | Aβ1‐40 | Aβ1‐40↑: differentiates between AD(+) and AD(−) individuals 18 , 19 , 99 | |
| Aβ1‐42 | Aβ1‐42↓: Aβ plaque formation 32 , 33 , 34 , 35 , 36 | ||
| Aβ42/40 | Aβ42/40↓: correlates with plaque status, amyloid‐PET; surpasses standalone CSF Aβ 32 , 33 , 34 , 35 , 36 , 137 , 138 | ||
| Brain | Amyloid‐PET | Detection of amyloid deposition 99 | |
| Tau | |||
| CSF | p‐tau181 | p‐tau181↑: the gold standard in evaluating phosphorylation, helps diagnose AD; differentiates between Aβ(+) and Aβ(−) patients 139 , 140 | |
| p‐tau205 | p‐tau205↑: the onset of neuronal dysfunction 60 , 139 | ||
| p‐tau217 (best performance) | p‐tau217↑: differentiates between Aβ(+) and Aβ(−) patients; predicts Aβ plaque formation; strong correlation with tau‐PET 139 , 140 , 141 , 142 | ||
| p‐tau231 | p‐tau231↑: differentiates between Aβ(+) and Aβ(−) patients 139 , 140 | ||
| p‐tau262 | p‐tau262↑: diagnostic value in AD; sensitive detection of NFTs 143 | ||
| p‐tau356 | p‐tau356↑: helps diagnose AD; correlates with NFTs 143 | ||
| MTBR‐tau species 243, 299, 354 | MTBR‐tau species 243, 299, 354↑: associate with tau‐PET imaging, cognitive decline 150 | ||
| Plasma | p‐tau181,231,217 | p‐tau181,231,217↑: promising diagnostic markers of AD; p‐tau217 owes the best diagnostic performance with accuracy on differentiation and early diagnosis 144 , 145 , 146 , 147 , 148 , 149 | |
| Brain | TAU‐PET | Localization and quantification of tau deposits; PET‐based Braak staging in clinical setting 68 , 69 , 151 , 152 , 153 , 154 , 155 | |
| Neurodegeneration | |||
| CSF, plasma | NfL | NfL↑: neuroaxonal damage; ↑with disease severity; more obvious upregulation happens due to Aβ and tau in advanced stages 79 , 80 , 81 , 139 , 156 | |
| CSF | BDNF | BDNF↓: impaired spatial memory, neuronal damage 157 , 158 , 159 | |
| CSF, plasma | t‐tau | t‐tau↑: neurodegeneration; dependent on Aβ‐related pathology 101 | |
| Brain | MRI brain volume | MRI↓brain volume: atrophy; neurodegeneration; early diagnosis of AD 83 , 84 | |
| Brain | 18FDG‐PET reactivity | ↓18F‐FDGPET reactivity:↓glucose metabolism; AD diagnostic indicator; differentiate between AD(+) and AD(−) patients 82 , 160 | |
| Neuroinflammation | |||
| CSF, plasma | GFAP | GFAP↑: astrocyte activation; correlates with Aβ pathology 97 , 161 , 162 , 163 , 164 , 165 , 166 , 172 , 173 | |
| YKL‐40 | YKL‐40↑: reflects glial activation, neuroinflammation; mediates Aβ‐induced tau pathology 99 , 173 , 174 , 175 , 176 | ||
| IBA‐1 | IBA‐1↑: microglia activation 99 , 177 | ||
| TREM2 | TREM2↑: microglia activation; associates with Aβ plaque deposition 177 , 178 , 179 , 180 , 181 , 184 , 185 , 186 , 187 , 188 , 189 , 190 , 191 | ||
Abbreviations: 18FDG‐PET, 18F‐fluorodeoxyglucose positron emission tomography; Aβ, amyloid‐beta; AD, Alzheimer's disease; BDNF, brain‐derived neurotrophic factor; CSF, cerebral spinal fluid; GFAP, glial fibrillary acidic protein; MRI, magnetic resonance imaging; MTBR, microtubule‐binding region; NfL, neurofilament light chain; NFT, neurofibrillary tangle; PET, positron emission tomography.
3.2. Targeting therapy via biomarker in AD: Unmet needs
The complexity and multifactorial nature of AD pose significant challenges for effective treatment. To date, only nine FDA‐approved drugs exist for AD. Among these, cholinesterase inhibitors—tacrine, donepezil, galantamine (capable of crossing the BBB), and rivastigmine—and the NMDA receptor antagonist memantine 195 are utilized to mitigate cognitive decline. Additionally, suvorexant and brexpiprazole are prescribed to manage non‐cognitive symptoms such as sleep disturbances and agitation, respectively. Despite their use, clinical trials have demonstrated limited efficacy in altering disease progression or improving MCI. 196 , 197 This has shifted the focus toward disease‐modifying therapies and immunotherapies targeting specific pathological products, marking a new era in AD treatment.
Therapeutic strategies mainly fall into two categories: one focuses on preventing the formation of pathological products to inhibit disease progression, while the other aims to facilitate the clearance of existing pathological products through pharmacological or other therapeutic interventions, accelerating their degradation. Immunotherapy has emerged as a promising strategy in this domain. Biomarkers are invaluable tools for disease targeting and monitoring of treatment efficacy, and a deeper understanding of the molecular mechanisms underlying AD will further clarify the significance of dynamic changes in these biomarkers.
3.2.1. Conformation‐specific Aβ immunotherapy: Efficacy and amyloid‐related imaging abnormalities risks
Therapeutic strategies targeting Aβ have undergone extensive investigation. Clinical studies have indicated that approaches aimed at interfering with β‐ and γ‐secretases, which are responsible for the production of Aβ peptides, have not yielded significant outcomes. Recently, Aβ‐targeted immunotherapies, such as aducanumab and lecanemab, which target soluble Aβ oligomers to insoluble plaques, received approval from the FDA in 2021 and 2023, respectively. Phase III clinical trials have demonstrated that these therapies effectively reduce amyloid plaque burden, with improvements in cognitive and functional outcomes. 198 For instance, aducanumab binds to aggregated forms of Aβ, reducing plaque deposition and slowing disease progression by approximately 27%. The ENGAGE and EMERGE clinical trials of aducanumab demonstrated 13%–16% reductions in plasma p‐tau181 levels in high‐ and low‐dose groups compared to placebo after 56 weeks of treatment. These results were consistent with amyloid‐PET findings, supporting the drug's efficacy in reducing amyloid pathology and the potential of biomarkers in monitoring treatment effects. 199
Similarly, the TRAILBLAZER‐ALZ randomized clinical trial led by Michael Pontecorvo showed that donanemab, targeting a specific form of Aβ, namely Aβ with pyroglutamate at the N‐terminal 3rd position (abbreviated as N3pG‐Aβ), significantly reduced plasma p‐tau217 levels by 24% and also decreased GFAP levels compared to placebo. These effects were in line with reductions in amyloid burden observed in Aβ‐PET imaging, as demonstrated in Sergey Shcherbinin's study. 200 , 201 In contrast, the phase III trial of solanezumab, a monoclonal antibody targeting monomeric amyloid, showed its ability to slow Aβ aggregation as observed using amyloid‐PET over 5 years but failed to influence tau propagation (measured by tau‐PET), cognitive decline, or brain atrophy (measured by MRI). 202 Despite these limitations, solanezumab provides insights into the selective targeting of Aβ pathology. In a recent phase III randomized clinical trial, both solanezumab and gantenerumab demonstrated reduction of Aβ fibrils, as well as lowering levels of CSF sTREM2, GFAP, and NfL in both blood and CSF, biomarkers associated with neuroinflammation and neurodegeneration. 203
It is important to note that Aβ immunotherapies carry significant risks. In phase III trials of Aducanumab, approximately one‐third of participants experienced amyloid‐related imaging abnormalities (ARIAs), 204 with ApoE4 carriers at heightened risk. 205 ARIAs include ARIA‐E (vasogenic edema or subarachnoid/subcortical effusion) and ARIA‐H (cerebral microbleeds or hemosiderin deposits). Although often asymptomatic, ARIAs can occasionally lead to severe or even fatal cerebral hemorrhage. Despite representing a significant milestone in the treatment of AD, immunotherapies carry serious side effects, necessitating the adoption of stringent clinical usage criteria.
3.2.2. Tau‐targeted therapies: From phosphorylation to clearance, triumphs, and translation gaps
p‐Tau has emerged as a valuable tool for monitoring therapeutic responses, as it reflects both tau and Aβ pathologies. 206 Meanwhile, tau is also highly promising as a therapeutic target since it directly associates with synaptic loss and neurodegeneration. 66
Glycogen synthase kinase‐3 (GSK‐3), a critical kinase involved in tau phosphorylation in AD, has been explored as a therapeutic target. Blocking GSK‐3 activity in transgenic mouse models reduced tau phosphorylation and mitigated axonal transport deficits from Aβ pathology. 207 Consequently, GSK‐3 is considered a highly promising therapeutic target. Currently, drugs targeting GSK‐3 are categorized into adenosine triphosphate (ATP) ‐competitive inhibitors, exhibiting high activity and well‐defined mechanisms but with limited kinase selectivity, and ATP‐noncompetitive inhibitors, offering high selectivity but possessing less‐understood mechanisms. 208 Among these agents, only lithium carbonate and tideglusib have advanced to clinical trials. Preclinical studies on tideglusib using double transgenic mice expressing human APP and tau reported its ability to lower tau phosphorylation, reduce amyloid deposition, attenuate neuronal loss, and improve cognitive function. 209 However, as an irreversible, time‐dependent inhibitor, 210 , 211 tideglusib showed limited efficacy in reducing cognitive and functional decline in a phase II trial conducted on patients with mild‐to‐moderate AD. 212
Another approach to reducing tau protein expression targets tau mRNA transcripts. For example, antisense oligonucleotides (ASOs) regulate tau mRNA through multiple mechanisms, including direct degradation of mRNA, modulation of mRNA splicing, regulation of mRNA stability, and inhibition of mRNA translation. 213 ASOs decrease the production of pathological tau and possibly reverse existing tau productions. 101 Besides, ASOs also effectively reduced human tau mRNA and protein levels in P301S mutant tau‐expressing mice and cynomolgus monkeys, achieving reductions in brain and CSF tau. 214 This was associated with lowered tau inclusions, reversal of p‐tau, prevention of MRI‐detected hippocampal atrophy, and cognitive improvement, which highlight the therapeutic potential of tau‐lowering ASOs for patients with tau‐positive inclusions, even in advanced stages of tau pathology. A phase 1b trial investigating ASOs targeting MAPT demonstrated their safety in humans and a dose‐dependent reduction in t‐tau and p‐tau181 levels. 215 In a Phase 1b trial involving participants with mild AD, the MAPT‐targeting ASO BIIB080 demonstrated favorable tolerability and efficacy. The treatment led to reductions in CSF t‐tau, p‐tau181, and tau PET signals, highlighting its potential to mitigate cognitive decline. 216 Evaluating the potential of this mechanism for AD treatment is crucially dependent on elucidating the role and extent of extracellular tau in pathological propagation, as well as demonstrating the efficacy of therapeutic interventions targeting this process. 217
As an alternative and highly competitive therapeutic approach, current immunotherapy primarily targets the N‐terminal and MTBR domains of tau protein, which are critical functional regions involved in tau's interaction with microtubules. 152 These regions can be conceptualized as “staplers” facilitating this binding process. Immunotherapy can be classified into active and passive immunization, both targeting the inhibition of tau propagation between neurons and the prevention of its aggregation. Active immunization strategies, including AADvac‐1, which targets the N‐terminal region of tau, and ACI‐35, which targets the phosphorylated tau epitope p‐tau396/404, have completed phase 1 clinical trials and are currently undergoing phase 2 trials. 218 Therapeutic approaches directed at the N‐terminal region, such as semorinemab, and those targeting the MTBR, including zagotenemab, have demonstrated reductions in CSF p‐tau levels during phase 2 clinical trials. 219 Specifically, semorinemab has exhibited limited effectiveness in the early stages of AD. However, none of these trials have shown significant reductions in tau‐PET signals, a key marker of pathological tau burden. The underlying mechanisms remain poorly understood, with several hypotheses suggesting that these therapies may fail to target the majority of pathological tau forms or adequately address the most toxic tau species. 152 Further research is needed to better delineate tau's pathophysiological role and optimize therapeutic strategies.
4. CHALLENGES IN BIOMARKER RESEARCH: BETWEEN HUMANS AND RODENTS
4.1. The biomarker dilemma in human AD: Latency, dynamics, and heterogeneity
The study of AD biomarkers in humans faces several limitations. First, the disease has a prolonged course with pathological changes likely initiating years before the onset of noticeable clinical symptoms, making early intervention challenging. 220 Furthermore, the invasive nature of CSF sampling in clinical practice raises ethical concerns and substantially affects patient compliance. Second, AD is an immensely complex disorder. Relying on a single biomarker provides limited insight due to constrained sensitivity and specificity. 221 Additionally, some biomarkers exhibit non‐linear dynamics during the disease course, plateauing in the later stages and reducing their accessibility for monitoring disease progression. 101 AD also displays marked heterogeneity across individuals, especially during the MCI stage, where patients may present with diverse clinical symptoms. 222 , 223 This high degree of variability may arise from the inherent complexity of AD itself or the overlapping contributions of multiple neurodegenerative and non‐neurodegenerative conditions, complicating biomarkers’ interpretation in clinical and research contexts. 101
Finally, the role of the APOE ε4 allele, an established risk factor for AD, introduces further complexity to AD by shaping the clinical phenotypes and imparting distinct patterns of regional brain vulnerability. 116 Therefore, incorporating APOE ε4 genotype information is critical for improving the diagnostic accuracy and risk prediction associated with biomarker analyses. Clinical research faces challenges such as low follow‐up rates due to participant mortality, compromising the completeness and reliability of the findings. Additionally, autopsy‐based brain tissue analysis provides only a static snapshot of the disease at a specific time point, restricting insights into the dynamic processes of disease progression.
4.2. The cross‐species challenges in rodents AD modeling: Temporal disconnect, pathological mismatch, and recapitulation failure
As the most commonly used and well‐established animal models in AD research, rodents have undergone various transgenic modifications to generate models exhibiting key phenotypes associated with AD. For example, rodents have been developed to accelerate amyloid pathology through the introduction of human APP or APP+PSEN1 mutations, to model tau pathology with human MAPT mutations, and to combine genetic factors in 3xTg models (APP/PS1/Tau transgenic) to capture multiple hallmarks of human AD. 224 Recently, models such as 5xFAD mice (carrying APP Swe, Flo, Lon, and PS1 M146L, L286V mutations), which exhibit rapid‐onset amyloidosis, neurodegeneration, and memory impairment, 225 as well as 3xTg‐AD models (expressing human APP695(Swe), MAPT4R0N(P301L), and knock‐in Psen1M146V mutations) that replicate the interplay between Aβ, tau pathology, and synaptic dysfunction, 226 have been used for studying AD pathology. With the advancement of study, transgenic models targeting the APOE genetic background of human AD have been developed. 227 , 228 These models simulate the genetic context of human APOE by expressing human APOEε3 or APOEε4 while knocking out the rodent APOE gene. Additionally, by knocking out the TREM2 gene, researchers can replicate the functional loss or specific mutations of TREM2 observed in human AD, thereby enabling in‐depth investigation into its role in the disease's pathogenesis. 229
Despite their significance, rodent models have an alarming failure rate as high as 99.6% when translating successful therapeutic strategies to humans during drug development, underscoring the immense challenges inherent in cross‐species research and raising significant doubts about their validity. 230 , 231 One of the inherent limitations of using rodent models in AD research is their inability to fully replicate the broader pathological and clinical characteristics observed in human AD. 232 For instance, while rodents can develop amyloid plaque formation, they often fail to exhibit the cognitive deficits typical in humans. 233 Additionally, rodents express fewer isoforms of key proteins, and their tau protein sequence lacks certain human‐specific variants or post‐translational modifications critical for the pathology of human AD. This discrepancy hinders the ability of rodent models to accurately reflect key aspects of tau protein phosphorylation and the resulting pathological folding and aggregation observed in humans. Another issue lies in the fact that rodent models exhibit pathological features not consistent with human AD, possibly due to overexpression of pathological markers that potentially lead to the mislocalization of amyloid plaques, 234 a phenomenon rarely observed in human AD.
A critical consideration in the evaluation of models is the interplay between amyloid and tau proteins, which, in human AD, progress in a closely interconnected and mutually influential manner. In rodents, these pathological progressions often remain relatively independent and disconnected, limiting their ability to fully represent human AD pathogenesis. 235 , 236 Introducing Aβ or tau proteins into rodents can accelerate pathological accumulation but offers little insight into initial mechanisms. Moreover, it is critical to note that over 95% of AD cases are sporadic with poorly understood pathology, 237 whereas the most commonly used transgenic rodent models carry mutations associated with early‐onset familial AD, which accounts for less than 5% of cases. 238 Future improvements in mouse models still have vast potential in experimental design, technological optimization, and applications.
5. UNLOCKING AD INSIGHTS: NON‐HUMAN PRIMATE MODELS FOR AD
Difficulty in developing effective therapies for AD underscores the urgent need for animal models that fully reflect disease pathology and progression while serving as platforms for exploring potential treatments. NHP models provide invaluable advantages for AD research due to their physiological congruence with humans. For instance, rhesus macaques share approximately 93% genomic similarity with humans, 239 making them an ideal model for studying molecular and cellular biological processes, immune responses, and signal transduction mechanisms. Moreover, NHPs show 100% human Aβ sequence homology, express both 3R and 4R tau isoforms, and exhibit robust large‐scale neurogenesis. 240 Advanced imaging enabled by their larger brains offers insights into lesion dynamics, while genetic engineering techniques further refine their relevance by replicating specific human AD pathologies.
Furthermore, NHPs exhibit behavior changes similar to humans, as has been systematically summarized in previous studies, such as a reduction in gait speed. These characteristics make NHPs an ideal model for studying brain pathology, as they allow for the MCI simultaneous evaluation of behavioral changes associated with AD and offer deeper insights into the potential connections between pathological changes and behavioral manifestations.
In AD research, the clinical relevance of biomarkers in diagnostic and treatment monitoring has been strongly supported by NHP models. These models demonstrate pathological features and behavioral changes within a biological and physiological context highly analogous to human conditions. These features and changes serve not only as diagnostic indicators but also as measures of pathological progression, thereby enhancing the predictive validity of preclinical studies. Over the past two decades, the application of NHP models in AD research has shown a steady increasing trend (Table 2). At the same time, the evaluation of biomarkers in NHP models has also witnessed increased utilization.
TABLE 2.
Summary of NHP models for AD
| Characteristic | Methods | Reference | Primate selection | A | T | N | NI | ATN(X) biomarkers | Other findings* |
|---|---|---|---|---|---|---|---|---|---|
| Aβ | |||||||||
| Sporadic | |||||||||
| Souder et al.,2021 241 | Rhesus monkeys (∼31 year) | – | – | – | – | None | Aβ plaques [temporal cortex]; mitochondrial distribution around plaques; lipofusin particles[brain]; IBA‐1↑; GFAP↑ | ||
| Stonebarger et al.,2020 242 | Rhesus monkeys (22–44 year, average 31.8 year) | – | – | – | – | None | Aβ plaques [Brodmann areas 32 & 46 of PFC]; NEUN+↓ | ||
| Zhang et al.,2019 243 | Aged rhesus monkeys (19–31year) | – | – | – | – | None | Aβ plaques [cerebral cortex]; IBA‐1↑, GFAP↑ [cerebral cortex] | ||
| Zhou et al.,2018 244 | Rhesus monkeys (> 30); cynomolgus monkeys (29–32 year) | – | – | – | – | None | Aβ plaques [cerebral cortex; hippocampus] | ||
| Lardenoije et al.,2018 245 | Caribbean vervets (12.2–32 year) | – | – | – | – | None | Aβ plaques [TC; hippocampus] | ||
| Poduri et al.,1994 246 | rhesus monkeys (3–15 year; 20–35 year) | – | – | – | – | None | Neuritic Aβ plaques [cortex; hippocampus]; Bergmann's astrocytes; APOE e4(PCR); APOE immunoreactivity [ERC; neocortex; hippocampus] | ||
| Bons et al.,1994 247 | Microcebus murinus (> 8y) | – | – | – | – | None | Amyloid deposits [cortical neuropil; walls of selected meningeal and cerebral vessels (CAA)] | ||
| Podlisny et al.,1991 248 | Cynomolgus monkeys | – | – | – | – | None | Aβ plaques[PFC]; CAA [cortex]; GFAP (around plaques); complete Amino Acid Homology to human | ||
| Injection | |||||||||
| AβOs(100 µg) once/3d for 24d(data last collected on day30) | Beckman et al.,2019 249 | Rhesus monkeys (female11–19 year; 22–28 year) | + | – | – | – | Aβ1‐42↑(CSF);AβOs↑(CSF) | Oligomeric Aβ[postsynaptic densities]; selective loss of highly plastic thin spines [area 46 of DLPFC]; IBA‐1↑(microglia activity, priming; ↑microglia volume); TNF‐α↑(CSF) | |
| Aβ fibrils+ lipopolysaccharide (LPS);(5months) | Philippens et al.,2017 250 | Marmosets (Callithrix jacchus) | – | – | – | – | None | Aβ plaques (61% Aβ1‐42 positive); IBA‐1+ [around plaques];↑CD95 and CD45RA expression on CD3+CD4+ cells(blood) | |
| Intracortical injections of Aβ (200 pg) | Sani et al.,2003 251 | Rhesus monkeys (5 year; 25–30 year) | – | – | – | – | None | Aβ deposits& plaques [cerebral cortex]; CAA [cerebral cortex; occipital cortice];plaque composition: Aβ40/Aβ42≈2.1 | |
| Tau | |||||||||
| Sporadic | |||||||||
| Leslie et al.,2021 252 | Rhesus monkeys (18–30 year) | – | – | – | – | None | WB: pSer235,396‐tau [brain] | ||
| Sharma et al.,2019 253 | Common marmosets (Callithrix jacchus; 0–5 year) | – | – | – | – | None | Immunoblotting: tau at pSer‐202,404,235,396; Thr231 [PFC] | ||
| Injection | |||||||||
| AAV1‐P301L/S320F (data collected monthly for 6 m) | Beckman et al.,2024 254 | Rhesus macaques (female 10–16 year) | – | + | + | + | pSer199, 396, pThr‐181, 231(CSF)↑; t‐tau(CSF)↑; sTrem2↑(CSF); NfL↑, BDNF↓(CSF) | pretangle states (NEUN+/AT8+/ThioS–); mature tangles (NEUN+/AT8+/ThioS+); IBA‐1↑, GFAP↑ [hippocampus, ERC]; NEUN+↓ [left CA3/Hilus] | |
| AAV‐P301L/S320F(data collected at1m, 2 m, and 3 m) | Beckman et al.,2021 255 | Rhesus macaques (10–15 year) | – | + | + | + | pSer‐199,396, pThr‐231(CSF)↑; t‐tau(CSF)↑; sTrem2↑(CSF); NfL↑(CSF; plasma); BDNF↓(CSF) | NFTs(ThioS+ in left hippocampus); IBA1+↑[left hippocampus]); NEUN+↓(hippocampus); TNF‐α, IL‐6↑(CSF, plasma) | |
| Aβ&Tau | |||||||||
| Sporadic | |||||||||
| Huang et al.,2024 256 | Cynomolgus monkeys with T2DM (17–20 year) | + | – | + |
‐ |
Aβ1‐40,42↓(plasma); NfL↑(CSF) | Aβ plaques [PFC; FC; TC; ERC]; NFTs [FC; TC; hippocampus]; IBA‐1+microglia↑, GFAP+ astrocytes↑; synaptic damage(↓PSD95 immunoreactivity in ERC); TNF‐α↑(peripheral blood) | ||
| Latimer et al.,2018 257 | African green monkeys (mean 11.2 year (middle‐aged); 21.7[aged]) | + | + | + | – | Aβ1‐42↓(CSF); p‐tau181↑(CSF); MRI volumes↓(right prefrontal, left inferior, left posterior temporal cortex); CMRg(↓18F‐FDGPET) | Aβ plaques[lateral temporal lobe]; PHF‐tau[temporal cortex]; gait speed↓ | ||
| Darusman et al.,2019,2014,2013 258 , 259 , 260 | Cynomolgus monkeys (9–15; > 20 year) | + | – | + | – | Aβ1‐42↓(CSF; compared to young); MRI atrophy (cerebral cortex; hippocampus) | Aβ1‐42 plaques [frontal, temporal, parietal, occipital lobe; hippocampus.]; CAA; tauopathy [p‐Tau231 in cytoplasm of the neuron cell]; granulovacuolar degeneration (GVD) | ||
| Paspalas et al.,2018 261 | Rhesus macaques (4.5–31 year) | – | – | – | – | None | Aβ1‐42 plaques [layer5(ERC)]; CAA; NFTs, PHF‐tau [DLPFC; ERC]; pSer214‐tau [layerII ERC cell]; Ca2+ dysregulation (pSer2808‐RyR2[DSR cisterns]) | ||
| Injection | |||||||||
| 7.5 µL hTau‐expressing AAVs into the hippocampus; 59 weeks | Jiang et al.,2024 262 | Rhesus macaques (7–15 year) | + | + | + | – | Aβ42 peptide↓(CSF); Aβ40/42↓; p‐tau181,231↑(CSF); tau‐PET (hippocampus to cerebellum); t‐tau↑(CSF); NfL↑; CMRg (↓18F‐FDG PET); MRI volume↓(hippocampus) | Aβ plaques; intra/extracellular Aβ deposits [hippocampus]; IBA‐1+microglia↑, GFAP+ astrocytes↑; CD68; NFTs[hippocampus]; ↓NEUN+ reactivity [hippocampal DG/CA2/CA3/CA4 regions]; cognitive decline (DR task; (DMTS/DNMTS) task) | |
| (0 month)tau aggregates purified from human AD brains(ERC) + (6th month)ICV injections of oligomeric Aβ(18 m (data collect)) | Darricau et al.,2024 263 | Rhesus macaques (14.7–15.2 year) | – | + | + | – | p‐tau181↑(CSF); t‐tau↑(CSF) | Aβ plaques[ERC]; CAA(exclusively in AβO injected monkeys); IBA+ microglia↑, GFAP+ astrocytes↑; neuropril threads; NFTs [ERC]; hippocampus];↓NEUN+ reactivity [CA1 layers(exclusively in AβO injected monkeys)] | |
| ICV or IT injection of 100‐µg AβO, Q3W*4 weeks(12 week(data collect)) | Wakeman et al.,2022 264 | African green monkeys (9.9+‐0.6 year) | – | – | + | – | MRI volume↓(hippocampus) | diffuse‐like plaques [6E10(MTL)]; p‐tau [hippocampus]; IBA+ microglia↑, GFAP+ astrocytes↑ [DG] | |
| Streptozotocin treated T1DM adult male vervet monkeys receiving BID exogenous insulin injections for 8–20 weeks | Morales‐Corraliza et al.,2016 265 | Vervet monkeys (Chlorocebus aethiops) (6.9+_0.2) years | – | – | – | – | None | soluble Aβ40, 42↑[hippocampus; TC]; ↓Aβ‐degrading enzyme neprilysin (NEP) [hippocampus]; p‐tau [CP13; PHF‐1]; tau‐kinase activity [↑ERK1/2 widespread in the brain] | |
| Transgenic | |||||||||
| PSEN1‐ΔE9(genomic deletion of PSEN1 exon9 by CRISPR/Cas9) | Li et al.,2024 266 | Cynomolgus monkeys | + | + | – | – | Aβ42↑(CSF); Aβ42/40↑(CSF); tau217/t‐tau↑(CSF); tau181/t‐tau↑(CSF); tau‐PET | Alterations in plasma inflammatory and immune molecules. | |
| Injection of human MAPT gene with Tau p301L mutation (0N4R P301L);(42month(last data)) | Tu et al.,2023 267 | Cynomolgus monkeys | + | + | + | – | Aβ42 Oligomers↑(spinal cord); Aβ1‐42↑(spinal cord); pThr231‐tau↑(CSF, plasma); NfL↑(CSF, plasma); t‐tau↑(CSF, plasma); MRI volume↓ (hippocampus); ↓glucose metabolism(PET([18F] FDG)‐MRI) | NFTs; ↓NEUN+ reactivity [hippocampus]; IBA+ microglia↑, GFAP+ astrocytes↑; cognitive dysfunction((1)delayed response[short‐term& long‐term memory tests]; (2)sleep behavior changes; (3)↓motor function[fine motor coordination test]; (4)anxiety‐like behaviors& ↓exploration activity)) | |
| APPtg (Swedish K670N/M671L+ Indiana V717F mutations (APPSWE/IND)); sacraficed at 10‐year‐old | Chan et al.,2022 268 | Rhesus monkeys | + | + | + | – | Aβ42↑(4y, CSF); p‐tau/Aβ42↓(4y, CSF); t‐tau↑(2–4 year, CSF) | Aβ plaques; CAA[occipital, parietal, and caudal temporal neocortices]; IBA+ microglia↑(slightly);↓Barrier Detour task at 16 months, Object‐in‐Place task(9 years of age);↑hostility(6years) |
Note: “+” and “−” to indicate the presence or absence of results. Detailed findings are demonstrated in the rightmost column: “ATN(X) biomarkers”.
The table headers A, T, N, and NI represent amyloid, tau, neurodegeneration, and neuroinflammation, respectively. *The “other findings” section includes biomarker results excluded from ATN(X), post mortem brain tissue‐related results, and behavioral tests.
Abbreviations: 18FDG‐PET, 18F‐fluorodeoxyglucose positron emission tomography; Aβ, amyloid‐beta; AβO, amyloid‐beta oligomer; AD, Alzheimer's disease; APP, amyloid precursor protein; ATN, amyloid/tau/neurodegeneration; BDNF, brain‐derived neurotrophic factor; CAA, cerebral amyloid angiopathy; CSF, cerebral spinal fluid; GFAP, glial fibrillary acidic protein; IBA‐1, ionized calcium‐binding adapter molecule 1; ICV, intracerebroventricular; MAPT, microtubule‐associated protein tau; MRI, magnetic resonance imaging; MTL, medial temporal lobe; NfL, neurofilament light chain; NFT, neurofibrillary tangle; PET, positron emission tomography; TC, temporal cortex; TREM2, triggering receptor expressed on myeloid cells 2.
6. BRIDGING BIOMARKERS: INSIGHTS FROM NHP TO HUMANS
Over the past two decades, NHP models of AD have demonstrated remarkable parallels with human pathology and biomarker profiles (Figure 3). These models now allow assessment using criteria applied in humans, such as the Braak staging. These advances have positioned NHPs as a valuable platform for studying neurodegenerative disease mechanisms.
FIGURE 3.

Overview of non‐human primate models in AD. The right side depicts the further pathological changes in the brain following amyloid plaque and neurofibrillary tangle formation. The left side shows the corresponding biomarker results in the monkey model, including plasma, CSF, and imaging findings. The letters A, T, and N denote amyloid, tau, and neurodegeneration, respectively. The numbered stages on the right correspond to specific pathological stages, with the associated biomarker results indicated on the left. AD, Alzheimer's disease; CSF, cerebrospinal fluid
6.1. Recapitulating amyloid cascade
6.1.1. Sporadic aging models
Aging is a critical risk factor for late‐onset AD. In aged rhesus monkeys (19–30 years), age‐related Aβ deposits and plaque appear in the brain, particularly in the cerebral cortex, prefrontal cortex (PFC), temporal cortex (TC), and hippocampus. 241 , 242 , 243 , 244 , 246 The plaques are mainly comprised of Aβ42, with some exhibiting APOE immunoreactivity, 246 and amyloid‐dependent microglial activity shown by a distribution pattern of IBA‐1+ microglia tightly surrounding Aβ deposits with alterations in both morphology and volume. In Souder's study, Aβ‐driven microglial priming associated with aging was characterized, uncovering remarkable parallels in the regional impact of amyloid pathology compared to late‐onset human AD. Furthermore, positive immunostaining of GFAP surrounding plaques was also observed, 241 highlighting the potential of using rhesus monkeys as a model for investigating neuroinflammation in AD.
Research on aged cynomolgus monkeys also identified the presence of senile plaques, 244 , 248 , 258 with microglial and astrocytic activation observed surrounding the plaques, revealing neuroinflammatory responses. 248 The aged monkeys also exhibited CAA within cortical regions, characterized by Aβ deposition in the walls of blood vessels. This vascular abnormality further underscores neuropathological features closely resembling those observed in human AD. 248 With nearly 100% amino acid homology to humans, cynomolgus monkeys are strong models for AD.
Other NHP species, such as vervets and microcebes, 245 , 247 demonstrated plaque burden within the TC and hippocampus. The microcebes, in particular, presented with CAA, while vervets showed potential in developing plaque pathology in the frontal cortex at the early age of 15 due to their propensity. Despite this early onset, their average life span is relatively short, typically not exceeding 30 years. This unique characteristic positions vervets as a valuable model for studying the early development of AD‐related pathologies and their potential progression over a shorter lifespan compared to other NHPs.
6.1.2. Experimental injection models
Injection‐based NHP models have been employed to study AD, particularly by introducing Aβ‐related pathological agents, which resemble the prion‐like propagation of Aβ in the human brain.
In Beckman's study, injection of oligomeric Aβ into rhesus monkeys 249 increases CSF Aβ1‐42, AβOs, and TNF‐α, accompanied by abnormal activation and hypertrophy of microglia in a persistently primed state. Selective loss of highly plastic thin spines in the dorsolateral prefrontal cortex (area 46) was also observed, implicating synaptic plasticity deficits.
Sepher Sani's research adopting intracortical Aβ injections in rhesus monkeys induces Aβ plaques in the cerebral cortex and CAA involving both major vessels and capillaries, primarily in the occipital cortex. 251 A higher burden of senile plaques (composed of both Aβ40 and Aβ42) was correlated with more severe CAA, with some plaques showing cholinesterase‐positivity, similar to human Aβ plaques, further supporting the compositional similarity of senile plaques between rhesus monkeys and humans. Additional injections of lipopolysaccharide (LPS) alongside Aβ fibrils 250 conducted by Ingrid showed that monkeys suffered from chronic neuroinflammation and intracerebral plaques, whereas those injected solely with Aβ fibrils did not exhibit significant pathological changes. None of the subjects showed overt behavioral abnormalities. Blood analysis in LPS‐treated monkeys indicated increased expression of CD95 and CD45RA on CD3+CD4+ T‐cells, suggesting potential implications for immune‐based therapeutic strategies.
These findings collectively highlight the utility of NHP models in studying the pathophysiology of AD, particularly in replicating aspects of human Aβ pathology and related inflammatory processes.
6.2. NHPs with tauopathy
6.2.1. Sporadic models
Marmosets, due to their shorter lifespan and ease of maintenance, are studied for tauopathy. Young marmoset brains from newborns to 5‐year‐olds react with tau antibodies AT8 (Ser202/Thr205), AT180 (Thr231/Ser235), and PHF‐1 (Ser396/Ser404) through immunoblotting but decrease with age, 253 suggesting that this species may not develop tau proteinopathies associated with these phosphorylation sites. Moreover, adult marmosets exclusively express 4R tau isoforms, and tau immunotherapy shows limited efficacy in these animals.
Studies conducted on aged rhesus monkeys identified pSer235 and pSer396 in the PFC, in addition to well‐known phosphorylation sites. 252 Overexpression of tau proves to upregulate GSK‐3b acetylation, 269 which further facilitates tau phosphorylation at Serine235 and 396, 270 , 271 leading to a vicious cycle. This offers insights into the pathogenic risk of these sites and guides clinical targeting of therapeutics.
6.2.2. Virus‐induced models
In a novel‐tau‐based study on rhesus monkey models, intracortical injections of adeno‐associated virus (AAV) vectors encoding the P301L/S320F tau mutation into the left ERC of 10‐ to 15‐year‐old rhesus macaques 255 triggered increased CSF t‐tau and several p‐tau species, including p‐tau199, p‐tau396, and p‐tau231 in the CSF, as well as NFTs in the left hippocampus and tau aggregates in ERC‐connected brain regions, including V1, V4, retrosplenial cortex (RSC), hippocampus, and the ERC itself. Concurrently, evidence of neuroinflammation and neurodegeneration markers was observed. However, no significant changes, in CSF Aβ42 or Aβ40 levels were noted.
In Beckman's subsequent study involving intracortical injections of AAV1‐P301L/S320F into the left ERC of female rhesus macaques, a transition of tau pathology in neuronal populations from pretangle states (NEUN+/AT8+/ThioS−) to mature tangles (NEUN+/AT8+/ThioS+), accompanied by a reduction in the number of healthy neurons (fewer NEUN+ cells) 254 were observed over 6 months. Additionally, the study demonstrated activated microglia since the early stages of tau pathology, whereas astrocyte involvement occurred later, coinciding with neuronal loss. The study further highlighted the prion‐like propagation of exogenous human 4R‐tau, which co‐aggregated with endogenous rhesus monkey 3R‐tau during transneuronal spread, underscoring the translational relevance of this NHP model for studying human tauopathies.
NHPs serve as a critical tool for studying tau isoforms and phosphorylation during aging. They allow both cross‐sectional comparisons of similarities and differences across various monkey species and longitudinal biomarker analyses with lower confounding influences, providing a clearer perspective for research. In contrast, experimentally induced NHP models closely replicate human tau pathology, allowing researchers to track disease progression through biomarkers at different time points over shorter periods and support the groundwork for therapeutic development.
6.3. NHPs with both Aβ and tau pathology
6.3.1. Sporadic models
In sporadic rhesus monkeys, tau pathologies progress systematically with advancing age, 261 beginning at 7–9 years with PKA‐phosphorylated tau (pSer214‐tau) in layer 2 of the ERC, also in dendrites and transporting endosomes, leading to dysregulated Ca2+ homeostasis. By their twenties, tau fibrils appear in ERC layer II, associated with cognitive deficits and matching Braak stage I/II. In their thirties, widespread fibrillar tau and mature NFTs manifest in layers 2 and 5, alongside progressive cognitive impairment, consistent with Braak stage III/IV. Additionally, amyloid pathology was noted in aged macaques over 30 years, characterized by Aβ1‐42 senile plaques within the ERC. Notably, microglial engulfment and lipofuscin observed in older monkeys indicate aging. The models mirror late‐onset AD, characterized by amyloid and tau pathology that closely replicates the sporadic progression of aggregate deposition observed in humans. A tau‐based staging framework aids cross‐species pathological comparison.
In aged cynomolgus monkeys exhibiting both amyloid and tau changes, CSF Aβ1‐42 decreases, and MRI detects atrophy in the ERC and hippocampus. 258 , 259 , 260 Post mortem brain analysis identified Aβ plaques, intracellular tauopathy, and granulovacuolar cytoplasmic changes, implying neurodegeneration.
In another study, aged cynomolgus monkeys with type 2 diabetes (T2DM) showed reduced plasma Aβ1‐40 and Aβ1‐42, elevated peripheral TNF‐α, and Aβ plaques in PFC, FC, TC, and ERC. 256 Tau pathology ranged from fibrils to mature tangles in the hippocampus, closely resembling the patterns observed in human AD. In addition to these findings, neuroinflammation and synaptic damage were also noted, although neurodegeneration was less prominent. These results highlight aged cynomolgus monkeys as preclinical AD models.
In vervet monkeys, Latimer's group identified smaller MRI‐measured brain volumes correlated with elevated CSF p‐tau181 levels and reduced gait speed with lower CSF Aβ1‐42 levels. 257 Additionally, Aβ plaque distribution was associated with reduced 18F‐FDG PET signals. These findings support vervets as preclinical AD models for biomarker studies and mechanistic explorations.
6.3.2. Injection‐based models
Diabetes increases the risk of AD by affecting the inflammatory response, protein metabolism, and cell death through disruptions in insulin signaling. 272 , 273 , 274 In streptozotocin (STZ) ‐induced type 1 diabetes vervet monkeys, early AD‐associated pathologies included increased soluble Aβ1‐40 and Aβ1‐42 in the hippocampus and TC, reduced Aβ‐degrading enzymes, hyperphosphorylated tau fibrils, and elevated tau kinase activity. 265
In African green monkeys, chronic Aβ oligomer injections resulted in diffuse MTL amyloid plaques, accelerated tau pathology, activated microglia and astrocytes, and hippocampal atrophy detected by longitudinal MRI, paralleling cognitive decline by behavioral tests. These results position Aβ oligomers as a critical upstream driver of AD pathology, providing an experimental platform for exploring preventive interventions. 264
Injections of extracted human tau aggregates into the ERC and hippocampus successfully replicated prion‐like tau spread and Braak stage 1–4 pathology in primates, NFTs, and elevated CSF p‐tau181. Co‐administration of Aβ oligomers significantly accelerated these pathological changes, promoting severe NFTs, heightened t‐tau levels, and overt neurodegeneration. This highlights Aβ’s role in advancing tau progression, showing human‐derived pathological substrates retain toxicity in primate brains. 263
Zhouquan Jiang's group established a rapid and comprehensive primate AD model in rhesus monkeys via a single injection of AAV‐tau in the bilateral hippocampus. 262 Within 2–3 months, models replicated key AD biomarkers: elevated CSF Aβ1‐42 levels, reduced Aβ40/42 ratios, increased p‐tau181, p‐tau231, and t‐tau, elevated CSF NfL, reduced glucose metabolism, and hippocampal atrophy observed by MRI. Histological analyses revealed widespread amyloid plaques, NFTs, and neuroinflammation, and behavioral assessments confirmed significant cognitive decline. Compared to earlier models, this model offers rapid, comprehensive AD pathology for translational research.
6.3.3. Transgenic models
Transgenic APP rhesus monkeys exhibit prominent AD‐related biomarkers by age 4: elevated CSF Aβ1‐42, and t‐tau, alongside diminished CSF p‐tau/Aβ42 ratios indicating early pathological changes. 268 By age 10, some display widespread CAA, plaque deposition, and microglial activation, similar to human AD but with earlier onset. Behavioral anomalies arise by 16 months, demonstrating the model's value for preclinical studies, preventative interventions, and disease‐modifying therapies.
PSEN1‐mutant cynomolgus monkeys, utilizing CRISPR/Cas9 to delete exon 9 of the PSEN1 gene, yield a live‐birth success rate of 50%. 266 This AD model recapitulates familial AD with key neuropathological biomarkers, including increased CSF Aβ42, p‐tau217, and plasma immune and inflammatory changes, strengthening the model's power in studying familial AD, evaluating early diagnostic biomarkers, and aiding therapeutic interventions.
In cynomolgus monkeys expressing the human MAPT gene with the pathogenic P301L mutation (0N4R P301L), tau promoted Aβ aggregation within the spinal cord, a phenomenon identified for the first time in this model. Models also exhibit accelerated tau aggregation, notable biomarker alterations, neuroinflammation, synaptic degeneration, and motor deficits, faithfully replicating critical aspects of AD. 267 This demonstrates a mechanistic tau‐Aβ link, offering a unique opportunity for the investigation of targeted therapies.
Transgenic NHP models of AD have significantly advanced our understanding of AD pathology and facilitated therapeutic innovations. These models accurately mimic human symptoms, providing a deeper understanding of the intricate and diverse nature of the disease. Consequently, they shed light on advancing the development of personalized and precision medicine approaches.
7. EXPLORING NHP MODELS: PROMISE AND CHALLENGES IN AD RESEARCH
7.1. Key questions for optimizing present NHP models
NHP models provide key advantages in AD research by bridging gaps left by rodent models, particularly regarding age‐related pathology onset, genetic gaps, and the reproducibility of AD phenotypes. To maximize their relevance, it is essential to thoroughly evaluate disease features in NHPs.
In natural aging models, pathological variability among individuals of comparable ages underscores the need to uncover underlying mechanisms. Characterizing key pathological markers, such as 3R and 4R tau isoforms, phosphorylation patterns, and Aβ structure, may help refine the models. Natural models may not fully mirror human disease progression due to species and genetic differences. Identifying optimal NHP species, like rhesus monkeys, and establishing species‐specific timelines for the earliest pathological changes are critical for model development. Induced models—using transgenic techniques or pathological material injection—require careful timing and duration to better simulate human‐like disease progression. Longitudinal studies tracking AD hallmarks—such as Aβ deposition and tau abnormalities—are crucial for replicating the decades‐long progression seen in humans. Recent advances in transgenic NHP models, including multigene modifications (APOE, Aβ, tau), show promise in improving disease fidelity. Chinese NHP cohorts have highlighted the importance of rigorous validation and reproducibility to ensure the reliability and broader applicability of findings.
Addressing these challenges may enhance the precision and utility of NHP models, supporting breakthroughs in understanding AD pathophysiology and expediting the development of effective therapies. Additionally, other models also warrant consideration, such as cholinergic nervous system injury models frequently employed to investigate the effects of damage to the cholinergic pathways and the STZ injection method in the context of studying diabetes and related neurodegenerative processes. 275 These models provide valuable insights into specific pathological mechanisms and contribute to a more comprehensive understanding of the underlying conditions.
7.2. Potential of integrating neuroinflammation biomarkers into NHP models
A growing body of recent studies has specifically evaluated GFAP and sTREM2 as key neuroinflammation biomarkers in AD. 161 , 165 , 166 , 192 Supporting these findings, NHP studies from Beckman's group have reported elevated levels of sTREM2 in the CSF. 254 , 255 Additionally, most NHP studies have observed increased numbers of IBA‐1+ microglia and GFAP+ astrocytes in brain regions affected by AD, further highlighting the role of neuroinflammation in disease pathology. 241 , 243 , 246 , 248 , 250 , 254 , 255 , 256 , 262 Importantly, clinical trials have begun to include neuroinflammation biomarkers in their therapeutic evaluation for reducing the cost and complement to traditional Aβ and tau measurements. 276 , 277 , 278 , 279 Despite advances in biomarker research, there still remains a significant gap in the assessment of neuroinflammation‐based biomarkers in NHP studies, with only two included studies reporting neuroinflammation‐related findings. 254 , 255 This limitation is likely due to the predominant focus on Aβ and tau pathology in most NHP research, which often overlooks the assessment of additional inflammatory markers. While some research has measured general inflammatory indicators such as TNF‐α, specific examination of CNS glial cells is still lacking. 256 This gap may, in part, reflect constraints associated with the disease stage represented in current models. This gap may, in part, reflect constraints associated with the disease stage represented in current models. To address these limitations, incorporating neuroinflammation biomarkers into NHP studies is crucial. This approach may help uncover novel disease mechanisms and improve the relevance of NHP models in human AD, highlighting their potential clinical value.
7.3. Current limitations to wider adoption of NHP models
Although NHP models are indispensable in AD research, they face significant challenges in faithfully replicating the complex pathology and cognitive symptoms seen in humans. In naturally aging NHPs, AD‐related pathological features are often only partially replicated, and these models rarely reproduce the full spectrum or progression of the disease, despite their high genetic similarity to humans. Additionally, behavioral signs of cognitive decline are inconsistently observed.
Genetically modified or experimentally induced NHPs—through the introduction of human AD‐related genes or pathological proteins—can better replicate specific pathological features and align more closely with human clinical biomarkers. However, such models still struggle to reproduce several key pathological characteristics in human AD, such as the expression of human‐specific tau isoforms, characteristic Aβ plaque distribution patterns, and the mechanisms of disease spreading within the brain. From a modeling perspective, although introducing key pathological factors can induce corresponding disease features under ideal conditions, technical challenges such as mosaicism during model creation, low breeding efficiency, and the inability to fully replicate human disease mutations significantly hinder the establishment of highly accurate disease models. 280 Currently, stricter ethical regulations, complex approval processes, 280 and high costs of model generation and maintenance due to longer lifespans further restrict large‐scale application, while the effectiveness of advanced gene‐editing technologies like CRISPR still requires large‐scale validation.
Addressing these challenges is essential for improving the reliability and translational relevance of NHP models. Advances such as employing gene‐editing technologies can more precisely recreate the human genetic context, while multifactorial induction strategies may produce composite models that better emulate the disease's pathological complexity. Furthermore, fostering interdisciplinary collaboration across neuroscience, genetics, and molecular biology will be crucial to refining NHP models, ultimately providing more robust and predictive tools for advancing AD research.
8. DISCUSSION
Biomarkers play a crucial role in the early diagnosis, staging, and therapeutic monitoring of AD. The adoption of the ATN framework has greatly advanced the clinical utility of biomarkers, enabling more precise early intervention. However, existing biomarkers and the ATN framework may insufficiently capture the complex, multifactorial nature of AD, especially in its earliest stages. Diagnostic sensitivity and specificity are still not optimal for all patient populations, and routine use remains limited by cost and accessibility. Building on this concept, this study proposes that neuroinflammation (X) might serve as a promising complement to the ATN(X) framework, as it is now recognized as a major pathological feature in AD and is closely intertwined with amyloid and tau pathology. Currently, neuroinflammatory biomarkers are less established due to their variability in detection techniques and limited longitudinal data. The complex relationship between neuroinflammation and AD core pathology still remains under‐explored, further inhibiting the broader use of neuroinflammation biomarkers. Further research should focus on standardizing neuroinflammation biomarker assays, establishing longitudinal correlations with disease progression, and integrating these markers in large‐scale clinical studies.
NHPs have gained increasing attention in recent years as promising animal models for AD research. Biomarker detection has demonstrated an increasing trend in the study of AD‐related NHP models. However, a standardized primate model equivalent to the widely used 5xFAD mouse model is still lacking. Optimizing pathological induction techniques and gene‐editing approaches may enhance reproducibility and efficiency. Given the evidence of neuroinflammation in monkey brain slices, future research should focus on addressing the complexity of AD by standardizing NHP models and incorporating neuroinflammatory biomarkers. This integration is crucial for capturing disease dynamics and improving model reliability.
In conclusion, we posit that biomarkers serve as a critical thread that links their applications in early diagnosis, therapeutic monitoring, and their utilization in NHP models. As our understanding of biomarkers and their applications in NHP models continues to expand, and as these models are further refined and developed, we anticipate that these combined efforts will significantly enhance our understanding of AD and provide a robust and effective strategy in the ongoing battle against this prolonged war.
AUTHOR CONTRIBUTIONS
Conceptualization: Zhihui Jin, Huifang Tang, and Huashun Cui. Data curation (100%): Zhihui Jin. Formal Analysis (100%): Zhihui Jin. Methodology (100%): Zhihui Jin. Visualization(100%): Zhihui Jin. Investigation(100%): Zhihui Jin. Writing—Original draft (100%): Zhihui Jin. Supervision: Yan Lu and Huashun Cui. Project administration: Yan Lu. Resources: Yan Lu. Writing‐Review & editing: Yan Lu, Huifang Tang, and Huashun Cui. Validation (100%): Huifang Tang. Funding acquisition: Huashun Cui.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. All authors approved the final manuscript. Dr. Zhihui Jin (daughter of the co‐corresponding author Dr. Huashun Cui) is the sole first author. To ensure objectivity, we declare the following content: (1) Search strategy was audited by Prof.Huifang Tang; (2) Final inclusion was approved by all co‐authors with no family ties; (3) Data sources were exclusively public; (4) No industry/pharmacy funding supported this review. Dr. Huashun Cui recused herself from inclusion/exclusion of studies related to Traditional Chinese medicine (her expertise), and quality assessment (using NIA‐AA guidelines). Author disclosures are available in the supporting information.
CONSENT STATEMENT
The consent was not necessary.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors appreciate Sihui Song (Washington University in St. Louis) and Yun Chen (Washington University in St. Louis) in writing assistance. The figures were created by Biorender. This work was supported by the National Natural Science Foundation of China (No. 82074535).
Jin Z, Lu Y, Tang H, Cui H. Integrating neuroinflammation biomarkers into the ATN(X) framework: Advances in Alzheimer's pathogenesis, diagnosis, and insights from non‐human primate models. Alzheimer's Dement. 2025;21:e70472. 10.1002/alz.70472
Contributor Information
Yan Lu, Email: sophieluo896@njucm.edu.cn.
Huifang Tang, Email: tanghuifang@zju.edu.cn.
Huashun Cui, Email: cuihuashun@shutcm.edu.cn.
REFERENCES
- 1. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577‐1590. doi: 10.1016/s0140-6736(20)32205-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barker WW, Luis CA, Kashuba A, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16(4):203‐212. doi: 10.1097/00002093-200210000-00001 [DOI] [PubMed] [Google Scholar]
- 3. 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;16(3):391‐460. doi: 10.1002/alz.12068 [DOI] [Google Scholar]
- 4. Feng L, Li J, Yu JT, et al. Editorial: prevention of Alzheimer's disease in Chinese populations: status, challenges and directions. J Prev Alzheimers Dis. 2018;5(2):90‐94. doi: 10.14283/jpad.2018.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jia J, Wei C, Chen S, et al. The cost of Alzheimer's disease in China and re‐estimation of costs worldwide. Alzheimers Dement. 2018;14(4):483‐491. doi: 10.1016/j.jalz.2017.12.006 [DOI] [PubMed] [Google Scholar]
- 6. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19(4):1598‐1695. doi: 10.1002/alz.13016 [DOI] [PubMed] [Google Scholar]
- 7. 2024 Alzheimer's disease facts and figures. Alzheimers Dement. 2024;20(5):3708‐3821. doi: 10.1002/alz.13809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pierce AL, Bullain SS, Kawas CH. Late‐onset Alzheimer disease. Neurol Clin. 2017;35(2):283‐293. doi: 10.1016/j.ncl.2017.01.006 [DOI] [PubMed] [Google Scholar]
- 9. Serrano‐Pozo A, Das S, Hyman BT. APOE and Alzheimer's disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20(1):68‐80. doi: 10.1016/s1474-4422(20)30412-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17(3):241‐250. doi: 10.1016/s1474-4422(18)30028-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sullivan PM. Influence of Western diet and APOE genotype on Alzheimer's disease risk. Neurobiol Dis. 2020;138:104790. doi: 10.1016/j.nbd.2020.104790 [DOI] [PubMed] [Google Scholar]
- 12. Hoffman JL, Faccidomo S, Kim M, et al. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg‐AD mouse model of Alzheimer's disease. Int Rev Neurobiol. 2019;148:169‐230. doi: 10.1016/bs.irn.2019.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caruso A, Nicoletti F, Mango D, Saidi A, Orlando R, Scaccianoce S. Stress as risk factor for Alzheimer's disease. Pharmacol Res. 2018;132:130‐134. doi: 10.1016/j.phrs.2018.04.017 [DOI] [PubMed] [Google Scholar]
- 14. Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer's disease. Mol Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willem M, Tahirovic S, Busche MA, et al. η‐Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature. 2015;526(7573):443‐447. doi: 10.1038/nature14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184‐185. doi: 10.1126/science.1566067 [DOI] [PubMed] [Google Scholar]
- 17. Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin‐1 required for presenilin endoproteolysis and gamma‐secretase activity. Nature. 1999;398(6727):513‐517. doi: 10.1038/19077 [DOI] [PubMed] [Google Scholar]
- 18. Kim J, Onstead L, Randle S, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27(3):627‐633. doi: 10.1523/jneurosci.4849-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurkinen M, Fułek M, Fułek K, Beszłej JA, Kurpas D, Leszek J. The amyloid cascade hypothesis in Alzheimer's disease: should we change our thinking?. Biomolecules. 2023;13(3):453. doi: 10.3390/biom13030453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goate A. Segregation of a missense mutation in the amyloid beta‐protein precursor gene with familial Alzheimer's disease. J Alzheimers Dis. 2006;9(3 Suppl):341‐347. doi: 10.3233/jad-2006-9s338 [DOI] [PubMed] [Google Scholar]
- 21. Chen GF, Xu TH, Yan Y, et al. Amyloid beta: structure, biology and structure‐based therapeutic development. Acta Pharmacol Sin. 2017;38(9):1205‐1235. doi: 10.1038/aps.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol. 2007;8(2):101‐112. doi: 10.1038/nrm2101 [DOI] [PubMed] [Google Scholar]
- 23. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333‐366. doi: 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 24. Sengupta U, Nilson AN, Kayed R. The role of amyloid‐β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42‐49. doi: 10.1016/j.ebiom.2016.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SJ, Nam E, Lee HJ, Savelieff MG, Lim MH. Towards an understanding of amyloid‐β oligomers: characterization, toxicity mechanisms, and inhibitors. Chem Soc Rev. 2017;46(2):310‐323. doi: 10.1039/c6cs00731g [DOI] [PubMed] [Google Scholar]
- 26. Ortiz‐Sanz C, Balantzategi U, Quintela‐López T, et al. Amyloid β /PKC‐dependent alterations in NMDA receptor composition are detected in early stages of Alzheimer´s disease. Cell Death Dis. 2022;13(3):253. doi: 10.1038/s41419-022-04687-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alberdi E, Sánchez‐Gómez MV, Cavaliere F, et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47(3):264‐272. doi: 10.1016/j.ceca.2009.12.010 [DOI] [PubMed] [Google Scholar]
- 28. Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid‐beta protein induce reversible synapse loss by modulating an NMDA‐type glutamate receptor‐dependent signaling pathway. J Neurosci. 2007;27(11):2866‐2875. doi: 10.1523/jneurosci.4970-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahmed M, Davis J, Aucoin D, et al. Structural conversion of neurotoxic amyloid‐beta(1‐42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17(5):561‐567. doi: 10.1038/nsmb.1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willbold D, Strodel B, Schröder GF, Hoyer W, Heise H. Amyloid‐type protein aggregation and prion‐like properties of amyloids. Chem Rev. 2021;121(13):8285‐8307. doi: 10.1021/acs.chemrev.1c00196 [DOI] [PubMed] [Google Scholar]
- 31. Roher AE, Lowenson JD, Clarke S, et al. beta‐Amyloid‐(1‐42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Nat Acad Sci USA. 1993;90(22):10836‐10840. doi: 10.1073/pnas.90.22.10836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chatterjee P, Pedrini S, Doecke JD, et al. Plasma Aβ42/40 ratio, p‐tau181, GFAP, and NfL across the Alzheimer's disease continuum: a cross‐sectional and longitudinal study in the AIBL cohort. Alzheimers Dement. 2023;19(4):1117‐1134. doi: 10.1002/alz.12724 [DOI] [PubMed] [Google Scholar]
- 33. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐head comparison of 8 plasma amyloid‐β 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78(11):1375‐1382. doi: 10.1001/jamaneurol.2021.3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hansson O, Lehmann S, Otto M, Zetterberg H, Lewczuk P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer's disease. Alzheimers Res Ther. 2019;11(1):34. doi: 10.1186/s13195-019-0485-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Janelidze S, Barthélemy NR, Salvadó G, et al. Plasma phosphorylated tau 217 and Aβ42/40 to predict early brain Aβ accumulation in people without cognitive impairment. JAMA Neurol. 2024;81(9):947‐957. doi: 10.1001/jamaneurol.2024.2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1‐40) and Abeta(1‐42) and the risk of dementia: a prospective case‐cohort study. Lancet Neurol. 2006;5(8):655‐660. doi: 10.1016/s1474-4422(06)70501-4 [DOI] [PubMed] [Google Scholar]
- 37. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595‐608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discovery. 2011;10(9):698‐712. doi: 10.1038/nrd3505 [DOI] [PubMed] [Google Scholar]
- 39. Rostami J, Mothes T, Kolahdouzan M, et al. Crosstalk between astrocytes and microglia results in increased degradation of α‐synuclein and amyloid‐β aggregates. J Neuroinflam. 2021;18(1):124. doi: 10.1186/s12974-021-02158-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tzioras M, Daniels MJD, Davies C, et al. Human astrocytes and microglia show augmented ingestion of synapses in Alzheimer's disease via MFG‐E8. Cell Rep Med. 2023;4(9):101175. doi: 10.1016/j.xcrm.2023.101175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palmqvist S, Schöll M, Strandberg O, et al. Earliest accumulation of β‐amyloid occurs within the default‐mode network and concurrently affects brain connectivity. Nat Commun. 2017;8(1):1214. doi: 10.1038/s41467-017-01150-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791‐1800. doi: 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 43. Levin J, Vöglein J, Quiroz YT, et al. Testing the amyloid cascade hypothesis: prevention trials in autosomal dominant Alzheimer disease. Alzheimers Dement. 2022;18(12):2687‐2698. doi: 10.1002/alz.12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Granzotto A, Sensi SL. Once upon a time, the Amyloid Cascade Hypothesis. Ageing Res Rev. 2024;93:102161. doi: 10.1016/j.arr.2023.102161 [DOI] [PubMed] [Google Scholar]
- 45. Kepp KP, Robakis NK, Høilund‐Carlsen PF, Sensi SL, Vissel B. The amyloid cascade hypothesis: an updated critical review. Brain. 2023;146(10):3969‐3990. doi: 10.1093/brain/awad159 [DOI] [PubMed] [Google Scholar]
- 46. Nussbaum JM, Schilling S, Cynis H, et al. Prion‐like behaviour and tau‐dependent cytotoxicity of pyroglutamylated amyloid‐β. Nature. 2012;485(7400):651‐655. doi: 10.1038/nature11060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97(6):1284‐1298.e7. doi: 10.1016/j.neuron.2018.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hurtado DE, Molina‐Porcel L, Iba M, et al. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol. 2010;177(4):1977‐1988. doi: 10.2353/ajpath.2010.100346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leroy K, Ando K, Laporte V, et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol. 2012;181(6):1928‐1940. doi: 10.1016/j.ajpath.2012.08.012 [DOI] [PubMed] [Google Scholar]
- 50. Roberson ED, Scearce‐Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science. 2007;316(5825):750‐754. doi: 10.1126/science.1141736 [DOI] [PubMed] [Google Scholar]
- 51. Bloom GS. Amyloid‐β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71(4):505‐508. doi: 10.1001/jamaneurol.2013.5847 [DOI] [PubMed] [Google Scholar]
- 52. Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. NeuroMol Med. 2002;2(2):151‐165. doi: 10.1385/nmm:2:2:151 [DOI] [PubMed] [Google Scholar]
- 53. Hernandez P, Lee G, Sjoberg M, Maccioni RB. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25‐35): involvement of lipid rafts. J Alzheimers Dis. 2009;16(1):149‐156. doi: 10.3233/jad-2009-0933 [DOI] [PubMed] [Google Scholar]
- 54. Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491‐1495. doi: 10.1126/science.1062097 [DOI] [PubMed] [Google Scholar]
- 55. Cho H, Choi JY, Lee HS, et al. Progressive tau accumulation in Alzheimer disease: 2‐year follow‐up study. J Nucl Med. 2019;60(11):1611‐1621. doi: 10.2967/jnumed.118.221697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallego‐Rudolf J, Wiesman AI, Pichet Binette A, Villeneuve S, Baillet S. Synergistic association of Aβ and tau pathology with cortical neurophysiology and cognitive decline in asymptomatic older adults. Nat Neurosci. 2024;27(11):2130‐2137. doi: 10.1038/s41593-024-01763-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Nat Acad Sci USA. 1975;72(5):1858‐1862. doi: 10.1073/pnas.72.5.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15(3):112‐119. doi: 10.1016/j.molmed.2009.01.003 [DOI] [PubMed] [Google Scholar]
- 59. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665‐704. doi: 10.1007/s00401-017-1707-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barthélemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26(3):398‐407. doi: 10.1038/s41591-020-0781-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Limorenko G, Lashuel HA. Revisiting the grammar of Tau aggregation and pathology formation: how new insights from brain pathology are shaping how we study and target Tauopathies. Chem Soc Rev. 2022;51(2):513‐565. doi: 10.1039/d1cs00127b [DOI] [PubMed] [Google Scholar]
- 62. Hoover BR, Reed MN, Su J, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067‐1081. doi: 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Spires‐Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 2014;82(4):756‐771. doi: 10.1016/j.neuron.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Adams JN, Maass A, Harrison TM, Baker SL, Jagust WJ. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. eLife. 2019;8:e49132. doi: 10.7554/eLife.49132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ferber D. Neurodegenerative disease. Using the fruit fly to model tau malfunction. Science. 2001;292(5524):1983‐1984. doi: 10.1126/science.292.5524.1983a [DOI] [PubMed] [Google Scholar]
- 66. Tracy TE, Madero‐Pérez J, Swaney DL, et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell. 2022;185(4):712‐728.e14. doi: 10.1016/j.cell.2021.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hoenig MC, Bischof GN, Seemiller J, et al. Networks of tau distribution in Alzheimer's disease. Brain. 2018;141(2):568‐581. doi: 10.1093/brain/awx353 [DOI] [PubMed] [Google Scholar]
- 68. Macedo AC, Tissot C, Therriault J, et al. The use of tau PET to stage Alzheimer disease according to the Braak Staging Framework. J Nucl Med. 2023;64(8):1171‐1178. doi: 10.2967/jnumed.122.265200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lowe VJ, Lundt ES, Albertson SM, et al. Tau‐positron emission tomography correlates with neuropathology findings. Alzheimers Dement. 2020;16(3):561‐571. doi: 10.1016/j.jalz.2019.09.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353‐356. doi: 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- 71. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9(1):119‐128. doi: 10.1016/s1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roberson ED, Halabisky B, Yoo JW, et al. Amyloid‐β/Fyn‐induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31(2):700‐711. doi: 10.1523/jneurosci.4152-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Haass C, Mandelkow E. Fyn‐tau‐amyloid: a toxic triad. Cell. 2010;142(3):356‐358. doi: 10.1016/j.cell.2010.07.032 [DOI] [PubMed] [Google Scholar]
- 74. Shah SB, Nolan R, Davis E, et al. Examination of potential mechanisms of amyloid‐induced defects in neuronal transport. Neurobiol Dis. 2009;36(1):11‐25. doi: 10.1016/j.nbd.2009.05.016 [DOI] [PubMed] [Google Scholar]
- 75. Zhang H, Wei W, Zhao M, et al. Interaction between Aβ and tau in the pathogenesis of Alzheimer's disease. Int J Biol Sci. 2021;17(9):2181‐2192. doi: 10.7150/ijbs.57078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Amadoro G, Corsetti V, Stringaro A, et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis. 2010;21(2):445‐470. doi: 10.3233/jad-2010-100120 [DOI] [PubMed] [Google Scholar]
- 77. van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid‐β‐independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21‐35. doi: 10.1038/s41583-019-0240-3 [DOI] [PubMed] [Google Scholar]
- 78. Harrison TM, La Joie R, Maass A, et al. Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Ann Neurol. 2019;85(2):229‐240. doi: 10.1002/ana.25406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Coppens S, Lehmann S, Hopley C, Hirtz C. Neurofilament‐light, a promising biomarker: analytical, metrological and clinical challenges. Int J Mol Sci. 2023;24(14):11624. doi: 10.3390/ijms241411624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Parnetti L, Gaetani L, Di Filippo M. Serum neurofilament light chain as a preclinical marker of neurodegeneration. Lancet Neurol. 2019;18(12):1070‐1071. doi: 10.1016/s1474-4422(19)30405-3 [DOI] [PubMed] [Google Scholar]
- 81. Sellebjerg F, Magyari M. The prognostic value of neurofilament light chain in serum. Lancet Neurol. 2022;21(3):207‐208. doi: 10.1016/s1474-4422(22)00034-5 [DOI] [PubMed] [Google Scholar]
- 82. Minoshima S, Cross D, Thientunyakit T, Foster NL, Drzezga A. (18)F‐FDG PET imaging in neurodegenerative dementing disorders: insights into subtype classification, emerging disease categories, and mixed dementia with copathologies. J Nucl Med. 2022;63(Suppl 1):2s‐12s. doi: 10.2967/jnumed.121.263194 [DOI] [PubMed] [Google Scholar]
- 83. Bobinski M, de Leon MJ, Convit A, et al. MRI of entorhinal cortex in mild Alzheimer's disease. Lancet. 1999;353(9146):38‐40. doi: 10.1016/s0140-6736(05)74869-8 [DOI] [PubMed] [Google Scholar]
- 84. Pan D, Zeng A, Yang B, et al. Deep learning for brain MRI confirms patterned pathological progression in Alzheimer's disease. Adv Sci. 2023;10(6):e2204717. doi: 10.1002/advs.202204717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Heneka MT, van der Flier WM, Jessen F, et al. Neuroinflammation in Alzheimer disease. Nat Rev Immunol. 2025;25(5):321‐352. doi: 10.1038/s41577-024-01104-7 [DOI] [PubMed] [Google Scholar]
- 86. Yang L, Zhou Y, Jia H, Qi Y, Tu S, Shao A. Affective immunology: the crosstalk between microglia and astrocytes plays key role?. Front Immunol. 2020;11:1818. doi: 10.3389/fimmu.2020.01818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Matejuk A, Ransohoff RM. Crosstalk between astrocytes and microglia: an overview. Front Immunol. 2020;11:1416. doi: 10.3389/fimmu.2020.01416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wyss‐Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid‐beta in vitro and in situ. Nat Med. 2003;9(4):453‐457. doi: 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- 89. Kim S, Chun H, Kim Y, et al. Astrocytic autophagy plasticity modulates Aβ clearance and cognitive function in Alzheimer's disease. Mol Neurodegener. 2024;19(1):55. doi: 10.1186/s13024-024-00740-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer's disease. J Neuroinflam. 2022;19(1):206. doi: 10.1186/s12974-022-02565-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol. 1992;84(2):117‐127. doi: 10.1007/bf00311383 [DOI] [PubMed] [Google Scholar]
- 92. Deng Q, Wu C, Parker E, Liu TC, Duan R, Yang L. Microglia and astrocytes in Alzheimer's disease: significance and summary of recent advances. Aging Dis. 2024;15(4):1537‐1564. doi: 10.14336/ad.2023.0907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481‐487. doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here?. Nat Rev Neurol. 2021;17(3):157‐172. doi: 10.1038/s41582-020-00435-y [DOI] [PubMed] [Google Scholar]
- 95. Plantone D, Pardini M, Righi D, Manco C, Colombo BM, De StefanoN. The role of TNF‐α in Alzheimer's disease: a narrative review. Cells. 2023;13(1):54. doi: 10.3390/cells13010054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ng A, Tam WW, Zhang MW, et al. IL‐1β, IL‐6, TNF‐ α and CRP in elderly patients with depression or Alzheimer's disease: systematic review and meta‐analysis. Sci Rep. 2018;8(1):12050. doi: 10.1038/s41598-018-30487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chatterjee P, Vermunt L, Gordon BA, et al. Plasma glial fibrillary acidic protein in autosomal dominant Alzheimer's disease: associations with Aβ‐PET, neurodegeneration, and cognition. Alzheimers Dement. 2023;19(7):2790‐2804. doi: 10.1002/alz.12879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Blazevic N, Rogic D, Pelajic S, et al. YKL‐40 as a biomarker in various inflammatory diseases: a review. Biochemia medica. 2024;34(1):010502. doi: 10.11613/bm.2024.010502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta‐analysis. Lancet Neurology. 2016;15(7):673‐684. doi: 10.1016/s1474-4422(16)00070-3 [DOI] [PubMed] [Google Scholar]
- 100. Biel D, Suárez‐Calvet M, Hager P, et al. sTREM2 is associated with amyloid‐related p‐tau increases and glucose hypermetabolism in Alzheimer's disease. EMBO Mol Med. 2023;15(2):e16987. doi: 10.15252/emmm.202216987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Self WK, Holtzman DM. Emerging diagnostics and therapeutics for Alzheimer disease. Nat Med. 2023;29(9):2187‐2199. doi: 10.1038/s41591-023-02505-2 [DOI] [PubMed] [Google Scholar]
- 102. Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by beta‐amyloid protein and interferon‐gamma. Nature. 1995;374(6523):647‐650. doi: 10.1038/374647a0 [DOI] [PubMed] [Google Scholar]
- 103. Carrano A, Hoozemans JJ, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJ. Neuroinflammation and blood‐brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis. 2012;10(1‐4):329‐331. doi: 10.1159/000334916 [DOI] [PubMed] [Google Scholar]
- 104. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217‐224. doi: 10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- 105. Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer's disease. Arch Med Res. 2008;39(1):1‐16. doi: 10.1016/j.arcmed.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 106. Gerrits E, Brouwer N, Kooistra SM, et al. Distinct amyloid‐β and tau‐associated microglia profiles in Alzheimer's disease. Acta Neuropathol. 2021;141(5):681‐696. doi: 10.1007/s00401-021-02263-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kim DW, Tu KJ, Wei A, et al. Amyloid‐beta and tau pathologies act synergistically to induce novel disease stage‐specific microglia subtypes. Mol Neurodegener. 2022;17(1):83. doi: 10.1186/s13024-022-00589-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Keren‐Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169(7):1276‐1290.e17. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 109. Chen Y, Colonna M. Microglia in Alzheimer's disease at single‐cell level. Are there common patterns in humans and mice?. J Exp Med. 2021;218(9):e20202717. doi: 10.1084/jem.20202717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458‐3483. doi: 10.1016/j.neuron.2022.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. 2018;217(2):459‐472. doi: 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bellaver B, Povala G, Ferreira PCL, et al. Astrocyte reactivity influences amyloid‐β effects on tau pathology in preclinical Alzheimer's disease. Nat Med. 2023;29(7):1775‐1781. doi: 10.1038/s41591-023-02380-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci. 2006;7(1):41‐53. doi: 10.1038/nrn1824 [DOI] [PubMed] [Google Scholar]
- 114. Uchida Y, Kan H, Furukawa G, et al. Relationship between brain iron dynamics and blood‐brain barrier function during childhood: a quantitative magnetic resonance imaging study. Fluids Barriers CNS. 2023;20(1):60. doi: 10.1186/s12987-023-00464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pollak TA, Drndarski S, Stone JM, David AS, McGuire P, Abbott NJ. The blood‐brain barrier in psychosis. Lancet Psychiatry. 2018;5(1):79‐92. doi: 10.1016/s2215-0366(17)30293-6 [DOI] [PubMed] [Google Scholar]
- 116. Ishii M, Iadecola C. Risk factor for Alzheimer's disease breaks the blood‐brain barrier. Nature. 2020;581(7806):31‐32. doi: 10.1038/d41586-020-01152-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Uchida Y, Kan H, Sakurai K, Oishi K, Matsukawa N. Contributions of blood‐brain barrier imaging to neurovascular unit pathophysiology of Alzheimer's disease and related dementias. Front Aging Neurosci. 2023;15:1111448. doi: 10.3389/fnagi.2023.1111448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood‐brain barrier. Nat Med. 2013;19(12):1584‐1596. doi: 10.1038/nm.3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106‐118. doi: 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Koutsodendris N, Nelson MR, Rao A, Huang Y. Apolipoprotein E and Alzheimer's disease: findings, hypotheses, and potential mechanisms. Annu Rev Pathol. 2022;17:73‐99. doi: 10.1146/annurev-pathmechdis-030421-112756 [DOI] [PubMed] [Google Scholar]
- 121. Uchida Y, Kan H, Sakurai K, et al. Iron leakage owing to blood‐brain barrier disruption in small vessel disease CADASIL. Neurology. 2020;95(9):e1188‐e1198. doi: 10.1212/wnl.0000000000010148 [DOI] [PubMed] [Google Scholar]
- 122. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133‐150. doi: 10.1038/nrneurol.2017.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood‐brain barrier. Cell. 2015;163(5):1064‐1078. doi: 10.1016/j.cell.2015.10.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chapleau M, Iaccarino L, Soleimani‐Meigooni D, Rabinovici GD. The role of amyloid pet in imaging neurodegenerative disorders: a review. J Nucl Med. 2022;63(Suppl 1):13s‐19s. doi: 10.2967/jnumed.121.263195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. The need for early detection and treatment in Alzheimer's disease. EBioMedicine. 2016;9:1‐2. doi: 10.1016/j.ebiom.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shaw LM, Korecka M, Clark CM, Lee VM, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discovery. 2007;6(4):295‐303. doi: 10.1038/nrd2176 [DOI] [PubMed] [Google Scholar]
- 127. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jack CR Jr, Andrews SJ, Beach TG, et al. Revised criteria for the diagnosis and staging of Alzheimer's disease. Nat Med. 2024;30(8):2121‐2124. doi: 10.1038/s41591-024-02988-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ebenau JL, Timmers T, Wesselman LMP, et al. ATN classification and clinical progression in subjective cognitive decline: the SCIENCe project. Neurology. 2020;95(1):e46‐e58. doi: 10.1212/wnl.0000000000009724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tan MS, Ji X, Li JQ, et al. Longitudinal trajectories of Alzheimer's ATN biomarkers in elderly persons without dementia. Alzheimer's research & therapy. 2020;12(1):55. doi: 10.1186/s13195-020-00621-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Allegri RF, Chrem Méndez P, Calandri I, et al. Prognostic value of ATN Alzheimer biomarkers: 60‐month follow‐up results from the Argentine Alzheimer's disease neuroimaging initiative. Alzheimers Dement. 2020;12(1):e12026. doi: 10.1002/dad2.12026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Altomare D, de Wilde A, Ossenkoppele R, et al. Applying the ATN scheme in a memory clinic population: the ABIDE project. Neurology. 2019;93(17):e1635‐e1646. doi: 10.1212/wnl.0000000000008361 [DOI] [PubMed] [Google Scholar]
- 133. Soldan A, Pettigrew C, Fagan AM, et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology. 2019;92(14):e1567‐e1579. doi: 10.1212/wnl.0000000000007248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Jack CR Jr, Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321(23):2316‐2325. doi: 10.1001/jama.2019.7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hampel H, Cummings J, Blennow K, Gao P, Jack CR Jr, Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021;17(9):580‐589. doi: 10.1038/s41582-021-00520-w [DOI] [PubMed] [Google Scholar]
- 136. Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. Lancet Neurol. 2003;2(10):605‐613. doi: 10.1016/s1474-4422(03)00530-1 [DOI] [PubMed] [Google Scholar]
- 137. Li Y, Schindler SE, Bollinger JG, et al. Validation of plasma amyloid‐beta 42/40 for detecting Alzheimer disease amyloid plaques. Neurology. 2022;98(7):e688‐e699. doi: 10.1212/WNL.0000000000013211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zicha S, Bateman RJ, Shaw LM, et al. Comparative analytical performance of multiple plasma Abeta42 and Abeta40 assays and their ability to predict positron emission tomography amyloid positivity. Alzheimers Dement. 2023;19(3):956‐966. doi: 10.1002/alz.12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76(7):791‐799. doi: 10.1001/jamaneurol.2019.0765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Suárez‐Calvet M, Karikari TK, Ashton NJ, et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer's continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med. 2020;12(12):e12921. doi: 10.15252/emmm.202012921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26(3):379‐386. doi: 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 142. Leuzy A, Janelidze S, Mattsson‐Carlgren N, et al. Comparing the clinical utility and diagnostic performance of CSF P‐Tau181, P‐Tau217, and P‐Tau231 assays. Neurology. 2021;97(17):e1681‐e1694. doi: 10.1212/wnl.0000000000012727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Islam T, Hill E, Abrahamson EE, et al. Phospho‐tau serine‐262 and serine‐356 as biomarkers of pre‐tangle soluble tau assemblies in Alzheimer's disease. Nat Med. 2025;31(2):574‐588. doi: 10.1038/s41591-024-03400-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Mattsson‐Carlgren N, Salvadó G, Ashton NJ, et al. Prediction of longitudinal cognitive decline in preclinical Alzheimer disease using plasma biomarkers. JAMA Neurol. 2023;80(4):360‐369. doi: 10.1001/jamaneurol.2022.5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Therriault J, Servaes S, Tissot C, et al. Equivalence of plasma p‐tau217 with cerebrospinal fluid in the diagnosis of Alzheimer's disease. Alzheimers Dement. 2023;19(11):4967‐4977. doi: 10.1002/alz.13026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26(3):387‐397. doi: 10.1038/s41591-020-0762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19(5):422‐433. doi: 10.1016/s1474-4422(20)30071-5 [DOI] [PubMed] [Google Scholar]
- 148. Laske C, Sohrabi HR, Frost SM, et al. Innovative diagnostic tools for early detection of Alzheimer's disease. Alzheimers Dement. 2015;11(5):561‐578. doi: 10.1016/j.jalz.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 149. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho‐tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772‐781. doi: 10.1001/jama.2020.12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Horie K, Salvadó G, Koppisetti RK, et al. Plasma MTBR‐tau243 biomarker identifies tau tangle pathology in Alzheimer's disease. Nat Med. 2025;31(6):2044‐2053. doi: 10.1038/s41591-025-03617-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ossenkoppele R, Rabinovici GD, Smith R, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2018;320(11):1151‐1162. doi: 10.1001/jama.2018.12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Ossenkoppele R, van der Kant R, Hansson O. Tau biomarkers in Alzheimer's disease: towards implementation in clinical practice and trials. Lancet Neurol. 2022;21(8):726‐734. doi: 10.1016/s1474-4422(22)00168-5 [DOI] [PubMed] [Google Scholar]
- 153. Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971‐982. doi: 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med. 2020;12(524):eaau5732. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27(5):871‐881. doi: 10.1038/s41591-021-01309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Quiroz YT, Zetterberg H, Reiman EM, et al. Plasma neurofilament light chain in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross‐sectional and longitudinal cohort study. Lancet Neurol. 2020;19(6):513‐521. doi: 10.1016/s1474-4422(20)30137-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gao L, Zhang Y, Sterling K, Song W. Brain‐derived neurotrophic factor in Alzheimer's disease and its pharmaceutical potential. Transl Neurodegener. 2022;11(1):4. doi: 10.1186/s40035-022-00279-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wang T, Li T, Hao S, Han Y, Cai Y. Association of plasma BDNF levels with different stages of Alzheimer's disease: a cross‐sectional study. Neurol Res. 2023;45(3):234‐240. doi: 10.1080/01616412.2022.2129760 [DOI] [PubMed] [Google Scholar]
- 159. Faria MC, Gonçalves GS, Rocha NP, et al. Increased plasma levels of BDNF and inflammatory markers in Alzheimer's disease. J Psychiatr Res. 2014;53:166‐172. doi: 10.1016/j.jpsychires.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 160. Hoffman JM, Hanson MW, Welsh KA, et al. Interpretation variability of 18FDG‐positron emission tomography studies in dementia. Invest Radiol. 1996;31(6):316‐322. doi: 10.1097/00004424-199606000-00002 [DOI] [PubMed] [Google Scholar]
- 161. Baiardi S, Quadalti C, Mammana A, et al. Diagnostic value of plasma p‐tau181, NfL, and GFAP in a clinical setting cohort of prevalent neurodegenerative dementias. Alzheimers Res Ther. 2022;14(1):153. doi: 10.1186/s13195-022-01093-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐β but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505‐3516. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry. 2021;11(1):27. doi: 10.1038/s41398-020-01137-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yakoub Y, Ashton NJ, Strikwerda‐Brown C, et al. Longitudinal blood biomarker trajectories in preclinical Alzheimer's disease. Alzheimers Dement. 2023;19(12):5620‐5631. doi: 10.1002/alz.13318 [DOI] [PubMed] [Google Scholar]
- 165. Prins S, de Kam ML, Teunissen CE, Groeneveld GJ. Inflammatory plasma biomarkers in subjects with preclinical Alzheimer's disease. Alzheimers Res Ther. 2022;14(1):106. doi: 10.1186/s13195-022-01051-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Shen XN, Huang SY, Cui M, et al. Plasma glial fibrillary acidic protein in the Alzheimer disease continuum: relationship to other biomarkers, differential diagnosis, and prediction of clinical progression. Clin Chem. 2023;69(4):411‐421. doi: 10.1093/clinchem/hvad018 [DOI] [PubMed] [Google Scholar]
- 167. Liu T, Zuo H, Ma D, Song D, Zhao Y, Cheng O. Cerebrospinal fluid GFAP is a predictive biomarker for conversion to dementia and Alzheimer's disease‐associated biomarkers alterations among de novo Parkinson's disease patients: a prospective cohort study. J Neuroinflam. 2023;20(1):167. doi: 10.1186/s12974-023-02843-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Morenas‐Rodríguez E, Alcolea D, Suárez‐Calvet M, et al. Different pattern of CSF glial markers between dementia with Lewy bodies and Alzheimer's disease. Sci Rep. 2019;9(1):7803. doi: 10.1038/s41598-019-44173-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Chouliaras L, Thomas A, Malpetti M, et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer's disease, frontotemporal dementia and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2022;93(6):651‐658. doi: 10.1136/jnnp-2021-327788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Yang Z, Wang KK. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015;38(6):364‐374. doi: 10.1016/j.tins.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Olabarria M, Putilina M, Riemer EC, Goldman JE. Astrocyte pathology in Alexander disease causes a marked inflammatory environment. Acta Neuropathol. 2015;130(4):469‐486. doi: 10.1007/s00401-015-1469-1 [DOI] [PubMed] [Google Scholar]
- 172. Jang H, Shin D, Yoo H, et al. Differential roles of Alzheimer's disease plasma biomarkers in stepwise biomarker‐guided diagnostics. Alzheimers Dement. 2025;21(2):e14526. doi: 10.1002/alz.14526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Pelkmans W, Shekari M, Brugulat‐Serrat A, et al. Astrocyte biomarkers GFAP and YKL‐40 mediate early Alzheimer's disease progression. Alzheimers Dement. 2024;20(1):483‐493. doi: 10.1002/alz.13450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Bonneh‐Barkay D, Bissel SJ, Wang G, et al. YKL‐40, a marker of simian immunodeficiency virus encephalitis, modulates the biological activity of basic fibroblast growth factor. Am J Pathol. 2008;173(1):130‐143. doi: 10.2353/ajpath.2008.080045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Horbinski C, Wang G, Wiley CA. YKL‐40 is directly produced by tumor cells and is inversely linked to EGFR in glioblastomas. Int J Clin Exp Pathol. 2010;3(3):226‐237. [PMC free article] [PubMed] [Google Scholar]
- 176. Connolly K, Lehoux M, O'Rourke R, et al. Potential role of chitinase‐3‐like protein 1 (CHI3L1/YKL‐40) in neurodegeneration and Alzheimer's disease. Alzheimers Dement. 2023;19(1):9‐24. doi: 10.1002/alz.12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Nordengen K, Kirsebom BE, Henjum K, et al. Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflam. 2019;16(1):46. doi: 10.1186/s12974-019-1399-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhang L, Xiang X, Li Y, Bu G, Chen XF. TREM2 and sTREM2 in Alzheimer's disease: from mechanisms to therapies. Mol Neurodegener. 2025;20(1):43. doi: 10.1186/s13024-025-00834-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441‐468. doi: 10.1146/annurev-immunol-051116-052358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359‐1369. doi: 10.1038/s41593-018-0242-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. 2017;127(9):3240‐3249. doi: 10.1172/jci90606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. McManus RM, Komes MP, Griep A, et al. NLRP3‐mediated glutaminolysis controls microglial phagocytosis to promote Alzheimer's disease progression. Immunity. 2025;58(2):326‐343.e11. doi: 10.1016/j.immuni.2025.01.007 [DOI] [PubMed] [Google Scholar]
- 183. Olah M, Menon V, Habib N, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nat Commun. 2020;11(1):6129. doi: 10.1038/s41467-020-19737-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Song WM, Colonna M. The identity and function of microglia in neurodegeneration. Nat Immunol. 2018;19(10):1048‐1058. doi: 10.1038/s41590-018-0212-1 [DOI] [PubMed] [Google Scholar]
- 185. Colonna M. The biology of TREM receptors. Nat Rev Immunol. 2023;23(9):580‐594. doi: 10.1038/s41577-023-00837-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Ulland TK, Song WM, Huang SC, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell. 2017;170(4):649‐663.e13. doi: 10.1016/j.cell.2017.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro‐inflammatory subset of disease‐associated‐microglia in Alzheimer's disease. Mol Neurodegener. 2018;13(1):24. doi: 10.1186/s13024-018-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Rao A, Chen N, Kim MJ, et al. Microglia depletion reduces human neuronal APOE4‐related pathologies in a chimeric Alzheimer's disease model. Cell stem cell. 2025;32(1):86‐104.e7. doi: 10.1016/j.stem.2024.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Chen C, Bao Y, Xing L, et al. Exosomes derived from M2 microglial cells modulated by 1070‐nm light improve cognition in an Alzheimer's disease mouse model. Adv Sci. 2023;10(32):e2304025. doi: 10.1002/advs.202304025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Ewers M, Franzmeier N, Suárez‐Calvet M, et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease. Sci Transl Med. 2019;11(507):eaav6221. doi: 10.1126/scitranslmed.aav6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Morenas‐Rodríguez E, Li Y, Nuscher B, et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal‐dominant Alzheimer's disease: a longitudinal observational study. Lancet Neurol. 2022;21(4):329‐341. doi: 10.1016/s1474-4422(22)00027-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Tan ZS, Beiser AS, Vasan RS, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007;68(22):1902‐1908. doi: 10.1212/01.wnl.0000263217.36439.da [DOI] [PubMed] [Google Scholar]
- 193. Temmerman J, Engelborghs S, Bjerke M, D'Haeseleer M. Cerebrospinal fluid inflammatory biomarkers for disease progression in Alzheimer's disease and multiple sclerosis: a systematic review. Front Immunol. 2023;14:1162340. doi: 10.3389/fimmu.2023.1162340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Dubois B, Villain N, Frisoni GB, et al. Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol. 2021;20(6):484‐496. doi: 10.1016/s1474-4422(21)00066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Farlow MR, Cummings JL. Effective pharmacologic management of Alzheimer's disease. Am J Med. 2007;120(5):388‐397. doi: 10.1016/j.amjmed.2006.08.036 [DOI] [PubMed] [Google Scholar]
- 196. Winblad B, Gauthier S, Scinto L, et al. Safety and efficacy of galantamine in subjects with mild cognitive impairment. Neurology. 2008;70(22):2024‐2035. doi: 10.1212/01.wnl.0000303815.69777.26 [DOI] [PubMed] [Google Scholar]
- 197. Tricco AC, Soobiah C, Berliner S, et al. Efficacy and safety of cognitive enhancers for patients with mild cognitive impairment: a systematic review and meta‐analysis. CMAJ. 2013;185(16):1393‐1401. doi: 10.1503/cmaj.130451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388(1):9‐21. doi: 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- 199. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of Aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9(2):197‐210. doi: 10.14283/jpad.2022.30 [DOI] [PubMed] [Google Scholar]
- 200. Pontecorvo MJ, Lu M, Burnham SC, et al. Association of Donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: a secondary analysis of the TRAILBLAZER‐ALZ randomized clinical trial. JAMA Neurol. 2022;79(12):1250‐1259. doi: 10.1001/jamaneurol.2022.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Shcherbinin S, Evans CD, Lu M, et al. Association of amyloid reduction after Donanemab treatment with tau pathology and clinical outcomes: the TRAILBLAZER‐ALZ randomized clinical trial. JAMA Neurol. 2022;79(10):1015‐1024. doi: 10.1001/jamaneurol.2022.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Sperling RA, Donohue MC, Raman R, et al. Trial of Solanezumab in preclinical Alzheimer's disease. N Engl J Med. 2023;389(12):1096‐1107. doi: 10.1056/NEJMoa2305032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Wagemann O, Liu H, Wang G, et al. Downstream biomarker effects of Gantenerumab or Solanezumab in dominantly inherited Alzheimer disease: the DIAN‐TU‐001 randomized clinical trial. JAMA Neurol. 2024;81(6):582‐593. doi: 10.1001/jamaneurol.2024.0991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Salloway S, Chalkias S, Barkhof F, et al. Amyloid‐related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79(1):13‐21. doi: 10.1001/jamaneurol.2021.4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Kane M. Lecanemab therapy and APOE genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kattman BL, Malheiro AJ, eds. Medical Genetics Summaries. National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- 206. Gonzalez‐Ortiz F, Kac PR, Brum WS, Zetterberg H, Blennow K, Karikari TK. Plasma phospho‐tau in Alzheimer's disease: towards diagnostic and therapeutic trial applications. Mol Neurodegener. 2023;18(1):18. doi: 10.1186/s13024-023-00605-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Vossel KA, Xu JC, Fomenko V, et al. Tau reduction prevents Aβ‐induced axonal transport deficits by blocking activation of GSK3β. J Cell Biol. 2015;209(3):419‐433. doi: 10.1083/jcb.201407065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Cheng Z, Han T, Yao J, et al. Targeting glycogen synthase kinase‐3β for Alzheimer's disease: recent advances and future prospects. Eur J Med Chem. 2024;265:116065. doi: 10.1016/j.ejmech.2023.116065 [DOI] [PubMed] [Google Scholar]
- 209. Serenó L, Coma M, Rodríguez M, et al. A novel GSK‐3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35(3):359‐367. doi: 10.1016/j.nbd.2009.05.025 [DOI] [PubMed] [Google Scholar]
- 210. Swinney ZT, Haubrich BA, Xia S, et al. A four‐point screening Method for Assessing Molecular Mechanism of Action (MMOA) identifies Tideglusib as a time‐dependent inhibitor of Trypanosoma brucei GSK3β. PLoS Negl Trop Dis. 2016;10(3):e0004506. doi: 10.1371/journal.pntd.0004506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Pandey MK, DeGrado TR. Glycogen synthase kinase‐3 (GSK‐3)‐targeted therapy and imaging. Theranostics. 2016;6(4):571‐593. doi: 10.7150/thno.14334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Lovestone S, Boada M, Dubois B, et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis. 2015;45(1):75‐88. doi: 10.3233/jad-141959 [DOI] [PubMed] [Google Scholar]
- 213. Collotta D, Bertocchi I, Chiapello E, Collino M. Antisense oligonucleotides: a novel Frontier in pharmacological strategy. Front Pharmacol. 2023;14:1304342. doi: 10.3389/fphar.2023.1304342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374):eaag0481. doi: 10.1126/scitranslmed.aag0481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Mummery CJ, Börjesson‐Hanson A, Blackburn DJ, et al. Tau‐targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer's disease: a phase 1b, randomized, placebo‐controlled trial. Nat Med. 2023;29(6):1437‐1447. doi: 10.1038/s41591-023-02326-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Edwards AL, Collins JA, Junge C, et al. Exploratory tau biomarker results from a multiple ascending‐dose study of BIIB080 in Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2023;80(12):1344‐1352. doi: 10.1001/jamaneurol.2023.3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312‐339. doi: 10.1016/j.cell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Hickman DT, López‐Deber MP, Ndao DM, et al. Sequence‐independent control of peptide conformation in liposomal vaccines for targeting protein misfolding diseases. J Biol Chem. 2011;286(16):13966‐13976. doi: 10.1074/jbc.M110.186338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Mullard A. Anti‐tau antibody failures stack up. Nat Rev Drug Discovery. 2021;20(12):888. doi: 10.1038/d41573-021-00187-4 [DOI] [PubMed] [Google Scholar]
- 220. Knopman DS, Amieva H, Petersen RC, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33. doi: 10.1038/s41572-021-00269-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270‐279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14(1):32. doi: 10.1186/s13024-019-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Ferrari C, Sorbi S. The complexity of Alzheimer's disease: an evolving puzzle. Physiol Rev. 2021;101(3):1047‐1081. doi: 10.1152/physrev.00015.2020 [DOI] [PubMed] [Google Scholar]
- 224. Jankowsky JL, Zheng H. Practical considerations for choosing a mouse model of Alzheimer's disease. Mol Neurodegener. 2017;12(1):89. doi: 10.1186/s13024-017-0231-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Oakley H, Cole SL, Logan S, et al. Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129‐10140. doi: 10.1523/jneurosci.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Oddo S, Caccamo A, Shepherd JD, et al. Triple‐transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409‐421. doi: 10.1016/s0896-6273(03)00434-3 [DOI] [PubMed] [Google Scholar]
- 227. Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau‐mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523‐527. doi: 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Zhong MZ, Peng T, Duarte ML, Wang M, Cai D. Updates on mouse models of Alzheimer's disease. Mol Neurodegener. 2024;19(1):23. doi: 10.1186/s13024-024-00712-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Götz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci. 2018;19(10):583‐598. doi: 10.1038/s41583-018-0054-8 [DOI] [PubMed] [Google Scholar]
- 230. Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late‐stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275(3):251‐283. doi: 10.1111/joim.12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. doi: 10.1186/alzrt269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Sasaguri H, Hashimoto S, Watamura N, et al. Recent Advances in the Modeling of Alzheimer's Disease. Front Neurosci. 2022;16:807473. doi: 10.3389/fnins.2022.807473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Oblak AL, Lin PB, Kotredes KP, et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: a MODEL‐AD Study. Front Aging Neurosci. 2021;13:713726. doi: 10.3389/fnagi.2021.713726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Nakai T, Yamada K, Mizoguchi H. Alzheimer's Disease Animal Models: elucidation of Biomarkers and Therapeutic Approaches for Cognitive Impairment. Int J Mol Sci. 2021;22(11):5549. doi: 10.3390/ijms22115549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Kosel F, Pelley JMS, Franklin TB. Behavioural and psychological symptoms of dementia in mouse models of Alzheimer's disease‐related pathology. Neurosci Biobehav Rev. 2020;112:634‐647. doi: 10.1016/j.neubiorev.2020.02.012 [DOI] [PubMed] [Google Scholar]
- 236. Bilkei‐Gorzo A. Genetic mouse models of brain ageing and Alzheimer's disease. Pharmacol Ther. 2014;142(2):244‐257. doi: 10.1016/j.pharmthera.2013.12.009 [DOI] [PubMed] [Google Scholar]
- 237. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early‐onset Alzheimer's disease revisited. Alzheimers Dement. 2016;12(6):733‐748. doi: 10.1016/j.jalz.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 238. Karch CM, Cruchaga C, Goate AM. Alzheimer's disease genetics: from the bench to the clinic. Neuron. 2014;83(1):11‐26. doi: 10.1016/j.neuron.2014.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Gibbs RA, Rogers J, Katze MG, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316(5822):222‐234. doi: 10.1126/science.1139247 [DOI] [PubMed] [Google Scholar]
- 240. Zhang R, Quan H, Wang Y, Luo F. Neurogenesis in primates versus rodents and the value of non‐human primate models. Natl Sci Rev. 2023;10(11):nwad248. doi: 10.1093/nsr/nwad248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Souder DC, Dreischmeier IA, Smith AB, et al. Rhesus monkeys as a translational model for late‐onset Alzheimer's disease. Aging Cell. 2021;20(6):e13374. doi: 10.1111/acel.13374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Stonebarger GA, Urbanski HF, Woltjer RL, et al. Amyloidosis increase is not attenuated by long‐term calorie restriction or related to neuron density in the prefrontal cortex of extremely aged rhesus macaques. Geroscience. 2020;42(6):1733‐1749. doi: 10.1007/s11357-020-00259-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Zhang J, Chen B, Lu J, et al. Brains of rhesus monkeys display Aβ deposits and glial pathology while lacking Aβ dimers and other Alzheimer's pathologies. Aging Cell. 2019;18(4):e12978. doi: 10.1111/acel.12978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Zhou FQ, Jiang J, Griffith CM, et al. Lack of human‐like extracellular sortilin neuropathology in transgenic Alzheimer's disease model mice and macaques. Alzheimers Res Ther. 2018;10(1):40. doi: 10.1186/s13195-018-0370-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Lardenoije R, van den Hove DLA, Havermans M, et al. Age‐related epigenetic changes in hippocampal subregions of four animal models of Alzheimer's disease. Mol Cell Neurosci. 2018;86:1‐15. doi: 10.1016/j.mcn.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Poduri A, Gearing M, Rebeck GW, Mirra SS, Tigges J, Hyman BT. Apolipoprotein E4 and beta amyloid in senile plaques and cerebral blood vessels of aged rhesus monkeys. Am J Pathol. 1994;144(6):1183‐1187. [PMC free article] [PubMed] [Google Scholar]
- 247. Bons N, Mestre N, Ritchie K, Petter A, Podlisny M, Selkoe D. Identification of amyloid beta protein in the brain of the small, short‐lived lemurian primate Microcebus murinus. Neurobiol Aging. 1994;15(2):215‐220. doi: 10.1016/0197-4580(94)90115-5 [DOI] [PubMed] [Google Scholar]
- 248. Podlisny MB, Tolan DR, Selkoe DJ. Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer's disease. Am J Pathol. 1991;138(6):1423‐1435. [PMC free article] [PubMed] [Google Scholar]
- 249. Beckman D, Ott S, Donis‐Cox K, et al. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc Nat Acad Sci USA. 2019;116(52):26239‐26246. doi: 10.1073/pnas.1902301116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Philippens IH, Ormel PR, Baarends G, Johansson M, Remarque EJ, Doverskog M. Acceleration of amyloidosis by inflammation in the amyloid‐beta marmoset monkey model of Alzheimer's disease. J Alzheimers Dis. 2017;55(1):101‐113. doi: 10.3233/jad-160673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Sani S, Traul D, Klink A, et al. Distribution, progression and chemical composition of cortical amyloid‐beta deposits in aged rhesus monkeys: similarities to the human. Acta Neuropathol. 2003;105(2):145‐156. doi: 10.1007/s00401-002-0626-5 [DOI] [PubMed] [Google Scholar]
- 252. Leslie SN, Kanyo J, Datta D, et al. Simple, single‐shot phosphoproteomic analysis of heat‐stable tau identifies age‐related changes in pS235‐ and pS396‐Tau Levels in non‐human primates. Front Aging Neurosci. 2021;13:767322. doi: 10.3389/fnagi.2021.767322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Sharma G, Huo A, Kimura T, et al. Tau isoform expression and phosphorylation in marmoset brains. J Biol Chem. 2019;294(30):11433‐11444. doi: 10.1074/jbc.RA119.008415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Beckman D, Diniz GB, Ott S, et al. Temporal progression of tau pathology and neuroinflammation in a rhesus monkey model of Alzheimer's disease. Alzheimers Dement. 2024;20(8):5198‐5219. doi: 10.1002/alz.13868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Beckman D, Chakrabarty P, Ott S, et al. A novel tau‐based rhesus monkey model of Alzheimer's pathogenesis. Alzheimers Dement. 2021;17(6):933‐945. doi: 10.1002/alz.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Huang X, Huang S, Fu F, Song J, Zhang Y, Yue F. Characterization of preclinical Alzheimer's disease model: spontaneous type 2 diabetic cynomolgus monkeys with systemic pro‐inflammation, positive biomarkers and developing AD‐like pathology. Alzheimers Res Ther. 2024;16(1):52. doi: 10.1186/s13195-024-01416-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Latimer CS, Shively CA, Keene CD, et al. A nonhuman primate model of early Alzheimer's disease pathologic change: implications for disease pathogenesis. Alzheimers Dement. 2019;15(1):93‐105. doi: 10.1016/j.jalz.2018.06.3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Darusman HS, Sajuthi D, Kalliokoski O, et al. Correlations between serum levels of beta amyloid, cerebrospinal levels of tau and phospho tau, and delayed response tasks in young and aged cynomolgus monkeys (Macaca fascicularis). J Med Primatol. 2013;42(3):137‐146. doi: 10.1111/jmp.12044 [DOI] [PubMed] [Google Scholar]
- 259. Darusman HS, Gjedde A, Sajuthi D, et al. Amyloid Beta1‐42 and the Phoshorylated Tau Threonine 231 in brains of aged cynomolgus monkeys (Macaca fascicularis). Front Aging Neurosci. 2014;6:313. doi: 10.3389/fnagi.2014.00313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Darusman HS, Agungpriyono DR, Kusumaputri VA, Sajuthi D, Schapiro SJ, Hau J. Granulovacuolar degeneration in brains of senile cynomolgus monkeys. Front Aging Neurosci. 2019;11:50. doi: 10.3389/fnagi.2019.00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Paspalas CD, Carlyle BC, Leslie S, et al. The aged rhesus macaque manifests Braak stage III/IV Alzheimer's‐like pathology. Alzheimers Dement. 2018;14(5):680‐691. doi: 10.1016/j.jalz.2017.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Jiang Z, Wang J, Qin Y, et al. A nonhuman primate model with Alzheimer's disease‐like pathology induced by hippocampal overexpression of human tau. Alzheimers Res Ther. 2024;16(1):22. doi: 10.1186/s13195-024-01392-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Darricau M, Dou C, Kinet R, et al. Tau seeds from Alzheimer's disease brains trigger tau spread in macaques while oligomeric‐Aβ mediates pathology maturation. Alzheimers Dement. 2024;20(3):1894‐1912. doi: 10.1002/alz.13604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Wakeman DR, Weed MR, Perez SE, et al. Intrathecal amyloid‐beta oligomer administration increases tau phosphorylation in the medial temporal lobe in the African green monkey: a nonhuman primate model of Alzheimer's disease. Neuropathol Appl Neurobiol. 2022;48(4):e12800. doi: 10.1111/nan.12800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Morales‐Corraliza J, Wong H, Mazzella MJ, et al. Brain‐wide insulin resistance, tau phosphorylation changes, and hippocampal neprilysin and amyloid‐β alterations in a monkey model of type 1 diabetes. J Neurosci. 2016;36(15):4248‐4258. doi: 10.1523/jneurosci.4640-14.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Li M, Guan M, Lin J, et al. Early blood immune molecular alterations in cynomolgus monkeys with a PSEN1 mutation causing familial Alzheimer's disease. Alzheimers Dement. 2024;20(8):5492‐5510. doi: 10.1002/alz.14046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Tu Z, Yan S, Han B, et al. Tauopathy promotes spinal cord‐dependent production of toxic amyloid‐beta in transgenic monkeys. Signal Transduct Target Ther. 2023;8(1):358. doi: 10.1038/s41392-023-01601-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Chan AWS, Cho IK, Li CX, et al. Cerebral Aβ deposition in an Aβ‐precursor protein‐transgenic rhesus monkey. Aging brain. 2022;2:100044. doi: 10.1016/j.nbas.2022.100044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Zhou Q, Li S, Li M, et al. Human tau accumulation promotes glycogen synthase kinase‐3β acetylation and thus upregulates the kinase: a vicious cycle in Alzheimer neurodegeneration. EBioMedicine. 2022;78:103970. doi: 10.1016/j.ebiom.2022.103970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5‐21. doi: 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- 271. Li T, Paudel HK. Glycogen synthase kinase 3beta phosphorylates Alzheimer's disease‐specific Ser396 of microtubule‐associated protein tau by a sequential mechanism. Biochemistry. 2006;45(10):3125‐3133. doi: 10.1021/bi051634r [DOI] [PubMed] [Google Scholar]
- 272. Shen S, Liao Q, Wong YK, et al. The role of melatonin in the treatment of type 2 diabetes mellitus and Alzheimer's disease. International journal of biological sciences. 2022;18(3):983‐994. doi: 10.7150/ijbs.66871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Baglietto‐Vargas D, Shi J, Yaeger DM, Ager R, LaFerla FM. Diabetes and Alzheimer's disease crosstalk. Neurosci Biobehav Rev. 2016;64:272‐287. doi: 10.1016/j.neubiorev.2016.03.005 [DOI] [PubMed] [Google Scholar]
- 274. Hardy J, de Strooper B, Escott‐Price V. Diabetes and Alzheimer's disease: shared genetic susceptibility?. Lancet Neurol. 2022;21(11):962‐964. doi: 10.1016/s1474-4422(22)00395-7 [DOI] [PubMed] [Google Scholar]
- 275. Li HW, Zhang L, Qin C. Current state of research on non‐human primate models of Alzheimer's disease. Animal Model Exp Med. 2019;2(4):227‐238. doi: 10.1002/ame2.12092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Parnetti L, Eusebi P. Cerebrospinal fluid biomarkers in Alzheimer's disease: an invaluable tool for clinical diagnosis and trial enrichment. J Alzheimers Dis. 2018;64(s1):S281‐s287. doi: 10.3233/jad-179910 [DOI] [PubMed] [Google Scholar]
- 277. Long H, Simmons A, Mayorga A, et al. Preclinical and first‐in‐human evaluation of AL002, a novel TREM2 agonistic antibody for Alzheimer's disease. Alzheimers Res Ther. 2024;16(1):235. doi: 10.1186/s13195-024-01599-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Lista S, Imbimbo BP, Grasso M, et al. Tracking neuroinflammatory biomarkers in Alzheimer's disease: a strategy for individualized therapeutic approaches?. J Neuroinflam. 2024;21(1):187. doi: 10.1186/s12974-024-03163-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Chandra A, Valkimadi PE, Pagano G, Cousins O, Dervenoulas G, Politis M. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer's disease and mild cognitive impairment. Hum Brain Mapp. 2019;40(18):5424‐5442. doi: 10.1002/hbm.24782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Schmidt JK, Jones KM, Van Vleck T, Emborg ME. Modeling genetic diseases in nonhuman primates through embryonic and germline modification: considerations and challenges. Sci Transl Med. 2022;14(634):eabf4879. doi: 10.1126/scitranslmed.abf4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information