

Abstract
An innovative replica exchange (parallel tempering) method called replica exchange with solute tempering (REST) for the efficient sampling of aqueous protein solutions is presented here. The method bypasses the poor scaling with system size of standard replica exchange and thus reduces the number of replicas (parallel processes) that must be used. This reduction is accomplished by deforming the Hamiltonian function for each replica in such a way that the acceptance probability for the exchange of replica configurations does not depend on the number of explicit water molecules in the system. For proof of concept, REST is compared with standard replica exchange for an alanine dipeptide molecule in water. The comparisons confirm that REST greatly reduces the number of CPUs required by regular replica exchange and increases the sampling efficiency. This method reduces the CPU time required for calculating thermodynamic averages and for the ab initio folding of proteins in explicit water.
Keywords: molecular dynamics, Monte Carlo, parallel tempering, protein solutions, rough energy landscapes
Sampling the conformation space of complex systems, such as proteins, is a notoriously difficult problem in structural biology and theoretical chemistry. The difficulty arises from the infrequent crossings of high-energy barriers between local energy minima, leading to local trapping for long times and concomitant quasi-ergodicity in the sampling. Many methods have been devised to overcome the problem of quasi-ergodicity. These methods include the multicanonical ensemble method (1–3), the simulated tempering method (4–6), and the parallel tempering or replica exchange method (REM) (7–9).
The first two methods require a non-Boltzmann weight factor arrived at by iteration. For systems with rough energy landscapes, such as proteins dissolved in explicit water, obtaining the weight factor is not a trivial process. Thus, the REM has been attracting more and more attention because the standard Boltzmann weight factor can be used. By using high-temperature replicas to overcome the energy barrier, the REM has proven to be a useful method for sampling phase space (10, 11).
For the standard REM, the number of replicas needed increases as O(f1/2), where f is the solution's total number of degrees of freedom (12). Even for a relatively small biomolecular system consisting of one β-hairpin protein molecule dissolved in water (4,342 atoms in all), 64 replicas were needed to cover the temperature range between 270 and 695 K with a nonvanishing acceptance ratio for replica exchange (13). This requirement severely restricts the applicability of REM to reasonably small systems, unless one has access to a massively parallel computer.
The main reason that a large number of replicas are required is that the overall Hamiltonian grows with system size. The acceptance probability for the exchange of configurations between two replicas at different temperatures is exp(ΔβΔE), a quantity that depends exponentially on the change in energy. For a larger system, one must choose smaller Δβ to obtain viable acceptance probabilities: that is, replica systems more closely spaced in temperature and, concomitantly, more replica systems to cover the same upper and lower temperatures. If we can devise a method that depends only on the change in energy of a small part of the system, we can achieve a sufficiently large acceptance probability even for replicas with widely separated temperatures, thus reducing the need for a large number of replicas.
Fukunishi et al. (12) devised a useful alternative to REM, the so-called Hamiltonian REM, by using a transformed Hamiltonian at each replica level. They showed for a simple transformed Hamiltonian that the sampling efficiency can be comparable or superior to the standard REM (12, 14). Their studies using Hamiltonian REM focused on biomolecules dissolved in implicit solvents or a vacuum. Transformations of the potential energy surface, albeit different ones, for sampling were also used earlier in a variant of simulated tempering (6).
It is often desirable or necessary to explicitly include water molecules. For example, the implicit solvent model cannot reproduce the folding free-energy landscape of a small peptide, such as a β-hairpin, in explicit water (13). Here, we propose a unique scheme based on a simple physical principle. We allow the potential energy to scale with temperature in such a way that the molecule of interest appears to get hotter, but the water stays cold as one climbs the replica ladder. We devise a rigorous transformation in which the acceptance probability for replica exchange scales only with the number of degrees of freedom of the biomolecule but not the number of water molecules. This transformation leads to a method that we call replica exchange with solute tempering (REST) for the efficient all-atom simulations in explicit water. Our basic approach can also be used in the usual potential energy-based Monte Carlo method, but for biological systems, the state-generating engine is usually molecular dynamics (MD) rather than Monte Carlo.
The REST method is described in Methods and is followed by an application to an alanine dipeptide dissolved in explicit water molecules, a system consisting of 1,558 atoms. We show that REST can sample the conformation space very effectively and is significantly more efficient than standard REM.
Methods
Replica exchange (or parallel tempering) involves running Monte Carlo (or constant temperature MD) for a certain number of passes (or time steps) in parallel on a set of replica systems, each at a different temperature, {T0, T1, T2,..., TN}, where the temperatures are ordered from the lowest T0 to the highest TN. At the end of this period, an attempt is made to exchange the configurations of a pair of neighboring replicas, and this exchange is accepted with a probability satisfying detailed balance. The process is then repeated. The highest temperature, TN, is chosen so that its replica can rapidly cross the potential energy barriers. Because configurations sampled at the high temperatures can eventually exchange with the low-temperature replicas, the low-temperature systems will experience jumps between potential basins separated by high barriers, something they would not be able to do easily in ordinary Monte Carlo or MD. Likewise, different replicas can have not only different temperatures but also different potential functions, {E0(X0), E1(X1), E2(X2),..., EN(XN)}, where Xn represents the configurational coordinates of the nth replica system. The potential functions can be tailored to specific problems. There is a long history of using deformed potentials in sampling (15). Fukunishi et al. (12) and Jang et al. (14), in particular, have applied this more general form of replica exchange to solutions in which the solvent is a continuum (or implicit) solvent or a vacuum. In this study, we apply a generalized replica exchange theory in a rigorous fashion to bypass the poor scaling with system size of ordinary replica exchange. The basic trick is to pick a good scaling of the potential function. Our approach aims to reduce the number of replicas required and thereby the time required to sample large systems, such as a protein molecule in explicit water solvent.
It is a simple exercise to derive the acceptance probability for the exchange of configurations between the nth and mth replicas [see Fukunishi et al. (12)],
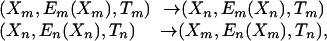 |
[1] |
where Xm, Em(Xm), and Tm are, respectively, the configuration, the energy, and the temperature of the mth replica just before an exchange of replicas is attempted (with corresponding expressions for other replicas). The equilibrium probability for this state is
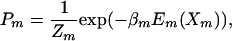 |
[2] |
where βm = 1/(kBTm) and Zm is the corresponding configurational partition function. Denoting the transition probability for the exchange i → f specified in Eq. 1 by T(i → f) and for the reverse exchange by T(f → i) and applying the detailed balance condition
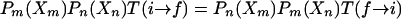 |
[3] |
gives the ratio of the transition probabilities
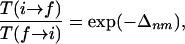 |
[4] |
where
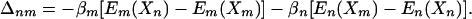 |
[5] |
If the Metropolis criteria is applied, the acceptance probability can be obtained as follows:
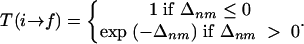 |
[6] |
The trick we use to improve the scaling with system size is to subdivide the system into two parts in a simple way. For a protein solution consisting of one large nondissociating protein molecule (labeled p) dissolved in a large number of water molecules (labeled w), we take the protein as one part (the central group) and the water as the other part (the bath). (Later, we show how we can achieve a speedup, albeit a smaller one, by taking the protein and its solvation shell as the central group and the rest of the water as the bath.) Thus, the system consists of a central part (p) and the bath (w). The potential energy of this solution is
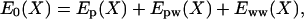 |
[7] |
where Ep, Epw, and Eww are, respectively, the internal energy of the protein, the interaction energy between the protein and water, and the interaction of the water molecules with each other. Under usual conditions, the first two terms depend on only a relatively small set of coordinates compared with the last term.
We take the lowest replica to be the protein solution with the potential surface given by Eq. 7 at temperature T0, and we label this replica by the index 0. As we go up the replica ladder, we rescale the potential surface as follows. The potential surface for replica m can be decomposed into three terms,
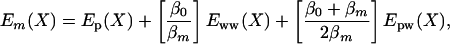 |
[8] |
where βm = 1/(kBTm) and the terms Ep, Eww, and Epw are the interactions within the central group, within the bath, and between the central group and the bath, respectively. When βm = β0 (T0 is the target temperature: e.g., 300 K), the original energy surface is recovered. In the case where the central group includes the entire system, REST reduces to REM. Substituting Eq. 8 into Eq. 5, one finds that
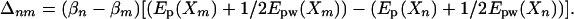 |
[9] |
It should be noted that Eww does not appear in this expression, because it cancels out in the algebra. This term causes the poor scaling with system size in the ordinary REM. The acceptance probability for the exchange is thus much larger for the scaled potentials, because |Ep + (1/2)Epw| ≪ |Ep + Epw + Eww|.
It should also be noted that the Boltzmann factors for the replicas are
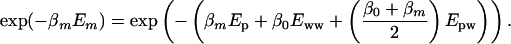 |
[10] |
Physically, our scheme is similar to but not the same as changing the temperature of the central group while the temperature of the surrounding group is kept at the same target temperature. For this reason, we named our scheme “replica exchange with solute tempering” (REST). Our method is very different from the partial REM or the local REM recently proposed by Cheng et al. (16). Their method is based on the assumption that the coupling between the central group and the surrounding group is weak and that the conformation of the surrounding group is hardly changed during the simulation. Generally, this assumption is not correct for many important processes, such as protein folding. Furthermore, they thermostated different parts of the system at different temperatures, thus sampling a steady-state distribution and not the true equilibrium distribution. By scaling the potential surface appropriately, our method does not introduce any approximation or assumptions and therefore is rigorous.
It is important to recognize that because the potential energy surfaces of all of the replicas other than the lowest one are deformed, this method only samples the correct distribution for replica 0. Nevertheless, because of the improved scaling with system size, we sample exchanges between replicas up and down the ladder of replicas more efficiently than in ordinary replica exchange.
In the present study, we chose the molecules we are interested in as the central group. Although we chose this particular partition, REST is applicable to any choice of partitions. However, if there is significant exchange of molecules between the central group and surrounding group (e.g., if the water molecules in the first solvation shell are also included in the central group), the groups should be updated periodically. During the update process, another Metropolis decision must be made to ensure that detailed balance is satisfied.
Results
As our first application, we apply REST to an aqueous solution consisting of one small peptide molecule, alanine dipeptide, dissolved in 512 TIP4P (17) water molecules. The potential model for alanine dipeptide is taken to be the OPLS-AA/L (all-atom optimized potentials for liquid simulations) force field (18, 19). We simulate the system with cubic periodic boundary conditions by using the P3ME (particle–particle particle–mesh Ewald) method (20–22) for calculating the electrostatic interactions. The internal geometries of water molecules and the bond lengths of the alanine dipeptide are constrained with the RATTLE algorithm (23). The system temperature is controlled by Nose–Hoover chain thermostats (24). We performed a REST simulation with the alanine dipeptide as the central group and solvent as the bath. The target temperature for REST was set to 300 K. Three replicas, with temperatures of 300, 420, and 600 K, were used to generate acceptance ratios ranging from 23% to 29%. For comparison, a regular replica exchange simulation was performed with 22 replica systems distributed roughly exponentially between 300 and 603 K, with corresponding nearest-neighbor acceptance ratios ranging from 22% to 29%.
The energy probability distributions obtained from regular replica exchange are shown in Fig. 1 Upper. If there were only three replicas, with 300, 417, and 603 K, essentially no exchanges would be accepted because there would be no overlap between those corresponding energy distributions. When REST is applied, however, there are sufficient overlaps between the distributions that, even with only three replicas, one gets the aforementioned large acceptance probabilities (≈22–29%), because the exchange is based on Ep + 0.5Epw rather than on the full potential energy, which includes Eww.
Fig. 1.

The probability distributions of potential energy for different temperatures for REM (Upper) and REST (Lower). For REST, only the “effective potential energy,” Ep + 0.5Epw, is considered, based on the formula-of-acceptance ratio (Eq. 9).
To test the accuracy of our scheme, we compare the Ramachandran plots, the population distributions of φ and ψ backbone dihedral angles of alanine dipeptide, of REST to those of REM at 300 K. As shown in Fig. 2, the population distributions of REM and REST are very similar. There are four pronounced peaks, regions PII, αR, β, and α′, ordered by population. However, for the regular REM, there is still a difference in the Ramachandran plots, even after a 10-ns replica exchange simulation, if we start from two quite different initial conformations, A and B. However, the final population distributions are almost identical for two different REST runs. Table 1 shows the populations for REM and REST in each region, defined in Fig. 2, for two independent simulations. Our simulation results based on the OPLS-AA/L/TIP4P force field seem to be able to generate population distributions in good agreement with experiments, which have a dominant region PII and two minor populated regions, β and αR, for alanine tripeptide (25, 26).
Fig. 2.

The Ramachandran plots of backbone dihedral angles of alanine dipeptide, φ and ψ, at 300 K. The two subplots in Upper are collected for 10 ns through standard REM simulation over 22 replicas. The plots in Lower are from 10-ns simulation of REST over three replicas. Left and Right are from two distinct initial configurations, A (in the region PII) and B (in the region αR), respectively. All of the plots are color-coded and follow the spectrum from blue (lowest probability) to red (highest probability).
Table 1. Comparison of the percent population of four regions.
| Population region
|
REM
|
REST
|
||
|---|---|---|---|---|
| A | B | A | B | |
| PII | 0.430 | 0.463 | 0.467 | 0.466 |
| αR | 0.233 | 0.214 | 0.212 | 0.214 |
| β | 0.182 | 0.181 | 0.183 | 0.183 |
| α′ | 0.155 | 0.142 | 0.138 | 0.137 |
The results are collected over 10 ns for standard REM and REST. The errors listed are the standard deviations of the percent population.
If a sampling procedure is ergodic, averages of any property of the system computed from two independent trajectories A and B should be equal. This condition of self-averaging must be satisfied if one is to equate trajectory averages with statistical averages over conformation space. Thirumalai et al. (27) and Whitfield et al. (28) have proposed a simple means for measuring the simulation length needed to guarantee self-averaging: the “ergodic measure.” They define the mean-square difference between the average of the property taken over the A trajectory and the average taken over the B trajectory summed over all atoms of the protein. This metric provides a measure of the convergence of the two averages. In an ergodic system, the mean-square difference will decay to zero at long times as 1/Dt, where D is the generalized diffusion constant, which provides a time scale for self-averaging in the simulation. The decay of the ergodic measure to zero at long times is a necessary condition for the system's average properties to correspond to equilibrium thermodynamic averages. Moreover, in the optimization of a computational algorithm, one may choose the optimum value of a variable parameter to maximize the generalized diffusion coefficient and the rate of phase space sampling. To check the convergence for both methods, REST and REM, we calculated the decay of the ergodic measure for different properties. To accomplish this, two simulations starting from two significantly different initial configurations A and B were performed for 10 ns for each method. The total simulation time was >0.5 μs.
The first ergodic measure we calculate is for the conformation change. For this purpose, the Ramachandran plot can be discretized by dividing the φ and ψ axes into n × m uniform cells of area ΔφΔψ = 4 π 2/nm. The population in the ijth cell is denoted Ri,j, and Ri,j can then be regarded as a population matrix. We define the ergodic measure of the population as
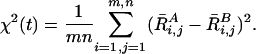 |
[11] |
For long simulations, the mean-square deviation χ2(t) should decay to zero if the sampling method is ergodic. Fig. 3 shows this ergodic measure as a function of CPU time (including the contributions from all of the replicas). Both methods, REM and REST, can give convergent results if the CPU time is long enough. However, it is obvious that the REST converges much faster than the regular REM. For example, for a variance (χ2) of 0.0005, REST is ≈10 times more efficient than ordinary REM.
Fig. 3.

The Ramachandran plot variance, defined in Eq. 1, as a function of CPU time. (Inset) The magnified plot in y direction.
As a further test of REST, we alter the potential function of alanine dipeptide to raise some of the energy barriers that separate the free-energy basins by including the following extra term:
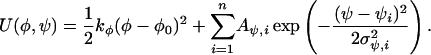 |
[12] |
The first term, ½kφ(φ – φ0)2, restrains the φ angle to be near φ0. The second term creates potential wells at a series of points ψi in y direction. Aψ,i characterizes the well depth, and σψ,i defines a spread of this potential well in the ψ direction. We apply two wells (n = 2) on the centers of rectangular boxes of conformation PII and αR, shown in Fig. 2, with the spread in both directions of 10° and a well depth of 3.0 kcal/mol. For each method, two independent simulations are run for 10 ns starting from these two distinct wells. For comparison, regular MD (neither REM nor REST) is performed for 10 ns as well. The cumulative simulation time is >0.5 μs.
In Fig. 4, we monitor the dihedral angle ψ, which characterizes which basin the system is in, as a function of CPU time. For the regular MD, during 20 units of CPU time (1 unit corresponds to 1 ns of regular MD simulation), only three transitions between these two wells were observed. In comparison, 19 transitions, on average, were made in REM within the same time window, and 189 transitions were made for REST during the same period. If the efficiency to sample the phase space can be measured by the number of transitions made in a time period, REST appears to be ≈10 times more efficient than REM and 60 times more efficient than the regular MD for this very small system.
Fig. 4.

The trajectories of ψ for MD (Top), REM (Middle), and REST (Bottom) for the alanine dipeptide with biased potentials. A and B correspond to the results from two different initial configurations, as described in the legend of Fig. 2.
Finally, we investigate the ergodic measure for the 1,4-pair distance metric d14(t), first introduced by Thirumalai,
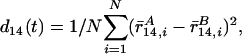 |
[13] |
to compare the convergence of REST and REM on the system with the modified potential (cf. Eq. 12). Here, N is the number of 1,4 pairs, and r̄14 is the average 1,4-pair distance over time. Fig. 5 shows the ergodic measure d14(t) for REST and REM. With the help of high-temperature replicas, both generate reasonable convergence, indicated by the monotonic decay for these two curves. If the value 0.05 is set as the satisfying criteria for the convergence of d14(t), REST appears to converge seven times faster than REM.
Fig. 5.

The 1,4-distance correlation function for the alanine dipeptide with biased potentials. The black and red lines correspond to the results from REM and REST simulations, respectively. (Inset) The magnified plot in y direction.
Discussion
We introduce REST, an innovative replica exchange algorithm for the efficient sampling of systems with rough potential energy surfaces, including biological systems such as a protein in water. In the algorithm, we partition the system into two groups: a central group and the bath group. REST builds on the idea that the sampling can be enhanced by using high-temperature replicas of the central group but low-temperature (target temperature) replicas of the bath. This idea guides us in defining a scaled potential energy surface for the replicas. By this scaling of the potential energy function, the number of replica systems required can be greatly reduced, because the acceptance probability for replica exchange becomes independent of the solvent–solvent interaction energy, which is the main factor leading to poor scaling with system size in ordinary replica exchange. Thus REST, unlike REM, scales well with system size and hence requires substantially fewer parallel processors than REM.
We have simulated alanine dipeptide in water. We simulated this system by using standard REM and REST for the alanine dipeptide system. The REST simulation of alanine dipeptide in water showed that even for a simple system as small as alanine dipeptide in 512 water molecules, the sampling in REST was 7–10 times more efficient than in REM, an impressive speedup for such a small system. The larger the system, the better will be the relative performance of REST to REM. A hand-waving argument suggests that the efficiency of REST versus REM will scale as 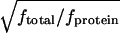 , where ftotal is the total number of degrees of freedom in the system (protein plus bath) and fprotein is the number of degrees of freedom in the protein. In the alanine dipeptide system the dipeptide consists of 45 degrees of freedom, and the water consists of 3,072 degrees of freedom, so that REST should be approximately
, where ftotal is the total number of degrees of freedom in the system (protein plus bath) and fprotein is the number of degrees of freedom in the protein. In the alanine dipeptide system the dipeptide consists of 45 degrees of freedom, and the water consists of 3,072 degrees of freedom, so that REST should be approximately 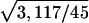 , or eight times more efficient than REM, in agreement with what was found in the simulation.
, or eight times more efficient than REM, in agreement with what was found in the simulation.
We note that REST can sample the phase space efficiently only at the target temperature because the Hamiltonians of the replicas at different temperatures are different from the real Hamiltonian. However, if one is interested in sampling biological systems at a certain temperature [for example, protein folding at body temperature (310 K) or room temperature (298 K)], REST is a powerful tool. Because the ratio between the size of the total system and the central group increases with increasing system size, the saving will be more pronounced for larger systems.
It is possible to include one or two shells of water with the protein as the central group, which we have done by storing a neighbor list of the protein. Because the water molecules originally in the central group will thus move out of the solvation shell of the protein as the configuration of a replica changes during its normal walks, and because the potential is different for the waters in the central group and the bath, it is necessary to invoke a Metropolis criterion for updating the neighbor lists. This requirement adds some overhead to the calculation. For example, choosing to include one shell of waters in the central group, we find that REST is only three to five times faster than REM for the alanine dipeptide system, rather than the 7- to 10-fold speedup of REST with only the protein as the central group. In this study, we use the simple version of REST in which all of the water molecules are treated as a bath.
The choice for the deformation of the energy given in Eq. 8 is a special case of the more general Em(X) = Ep(X) + amEww(X) + bmEpw, where we choose am = β0/βm and bm = (β0 + βm)/2βm. The only conditions required to get the correct Boltzmann distribution at T0 are am → 1 and bm → 1 as m → 0. The specific form we chose for am was dictated by the desire to have terms arising from Eww cancel in the acceptance probability. The choice for bm is less obvious. Our choice is only one of many possible choices. We could have just as well chosen bm = (β0/βm)1/2 or bm ≡ 1. In this study, we made no effort to optimize bm but chose it to correspond to a temperature intermediate between T0 and Tm.
To get some feeling for the magnitudes of the above effects of our choice of bm, we note that for the highest temperature replica in the dipeptide simulation (T0 = 300 K and Thighest = 600 K), am = 2 and bm = 1.5. For this energy surface, the water–water interaction is twice as strong as the “true” interaction, and the protein–water interaction is 1.5 times as strong as the true interaction, which has the effect of making the solvent stiffer and the protein–water repulsions and attractions stronger. From Eq. 10 we see that, despite this, the waters will move no more slowly than they normally would on the ground-state surface, albeit with a somewhat stronger protein–water interaction, the latter being adjustable by a different choice of bm.
One very feature of REST that may prove useful in the study of more complex systems is that the solvent (or whatever we consider to be the bath) will be unlikely to undergo an extreme structural transformation (like a phase transformation), even at the highest temperatures used. The contrary is true in REM, where, at high temperatures, it is possible to have large-scale configurational changes. Should that happen, the acceptance probability for swapping the configurations of a replica lower in temperature with one above this temperature will yield a vanishing acceptance probability and will thus destroy the ergodicity of REM.
REST should be useful for complex systems containing a large molecule of interest embedded in a matrix dispersed or dissolved in water. Because the upper temperature is chosen to surmount barriers, it might be sufficiently high that the matrix would collapse. Taking the water and matrix as the bath and the protein as the system, REST will leave the matrix intact, whereas REM might lead to the destruction of the matrix. An example would be a flexible molecule embedded in a detergent micelle or a protein embedded in a membrane and surrounded by a salt solution. One might then take the bath to be the micelle (or membrane) plus water and the system to be the flexible molecule (or protein). In REM, the micelle (or membrane) would fall apart at high temperatures but not in REST.
We believe that REST will prove to be a useful sampling methodology for certain complex biochemical systems because it reduces the number of processors required in REM, it gives enhanced barrier-crossing like REM does, and, for the high-temperature replicas, it avoids structural transitions in the solvent or surrounding matrix, thereby enabling simulations of complex systems (like membranes) that are not amenable to application of ordinary REM.
Acknowledgments
We thank Drs. Thomas Young, Ajay Royyuru, Ruhong Zhou, Gustavo Stolovitsky, Robert Germain, and Michael Pitman, and Mr. Morten Hagen for useful discussions and helpful comments. This work was supported by National Science Foundation Grant CHE-03-16896 (to B.J.B.) and National Institutes of Health Grants GM43340 (to B.J.B.) and GM52018 (to R.A.F.). This work was also supported by National Computational Science Alliance Grant MCA95C007N for the Xeon Linux Cluster and by a Shared University Research grant from IBM (White Plains, NY) for the purchase of an IBM Linux Cluster.
Author contributions: B.J.B. designed research; P.L. and B.K. performed research; P.L. and B.K. analyzed data; and P.L., B.K., R.A.F., and B.J.B. wrote the paper.
Abbreviations: REST, replica exchange with solute tempering; REM, replica exchange method; MD, molecular dynamics.
References
- 1.Berg, B. A. & Neuhaus, T. (1991) Phys. Lett. B 267, 249–253. [Google Scholar]
- 2.Sugita, Y. & Okamoto, Y. (2000) Chem. Phys. Lett. 329, 261–270. [Google Scholar]
- 3.Xu, H. & Berne, B. J. (1999) J. Chem. Phys. 110, 10299–10306. [Google Scholar]
- 4.Marinari, E. & Parisi, G. (1992) Europhys. Lett. 19, 451–458. [Google Scholar]
- 5.Lyubartsev, A. P., Martinovski, A. A., Shevkunov, S. V. & Vorontsov-Velyaminov, P. N. (1992) J. Chem. Phys. 96, 1776–1783. [Google Scholar]
- 6.Stolovitzky, G. & Berne, B. J. (2000) Proc. Natl. Acad. Sci. USA 97, 11164–11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swendsen, R. H. & Wang, J. S. (1986) Phys. Rev. Lett. 57, 2607–2609. [DOI] [PubMed] [Google Scholar]
- 8.Hukushima, K. & Nemoto, K. (1996) J. Phys. Soc. Jpn. 65, 1604–1608. [Google Scholar]
- 9.Tesi, M. C., van Rensburg, E. J. J., Orlandini, E. & Whittington, S. G. (1996) J. Stat. Phys. 82, 155–181. [Google Scholar]
- 10.Okamoto, Y. (2004) J. Mol. Graphics Model. 22, 425–439. [DOI] [PubMed] [Google Scholar]
- 11.Paschek, D. & Garcia, A. E. (2004) Phys. Rev. Lett. 93, 238105. [DOI] [PubMed] [Google Scholar]
- 12.Fukunishi, H., Watanabe, O. & Takada, S. (2002) J. Chem. Phys. 116, 9058–9067. [Google Scholar]
- 13.Zhou, R. & Berne, B. (2002) Proc. Natl. Acad. Sci. USA 99, 12777–12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang, S., Shin, S. & Pak, Y. (2003) Phys. Rev. Lett. 91, 058305. [DOI] [PubMed] [Google Scholar]
- 15.Berne, B. J. & Straub, J. E. (1997) Curr. Opin. Struct. Biol. 7, 181–189. [DOI] [PubMed] [Google Scholar]
- 16.Cheng, X., Cui, G., Hornak, V. & Simmerling, C. (2005) J. Phys. Chem. B 109, 8220–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) J. Chem. Phys. 79, 926–935. [Google Scholar]
- 18.Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. (1996) J. Am. Chem. Soc. 118, 11225–11236. [Google Scholar]
- 19.Kaminski, G. A., Friesner, R. A., Tirado-Rives, J. & Jorgensen, W. L. (2001) J. Phys. Chem. B 105, 6474–6487. [Google Scholar]
- 20.Hockney, R. W. & Eastwood, J. W. (1988) Computer Simulations Using Particles (Institute of Physics, Bristol, U.K.).
- 21.Deserno, M. & Holm, C. (1998) J. Chem. Phys. 109, 7678–7693. [Google Scholar]
- 22.Zhou, R., Harder, E., Xu, H. & Berne, B. J. (2001) J. Chem. Phys. 115, 2348–2358. [Google Scholar]
- 23.Andersen, H. C. (1983) J. Comput. Phys. 52, 24–34. [Google Scholar]
- 24.Martyna, G., Klein, M. & Tuckerman, M. (1992) J. Chem. Phys. 97, 2635–2643. [Google Scholar]
- 25.Woutersen, S., Pfister, R., Hamm, P., Mu, Y., Kosov, D. S. & Stock, G. (2002) J. Chem. Phys. 117, 6833–6840. [Google Scholar]
- 26.Schweitzer-Stenner, R. (2002) Biophys. J. 83, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thirumalai, D., Mountain, R. D. & Kirkpatrick, T. R. (1989) Phys. Rev. A 39, 3563–3574. [DOI] [PubMed] [Google Scholar]
- 28.Whitfield, T., Bu, L. & Straub, J. (2002) Physica A 305, 157–171. [Google Scholar]