

Abstract
We report the analysis of 335 microsatellite markers genotyped in 110 multiplex families with autism. All families include at least two “affected” siblings, at least one of whom has autism; the remaining affected sibs carry diagnoses of either Asperger syndrome or pervasive developmental disorder. Affected sib-pair analysis yielded multipoint maximum LOD scores (MLS) that reach the accepted threshold for suggestive linkage on chromosomes 5, X, and 19. Nominal evidence for linkage (point-wise P<.05) was obtained on chromosomes 2, 3, 4, 8, 10, 11, 12, 15, 16, 18, and 20, and secondary loci were found on chromosomes 5 and 19. Analysis of families sharing alleles at the putative X chromosomal linked locus and one or more other putative linked loci produced an MLS of 3.56 for the DXS470-D19S174 marker combination. In an effort to increase power to detect linkage, scan statistics were used to evaluate the significance of peak LOD scores based on statistical evidence at adjacent marker loci. This analysis yielded impressive evidence for linkage to autism and autism-spectrum disorders with significant genomewide P values <.05 for markers on chromosomes 5 and 8 and with suggestive linkage evidence for a marker on chromosome 19.
Introduction
Autism [MIM 209850] is a neuropsychiatric disorder characterized by severe social and communicative deficits, together with a pattern of restricted or repetitive behaviors or interests. Developmental abnormalities associated with autism typically manifest in the first 3 years of life and persist into adulthood. Autism is typically characterized as part of a spectrum of disorders that includes Asperger syndrome (AS) and other pervasive developmental disorders (PDD). AS is distinguished from autistic disorder by the lack of a clinically significant delay in language development (phrase use at age ⩽3 years; single words at age ⩽2 years) in the presence of the impaired social interaction and restricted-repetitive behaviors, interests, and activities that characterize the autism-spectrum disorders (ASDs). PDD is used to categorize children who do not meet the strict criteria for autism but who come close, either by manifesting atypical autism or by nearly meeting diagnostic criteria in two or three of the key areas.
Until fairly recently, autism was considered a rare disorder with an estimated prevalence of 4–5 patients per 10,000 individuals. More-recent studies have led to estimates of 10–12 patients per 10,000 individuals (Gillberg and Wing 1999). We do not yet know whether this difference reflects an increased prevalence of autism, a gradual change in diagnostic criteria, a recognition of greater variability of disease expression, or a heightened awareness of the disorder. The estimated prevalence of autism plus related spectrum disorders, excluding AS, is approximately double that of autism alone (Wing and Gould 1979). It is well documented that autism occurs three to four times more frequently in males than in females.
Evidence from twin and family studies clearly establishes the importance of genetic factors in the development of autism and ASDs (Folstein and Rutter 1977; Bailey et al. 1995). In the largest study to date, 15 (60%) of 25 monozygotic twins were concordant for autism, compared with 0 of 20 dizygotic pairs. In the same study, >90% of monozygotic twins were concordant for social and cognitive abnormalities that meet criteria for ASD (Bailey et al. 1995). These data suggest an unusually high degree of heritability for susceptibility to autism and ASDs; >90% for the latter, when a multifactorial threshold model for disease liability is assumed. The sibling recurrence risk for autism is estimated to be ∼2%–4% (Jorde et al. 1991; Szatmari et al. 1998). Thus, when a population prevalence of 0.05%–0.1% is assumed, the estimated relative risk for autism in a sibling is 45–90 times greater than the population risk. The incidence of autism falls significantly with decreasing degree of relatedness to an affected individual, an observation consistent with the involvement of multiple interacting genetic loci. Thus, autism and related spectrum disorders present as complex genetic disorders and most likely result from the combined effects of multiple susceptibility alleles in concert with environmental factors or other nongenetic factors.
Autism appears to be both clinically and genetically heterogeneous. Approximately 10%–25% of patients with autism present with other medical disorders, principal among which are fragile X syndrome (MIM 309550) (Feinstein and Reiss 1998) and tuberous sclerosis (MIM 191100) (Smalley 1998). In addition, case studies suggest that a small but significant portion of autism cases are associated with a broad spectrum of chromosomal abnormalities, most frequent among which are chromosome 15 structural anomalies (15q11-q13) (Christian et al. 1999). Interest in this region is further enhanced by evidence for imprinting among several resident genes and by the mapping of Prader-Willi syndrome (PWS) and Angelman syndrome to this region. Both disorders are affected by imprinted genes, and a subset of patients with both PWS and Angelman syndrome reportedly manifest autismlike symptoms (Reik and Walter 1998). It has recently been reported that a disproportionate subset of patients with Turner syndrome (single X chromosome) develop autismlike symptoms (Skuse et al. 1997). Among patients with Turner syndrome, those who inherit a single maternal X chromosome are apparently at significantly greater risk of developing autism than are their counterparts who inherit a paternal chromosome, again suggesting an imprinting mechanism (Skuse 2000).
Although there are a number of biological and neuroanatomical clues that might inform the genetic study of autism, most findings are not consistent or robust from study to study, probably because of the heterogeneous etiology of the disorder. Among the most consistent findings are reports of higher numbers of platelets and/or higher levels of serotonin in urine in patients with autism than in control subjects (Anderson et al. 1987). Other studies indicate that serotonin reuptake inhibitors improve autism symptoms in an as yet undefined subset of patients (Brodkin et al. 1997; Fatemi et al. 1998). Both linkage and association studies appear to hint at a role for the serotonin transporter gene; however, the evidence remains unclear (Cook et al. 1997; Maestrini et al. 1999; Zhong et al. 1999).
Several independent groups have recently reported full-genome scans in which they used microsatellite markers to detect linkage to autism or ASDs. The results of four such studies are summarized in a recent review article (Lamb et al. 2000). In the first study, which was conducted by the International Molecular Genetic Study of Autism Consortium (IMGSAC) (1998), six chromosomal regions were reported to yield maximum multipoint LOD scores >1.0 (chromosomes 4, 7, 10, 16, 19, and 22). The highest positive LOD score resulting from this analysis was 2.53 on chromosome 7q32-35. Subsequent investigation of this region with a dense set of microsatellite markers and 26 additional families increased the multipoint LOD score to 3.63 (Maestrini and IMGSAC 1999). The Paris Autism Research International Sibpair Study (Philippe et al. 1999) reported hints of linkage (P values <.05) at 11 chromosome regions, including chromosomes 2, 4, 5, 6, 7, 10, 15, 16, 18, 19, and X. Four chromosomal regions—2q, 7q, 16p, and 19p—appear to overlap with results from the IMGSAC study. The highest positive LOD score from this second genome scan was 2.23, on the long arm of chromosome 6.
A third full-genome scan (Risch et al. 1999) yielded maximum likelihood scores >1.0 on chromosomes 17p, 7p, and 18q, with a highest genomewide score of 2.15 on chromosome 1p. The authors build a case that their data are most consistent with a disease model specifying ⩾15 loci (Risch et al. 1999). Finally, the Collaborative Linkage Study of Autism (CLSA) (Barrett et al. 1999) produced maximum multipoint heterogeneity LOD scores (MMLS/het) of 3.0 at D13S100 and 2.3 between markers D13S217 and D13S1229. The third highest score from this analysis was 2.2, at D7S1813 (7q), under the recessive model. In follow-up studies of the chromosome 7 findings, Buxbaum et al. (1999) and Ashley-Kock et al. (1999) independently report moderate evidence for linkage to chromosome 7q, after analyses of sample sets of 60 families and 76 families, respectively.
In the present study, we report the whole-genome microsatellite marker analysis of 110 multiplex families affected by autism. We use a battery of 335 microsatellite markers and analyze the data with an affected sib-pair (ASP) approach and a novel approach that employs scan statistics.
Families and Methods
Family Recruitment and Diagnostic Assessments
The Cure Autism Now (CAN) Foundation and Human Biological Data Interchange have created a large central repository of family DNA samples, known as the Autism Genetic Resource Exchange (AGRE), for genetic studies of autism. Families are recruited nationwide through advertisement sponsored by the CAN national support group and by referrals from clinicians. The initial selection criteria require that at least two family members have a diagnosis of autism or ASD (i.e., autism, PDD, or AS). Prospective families are then contacted, and an in-home visit is arranged with a trained diagnostician. During these visits, one or both parents are interviewed regarding the affected children in the family, using the Autism Diagnostic Interview-Revised (ADI-R) protocol (Lord et al. 1994). The ADI-R is a scored, semistructured interview that is based on the criteria for diagnosis of autism described in the International Statistical Classification of Diseases and Related Health Problems, 10th revision, and the Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Diagnostic assessments are videotaped and subjected to independent reliability checks by other trained interviewers. Individuals scored as affected in the current linkage analysis had to satisfy the prespecified cutoff scores in all three symptom areas of the ADI-R (social impairment, language and communication impairment, and unusual or restricted interests), as well as have an age at onset of <3 years. In addition, other pediatric, neurodevelopmental, and psychiatric information are being collected from these families, using a uniform medical examination, the Autism Diagnostic Observation Scale, as well as other measures.
In this study we have chosen two phenotypes for genetic analysis; first, a narrow diagnostic category that includes only autism, and second, a broader category that includes autism plus AS and PDD. As described in the report of the IMGSAC study (1998), genetic susceptibility to autism extends to AS and PDD (Bolton et al. 1994; Bailey et al. 1995), but the relatively low population prevalence of autism (Bailey et al. 1996) minimizes the likelihood that significant genetic heterogeneity will be introduced as a consequence of including relative pairs with autism plus either AS or PDD.
Laboratory Procedures
Blood was collected from all affected individuals and, when available, from parents and unaffected siblings. Blood samples were collected into Vacutainer tubes containing trisodium citrate, citric acid, and dextrose. Lymphocytes were immortalized by standard transformation protocols with Epstein-Barr virus (Anderson and Gusella 1984). DNA was extracted from both whole blood and immortalized lymphoblast cell lines by standard proteinase potassium-digestion and salting-out protocols. A complete description of each sample is available through AGRE. PCR amplification of microsatellite markers was performed as described elsewhere (Aita et al. 1999). In brief, multiplex PCR was performed in 384-well PCR plates (Marsh), using PTC 225 thermocyclers (MJ Research) with a total volume of 10 μl containing 50 ng of genomic DNA, 0.15-0.2 mM MgCl2, 0.2 mM dNTPs, and 0.5 units of Taq Platinum polymerase (Gibco Life Tech). On average, each multiplex PCR contains four microsatellite markers with varying amounts of primer (1–50 pmol), to achieve roughly even amplification of each marker. PCR products from two to three multiplex PCR reactions were pooled and genotyped using ABI model 377 DNA sequencers. For all markers, the last nucleotide of the reverse, nonfluorescent primer was modified to a guanine residue to promote the nontemplated addition of adenine by Taq DNA polymerase onto the complementary, fluorescence-labeled strand. This method has been reported to consistently shift the allelic profile of PCR products toward the allele plus and enzymatically added adenine residue, which allows for more accurate allele calling (Magnuson et al. 1996). PCR products were resolved using the PRISM 377XL data collection software and were sized by application of the GENESCAN 2.1 and GENOTYPER v.1.1.1 software packages (Applied Biosystems). The computer-generated genotypes were checked by two independent researchers who were blinded to disease diagnosis. The genotypes were then imported to the LABMAN genotyping database (Adams 1994), for allele binning and Mendelization checking. CEPH control individuals (1330-1 and 1330-2) were used to standardize the sizes of PCR products for each marker.
Genetic Markers
A total of 335 microsatellite markers were used in the full-genome scan. The majority of markers were selected from version 8.0 of the Marshfield fluorescence-labeled genome screening set (Center for Medical Genetics, Marshfield Medical Research Foundation). The average heterozygosity of markers used in this study is 0.77. Map distances (in Kosambi centimorgans) were obtained from Marshfield (Broman et al. 1998).
Statistical Analysis
Multipoint ASP analysis of genotype data was performed using the MAPMAKER/SIBS program (v2.1) (Kruglyak and Lander 1995). Allele frequencies were estimated from the family data, using the PEDMANAGER program. The main approach of ASP analysis is the likelihood-ratio method of Risch (1990a; 1990b), which maximizes the likelihood of incompletely polymorphic genotype data with respect to the probabilities of sharing zero, one, or two alleles identical by descent (IBD). Holmans (1993) and Faraway (1993) independently suggested restriction of the maximization to the set of sharing probabilities consistent with possible genetic models, denoted the “possible triangle”; defined by z1⩽0.5 and z0⩽0.5×z1, where z1 and z0 are the sharing of one and zero alleles IBD, respectively. It was shown that the power of the likelihood-ratio test can be increased by restricting maximization to this possible triangle (Holmans 1993; Holmans and Clayton 1995) Therefore, the present study calculated all maximum likelihoods by restricting to the possible triangle. We note that maximized LOD scores (MLS) obtained in this way are asymptotically distributed as mixtures of χ2 distributions with 1 and 2 df; hence, to obtain the nominal point-wise threshold of P<.05 an MLS > 0.74 is required, where
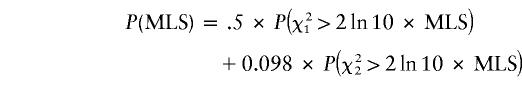 |
(Holmans 1993; Knapp 1997; Sham 1998; Nyholt 2000). However, evidence—based on an infinitely dense map—of genomewide suggestive (P<7.4×10-4) and significant (P<2.2×10-5) linkage (Lander and Kruglyak 1995) is reached at MLS values of 2.45 and 3.93, respectively.
Cordell et al. (1995) extended Holmans’ method to X-linked data, showing it was not appropriate to combine data from brother-brother, sister-sister, and brother-sister pairs; instead, they introduced an X-MLS statistic, which considers the three groups separately by restricting the maximization to the following genetically valid values: 0⩽z1bb⩽0.5, 0⩽z1ss⩽0.5, and 0⩽z1bs⩽0.5, where z1 represents IBD sharing of maternal alleles. The sum of these three MLS statistics, termed X-MLS, is a mixture of χ2 distributions with 1, 2, and 3 df (Cordell et al. 1995; Nyholt 2000); hence, to obtain the nominal threshold of P<.05, an X-MLS>1.18 is required. however, evidence—based on an infinitely dense map—for suggestive and significant linkage is reached at X-MLS values of 3.06 and 4.62 respectively, where
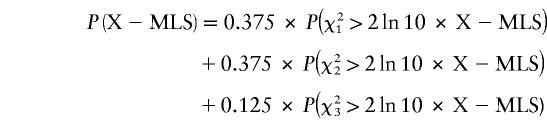 |
Additionally, we analyzed data for paternal and maternal contributions to IBD sharing, using the ASPEX (sib-phase) program (Hauser et al. 1996). We note, however, that the overall ASPEX results will differ from those obtained using MAPMAKER/SIBS, as ASPEX uses only the first sib of an affected pair and all unaffected sibs for reconstruction of missing parental genotypes, but MAPMAKER/SIBS uses all inheritance information. Furthermore, MAPMAKER/SIBS performs complete multipoint analysis, using all markers across the chromosome, but ASPEX simply finds the nearest informative markers flanking a particular locus, to help determine phase (IBD status).
To aid interpretation of the linkage analysis results, we used simulation to calculate sample-specific empirical genomewide significance values for our data sets. For both the broad and narrow data sets, 1,000 replicates were simulated under the assumption of no linkage, using the SIMULATE program (Terwilliger and Ott 1994). Each replicate was analyzed using MAPMAKER/SIBS. Genomewide significance values were obtained by counting the number of times an MLS (or equivalent X-MLS) was reached in the 1,000 replicates.
To increase power for localizing disease loci, we applied a novel approach that uses scan statistics to evaluate the significance of peak LOD scores on the basis of the LOD scores of the markers surrounding the peak (Hoh and Ott 2000). This approach is based on the fact that, in a genome screen, true peaks tend to be wider than false peaks (Terwilliger et al. 1997). Scan statistics are currently implemented for dichotomous data. From the given family observations, dichotomous data were obtained in the following two ways. (1) Two kinds of independent sib pairs were created, that is, affectedaffected (AA) and affected-unaffected (AU) pairs, by pairing offspring 1 with each of the subsequent offspring in turn. For the broad classification scheme, this resulted in 86 AA pairs and 91 AU pairs. (2) For undivided families, marker genotypes were generated by computer simulation, under the assumption of no linkage, such that observed data can be compared with “null” data. For the broad classification scheme, this resulted in 108 observed families and 432 computer-generated families. For each unit (sib-pair or family) of the two types of dichotomous data, nonparametric LOD scores were computed at each marker by the ALLEGRO program (Gudbjartsson et al. 2000). Analyses with scan statistics were performed only for the broad classification scheme, because it appeared to provide more linkage information than a narrow diagnostic scheme. Genomewide empirical levels, Pglobal, were computed with Monte Carlo permutation tests with sample sizes of 20,000 replicates (Hoh and Ott 2000)
Results
Quality of Genotyping Data
At the onset of this project, the AGRE sample consisted of 132 multiplex families, including 14 families with identical twins (12 autism:autism twins, 1 autism:PDD, and 1 autism:AS). Although these families were excluded from linkage analysis, they provide an internal control for estimation of genotyping errors. The 28 samples were configured randomly throughout the microtiter plates used for genotyping the entire sample. Researchers were blind to their identity. Three hundred thirty-five markers were genotyped against the 28 samples to yield ∼9,000 genotypes. Direct comparison of genotypes between identical twins identified seven allelic mismatches, giving an estimated error rate of 0.0007 (7/9,000). During the generation of ∼177,000 genotypes for the entire data set, 157 Mendelian errors were detected and corrected, giving an observed error rate of 0.00089 (157/177,215). Detectable Mendelian errors are expected to account for the majority of genotyping errors, although errors that do not violate transmission laws will certainly go undetected in the small nuclear families that constitute the majority of our sample.
Statistical Analyses
When we used the broad affection criteria, that is, autism, AS, or PDD—and after we removed an identical twin from 14 families—110 of the 132 families had two or more affected siblings, providing a total of 118 ASP comparisons. Analogously, our use of the narrow criteria (that is, strict autism) resulted in 72 families with two or more affected siblings and a total of 75 ASP comparisons. Of the 110 families, 40 include one unaffected sibling; 10 families include two, and 1 family includes four unaffected siblings. One hundred five families include both parents; the remaining five families include one parent. Phenotypic information is not available for parents. Four families included three affected sibs, and for the majority (71 families) of the remaining families with two affected sibs, both sibs are diagnosed with autism.
Results of multipoint ASP analysis of these families are presented in figure 1. We note that the broad and narrow data sets are not independent, with the narrow data set making up ∼64% (75/118) of the broad data set; however, a suitable correction is not clear. Subsequently, the analysis results are presented concurrently and without correction, to highlight the validity of using either diagnostic scheme, and their statistical interpretation is left to our readers.
Figure 1.


Chromosome-wise results from MAPMAKER/SIBS multipoint ASP analysis. Solid lines represent results based on use of the narrow diagnosis, and dashed lines represent results based on use of the broad diagnosis.
Linkage analysis under the broad-diagnosis scheme produced MLS peaks with significance below the nominal threshold of P<.05 on 13 chromosomes (2, 4, 5, 8, 10, 11, 12, 15, 16, 18, 19, 20, and X). The region of greatest significance was on chromosome 5 at marker D5S2494, which surpassed the recommended genomewide threshold for suggestive linkage, producing an MLS of 2.55 (P=.00058) (Lander and Kruglyak 1995). An MLS ⩾2.55 was obtained 197 times in the 1,000 simulated replicates, resulting in an occurrence expectation (OE) of 0.197 times in a genome scan; that is, we would expect to get a score of this magnitude once in every 5.08 (1/OE) genome scans. The next-most-significant region was on chromosome X at marker DXS1047, with an X-MLS of 2.56 (P=.0023), which also surpassed the suggestive linkage threshold, producing an OE of 0.887. Other regions of interest include peaks near D5S2488 with an MLS of 1.90 (P=.0028), which had a borderline suggestive linkage OE of 1.078 at D19S714 (MLS=1.72, P=.0043, and OE=1.646), D8S1179 (MLS=1.66, P=.005, and OE=1.897) and D16S2619 (MLS=1.46, P=.0081, and OE=3.038); all other loci produced MLS values <1.4 (P>.01).
Linkage analysis under the narrow diagnosis scheme produced MLS peaks with significance below P=.05 on seven chromosomes (3, 5, 10, 16, 18, 19, and X). The region of greatest significance was on chromosome 19, near marker D19S714, which also surpassed the accepted genomewide threshold for suggestive linkage, producing an MLS of 2.53 (P=.00061 and OE = 0.325). The next-most-significant region, also reaching the suggestive linkage threshold, was again on chromosome X at marker DXS1047 with an X-MLS of 2.67 (P=.0018 and OE=0.803). Other regions of interest include peaks near D16S2619 (MLS=1.93, P=.0026, and OE=1.125), D5S2488 (MLS=1.63, P=.0054, and OE=2.162), D5S2494 (MLS=1.41, P=.0092, and OE=2.504) and a secondary peak on chromosome 19, midway between D19S587 and D19S601 (MLS=1.70, P=.0045, OE=1.863); all other loci produced MLS values with P>.01.
Examination of the multipoint MLS plots in figure 1shows high concordance between results obtained using the broad and narrow diagnostic schemes. However, although some regional peaks differ considerably in size (∼1 MLS unit)—for example, the peaks at D5S2494, D8S1179, and D19S714—the MLS pattern is conserved under both diagnostic schemes, thus indicating that the differences are due to heterogeneity between the two data sets and not necessarily to differences between the broad and narrow autism phenotypes.
Paternal and maternal contributions to increased allele sharing for loci significant at P<.05, in both the broad and narrow data sets, are presented in table 1. Examination of these contributions shows some striking partitioning between parental contributions. In particular, the majority of IBD sharing at D5S2488 seems to originate from the paternal side, and the majority of sharing at D5S2494 (59 cM away) is from the maternal side, which suggests that two distinct loci may be present on this chromosome. Similarly, the majority of IBD sharing at D19S714 is maternally contributed, but the sharing at D19S587-D19S601 (30 cM away) is paternally contributed, which again suggests the presence of two separate loci. In any event, the breakdown of parental contributions by sex provides further suggestive evidence for the role of imprinting in the genetics of autism.
Table 1.
Summary of Parental Contributions to IBD Sharing in ASPs[Note]
|
ASPEX MLS of IBD Sharing in |
||||||||
| Broad ASPs |
Narrow ASPs |
|||||||
| Chromosome | cM | Locus | Paternal | Maternal | Overall | Paternal | Maternal | Overall |
| 5 | .0 | D5S2488 | 1.75 | .08 | 1.77 | 1.02 | .46 | 1.44 |
| 5 | 58.9 | D5S2494 | .38 | .86 | 1.23 | −.28 | 1.07 | .79 |
| 16 | 20.0 | D16S2619 | .78 | .66 | 1.44 | .40 | 1.38 | 1.76 |
| 18 | .0 | D18S59 | .44 | .25 | .68 | .68 | .47 | 1.15 |
| 19 | 32.0 | D19S714 | .20 | .94 | 1.15 | .62 | 1.25 | 1.90 |
| 19 | 61.5 | D19587- D18601 | .36 | .00 | .37 | 1.12 | −.31 | .79 |
Note.— Locus positions are indicated as centimorgan distance from a start point = 0, at pter. ASPs are categorized according to diagnostic criteria described in Families and Methods. Maternal and paternal contributions to IBD sharing were obtained using the ASPEX (sib-phase) program, as described in Statistical Analysis. MLS are depicted for multiplex families partitioned according to the likelihood (<50% vs. ⩾50%) of sharing maternal alleles at DXS1047.
The presence of a strong sex effect was also noted by Ashley-Koch et al. (1999). In their study of 62 ASPs and nine markers spanning 35 cM on chromosome 7q, a categorization of linkage results according to parental contribution indicated significant paternal (P=.007), but not maternal (P=.75), IBD sharing. Subsequently, given our positive X-chromosome linkage results coupled with the observed paternal/maternal partitioning of IBD sharing, we decided to further investigate this apparent sex effect by partitioning our data on the basis of observed allele-sharing on the X chromosome. Specifically, both the narrowly and broadly characterized families were separated on the basis of the likelihood (<50% vs. ⩾50%) of sharing maternal alleles at DXS1047. The resulting data sets consisted of 39 “X-linked” and 36 “X-unlinked” narrow ASPs, and 56 “X-linked” and 62 “X-unlinked” broad ASPs. Linkage results for these data sets are summarized in table 2.
Table 2.
Summary of Linkage Results after Partitioning According to IBD Sharing at DXS1047[Note]
| MAPMAKER/SIBS MLS |
||||||
| Broad ASPs |
Narrow ASPs |
|||||
| Chromosome | cM | Locus | <50% | ⩾ 50% | <50% | ⩾50% |
| 1 | 251.3 | D1S547 | 2.22 | .0 | .37 | .0 |
| 5 | .0 | D5S2488 | .81 | 1.11 | 1.02 | .77 |
| 5 | 58.9 | D5S2494 | 1.02 | 1.54 | .29 | 1.28 |
| 7 | 152.8 | D1S219-D1S3058 | 1.21 | .0 | .56 | .13 |
| 16 | 20.0 | D16S2619 | .08 | 1.94 | .31 | 2.19 |
| 18 | .0 | D18S59 | .10 | .81 | .20 | 1.33 |
| 19 | 32.0 | D19S714 | .07 | 2.16 | .07 | 3.59 |
| 19 | 61.5 | D19587−D19601 | 1.00 | .28 | 1.53 | .32 |
| 19 | 73.4 | D19S601 | 1.31 | .01 | 2.52 | .0 |
Note.— ASPs are categorized according to diagnostic criteria described in Families and Methods.
When linkage analyses are conditioned on sharing at DXS1047, they provide further evidence for a strong sex effect in our populations. Of particular note is the apparent interaction between sharing at DXS1047 and D19S714, with alleles at these loci tending to be maternally coinherited. Furthermore, the peak at D19S587-D19S601 is predominantly inherited from the paternal side in families with no increased sharing at DXS1047. Similarly, sharing at D1S547 and D7S2195-D7S3058 is restricted to families not sharing alleles at DXS1047, and sharing at D16S2619 and D18S59 is concentrated in families with increased sharing at DXS1047. Our data clearly hint at some interaction between loci on the X and autosomal chromosomes that may help to explain the skewed sex ratio in autism.
Application of scan statistics (Hoh and Ott 2000) to quasi-independent sib-pair data yielded the results presented in table 3 (only the broad classification is presented because the narrow classification shows no interesting results). For each marker, the difference in total LOD score between AA and AU pairs was computed, which represented the marker-specific statistic, Xi, for the i-th marker. Then, sums of such single-marker statistics for L consecutive markers were formed. The most significant of these is the scan statistic, SL, for given length L. Conventional analysis of one marker at a time (SL = 1) revealed D8S1136 on chromosome 8 as the single best marker, with an associated empirical significance level of Pglobal=.131. On the other hand, the best scan statistic consisted of the sum of the marker-specific statistics for the six contiguous markers surrounding D8S1136 (i.e., D8S1477-D8S1119). The associated genomewide significance level was P=.015. An allowance for multiple testing of various lengths of scan statistics reduced this significance to Pglobal=.047. When scan statistics were applied to the markers on one chromosome at a time, none of the chromosomes (other than chromosome 8) showed results significant at the 5% level. Application of scan statistics to all possible AA and AU sib-pairs (without different weighting depending on sibship size) furnished analogous, but nonsignificant, results.
Table 3.
Scan Statistics SL of Varying Lengths L for Sib-Pair Data (Broad Diagnosis)[Note]
| L | Varianta | SLb | Pc |
| 1 | D8S1136 | 3.50 | .131 |
| 2 | D8S1136 | 6.91 | .050 |
| 3 | D8S1113 | 9.69 | .034 |
| 4 | D8S1113 | 11.85 | .030 |
| 5 | D8S1477 | 14.27 | .021 |
| 6d | D8S1477 | 16.42 | .015 |
| 7 | D8S382 | 17.76 | .016 |
| 8 | D8S382 | 18.58 | .020 |
| 9 | D8S1145 | 19.17 | .024 |
| 10 | D8S1106 | 18.94 | .040 |
Note.— ASPs are categorized according to diagnostic criteria described in Families and Methods.
Variant = first element of SL.
Scan statistics SL of varying lengths L for sib-pair data resulting in a statistic of Pmin=.015.
Overall significance level is Pglobal=.047.
Best scan statistic.
Although contrasting AA pairs (positive LOD scores expected with linkage) with AU pairs (negative LOD scores expected with linkage) is expected to be powerful, results somewhat depend on the choice of offspring for the first sibling when quasi-independent sib-pairs are formed (sib 1 vs. sib 2, sib 1 vs. sib 3, etc.). Therefore, we also applied scan statistics to entire families, even though their LOD scores can only be compared with an expected value of zero for no linkage. The marker-specific statistic of interest was the LOD score achieved for the given marker, and sums of these statistics for contiguous markers were calculated as described above. As noted above, empirical genomewide significance levels, Pglobal, were estimated through permutation samples (which generated four times more “null” results than did observed data). The strongest result was obtained for marker D5S2488 (Pglobal=.0104, which, according to Lander and Kruglyak [1995], corresponds to a maximum LOD score of Zmax=4.38). Marker D8S1179 also showed a significant result with Pglobal=.0162 (corresponding to Zmax=4.17). Finally, suggestive linkage evidence was obtained for D19S714 with Pglobal=.3426 (corresponding Zmax=2.60).
Discussion
Table 4 summarizes linkage findings from the current and previous whole-genome linkage studies. Among the five whole-genome scans performed to date, 14 LOD scores ⩾1.0 have been reported from 10 different chromosomes. Five of the putative linked regions are from the present study, including three findings on chromosomes 5q, 8q, and Xqter that have not been previously reported. Two of the findings from the present study, those for chromosomes 16p and 19p, were also identified in the scans reported elsewhere. Among the three strongest findings to emerge from our study (chromosomes 5q, 19q, and Xqter), only the region on chromosome 19q has previously been implicated in autism linkage studies. Interpretation of such putative linkage findings is complicated, a point that is well illustrated in the analytical section of the report by Risch and colleagues (1999). Given the numerous complications that affect interpretation of complex linkage studies, it is of interest that data from five genomewide scans and several partial scans indicate a convergence of evidence for several regions, among which chromosome 7 appears to be prominent. A similar convergence of data, implicating a section of chromosome 10 as harboring a gene for Alzheimer disease, was recently reported by three groups (Bertram et al. 2000; Ertekin-Taner et al. 2000; Myers et al. 2000).
Table 4.
Summary of Linkage Evidence for Autism[Note]
|
IMGSA (99 Families) |
Philippe et al.(51 Sib Pairs) |
Risch et al.(90 Families) |
Barrett et al. (75 Families) |
Present Study(110 Families) |
||||||||||||
| Chromosome | Locus | Position(cM) | MMLSa | Locus | Position(cM) | MMLSb | Locus | Position(cM) | MMLSa | Locus | Position(cM) | MMLSc | Locus | Position(cM) | MMLSb | ScanStatisticd |
| 1p | … | … | … | … | … | … | D1S1675 | 149 | 2.15 (N) | … | … | … | … | … | … | … |
| 2q | D2S142- D2S326 | 161–177 | .52 (B) | D2S382- D2S364 | 169–186 | .64 (N) | … | … | … | … | … | … | … | … | … | … |
| 5q | … | … | … | … | … | … | … | … | … | … | … | … | D5S2494 | 45–69 | 2.55 (B) | 4.38 (B) |
| 6q | … | … | … | D6S283- D6S261 | 109–120 | 2.23 (N) | … | … | … | … | … | … | … | … | … | … |
| 7q | D7S530- D7S684 | 134–147 | 2.53 (B) | D7S486 | 124 | .83 (N) | D7S1804 | 137 | .93 (N) | D7S1813 | 104 | 2.2 (N) | D7S523 (N) D7S483 (N) | 123 165 | 1.02e 2.13e | … |
| 8q | … | … | … | … | … | … | … | … | … | … | … | … | D8S1179 | 134 | 1.66 (B) | 4.17 (B) |
| 13q | … | … | … | … | … | … | D13S800 | 55 | .68 (N) | D13S800 | 55 | 3 (N) | … | … | … | … |
| … | … | … | … | … | … | … | … | … | D13S217- D13S1229 | 19 | 2.3 (N) | … | … | … | … | |
| 16p | D16S407- D16S3114 | 18–23 | 1.51(B) | D16S3075- D16S3036 | 23–39 | .74 (N) | … | … | … | … | … | … | D16S2619 | 28 | 1.91 (N) | ND |
| 18q | … | … | … | D18S68 | 96 | .62 (N) | D18S878 | ND | 1 (N) | … | … | … | … | … | … | … |
| 19p | D19S221- D19S49 | 36–51 | .99 (B) | D19S226 | 42 | 1.37 (N) | … | … | … | … | … | … | D19S433 | 52 | 2.46 (N) | ND |
| Xq | … | … | … | … | … | … | … | … | … | … | … | … | DXS1047-q tel | 82 | 2.67 (N) | ND |
Note.— Map positions are derived from the Marshfield Genetic Laboratory’s genetic map. Letters in parentheses indicate diagnostic model used for linkage analysis: N = narrow; B = broad (see Families and Methods).
Multipoint maximum LOD score as determined by ASPEX.
Multipoint maximum LOD score as determined by MAPMAKER/SIBS.
Maximum multipoint heterogeneity LOD score.
Scan Statistics based on whole families based were calculated only for the broad phenotype.
Analysis includes the 110 families described in this study plus 50 additional nuclear families.
Given the interest in chromosome 7, we performed a follow-up analysis of the chromosomal region implicated by previous studies (position 115 to the telomere). Figure 2 depicts the six microsatellite markers used for the genome scan, plus 28 additional microsatellite markers employed in the “dense” follow-up analysis. The follow-up analysis includes the original 110 families described in this study, plus an additional 50 AGRE families of composition very similar to those of the initial collection. As is shown in figure 2, the dense-marker follow-up analysis reveals a peak LOD score of 1.0 at position 125 (marker D7S523) and another peak of 2.13 at position 165 (marker D7S483). The peak around position 125 might be construed as supportive of studies reported elsewhere, but the second peak at position 165 appears to be novel.
Figure 2.

LOD scores from multipoint ASP analysis of 160 nuclear families. Microsatellite markers used in the original genome scan are shown as unfilled triangles along the X axis. New microsatellite markers not included in the original scan are depicted as blackened triangles. Solid lines represent results based on use of the narrow diagnosis, and dashed lines represent results based on use of the broad diagnosis.
The second-most-significant finding from the current study derives from marker DXS1047 with an X-MLS of 2.56 (P=.0023). It is noteworthy that previous whole-genome scans have not detected evidence for loci on the X chromosome that predispose to ASD, despite several lines of evidence that suggest an X chromosomal contribution. The most compelling evidence for sexrelated differences in susceptibility to autism comes from the study of women with Turner syndrome (single X chromosome). Creswell and Skuse (1999) recently showed that 0 of 65 individuals with Turner syndrome who harbored complete paternal X chromosomes developed autism, compared with 10 of 156 individuals who had complete maternal X chromosomes. Furthermore, individuals in whom Turner syndrome is characterized by cognitive defects are more likely to inherit maternal X chromosomes than are individuals whose cognitive function appears to be less impaired by Turner syndrome (Skuse et al. 1997). These data led Skuse et al. to propose that a gene related to social behavior maps to the X chromosome and is subject to imprinting. Furthermore, Gillberg (1989) noted that, although severely disabled mentally retarded patients with ASD are distributed at a 1:1 sex ratio, higher functioning patients with ASD exhibit a 10:1 male:female ratio.
In view of this evidence and our data suggesting sex-specific parental contributions to IBD-sharing in ASPs (table 1), we performed an additional analysis, wherein we conditioned linkage data on maternal-allele sharing at the DXS1047 locus. ASPs that share predominantly maternal alleles at both D19S714 and DXS1047 yield an MLS score of 3.59. Our data suggest that a putative X chromosomal ASD locus might function optimally in concert with other autosomal loci, thereby providing a possible explanation for the failure of previous linkage studies to identify ASD susceptibility loci on the X chromosome.
In the present study, we have supplemented our statistical genetic analyses with the recently developed scan-statistic method of human gene mapping (Hoh and Ott 2000). The method exploits information contained in marker loci that flank the marker with a given peak LOD score (or other peak statistic). In the sib-pair data, for chromosome 8 markers, the genomewide significance level for a susceptibility locus at or near D8S1136 is Pglobal=.131, on a marker-by-marker basis. When flanking markers are taken into account through the scan statistic, the corresponding significance level drops to <5%, a value that is generally considered significant. The scan statistic does not improve the localization of susceptibility genes, but it tends to make results more significant, that is, it improves power. The use of scan statistics in the analysis of entire families provided stronger significance for markers D5S2488 and D8S1179 than was obtained with independent sib pairs. It also confirmed the results for marker D19S714 obtained with multipoint analysis.
A major concern in the mapping of complex trait loci is how best to interpret evidence for linkage between marker alleles and phenotype. In the Results section of this report, we refer to suggested thresholds for acceptance of genomewide suggestive and significant linkage; however, these figures are known to be conservative, as they assume complete (100%) marker informativeness and an infinitely dense map. Although Lander and Kruglyak (1995) clearly state this, it is difficult to extend their thresholds to real data sets. For example, although they give estimates for how much the significant LOD score threshold would decrease if map densities were 10 cM (∼20%), they mention that this decrease in threshold would be appropriate only if one performed two-point analyses. Measured according to the Marshfield map (Broman et al. 1998), the 335 markers used in the present study span ∼4,400 cM and thus result in an average map density of ∼13 cM. If we liberally apply the proposed 10-cM decrease of 20% to the standard (parametric) significant linkage threshold of LOD>3.3 (P<4.9×10-5), we generate a lower significant linkage threshold of LOD>2.64 (P<2.4×10-4). By the same reasoning, when we apply possible-triangle thresholds, we generate MLS thresholds >2.91 and X chromosome MLS thresholds >3.55. However, because we report multipoint LOD scores with an average of 81% marker informativeness (calculated using the multipoint Infomap function in MAPMAKER/SIBS), it is difficult to interpret our results according to the proposed metrics.
To mitigate such difficulties in interpretation of the linkage-analysis results, we used simulation to calculate sample-specific empirical genomewide significance values for our data sets. Put simply, suggestive and significant linkages correspond to statistical evidence expected to occur (i.e., an OE) of 1 and 0.05 times by chance per genome scan, respectively (Lander and Kruglyak 1995). The improvement observed in genomewide significance was substantial, with loci on chromosomes 5, 19, and X reaching the accepted threshold for suggestive linkage. If we strictly used the LOD score thresholds of Lander and Kruglyak (1995), only the loci on chromosomes 5 and 19 would reach these criteria. Instead, the simulation results produced genomewide values that were many times more significant; for example, the chromosome 5 result is five times more significant, and the chromosome 19 result is more than three times more significant. As a result, we urge the use of caution when interpreting genome-scan results in terms of the strict genomewide significance values of Lander and Kruglyak (1995) and, instead, strongly recommend that simulations be used to interpret genomewide significance.
Acknowledgments
We thank Cure Autism Now (CAN), the Autism Genetic Resource Exchange (AGRE), and Dr. Judith P. Sulzberger, for support (to J.L. and T.C.G.) for this research. We also gratefully acknowledge support (to E.P. and P.P.) from Telethon, Italy (grant E.911). We thank Adina Grun, Xiaomei Tong, and Miguel Brito, for technical support. Most importantly, we thank the families who have participated in and contributed to these studies.
Appendix: Members of the AGRE Consortium
Daniel H. Geschwind, University of California at Los Angeles, Los Angeles; Maya Bucan, University of Pennsylvania, Philadelphia; W. Ted Brown, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY; Joseph Buxbaum, Mt. Sinai School of Medicine, New York; Edwin H. Cook, Jr., University of Chicago; T. Conrad Gilliam, Columbia Genome Center, New York; David A. Greenberg, Mt. Sinai Medical Center, New York; David H. Ledbetter, University of Chicago, Chicago; Bruce Miller, University of California at San Francisco, San Francisco; Stanley F. Nelson, University of California at Los Angeles School of Medicine, Los Angeles; Jonathan Pevsner, Kennedy Krieger Institute, Baltimore; Jerome I. Rotter, Cedars-Sinai Medical Center, Los Angeles; Gerard D. Schellenberg, University of Washington, Seattle; Carol A. Sprouse, Children’s National Medical Center, Baltimore; Rudolph E. Tanzi, Massachusetts General Hospital, Boston; Kirk C. Wilhelmsen, University of California at San Francisco, San Francisco; and Jeremy M. Silverman, Mt. Sinai Medical School, New York.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Autism Genetic Resource Exchange, http://www.agre.org (for DNA samples)
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/ (for fluorescence-labeled genome screening set)
- Cure Autism Now Foundation, http://www.canfoundation.org (for recruitment of families affected by autism)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for autism [MIM209850], fragile X syndrome [MIM 309550] and tuberous sclerosis [MIM191100])
References
- Adams P (1994) LABMAN and LINKMAN: a data management system specifically designed for genome searches of complex diseases. Genet Epidemiol 11:87–98 [DOI] [PubMed] [Google Scholar]
- Aita VM, Liu J, Knowles JA, Terwilliger JD, Baltazar R, Grunn A, Loth JE, Kanyas K, Lerer B, Endicott J, Wang Z, Penchaszadeh G, Gilliam TC, Baron M (1999) A comprehensive linkage analysis of chromosome 21q22 supports prior evidence for a putative bipolar affective disorder locus. Am J Hum Genet 64:210–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GM, Freedman DX, Cohen DJ, Volkmar FR, Hoder EL, McPhedran P, Minderaa RB, Hansen CR, Young JG (1987) Whole blood serotonin in autistic and normal subjects. J Child Psychol Psychiatry 28:885–900 [DOI] [PubMed] [Google Scholar]
- Anderson MA, Gusella JF (1984) Use of cyclosporin A in establishing Epstein-Barr virus–transformed human lymphoblastoid cell lines. In Vitro 20:856–858 [DOI] [PubMed] [Google Scholar]
- Ashley-Koch A, Wolpert CM, Menold MM, Zaeem L, Basu S, Donnelly SL, Ravan SA, Powell CM, Qumsiyeh MB, Aylsworth AS, Vance JM, Gilbert JR, Wright HH, Abramson RK, DeLong GR, Cuccaro ML, Pericak-Vance MA (1999) Genetic studies of autistic disorder and chromosome 7. Genomics 61:227–236 [DOI] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25:63–77 [DOI] [PubMed] [Google Scholar]
- Bailey A, Phillips W, Rutter M (1996) Autism: towards an integration of clinical, genetic, neuropsychological, and neurobiological perspectives. J Child Psychol Psychiatry 37:89–126 [DOI] [PubMed] [Google Scholar]
- Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, Childress D, et al. (1999) An autosomal genomic screen for autism: collaborative linkage study of autism. Am J Med Genet 88:609–615 [DOI] [PubMed] [Google Scholar]
- Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE (2000) Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science 290:2302–2303 [DOI] [PubMed] [Google Scholar]
- Bolton P, Macdonald H, Pickles A, Rios P, Goode S, Crowson M, Bailey A, Rutter M (1994) A case-control family history study of autism. J Child Psychol Psychiatry 35:877–900 [DOI] [PubMed] [Google Scholar]
- Brodkin ES, McDougle CJ, Naylor ST, Cohen DJ, Price LH (1997) Clomipramine in adults with pervasive developmental disorders: a prospective open-label investigation. J Child Adolesc Psychopharmacol 7:109–121 [DOI] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Kilifarski MA, Reichert JG, Song CY, Zhou G, Lawlor BA, Fitzgerald M, Galvin P, Whiting K, Greenberg DA, Davis KL (1999) A genome-wide linkage study in autism. Mol Psychiatry Suppl 4:S13 [Google Scholar]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH (1999) Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet 8:1025–1037 [DOI] [PubMed] [Google Scholar]
- Cook EH Jr, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A, Haas R, Courchesne E, Leventhal BL (1997) Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry 2:247–250 [DOI] [PubMed] [Google Scholar]
- Cordell HJ, Kawaguchi Y, Todd JA, Farrall M (1995) An extension of the maximum LOD score method to X-linked loci. Ann Hum Genet 59:435–449 [DOI] [PubMed] [Google Scholar]
- Creswell C, Skuse D (1999) Autism in association with Turner syndrome: genetic implications for male vulnerability to pervasive developmental disorders. Neurocase 5:111–118 [Google Scholar]
- Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG (2000) Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science 290:2303–2304 [DOI] [PubMed] [Google Scholar]
- Faraway JJ (1993) Improved sib-pair linkage test for disease susceptibility loci. Genet Epidemiol 10:225–233 [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Realmuto GM, Khan L, Thuras P (1998) Fluoxetine in treatment of adolescent patients with autism: a longitudinal open trial. J Autism Dev Disord 28:303–307 [DOI] [PubMed] [Google Scholar]
- Feinstein C, Reiss AL (1998) Autism: the point of view from fragile X studies. J Autism Dev Disord 28:393–405 [DOI] [PubMed] [Google Scholar]
- Folstein S, Rutter M (1977) Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry 18:297–321 [DOI] [PubMed] [Google Scholar]
- Gillberg C (1989) Asperger syndrome in 23 Swedish children. Dev Med Child Neurol 31:520–531 [DOI] [PubMed] [Google Scholar]
- Gillberg C, Wing L (1999) Autism: not an extremely rare disorder. Acta Psychiatr Scand 99:399–406 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Hauser ER, Boehnke M, Guo SW, Risch N (1996) Affected-sib-pair interval mapping and exclusion for complex genetic traits: sampling considerations. Genet Epidemiol 13:117–137 [DOI] [PubMed] [Google Scholar]
- Hoh J, Ott J (2000) Scan statistics to scan markers for susceptibility genes. Proc Natl Acad Sci USA 97:9615–9617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmans P (1993) Asymptotic properties of affected-sib-pair linkage analysis. Am J Hum Genet 52:362–374 [PMC free article] [PubMed] [Google Scholar]
- Holmans P, Clayton D (1995) Efficiency of typing unaffected relatives in an affected-sib-pair linkage study with single-locus and multiple tightly linked markers. Am J Hum Genet 57:1221–1232 [PMC free article] [PubMed] [Google Scholar]
- International Molecular Genetic Study of Autism Consortium (1998) A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet 7:571–578 [DOI] [PubMed] [Google Scholar]
- Jorde LB, Hasstedt SJ, Ritvo ER, Mason-Brothers A, Freeman BJ, Pingree C, McMahon WM, Petersen B, Jenson WR, Mo A (1991) Complex segregation analysis of autism. Am J Hum Genet 49:932–938 [PMC free article] [PubMed] [Google Scholar]
- Knapp M (1997) The affected sib pair method for linkage analysis. In: Pawlowitzki IH, Edwards JH, Thompson EA (eds) Genetic mapping of disease genes. Academic Press, London, pp 150–151 [Google Scholar]
- Kruglyak L, Lander ES (1995) Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am J Hum Genet 57:439–454 [PMC free article] [PubMed] [Google Scholar]
- Lamb JA, Moore J, Bailey A, Monaco AP (2000) Autism: recent molecular genetic advances. Hum Mol Genet 9:861–868 [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A (1994) Autism Diagnostic Interview—Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 24:659–685 [DOI] [PubMed] [Google Scholar]
- Maestrini E, International Molecular Genetic Study of Autism Consortium (1999) Search for autism susceptibility loci: genome screen follow-up and fine mapping of a candidate region on 7q. Am J Hum Genet Suppl 65:A106 [Google Scholar]
- Maestrini E, Lai C, Marlow A, Matthews N, Wallace S, Bailey A, Cook EH, Weeks DE, Monaco AP (1999) Serotonin transporter (5-HTT) and gamma-aminobutyric acid receptor subunit β3 (GABRB3) gene polymorphisms are not associated with autism in the IMGSA families. The International Molecular Genetic Study of Autism Consortium. Am J Med Genet 88:492–496 [DOI] [PubMed] [Google Scholar]
- Magnuson VL, Ally DS, Nylund SJ, Karanjawala ZE, Rayman JB, Knapp JI, Lowe AL, Ghosh S, Collins FS (1996) Substrate nucleotide-determined non-templated addition of adenine by Taq DNA polymerase: implications for PCR-based genotyping and cloning. Biotechniques 21:700–709 [DOI] [PubMed] [Google Scholar]
- Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM (2000) Susceptibility locus for Alzheimer's disease on chromosome 10. Science 290:2304–2305 [DOI] [PubMed] [Google Scholar]
- Nyholt DR (2000) All LODs are not created equal. Am J Hum Genet 67:282–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, Penet C, Feingold J, Brice A, Leboyer M, van Malldergerme L (1999) Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet 8:805–812 [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J (1998) Imprinting mechanisms in mammals. Curr Opin Genet Dev 8:154–164 [DOI] [PubMed] [Google Scholar]
- Risch N (1990a) Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am J Hum Genet 46:229–241 [PMC free article] [PubMed] [Google Scholar]
- Risch N (1990b) Linkage strategies for genetically complex traits. III. The effect of marker polymorphism on analysis of affected relative pairs. Am J Hum Genet 46:242–253 [PMC free article] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, et al (1999) A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet 65:493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham P (1998) Statistics in human genetics. Arnold, London, pp 110 [Google Scholar]
- Skuse DH (2000) Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatr Res 47:9–16 [DOI] [PubMed] [Google Scholar]
- Skuse DH, James RS, Bishop DV, Coppin B, Dalton P, Aamodt-Leeper G, Bacarese-Hamilton M, Creswell C, McGurk R, Jacobs PA (1997) Evidence from Turner's syndrome of an imprinted X-linked locus affecting cognitive function. Nature 387:705–7089192895 [Google Scholar]
- Smalley SL (1998) Autism and tuberous sclerosis. J Autism Dev Disord 28:407–414 [DOI] [PubMed] [Google Scholar]
- Szatmari P, Jones MB, Zwaigenbaum L, MacLean JE (1998) Genetics of autism: overview and new directions. J Autism Dev Disord 28:351–368 [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore, pp 245–250 [Google Scholar]
- Terwilliger JD, Shannon WD, Lathrop GM, Nolan JP, Goldin LR, Chase GA, Weeks DE (1997) True and false positive peaks in genomewide scans: applications of length-biased sampling to linkage mapping. Am J Hum Genet 61:430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing L, Gould J (1979) Severe impairments of social interaction and associated abnormalities in children: epidemiology and classification. J Autism Dev Disord 9:11–29 [DOI] [PubMed] [Google Scholar]
- Zhong N, Ye L, Ju W, Brown WT, Tsiouris J, Cohen I (1999) 5-HTTLPR variants not associated with autistic spectrum disorders. Neurogenetics 2:129–131 [DOI] [PubMed] [Google Scholar]