

Abstract
The European Heart Rhythm Association Practical Compendium of Anti-arrhythmic Drugs (AADs) offers advice on these drugs, focusing on their clinical use and the global impact of cardiac arrhythmias. This document aims to provide practical instructions to clinicians in arrhythmia management through pharmacological strategies. The compendium highlights persistent challenges in arrhythmia treatment, including clinical constraints, procedural risks, and the complexity of certain arrhythmias. Notably, atrial fibrillation is highly prevalent, and the demand for invasive treatment often surpasses the capacity of existing healthcare systems. As a result, pharmacological management remains essential. This is particularly relevant for patients with cardiac implantable electronic devices or channelopathies, where ablation is often not a suitable option. Anti-arrhythmic drugs play a pivotal role in these scenarios. The compendium introduces the ABC framework for AAD therapy: A (Appropriate therapy), for patients in whom AADs are the best therapeutic option; B (Backup therapy), as adjunctive treatment to invasive procedures, such as catheter ablation; and C (Complementary therapy), in combination with other therapies. The document provides detailed insights into the mechanisms of action, efficacy, safety profiles, and drug interactions of each class of AADs. Additionally, the compendium covers practical considerations, including initiation, combination strategies, monitoring, follow-up, special populations, and adverse effect management, with an emphasis on pro-arrhythmia risk mitigation. It also explores the integration of AADs with other therapeutic modalities, promoting a synergistic approach to optimize patient outcomes. In summary, this compendium serves as an indispensable resource for clinicians, offering practical advice and evidence-based insights to navigate the complexities of arrhythmia management effectively.
Keywords: Adverse drug reactions, Anti-arrhythmic drugs, Anti-arrhythmic drug combinations, Arrhythmia, Atrial fibrillation, Mechanisms, Pharmacology, Drug interactions, Ventricular arrhythmias
Graphical Abstract
Graphical Abstract.

Table of contents
1 Introduction
2 Summary of advice
3 Definition and principles of anti-arrhythmic drug mechanisms
3.1 Mechanism of action of anti-arrhythmic drugs
3.2 Ion channel kinetics in cardiomyocyte membranes: fundamental states and use dependence effects of anti-arrhythmic drugs
3.2.1 Fundamental states of ion channels
3.2.2 Ion channel kinetics in cardiomyocyte membranes
3.2.3 Use dependence and reverse use dependence
3.2.4 Anti-arrhythmic drug binding kinetics
3.3 Cardiac and systemic specificities of anti-arrhythmic drugs
3.4 Pharmacokinetics of anti-arrhythmic drugs
3.4.1 Intestinal absorption
3.4.2 First-pass hepatic metabolism
3.4.3 Distribution
3.4.4 Renal and hepatic excretion
3.5 Genetics and anti-arrhythmic drugs
3.6 Classification of anti-arrhythmic drugs
3.6.1 Class 0
3.6.1.1 Ivabradine
3.6.2 Class Ia
3.6.2.1 Quinidine
3.6.2.2 Disopyramide and ajmaline
3.6.2.3 Procainamide
3.6.3 Class Ib
3.6.3.1 Lidocaine
3.6.3.2 Mexiletine
3.6.3.3 Phenytoin
3.6.4 Class Ic
3.6.4.1 Flecainide and propafenone
3.6.4.2 Other: cibenzoline, pilsicainide, and antazoline
3.6.5 Class Id
3.6.5.1 Ranolazine
3.6.6 Class III
3.6.7 Class IIIa
3.6.7.1 Amiodarone
3.6.7.2 Dronedarone
3.6.7.3 Sotalol
3.6.7.4 Dofetilide
3.6.7.5 Ibutilide
3.6.7.6 Vernakalant
3.6.8 Class IIIb
3.6.8.1 Nicorandil
3.6.9 Class IIa
3.6.9.1 Bisoprolol, metoprolol, carvedilol, nadolol, and propranolol
3.6.9.2 Other (nebivolol, esmolol, and landiolol)
3.6.10 Class IIb
3.6.10.1 Isoprenaline (isoproterenol)
3.6.11 Class IIc
3.6.11.1 Atropine
3.6.12 Class IId
3.6.12.1 Digoxin
3.6.13 Class IIe
3.6.13.1 Adenosine
3.6.14 Class IV
3.6.14.1 Verapamil and diltiazem
3.6.14.2 Bepridil
4 Treatment by arrhythmia
4.1 General
4.2 Arrhythmia prevention
4.2.1 Atrial arrhythmias
4.2.1.1 Premature atrial contractions and focal atrial tachycardia
4.2.1.2 Inappropriate sinus tachycardia
4.2.1.3 Multi-focal atrial tachycardia
4.2.1.4 Atrial flutter/macro re-entrant atrial tachycardia
4.2.1.5 Atrial fibrillation
4.2.1.6 Atrial fibrillation after cardiac surgery
4.2.1.7 Autonomic atrial fibrillation
4.2.1.8 Aberrant conduction vs. ventricular proarrhythmia
4.2.1.9 Anti-arrhythmic drugs for atrial fibrillation
4.2.2 Paroxysmal supraventricular tachycardias
4.2.2.1 Atrioventricular nodal re-entrant tachycardia
4.2.2.2 Atrioventricular re-entrant tachycardia
4.2.3 Ventricular arrhythmias
4.2.3.1 Idiopathic premature ventricular contractions and ventricular tachycardia
4.2.3.2 Premature ventricular contractions and structural heart disease
4.2.3.3 Ventricular tachycardia and structural heart disease
4.2.3.4 Ventricular fibrillation
4.3 Tachycardia termination
4.3.1 Atrial fibrillation (oral–pill-in-the-pocket)
4.3.2 Atrial fibrillation (intravenous)
4.3.3 Atrial flutter
4.3.4 Paroxysmal supraventricular tachycardia
4.3.5 Focal atrial tachycardia
4.3.6 Junctional ectopic tachycardia
4.3.7 Ventricular tachycardia–non-structural heart disease
4.3.8 Ventricular tachycardia–structural heart disease
4.3.9 Polymorphic ventricular tachycardia and ventricular fibrillation
5 Practical aspects
5.1 Initiation of anti-arrhythmic drug
5.2 Follow-up and monitoring of patients on anti-arrhythmic drugs
5.3 Electrocardiogram anti-arrhythmic drug effects
5.4 Anti-arrhythmic drug tests for electrophysiological evaluation
5.5 Pro-arrhythmia
5.5.1 Sinus bradycardia and arrest
5.5.2 Atrioventricular block
5.5.3 New-onset, sustained, monomorphic ventricular tachycardia
5.5.4 Increased frequency of sustained ventricular tachycardia
5.5.5 Incessant ventricular tachycardia
5.5.6 Torsades de pointes
5.5.7 Atrial pro-arrhythmia
5.5.8 Brugada mechanism
5.6 Toxicity and adverse effects
5.6.1 Amiodarone-induced thyroid dysfunction
5.6.2 Amiodarone-induced pulmonary and other systemic toxicities
5.6.3 Quinidine systemic toxicities
5.6.4 Other anti-arrhythmic drug systemic toxicities
5.7 Pro-arrhythmia and anti-arrhythmic drug toxicity management
5.7.1 General aspects
5.7.2 Torsades de pointes management
5.7.3 Drug-specific aspects
5.8 Contraindications and precautions
5.8.1 Flecainide
5.8.2 Propafenone
5.8.3 Amiodarone
5.8.4 Dronedarone
5.8.5 Sotalol and dofetilide
5.8.6 Verapamil and diltiazem
5.9 Anti-arrhythmic drug plasma concentration
5.10 Drug–drug interactions
5.10.1 Anti-arrhythmic drug–drug interactions
5.10.2 Drug–herb and drug–food interactions involving antiarrhythmic drugs
5.11 Anti-arrhythmic drug switch and combinations
6 Anti-arrhythmic drug in special situations
6.1 Pregnancy
6.2 Children
6.3 Foetal arrhythmias
6.4 Elderly
6.5 Athletes
6.6 Heart failure
6.6.1 Reduced ejection fraction
6.6.2 Preserved ejection fraction
6.7 Cardiomyopathies
6.7.1 Hypertrophic cardiomyopathy
6.7.2 Arrhythmogenic right ventricular cardiomyopathy
6.8 Renal and liver failure
6.9 Congenital heart disease
6.10 Channelopathies
6.10.1 Long QT and short QT syndromes
6.10.2 Brugada syndrome
6.10.3 Catecholaminergic polymorphic ventricular tachycardia
6.10.4 Early repolarization syndrome
6.11 Anticoagulation
6.12 Anti-arrhythmic drug and non-pharmacological antiarrhythmic therapies
6.12.1 Anti-arrhythmic drugs and pacemakers
6.12.2 Anti-arrhythmic drugs in patients with implantable cardioverter defibrillators
6.12.3 Anti-arrhythmic drugs following ablation therapy
6.12.4 Anti-arrhythmic drugs: effects on direct current cardioversion and defibrillation
7 Anti-arrhythmic drugs under development
7.1.1 Etripamil
7.1.2 Inhaled flecainide
7.1.3 Small-conductance calcium-activated potassium channel inhibitors
7.1.4 Sulcardine (HBI-3000)
7.1.5 Doxapram
7.1.6 Bucindolol
7.1.7 Budiodarone
7.1.8 Histone deacetylase 6 inhibitors
8 Areas of uncertainty and gaps of knowledge
9 Conclusions
10 Tables of advice
Supplementary material
Funding
Data availability
Abbreviations and acronyms
- AADs
Anti-arrhythmic drugs
- ACC
American College of Cardiology
- ACE
Angiotensin-converting enzyme
- ACS
Acute coronary syndrome
- AF
Atrial fibrillation
- AFL
Atrial flutter
- AHA
American Heart Association
- AIH
Amiodarone-induced hypo-thyroidism
- AIT
Amiodarone-induced thyrotoxicosis
- AIT 1
Type 1 amiodarone-induced thyrotoxicosis
- AIT 2
Type 2 amiodarone-induced thyrotoxicosis
- cAMP
Cyclic adenosine monophosphate
- AP
Action potential
- APD
Action potential duration
- APD90
Action potential duration at 90% repolarization
- Arg
Arginine
- ARVC
Arrhythmogenic right ventricular cardiomyopathy
- AT
Atrial tachycardia
- ATP
Adenosine triphosphate
- AV
Atrioventricular
- AVNRT
Atrioventricular nodal re-entrant tachycardia
- AVRT
Atrioventricular re-entrant tachycardia
- β-Blocker
Beta-blocker
- BBB
Bundle branch block
- BrS
Brugada syndrome
- Ca2+
Calcium
- CA
Cardiac arrest
- CAD
Coronary artery disease
- Cav
Calcium channel
- CCB
Calcium channel blocker
- CI
Confidence interval
- CNS
Central nervous system
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- CrCl
Creatinine clearance
- CVD
Cardiovascular disease
- CYP
Cytochrome P450
- DADs
Delayed afterdepolarizations
- DC
Direct current
- DFT
Defibrillation threshold
- DOAC
Direct oral anticoagulant
- EADs
Early afterdepolarizations
- ECG
Electrocardiogram
- EMA
European Medicines Agency
- EP
Electrophysiology
- ERP
Effective refractory period
- ERS
Early repolarization syndrome
- ES
Electrical storm
- ESC
European Society of Cardiology
- FDA
Food and Drug Administration
- GDMT
Guideline-directed medical therapies
- Gly
Glycine
- GMP
Guanosine monophosphate
- GTP
Guanosine triphosphate
- HCM
Hypertrophic cardiomyopathy
- HCN
Hyperpolarization and cyclic nucleotide gated
- HDAC6
Histone deacetylase 6
- hERG
Human ether-a-go-go-related gene
- HF
Heart failure
- HFpEF
Heart failure preserved ejection fraction
- HFrEF
Heart failure reduced ejection fraction
- HR
Hazard ratio
- HRS
Heart Rhythm Society
- IAST
Inappropriate sinus tachycardia
- ICD
Implantable cardioverter defibrillator
- ICa,L
L-type calcium current
- If
Funny current
- IK,ACh
Acetylcholine-activated potassium current
- IKr
Rapid delayed rectifier potassium current
- IKs
Slow delayed rectifier potassium current
- INa
Sodium current
- INa,L
Late sodium current
- INa,P
Peak sodium current
- Ito
Transient outward potassium current
- i.v.
Intravenous
- IVF
Idiopathic ventricular fibrillation
- JET
Junctional ectopic tachycardia
- K+
Potassium
- KATP
ATP-dependent potassium
- Kv
Potassium channel
- LBBB
Left bundle branch block
- LQTS
Long QT syndrome
- LVEF
Left ventricular ejection fraction
- LVH
Left ventricular hypertrophy
- MI
Myocardial infarction
- Na+
Sodium
- Nav
Sodium channel
- NO
Nitric oxide
- NSAT
Non-sustained atrial tachycardia
- NYHA
New York Heart Association
- P
P-value
- PFTs
Pulmonary function tests
- P-gp
P-glycoprotein
- PAC
Premature atrial contraction
- PD
Pharmacodynamics
- PITP
Pill-in-the-pocket
- PK
Pharmacokinetics
- PKA
Protein kinase A
- PM
Pacemaker
- PO
Per os, oral
- PSVT
Paroxysmal supraventricular tachycardia
- PV
Pulmonary veins
- PVC
Premature ventricular contraction
- PVT
Polymorphic ventricular tachycardia
- QRS
QRS complex
- QT
QT interval
- QTc
Corrected QT interval
- RMP
Resting membrane potential
- RyR2
Ryanodine receptor 2
- RVOT
Right ventricular outflow tract
- SA
Sinoatrial
- SERCA2a
Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a
- SCD
Sudden cardiac death
- SCN5A
Sodium channel protein type 5 sub-unit alpha
- SGLT2i
Sodium–glucose co-transporter-2 inhibitors
- SHD
Structural heart disease
- SK or KCa2
Small-conductance calcium-activated potassium channel
- SN
Sinus node
- SND
Sinus node dysfunction
- SQTS
Short QT syndrome
- SR
Sinus rhythm
- ST
ST-segment
- SVT
Supraventricular tachycardia
- T2DM
Type 2 diabetes mellitus
- T3
Triiodothyronine
- T4
Thyroxine
- TASK1 or K2P3.1
TWIK-related acid-sensitive potassium channel 1
- TdP
Torsade de pointes
- TSH
Thyroid-stimulating hormone
- VA
Ventricular arrhythmia
- VF
Ventricular fibrillation
- VT
Ventricular tachycardia
- VW
Vaughan Williams
- WPW
Wolff–Parkinson–White
Note: Supplementary material online, Table S1 provides a list of acronyms and summarized findings of the main trials on AADs.
What's new?
Introduction of the ABC framework: A novel conceptual model organizes AAD use into Appropriate, Backup, and Complementary therapies. This strategic framing helps clinicians contextualize pharmacological choices in relation to procedural options like ablation or device therapy, promoting a practical, tiered approach to rhythm control.
Emphasis on practical use beyond classical guidelines: Unlike prior documents, this compendium places high importance on real-world, scenario-based use of anti-arrhythmic drugs (AADs)—including drug initiation protocols, patient-specific adjustments, and drug switching strategies—especially where invasive procedures are not feasible or effective.
Expanded drug classifications and updates on emerging agents: It incorporates, in a simplified manner, modern updates to the Vaughan Williams (VW) classification, including the Oxford 2018 expansion (e.g. Class 0 for ivabradine) and introduces new and upcoming agents like small-conductance calcium-activated potassium channel inhibitors, etripamil, and budiodarone, positioning the document at the cutting edge of clinical pharmacology.
Focus on integration with other therapies: The document provides in-depth advice on how to synchronize AAD use with other interventions—including ablation, anticoagulation, device therapy, and even electrical cardioversion—offering a more integrative and patient-specific treatment model than prior texts.
1. Introduction
Cardiac arrhythmias significantly impact global health. A definitive cure by invasive procedures has been pursued in the last decades. However, despite advances in invasive management by catheter ablation, challenges remain, such as anatomical limitations, procedural risks, and complex arrhythmias. In addition, the prevalence of some arrhythmic disorders limits the generalizability of invasive arrhythmia management. For example, atrial fibrillation (AF), the most common sustained arrhythmia, affects 1–2% of the population. Presently, the demand for its invasive treatment commonly surpasses healthcare system capacity. In developed countries, only about 1% of AF patients currently receive ablation, with projections of it reaching only 10% in the foreseeable future due to limited resources and personnel.
Moreover, pharmacological management remains crucial for certain patients, either due to ablation failure or as part of peri-procedural care. Anti-arrhythmic drugs (AADs) are continued in ∼50% of patients following index ablation, while one in six undergoes repeat ablation, with most receiving concomitant AAD therapy thereafter. These findings highlight that, in current clinical practice, rhythm control often relies on a combined approach integrating catheter ablation and AADs.1 This is also especially relevant for patients with cardiac implantable electronic devices (CIEDs), who experience recurrent arrhythmias, where AADs play a critical role in prevention. Conditions such as channelopathies, which are often unsuitable for ablative therapy, also necessitate the use of AADs. Additionally, the acute management of arrhythmias in emergency settings underscores the crucial role of these medications. The current indications for AADs can be summarized by the acronym ABC, as shown in Box 1.
Box 1 ABC indications for the current use of AAD.
Appropriate therapy: AADs are often the appropriate and, in many cases, the sole therapy required for managing cardiac arrhythmias, including terminating arrhythmias during their initial presentation, addressing acute or incessant episodes, and treating patients who respond well to pharmacological treatment and prefer it over invasive procedures
Backup therapy: AADs are used as a backup therapy when other primary treatments, such as ablation or CIEDs, are unavailable, poorly tolerated, particularly risky, contraindicated, or ineffective in preventing or terminating arrhythmia episodes or their consequences
Complementary therapy: AADs serve as a valuable complement to other therapies, such as catheter ablation or CIEDs, by providing support during waiting periods, preparatory or post-operative phases, or by supplementing and enhancing their overall efficacy
Abbreviations: AAD, anti-arrhythmic drug; CIED, cardiac implantable electronic device.
Given these complexities, there is a clear need for appropriate, backup, and complementary strategies, placing AADs at the forefront as essential components in managing arrhythmias. To address this, the European Heart Rhythm Association (EHRA) gathered international experts to create a practical compendium on AAD use, overseen by two chairs. The chairs planned the outline of the compendium, and each expert was tasked with reviewing the medical literature of a specific section. These reviews were later discussed by the entire group, and the final text underwent an external review by an independent group of experts.
This practical compendium systematically navigates the intricate landscape of AADs, elucidating their mechanisms of action, efficacy, and safety profiles within the general population of patients with arrhythmias. Special attention is directed towards sub-populations with specific arrhythmia mechanisms or characteristics that may influence AAD efficacy and safety. The compendium aims to provide clinicians with a comprehensive understanding of these mechanisms, empowering them to make informed decisions in the complex arena of cardiac arrhythmias. Furthermore, this compendium offers practical advice, providing insights into the judicious integration of these drugs into clinical practice. It highlights how AADs interact with other treatments like cardiovascular drugs, ablation, electrical cardioversion, and implantable devices. This unveils a synergistic approach that optimizes patient outcomes, ensuring a holistic and evidence-based strategy for rhythm management.
2. Summary of advice
The EHRA Practical Compendium of AADs offers detailed advice on the usage, monitoring, and management of these medications in clinical practice. Key advice from the document includes the following:
-
Initiation of AADs
In-hospital initiation is preferred for Class Ia AADs and some Class III drugs. Outpatient initiation with appropriate monitoring in patients without structural heart disease (SHD) is suitable for Class Ic agents, amiodarone, dronedarone, and ranolazine.
-
Monitoring and follow-up
Regular electrocardiography (ECG) monitoring is advised, especially in the first hours of AAD use, to detect rhythm disturbances, particularly with Class Ia and some Class III drugs.
Baseline and routine assessments, for example, visual, thyroid, liver, and pulmonary function tests (PFTs), are advised for amiodarone.
-
Pro-arrhythmia risk management
There is increasing awareness of pro-arrhythmic risks, particularly with Class I and III drugs. Monitoring for QT interval (QT) prolongation and avoiding concomitant use of QT-prolonging agents are essential.
It is important to educate patients about warning symptoms such as worsening palpitations, dizziness, or chest pain and to provide guidance on lifestyle modifications to help avoid triggers, such as electrolyte imbalance.
-
Special populations
Specific advice is provided for the use of AADs in patients with SHD, pregnant women, and paediatric patients. For instance, β-blockers are preferred during pregnancy, while it is advised to avoid some drugs like amiodarone and dronedarone due to potential foetal harm.
-
Combination therapy
Specific combinations, such as sotalol with flecainide or amiodarone with β-blockers, may be appropriate for resilient cases with careful monitoring of drug effects.
Combining AADs with other therapies such as ablation or CIEDs is advised to enhance efficacy and manage complex cases.
-
Patient involvement and education
Engaging patients in their treatment plan by educating them about the potential side effects and importance of adherence to therapy.
It is advised to integrate nurses and other healthcare professionals into the care team to support the safe administration and monitoring of AADs.
Overall, the compendium emphasises a tailored approach to AAD therapy, considering individual patient characteristics, underlying conditions, and potential risks to optimize outcomes in arrhythmia management.
3. Definition and principles of anti-arrhythmic drug mechanisms
Anti-arrhythmic drugs are pharmacological agents designed to prevent or correct cardiac arrhythmias by modulating the heart’s electrical activity. This section explores their mechanisms of action, including their effects on ion channels, tissue specificity, and pharmacokinetics (PK), while also examining the role of genetics in influencing their efficacy and safety.
3.1. Mechanism of action of anti-arrhythmic drugs
Arrhythmias primarily manifest through three key mechanisms: automatism, triggered focal activity due to early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs), and re-entry (Supplementary material online, Figure S1). Among these, re-entry stands out as the most prevalent. This latter mechanism hinges on three main determinants that are crucial for its manifestation. First, a trigger is essential to initiate the re-entrant electrical activity. This trigger could be an ectopic beat originating from a specific heart location not necessarily linked to the re-entrant circuit. Second, a re-entrant circuit is necessary, representing a pathway that allows the electrical impulse to circulate within the heart tissue, perpetuating the abnormal rhythm. Re-entry within the circuit is promoted by shortened refractoriness, slowed conduction (or a combination of the two), and unidirectional block. Lastly, the overall autonomic status plays a significant role in modulating the susceptibility to re-entry mechanisms. The inter-play of sympathetic and parasympathetic influences on the heart’s electrical properties can either enhance or mitigate the likelihood of arrhythmic events. Knowledge of these fundamental mechanisms and their interdependencies is paramount to understanding the effect of AADs. It forms the basis for targeted interventions and tailored therapeutic strategies aimed at addressing the specific mechanisms underlying each patient’s arrhythmic presentation. However, a comprehensive review of them is beyond the scope of this practical compendium.2–4
Anti-arrhythmic drugs exert their anti-arrhythmic effect by modulating the electrophysiological determinants of automaticity, triggered activity and re-entry. Class I AADs (see the section ‘3.6 Classification of AADs’ below) block cardiac Na+ channels (Nav), reducing myocardial excitability and decreasing the likelihood of ectopic (triggered) activity.5 They may also extend the effective refractory period (ERP) by delaying cardiomyocyte recovery after repolarization, known as post-repolarization refractoriness. Some Class I AADs additionally prolong ERP through inhibition of rapid delayed rectifier potassium current (IKr) and other repolarization currents, causing action potential duration (APD) prolongation. Inhibition of IKr that leads to APD prolongation is also the primary mechanism of action of Class III AADs.5 At the same time, the prolonged ERP will reduce the likelihood that triggering events encounter excitable tissue to initiate arrhythmias, decreasing the vulnerable substrate, thus explaining the role of these AADs in secondary prevention of both atrial and ventricular arrhythmias (VAs). Class III AADs work mainly by inhibiting IKr, which prolongs APD. This extends ERP, making re-entry less stable and reducing the chance of persistent arrhythmias, justifying the use of Class I and III drugs for cardioversion.
Class II AADs have numerous indirect electrophysiological effects by reducing the β-adrenoceptor-dependent phosphorylation of numerous ion channels, Ca2+-handling, and myofilament proteins. The resulting reduction in ryanodine receptor 2 (RyR2) activity together with a smaller L-type calcium current (ICa,L) decreases the likelihood of DADs and EADs and thus the likelihood of ectopic (triggered) activity.6 Moreover, inhibition of β-adrenoceptor-mediated regulation of hyperpolarization and cyclic nucleotide-gated (HCN) channels and L-type Ca2+ channels reduces automaticity in sinoatrial (SA) cells, providing a rationale for the use of Class II AADs for sinus tachycardia.6 Similarly, inhibition of L-type Ca2+ channels, either indirectly by Class II AADs or directly by Class IV AADs, slows atrioventricular (AV) conduction, providing control of ventricular rate in atrial arrhythmias. Finally, the reduction in intra-cellular Ca2+ cycling due to ICa,L inhibition, which underlies the negative inotropic effects of Class II and Class IV AADs, is also expected to reduce the likelihood of DADs. Thus, the primary mechanisms of action of AADs are inhibition of ectopic (triggered) activity (primarily Class I and II AADs), reduction of the likelihood of re-entry (primarily Class I and III AADs), and modulation of impulse generation and conduction by the SA and AV nodes (primarily Class II and IV AADs).
3.2. Ion channel kinetics in cardiomyocyte membranes: fundamental states and use dependence effects of anti-arrhythmic drugs
Ion channels in cardiomyocyte membranes are essential for regulating cardiac action potentials and overall heart function. The kinetics of these ion channels and their fundamental states, along with the phenomena of use dependence and reverse use dependence, are key to understanding cardiac electrophysiology (EP) and the effects of AADs.
3.2.1. Fundamental states of ion channels
Ion channels in cardiomyocytes typically exist in three primary states: resting (closed), activated (open), and inactivated closed (Figure 1).7
Figure 1.

Schematic representation of the three main states (resting, activated, and inactivated) of an ionic channel in the cellular surface membrane of a cardiomyocyte. During the resting phase (left panel), the influx of ions into the cell is not possible (short arrow) because the channel remains closed (horizontal rectangles). Once the channel is activated (central panel), ions can enter the cell (long arrow) through the open channel (small oblique rectangles). Following activation, the channel transitions to an inactivated state (right panel, inferior horizontal rectangle), preventing further ion influx. Different anti-arrhythmic drugs (AADs) (e.g. flecainide) exhibit specific affinity and preferentially bind to particular states of the channel (e.g. the activated state).
Resting state: In the resting state, ion channels are closed, preventing ion flow across the membrane. This state is crucial for maintaining the resting membrane potential (RMP) of the cardiomyocyte.
Activated state: Upon depolarization, ion channels transition from the resting state to the activated state. In this state, the channels are open, allowing the influx or efflux of specific ions, which contributes to the rapid depolarization phase of the AP. For instance, the rapid influx of Na+ through voltage-gated Na+ channels is essential for the initial upstroke of the AP in atrial and ventricular cells, while the slow influx of Ca2+ through L-type voltage-gated channels is essential for the initial upstroke of the action potential in SA and AV nodal cells.
Inactivated state: Following activation, ion channels enter the inactivated state, during which they are closed but not capable of opening again immediately. This inactivation is vital for the refractory period, ensuring that the cell cannot be prematurely re-excited and facilitating a normal cardiac rhythm. After cellular repolarization, inactivated channels return to the resting state, making them ready for reactivation by a new stimulus. The movement from the inactivated to the resting state is termed channel reactivation.
3.2.2. Ion channel kinetics in cardiomyocyte membranes
Ion channel kinetics refer to the rates at which ion channels transition between their fundamental kinetic states: resting (closed), activated (open), and inactivated (closed but unresponsive to immediate reopening). These transitions can occur rapidly or slowly, depending on the type of ion channel and its physiological role.
Fast kinetics:
Sodium channels (Nav): Voltage-gated Na+ channel exhibit fast kinetics, with rapid transitions between states. Upon depolarization, these channels quickly move from the resting to the activated state, allowing a swift influx of Na+ ions, which is crucial for the rapid upstroke of the cardiac AP. The inactivation of Na+ channel also occurs quickly. Drugs with slow binding kinetics (e.g. Class Ic agents) accumulate within the channel during tachycardia, prolonging QRS complex (QRS) duration due to their persistent Na⁺ blockade, whereas those with fast binding kinetics (e.g. Class Ib agents) dissociate quickly, limiting their effects at normal heart rates.
(2) Slow kinetics:
Calcium channels (Cav): Voltage-gated Ca2+ channels, particularly L-type Ca2+ channels, display slower kinetics. These channels open more gradually in response to depolarization, allowing a sustained influx of Ca2+ ions. This prolonged entry of Ca2+ is vital for the plateau phase of the cardiac AP and is instrumental in triggering Ca2+-induced calcium release from the sarcoplasmic reticulum, leading to muscle contraction.
Potassium channels (Kv): Some K+ channels, like the delayed rectifier K+ channel, also exhibit slow kinetics. They gradually activate and contribute to the repolarization phase of the AP, restoring the RMP.
3.2.3. Use dependence and reverse use dependence
Anti-arrhythmic drugs interact with ion channels in different states, depending on the frequency of cardiac action potentials, resulting in use dependence and reverse use dependence (Figure 2).8
Figure 2.

Schematic representation of the effects of flecainide (A and B) and sotalol (C) on the transmembrane action potential during sinus rhythm (SR) (A), atrial fibrillation (AF) (B), and sinus bradycardia (C). The figure also illustrates their potential anti-arrhythmic and pro-arrhythmic effects on AF (ECG in panel B) and sinus bradycardia (ECG in panel C), respectively. Flecainide (green polygon) binds to the sodium channel (Na+ Ch) primarily in its activated (slightly separated red rectangles) and inactivated (closely aligned grey rectangles) states. Its maximal effect is observed during tachycardia, as the shortened action potential duration keeps the sodium channel in these states more frequently. This use-dependent property enables flecainide to effectively block the activation front, contributing to the termination of atrial fibrillation (AF). Additionally, its very slow dissociation kinetics and strong binding to the inactivated state play a crucial role in prolonging post-repolarization refractoriness—a key mechanism underlying its anti-arrhythmic efficacy but also a potential contributor to pro-arrhythmia. In contrast, sotalol (red polygon) binds to several potassium channels (K+Ch) mostly during its resting state (closely aligned blue rectangles). Its maximum effect occurs in bradycardia, where the channel remains in this state for a longer duration. This reverse use-dependent effect leads to prolonged action potential duration and QT interval prolongation, which can trigger early afterdepolarizations (EADs) and ventricular tachycardia, including torsades de pointes (TdP). Downward curved arrows represent anti-arrhythmic drug (AAD) binding to the ion channel, while upward curved arrows indicate the absence of binding.
Use dependence: Use dependence refers to the increased blocking effect of certain AADs on ion channels with increased frequency of APs. This is often observed with Class I AADs, such as flecainide, which block Na+ more effectively at higher heart rates (Figure 2B). The mechanism involves the drug preferentially binding to the activated and/or inactivated states of the channel, which are more prevalent at higher rates of depolarization. Consequently, the therapeutic effect of the drug is enhanced during tachycardia, providing a targeted approach to suppressing tachyarrhythmias. Use-dependent effects are less pronounced for slow-kinetic channels since their activation is not significantly increased by higher heart rates.
Reverse use dependence: In contrast, reverse use dependence describes the phenomenon where the effectiveness of a drug is greater at lower heart rates (Figure 2C). This is the case with some Class III AADs, such as sotalol, which block K+ channels. The binding affinity of these drugs to the ion channel is enhanced during the resting state, particularly at slower heart rates. Consequently, the drug exerts a more pronounced effect on prolonging the AP duration and refractory period during bradycardia. While this mechanism can aid in maintaining sinus rhythm (SR) and preventing arrhythmias, it also raises the potential risk of pro-arrhythmia, especially at slower heart rates.
3.2.4. Anti-arrhythmic drug binding kinetics
The effectiveness of AADs depends on their binding kinetics, which determine how quickly they attach to and dissociate from ion channels. Drugs like flecainide (Class Ic) have slow-on, slow-off kinetics, leading to cumulative Nav blockade at higher heart rates, prolonging QRS duration. In contrast, lidocaine (Class IB) binds and dissociates quickly (fast-on, fast-off), minimizing effects on conduction at normal heart rates.
For slow-kinetic K+ channels, such as IKr (the rapid component of the delayed rectifier K+ current), different Class III AADs exhibit distinct binding kinetics, which influence their clinical effects. Ibutilide, for instance, has very fast kinetics (rapid-on, moderate-off), making it effective for acute AF termination due to its use-dependent effect. In contrast, dofetilide and sotalol exhibit fast-on but slow-off kinetics, meaning their blocking effect is stronger at slower heart rates, leading to reverse use dependence, where QT prolongation becomes more pronounced with longer diastolic pauses.
Conversely, amiodarone and dronedarone display very slow binding kinetics (slow-on, very slow-off for amiodarone; slow-on, slow-off for dronedarone), resulting in weaker reverse use dependence. Additionally, their multi-channel blocking effects (IKr, IKs, INa, ICa, and β-blockade) further reduce the risk of bradycardia-induced pro-arrhythmia, making them safer options for patients with low heart rates.
Ultimately, the interaction between AAD binding affinity, channel kinetics, and heart rate dependence influences drug efficacy and pro-arrhythmic risk, highlighting the need for tailored anti-arrhythmic therapy.
3.3. Cardiac and systemic specificities of anti-arrhythmic drugs
Anti-arrhythmic drugs exert distinct effects on different regions of cardiac tissue. Class II and IV agents primarily slow conduction and prolong refractoriness in the sinus and AV nodes, while Class I and III agents predominantly affect the working myocardium at both atrial and ventricular levels. Another difference is the degree of ventricular myocardial contractility depression, which is most pronounced with Class Ic and Class IV agents but less significant with quinidine or oral amiodarone9 (i.v. amiodarone can cause acute hypotension and myocardial depression, mainly due to its solvent polysorbate 80 and benzyl alcohol). Recognizing these region-specific effects is essential for selecting the appropriate drug for each patient.
3.4. Pharmacokinetics of anti-arrhythmic drugs
Anti-arrhythmic drugs often exhibit a narrow therapeutic window, underscoring the critical role of PK in optimizing their efficacy and minimizing safety risks. A comprehensive understanding of their absorption, metabolism, distribution, and excretion processes enables clinicians to tailor therapies effectively and mitigate adverse effects (Figure 3).
Figure 3.

Schematic representation of intestinal absorption, tissue storage, and hepatic and renal excretion pathways for commonly affected anti-arrhythmic drugs (AADs). Box A: Intestinal absorption occurs through epithelial cells (enterocytes) of the small intestine. However, P-glycoprotein (P-gp) in enterocytes actively effluxes a portion of certain drugs back into the intestinal lumen, reducing systemic absorption. Box B: Lipophilic drugs tend to accumulate in fat-rich tissues and organs, such as the lungs, liver, thyroid, and adipose tissue (primary tissues of accumulation listed in brackets). Box C: Hydrophilic drugs exhibit minimal or no tissue accumulation and distribute predominantly in the extracellular fluid,—except for digoxin, which primarily accumulates in cardiac muscle. Box D: Drugs are metabolized by the liver and excreted via bile into faeces. Box E: Renal clearance eliminates drugs or their metabolites through the kidneys. Approximate percentages of drug efflux and elimination are indicated in the respective boxes. Flecainide is partially eliminated by the kidneys (~30–40%), and impaired renal function can lead to drug accumulation and increased risk of proarrhythmia. CCB, calcium channel blocker.
3.4.1. Intestinal absorption
Orally administered AADs rely on efficient intestinal absorption to achieve therapeutic plasma concentrations. Factors such as gastrointestinal pH, motility, and the presence of food can significantly influence drug absorption. For instance, the absorption of short-acting β-blockers like propranolol is enhanced when taken with food, attributed to delayed gastric emptying and prolonged intestinal transit time. Similarly, Class I agents, including flecainide and propafenone, depend on optimal gastrointestinal function to maintain steady plasma levels. Impaired gastric function or reduced intestinal absorption can reduce the amount of drug reaching the systemic circulation, leading to sub-therapeutic levels. Diarrhoea can lead to variable absorption and fluctuating plasma levels. Other AADs, such as metoprolol, verapamil, and dronedarone, are advised to be taken with meals to improve absorption and reduce gastrointestinal side effects. Verapamil’s absorption is slowed with food intake, decreasing the risk of adverse effects like dizziness or hypotension. Dronedarone’s bioavailability is substantially increased when taken with food, leading to more consistent plasma concentrations. Conversely, certain extended-release formulations may exhibit reduced sensitivity to food timing, and in some cases, administering these medications on an empty stomach prevents unpredictable absorption variations caused by food presence.
3.4.2. First-pass hepatic metabolism
Many AADs undergo significant first-pass metabolism in the liver, which can markedly reduce their bioavailability. Propranolol, for example, may exhibit up to a 10-fold variation in plasma levels for the same administered dose, primarily due to extensive hepatic metabolism before reaching the systemic circulation. Other AADs subject to notable first-pass metabolism include lidocaine—administered intravenously to bypass this effect—ibutilide, propafenone, and, to a lesser extent, flecainide. This variability underscores the necessity for meticulous dose titration and monitoring. The cytochrome P450 (CYP) enzyme system predominantly facilitates this metabolism, rendering AADs susceptible to drug–drug interactions. Individual differences in CYP enzyme activity can lead to significant interpatient variability in drug metabolism, influenced by genetic factors, environmental exposures, and concurrent disease states.
3.4.3. Distribution
After absorption and first-pass metabolism in the liver, AADs distribute throughout the body, with lipophilic agents like amiodarone achieving extensive tissue penetration. Amiodarone can accumulate in various tissues, including adipose tissue, liver, and lungs, resulting in a large volume of distribution and an extended half-life, sometimes exceeding 50 days. In contrast, hydrophilic agents such as sotalol have a more limited distribution, predominantly remaining within the extracellular fluid compartment.
3.4.4. Renal and hepatic excretion
The elimination pathways of AADs vary, with many Class I agents primarily undergoing hepatic clearance, while others like sotalol and nadolol are chiefly excreted renally (see the below section ‘6.8 Renal and liver failure’). Patients with impaired hepatic function may experience elevated plasma concentrations and heightened toxicity from hepatically metabolized drugs. Similarly, individuals with renal insufficiency may exhibit reduced clearance, prolonging the half-lives of renally excreted medications. Consequently, dosage adjustments based on organ function are often necessary to maintain therapeutic efficacy and prevent adverse effects.
In summary, the PK of AADs—including aspects of intestinal absorption, first-pass hepatic metabolism, tissue distribution, and renal or hepatic excretion—is crucial for therapeutic effectiveness and safety. Clinicians have to consider these factors, along with individual patient variability, to tailor anti-arrhythmic therapy appropriately and reduce the potential for adverse outcomes.
3.5. Genetics and anti-arrhythmic drugs
The influence of genetics on AADs is a critical aspect of pharmacogenetics, as genetic variations can significantly impact drug efficacy, metabolism, and the risk of adverse effects.10,11 The effectiveness and safety of AADs vary significantly among individuals due to genetic differences affecting their metabolism, transport, and pharmacodynamics (PD). Drug metabolism genes, such as CYP2D6 and CYP3A4, influence how AADs like flecainide and propafenone are processed, impacting drug levels and toxicity risks. Ion channel genes (e.g. SCN5A, KCNH2) affect drug binding and can predispose individuals to arrhythmias, while drug transporter genes (e.g. ABCB1 [encoding P glycoprotein]) modify AAD absorption and distribution. Variants in pharmacodynamic genes (e.g. ADRB1, CACNA1C) alter drug response, potentially affecting treatment success. Additionally, certain genetic mutations, such as those linked to long QT syndrome (LQTS) (KCNQ1, KCNH2, SCN5A), increase the risk of drug-induced arrhythmias like torsades de pointes (TdP). Disease-specific mutations in conditions like Brugada syndrome (BrS) or AF further influence drug selection. While pharmacogenetic testing is emerging in clinical practice, broader adoption requires further research and validation.
3.6. Classification of anti-arrhythmic drugs
In the early 1970s, the AADs known at that time were grouped into three classes based on their functional and electrophysiological effects by Vaughan, Williams (VW) and Singh: Class I drugs reduce myocardial excitability, Class II drugs (β-blockers) have sympatholytic effects, and Class III drugs prolong repolarization duration.12,13 The electrophysiological effects of Class I and Class III drugs were later attributed to inhibition of INa and potassium (K+) currents, respectively. The discovery of the anti-arrhythmic potential of verapamil, a calcium channel blocker (CCB), gave rise to Class IV. In addition, the distinct effects of different Class I AADs on repolarization duration, largely attributed to different binding and dissociation kinetics from the Na+ channel, resulted in a further sub-division into Classes Ia, Ib, and Ic. The strength of this classification lies in the clinical importance of the pharmacological properties on which it relies, resulting in electrophysiological actions, indications, and adverse effects that are typical for each group of drugs.14 However, subsequent research revealed that virtually all AADs affect multiple targets in cardiomyocytes, resulting in complex condition-specific electrophysiological effects that cannot be readily captured in the VW classification (see Supplementary material online, Table S2).14 Amiodarone and dronedarone are prime examples of AADs with pronounced multi-channel inhibitory effects. Although both are traditionally considered Class III AADs, they affect a range of cardiac currents, e.g. Nav, Kv, and Cav, along with α- and β-adrenoceptor blockade, thereby exhibiting effects of all four VW classes.15,16 Moreover, other compounds with anti-arrhythmic effects have been identified that do not fit into the VW classifications. These include, among others, magnesium sulfate for the treatment of TdP VA,17 and ivabradine, an HCN channel blocker primarily developed for lowering heart rates in patients with coronary artery disease (CAD), which has also been used to treat inappropriate sinus tachycardia (IAST) and may be effective against some VAs.18
The limitations of the traditional VW classification have fostered many attempts to improve the classification of AADs. The Sicilian Gambit was proposed in the early 1990s to integrate the multiple mechanistic actions of AADs with their clinical effects.19 Although not intended as an AAD classification,20 it accurately captures the complexity of AADs. However, the Sicilian Gambit has not been able to replace the VW classification in everyday clinical use of AADs. Subsequently, several extensions of the VW classification have been proposed to accommodate recent AADs as well as various compounds still under development. The most recent and most extensive of these is the 2018 Oxford AAD classification.21 This classification maintains the four VW classes but extends Class I with sub-class Id for late Na+ current (INa,L) blockers, further sub-divides Classes II and III, and expands Class IV with other regulators of intra-cellular Ca2+ handling, including RyR2 inhibitors, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) activators, and Na+–Ca2+ exchanger inhibitors. Furthermore, this classification adds Classes 0 (HCN channel blockers), V (mechanosensitive channel blockers), VI (gap junction channel blockers), and VII (upstream therapy). It should be noted that for many of these new (sub)classes, there are no clinically approved AADs available. Conversely, most clinically available AADs belong to multiple sub-classes due to their multi-channel blocking effects, including targeting of some elements of these new (sub)classes. Table 1 shows a simplfied 2018 Oxford AAD classification, and Table 2 summarizes the most widely available market formulations and advised dosing regimens for commonly used AADs. The different agents are described below, with Class III following Class I and Class IV following Class II, reflecting a grouping based on their predominant targets and clinical applications—Class I and III are primarily used to modify atrial and ventricular myocardium activity, while Class II and IV are mainly chosen for their effects on the sinus and AV nodes.
Table 1.
Simplified updated classification of anti-arrhythmic agents
| Class | Sub-class | Primary pharmacological target/action | Example of drugs |
|---|---|---|---|
| HCN channel blockers | |||
| 0 | HCN channel-mediated pacemaker current (If) | Ivabradine | |
| Na+ channel blockers | |||
| Ia | Ia | Nav1.5 (INa) open-state (intermediate dissociation) | Ajmaline, disopyramideb, procainamideb, quinidine/hydroquinidineb,c,d |
| Ib | Nav1.5 (INa) inactivated state (rapid dissociation) | Lidocaine, mexiletinec,d, phenytoin | |
| Ic | Nav1.5 open/inactivated state (slow dissociation) | Antazolinee, cibenzoline, flecainidef, pilsicainide, propafenonef | |
| Id | Late Na+ current | Ranolazine | |
| Inhibitors and activators of the autonomic nervous system | |||
| II | IIa | β-adrenoceptor antagonists | β1-blockers: atenolol, bisoprolol, esmolol, landiolol, metoprolol, nebivolol β1- and β2-blockers: nadolol, propranolol β1-, β2-, and α1-blockers: carvedilol, labetalol |
| IIb | β-adrenoceptor agonists | Isoprenaline | |
| IIc | Muscarinic M2 receptor inhibitors | Atropine | |
| IId | Vagal nerve/ACh release activators | Digoxin, digitoxin | |
| IIe | Adenosine A1 receptor activators | Adenosine | |
| K+ channel blockers and openers | |||
| IIIg | IIIa | Non-selective K+ channel blockers | Amiodaroneg, dronedaroneg, sotalolh, bretylium |
| Kv11.1 (hERG) K+ channel blockers | Dofetilidei, ibutilidej, nifekalant | ||
| Kv1.5 (IKur) K+ channel blockers | Vernakalanti,k | ||
| IIIb | Kir6.2 (KATP) K+ channel openers | Nicorandili | |
| IIIc | GIRK1 and GIRK4 (IKACh) blockers | No approved medications | |
| Ca2+ channel modulators | |||
| IV | IVa | Surface membrane non-selective and Cav1.2 and Cav1.3 channel-mediated L-type Ca2+ current (ICaL) blockers | Bepridil, diltiazem, etripamil, verapamil |
| IVb | Intra-cellular sarcoplasmic reticulum RyR2-Ca2+ channel blockers | No approved medications | |
| Mechanosensitive channel blockers | |||
| V | Transient receptor potential channel (TRPC3/TRPC6) blockers | No approved medications | |
| Gap junction channel blockers | |||
| VI | Cx (Cx40, Cx43, Cx45) blockers | No approved medications | |
| Upstream target modulators | |||
| VII | ACEI, ARNI, mineralocorticoid receptor antagonists, SGLT2 inhibitors, statins | Atorvastatin, enalapril, lisinopril, losartan, candesartan, sacubitril, spironolactone, etc. | |
Abbreviations: AAD, anti-arrhythmic drug; ACEI, angiotensin-converting enzyme inhibitors and receptor blockers; ACh, acetylcholine; ARNI, angiotensin receptor-neprilysin inhibitor; Cav, calcium channel; HCN, hyperpolarization and cyclic nucleotide gated; Nav, sodium channel; Kv, potassium channel; TdP, torsades de pointes.
aNav1.5 Na+ blockers differ based on their binding state and dissociation kinetics, which influence their therapeutic roles and effects on the cardiac action potential. Open-/inactivated state (slow dissociation kinetic) blockers, such as flecainide and propafenone, preferentially bind to open and inactivated Na+ channels and dissociate slowly. They significantly reduce conduction velocity, particularly during tachycardia, making them effective for atrial and ventricular arrhythmias (Class Ic). Open-state (rapid dissociation) blockers, such as lidocaine and mexiletine, bind to open channels but dissociate quickly, allowing selective targeting of ischaemic or depolarized tissues without affecting normal conduction (Class Ib). Inactivated state (intermediate dissociation kinetic) blockers, such as quinidine and amiodarone, bind tightly to the resting state of Na+ channels, prolonging the refractory period and reducing re-entrant arrhythmias (Class Ia and multi-class effects for amiodarone). Late Na+ current inhibitors, such as ranolazine, target persistent Na+ ion influx during the plateau phase, reducing Ca2+ overload and afterdepolarizations. This mechanism is particularly beneficial for ischaemic conditions and preventing arrhythmias like TdP.
bClass Ia AADs possess secondary anti-cholinergic activity (Class IIc), which is significant for disopyramide, moderate for quinidine, and mild for procainamide. This anti-cholinergic effect can lead to an accelerated sinus node rate by reducing parasympathetic influence on the heart.
cQuinidine and mexiletine also exhibit a secondary K+ channel blockade effect (Class III), which contributes to their ability to prolong repolarization and modulate action potential duration, enhancing their anti-arrhythmic efficacy in certain conditions.
dQuinidine and mexiletine also exhibit a secondary α-adrenergic blockade effect, which can potentially lead to hypotension, especially when used at higher doses or in sensitive patients.
eAntazoline also inhibits specific K+ channels, particularly HERG channels (IKr), which may result in QT interval prolongation on the ECG, thereby increasing the risk TdP. Additionally, it exhibits a mild blocking effect on L-type Ca2+ channels.
fFlecainide and propafenone also exhibit a secondary intra-cellular sarcoplasmic reticulum RyR2-Ca2+ channel–blocking effect (Class IVB), which is particularly relevant in specific arrhythmias like CPVT. This mechanism may be less relevant in their typical clinical use for common forms of arrhythmias such as AF or VT.
gAmiodarone and dronedarone also exhibit secondary effects, including Na+ channel blockade (Class I), Ca2+ channel blockade (Class IV), α-adrenoceptor blockade, and non-selective β-adrenoceptor blockade (Class II). These additional mechanisms enhance their anti-arrhythmic efficacy by slowing conduction, reducing automaticity, and mitigating sympathicus-driven arrhythmias.
hSotalol also exhibits a secondary non-selective β-adrenergic receptor antagonist effect (Class IIA), which becomes more prominent at lower doses of the drug.
iKv11.1 (hERG) K+ channel blockers, such as dofetilide, act on the IKr in both atria and ventricles, prolonging repolarization and the QT interval. They are used broadly for arrhythmia management but carry a significant risk of TdP due to excessive QT prolongation. In contrast, Kv1.5 (IKur) blockers, such as vernakalant, target atrial-specific repolarization, making them highly effective for AF with minimal risk of ventricular pro-arrhythmia. Kir6.2 (KATP) channel openers, like nicorandil, regulate K+ efflux in response to metabolic stress, shortening the action potential duration and providing protective effects during ischaemia. While their primary use is in ischaemic protection and vasodilation, excessive opening can lead to hypotension or re-entrant arrhythmias.
jIbutilide also enhances late inward Na+ current (INa), prolonging the action potential duration.
kVernakalant is a potent open-state blocker of Na+ channels, with rapid dissociation kinetics, with no major effects on K+ currents in the human ventricles.
Table 2.
Typical market formulations and dosing of commonly used AADs and anti-arrhythmic agents (for detailed information, please refer to Supplementary material online, Table S7)a
| Modified VW class | AAD | Intravenous bolus | Intravenous infusion | Oral loading | Oral maintenance |
|---|---|---|---|---|---|
| 0 | Ivabradine (5 and 7.5 mg tablets) | No i.v. formulation available | No i.v. formulation available | No oral loading dose is specified | 5–7.5 mg/12 h |
| Ia | Ajmaline (50 mg vials) | 1 mg/kg in 10 min (max 100 mg) | – | No oral formulation available | No oral formulation available |
|
Quinidine (Sulphate: 200 and 300 mg tablets. Gluconate: 800 mg vials. 324 mg tablets) |
Gluconate: <5 mg/kg at 0.25 mg/kg/min (max 10 mg/kg) | – | 200 mg/3 h (max 3 g in 1 d) |
|
|
| Procainamide (1 g vials. 250 mg capsules) | 100 mg; can be repeated every 5 min (max 500–750 mg, 50 mg/min) | 2–6 mg/min (max 1 g/day) | 500–1000 mg | 250 mg/6 h | |
| Disopyramide (50 mg vials. 100 and 150 mg ER capsules) | 2 mg/kg in 10 min | 400 μg/kg/h | No loading dose specified | 100–150 mg IR/6 h or 200–300 mg ER/12 h (max 750 mg/day) | |
| Ib | Lidocaine (50 and 100 mg vials) | 100 mg (1–1.5 mg/kg); can repeat 50 mg (0.5–0.75 mg/kg) in 5–10 min (max 3 mg/kg) | 1–4 mg/min (max 3 mg/kg) | No oral formulation available | No oral formulation available |
| Mexiletine (50, 100, 150, 167, 200, and 250 mg capsules) | No i.v. formulation available | No i.v. formulation available | 400 mg followed by 300 mg 2–3 times (max 1.2 g in 1 d) | 167 mg/day (max 500 mg) | |
| Phenytoin (100 mg vials. 30, 100, 200 and 300 mg ER capsules) | 50–100 mg every 10–15 min (max 1 g) | – | – | 300–400 mg/day orally in divided doses 1–4 times/day (max 600 mg/day) | |
| Ic | Flecainide (150 mg vial. 50, 100, and 150 mg IR tablets. 100, 150, and 200 mg ER capsules) | 2 mg/kg in 10 min | 50 mg/h (max 1 g/day) | 300 mgb | 100 mg/12 h or 200 mg ER/day (max 300 mg/dayc) |
| Propafenone (70 mg vial. 150, 225, and 300 mg IR tablets. 225, 325, and 425 mg ER tablets) | 2 mg/kg in 10 min | 7 mg/kg in 1 d | 600 mgb | 150–300 mg IR/8 h or 225–425 mg ER/12 h (max 900 mg/day) | |
| Antazoline (100 mg vial) | 100 mg in 1 min; 50 mg can be repeated every 5 min (max 300 mg) | 100 mg over 60 min (30–50 mg/min) | – | – | |
| Pilsicainide (50 mg vial. 25 and 50 mg capsules) | 0.75 mg/kg | – | 150 mg | 50 mg/8 h (max 225 mg/day) | |
| Cibenzoline (75 mg vial. 50 and 100 mg tablets) | 1 mg/kg | – | No oral loading dose is specified | 100 mg/8 h | |
| Id | Ranolazine (375, 500, 750, and 1000 mg ER tablets) | No i.v. formulation available | No i.v. formulation available | 2 gb | 500–750 mg /12 h (with food) (max 1 g/12 h) |
| IIa | Atenolol (5 mg vials. 25, 50, and 100 mg tablets) | 2.5 mg in 2.5 min (1 mg/min) repeated at 5 min intervals (max 10 mg) | 0.15 mg/kg bodyweight may be administered over a 20 min period and repeated every 12 h | No oral loading dose is specified. | 25–50 mg/day (max 100 mg/day) |
| Bisoprolol (1.25, 2.5, 5, and 10 mg tablets) | No i.v. formulation available | No i.v. formulation available | No loading dose is specified | 1.25–5 mg/day (max 20 mg/day) | |
| Carvedilol (3.125, 6.25, 12.5 and 25 mg tablets) | No i.v. formulation available | No i.v. formulation available | Initially 3.125 mg/12 h | 25 mg/12 h (max 100 mg/day) | |
| Metoprolol (5 mg vial. 25, 37.5, 50, 75, and 100 mg tablets) | 5 mg in 2 min (max 15 mg) | No dose is specified for prolonged infusion | No loading dose specified | 25–100 mg 12 h (metoprolol tartrate) or 50–200 mg/day (metoprolol XL succinate) | |
| Nebivolol (2.5, 5, 10, and 20 mg tablets) | No i.v. formulation available | No i.v. formulation available | No loading dose specified | 2.5–10 mg/day (max 20 mg/day) | |
| Propranolol (1, 5 and 10 mg vials. 10, 20, 40, 60, and 80 mg tablets. 60, 80, 120, and 160 mg ER tablets) | 1–3 mg in 1 min; repeat every 2–5 min if needed up to 5 mg (max 0.2 mg/kg) | – | No loading dose specified | 20–40 mg IR/6 h, 80–160 mg ER/day (max 240 mg/day) (with food) | |
| Nadolol (20, 40 and 80 mg tablets) | – | – | No loading dose specified | 40–80 mg/day (max 320 mg/day) | |
| Esmolol (100 mg vial) | 0.5 mg/kg in 1 min | 0.05–0.2 mg/kg/min (max 0.3 mg/kg/min) | No oral formulation available | No oral formulation available | |
| Landiolol (288 mg vial) | 0.1 mg/kg in 1 min (only if haemodinamically stable) | 10–40 μg/kg/min (max 57.6 mg/kg/day) or 1–10 μg/kg/min if LV dysfunction | No oral formulation available | No oral formulation available | |
| IIb | Isoprenaline (0.2 mg ampoules) | 10 μg | 2–20 μg/min | ||
| IIc | Atropine (0.4, 0.8 and 1 mg ampoules) | 1 mg followed by additional doses up to 3 mg (0.04 mg/kg) | No prolonged infusion advised | No oral formulation available | No oral formulation available |
| IId | Digoxin (0.25 mg ampoules. 0.125 and 0.25 mg tablets. 0.1–0.25 mg/mL solution | 0.25–0.5 mg followed by additional doses (max 1.5 mg/day) | 0.25 mg/day. No prolonged infusion advised | 0.5–0.75 mg in 2 doses 6 h apart (max 1.5 mg/day) | 0.25 mg/day (adjust to blood levels and CrCl) |
| Digitoxin (0.07 mg ampoules. 0.0625, 0.125, and 0.25 mg tablets) | 0.5 mg followed by additional doses (max 1.5 mg/day) | 0.1 mg/day. No prolonged infusion advised | 0.6–1.2 mg given in divided doses over 1 day | 0.05–0.1 mg/day (adjust to blood levels and CrCl) | |
| IIe | Adenosine (6, 12, 30, 60, 90 and 100 mg vials) | 6, 12, and 18 mg boluses | No prolonged infusion advised | No oral formulation available | No oral formulation available |
| III | Amiodarone (150 and 300 mg vials. 100, 200, and 400 mg tablets) | 150 mg in 10 min or 300 mg over 30 min followed by 900–1200 mg i.v. over 24 hd (max 2200 mg/day) | 600–1200 mg/day for 8–10 dayse | Standard: 600 mg/day in 2–4 weeks Accelerated: 1200 mg/day in 3 doses for 2 weekse (total ≈10 g) |
200 mg/day (max 600 mg/day) |
| Dronedarone (400 mg tablets) | No i.v. formulation available | No i.v. formulation available | – | 400 mg/12 h (with food) | |
| Dofetilide (125, 250 and 500 mg capsules) | No i.v. formulation available | No i.v. formulation available | No loading dose specified | 125–500 μg/12 h (specific algorithm followed based on QT and CrCl) | |
| Ibutilide (1 mg vial) | 1 mg over 10 min if ≥60 kg (0.01 mg/kg if <60 kg); can repeat 1 mg once if needed | – | No oral formulation available | No oral formulation available | |
| Sotalol (150 mg vials. 80, 120, 160, and 240 mg tablets and capsules) | 1 mg/kg in 10 min; can be repeated after 6 h (Adjust dose based on CrCl) (max 450 mg/day) | 75 mg/12 h | No oral loading dose specified | 80–160 mg/12 h (max 480 mg/day) | |
| Vernakalant (500 mg vials) | 3 mg/kg in 10 min followed in 15 min by 2 mg/kg in 10 min if needed | No prolonged infusion advised | No oral formulation available | No oral formulation available | |
| IV | Verapamil (5 mg ampoule. 40, 80, 120 mg IR tablets. 100, 120, 180, 240, 300, and 360 mg ER tablets) | 2.5–5 mg in 2 min followed in 15 min by 5–10 mg if needed | 2–4 mg/h (max 100 mg/day) | No loading dose specified | 80–120 mg IR/8 h or 180–240 mg ER/day (max 480 mg/day) |
| Diltiazem (25, 50, 75, 100 and 125 mg vials. 30, 60, 90 and 120 mg IR tablets and capsules. 120, 180, 200, 240 and 300 mg ER tablets and capsules) | 0.25 mg/kg in 2 min followed by 0.35 mg/kg if needed | 5–15 mg/h | No loading dose specified | 60 mg/8 h or 120–360 mg ER/24 h (max 480 mg/day) | |
| Bepidril (100 and 200 mg tablets) | – | – | No loading dose specified | 200 mg/day (max 400 mg/day) | |
| Other agents | Magnesium (1.5 g ampoules) | 1–2 g in 5 min | 0.5–1 g/h | No loading dose specified | 350 mg/day |
Abbreviations: AAD, anti-arrhythmic drug; CVC, central venous catheter; CrCl, creatinine clearance; EMA, European Medicines Agency; ER, extended release; FDA, Food and Drug Administration; IR, immediate release; LV, left ventricle; VT, ventricular tachycardia.
aSome of the drugs listed have varying availabilities and approval statuses for the treatment of arrhythmias. Ranolazine is approved by both the EMA and the U.S. FDA for the treatment of chronic angina, but not specifically for arrhythmias. Vernakalant is approved by the EMA for the rapid conversion of recent-onset AF to sinus rhythm in adults, but it has not received FDA approval. Conversely, dofetilide is approved by both the FDA and EMA for maintaining sinus rhythm in patients with AF or flutter; however, it is marketed only in the USA and not in Europe. Additionally, certain dosage formulations may not be available in all countries.
bSee Box 6.
cThe maximum advised dose in the USA for the treatment of VT is 400 mg/day.
dIt is advised to dilute the drug in 5% dextrose (glucose) to a concentration not exceeding 2 mg/mL. This dilution is advised to be administered via a CVC to minimize the risk of thrombophlebitis.
eGoal to achieve cumulative doses of 5–10 g by i.v. loading and 10–15 g by oral loading.
3.6.1. Class 0
3.6.1.1. Ivabradine
Ivabradine is a selective inhibitor of the SA node current or funny current (If). This current was originally identified in the SA node, but it has also been found in the specialized conduction system, including the AV node and Purkinje fibres. The If is a mixed Na+ and K+ current that plays a pivotal role in the spontaneous depolarization of the SA node. By specifically targeting this current, ivabradine reduces the rate of spontaneous depolarization in the SA node, consequently slowing the heart rate without significantly affecting contractility or AV conduction. Unlike traditional β-blockers or CCBs, which exert their effects on the entire myocardium, ivabradine’s selectivity for If allows for heart rate control without side effects on other heart functions. This specificity is particularly advantageous in patients with conditions such as heart failure with reduced ejection fraction (HFrEF). Ivabradine may be used to reduce heart rate and symptoms in patients with IAST. More recently, it has been proposed to reduce heart rate in AF but with a milder effect than digoxin (11.6 vs. 19.6 b.p.m. mean daytime heart rate decrease, P < 0.01)22 and for junctional ectopic tachycardia (JET).23 However, ivabradine is not advised for patients with paroxysmal AF, as it may promote arrhythmic episodes.
3.6.2. Class Ia
3.6.2.1. Quinidine
Quinidine, the D-isomer of the anti-malarial drug quinine, is one of the oldest known AAD.24 It is traditionally classified as a Class Ia AAD, inhibiting cardiac INa with high affinity for the open-state and intermediate dissociation kinetics (time constants of 1–5 s) from the Na+ channel, reducing both cardiac excitability and conduction velocity. The effects of quinidine are rate dependent, with more pronounced inhibition of INa at fast rates (use dependence). In addition to INa, quinidine inhibits a wide range of other currents, including repolarizing K+ currents (notably IKr, IKs, and Ito), as well as ICa,L and INa,L.25,26 Together, these effects result in significant quinidine-induced prolongation of repolarization duration, visible as QT interval prolongation on the ECG, particularly at slow rates (i.e. exhibiting reverse use dependence) (Figure 4). Quinidine also decreases the automaticity of the SA node and Purkinje cells but increases the sinus rate in vivo due to a combination of its anti-cholinergic (due to inhibition of muscarinic receptors) and haemodynamic effects.24 In particular, quinidine-mediated inhibition of α-adrenoceptors promotes peripheral vasodilation, hypotension, and subsequent reflex sinus tachycardia. This effect is most pronounced with intravenous (i.v.) quinidine administration or when combined with β-blockers or verapamil.
Figure 4.

Twelve-lead electrocardiograms (ECGs) illustrating the effect of quinidine on the QT interval in a female patient with no structural heart disease and a history of atrial fibrillation. (A) Baseline ECG recorded prior to quinidine administration, showing a normal QTc interval duration (two-arrowhead line). (B) ECG following quinidine administration, revealing marked QT interval prolongation, indicative of its effect on ventricular repolarization. This underscores the potential for pro-arrhythmic effects, even in the absence of structural heart disease.
The effects of quinidine after oral administration start 1–3 h after intake and remain for 6–8 h (see Supplementary material online, Table S3).24 Quinidine has a bioavailability of 60–80% and is 80–88% protein-bound in serum. Its concentration is 4–10 times higher in the heart than in the circulation. Quinidine is primarily eliminated by hepatic metabolism through the cytochrome P450 system (CYP3A4), resulting in hydroxylated metabolites, some of which have anti-arrhythmic effects.24 About 20% of quinidine is excreted unchanged via the kidneys. Quinidine is itself a potent inhibitor of CYP2D6 and P-glycoprotein (P-gp), potentially affecting effective concentrations of other drugs (see Supplementary material online, Tables S4 and S5). For example, a potentially hazardous interaction between quinidine and digoxin may occur due to quinidine-induced reduction in the renal tubular secretion of digoxin, thereby increasing its toxicity and the risk of cardiac arrhythmias (see the section ‘5.11 Anti-arrhythmic drug switch and combinations’ below).27
Quinidine was initially used for SR maintenance in AF patients and prevention of recurrences of VA by reducing ectopic activity and prolonging repolarization duration, thereby reducing the likelihood of re-entry. However, its prominent adverse effects and the availability of new anti-arrhythmic therapies with higher efficacy and/or better safety profiles have made quinidine obsolete for the treatment of AF.24 Quinidine is currently used for the treatment of several inherited arrhythmogenic disorders. Brugada syndrome is an inherited channelopathy resulting in a typical ECG pattern and increased risk of ventricular tachycardia (VT), with the epicardium of the right ventricular outflow tract region as the primary source of electrophysiological abnormalities. BrS is often associated with decreased INa, potentially resulting in an imbalance between depolarizing INa and early repolarizing K+ currents, including the transient outward K+ current (Ito), which is highly expressed in the epicardium of the right ventricular outflow tract. Inhibition of Ito normalizes the BrS ECG pattern, and the clinical efficacy of quinidine in BrS patients28 has been primarily attributed to its inhibition of Ito.24 Similarly, data from small cohorts suggest that quinidine may represent a potential treatment option for short QT syndrome (SQTS) due to its repolarization-prolonging effects, as well as in patients with idiopathic ventricular fibrillation (IVF), particularly those with contraindications for implantable cardioverter defibrillator (ICD) treatment.24,29 However, given the low prevalence of these rare arrhythmogenic conditions and the low price of quinidine, it has been considered economically unfavourable to widely distribute quinidine.30 As a result, quinidine is currently no longer available in many countries.30
Syncopal events (‘quinidine syncope’), first attributed to drug-induced TdP arrhythmias in 1964,31 are the most serious adverse effects associated with quinidine (see Supplementary material online, Table S6 and see sections ‘Pro arrhythmia’ and ‘Toxicity and adverse effects’ below). This pro-arrhythmia is typically the result of excessive, heterogeneous quinidine-induced repolarization prolongation, promoting the genesis of EADs initiating potentially life-threatening re-entrant VA. Quinidine has also been associated with increased mortality.32 In addition, quinidine has pronounced adverse gastrointestinal effects, typically diarrhoea, causing drug discontinuation in many patients.24 The electrolyte disturbances promoted by quinidine-induced diarrhoea can further increase the risk of VA. Other dose-related and reversible side effects, usually defined as cinchonism, include tinnitus, headache, dizziness, visual disturbances, nausea, and decreased hearing.24
3.6.2.2. Disopyramide and ajmaline
Although classified as Class I AADs, ajmaline and disopyramide are not commonly used for anti-arrhythmic therapy. Ajmaline was initially used to treat AF in patients with pre-excitation, but it was later replaced by flecainide or propafenone for this purpose. Currently, ajmaline is primarily used to unmask concealed arrhythmogenic phenotypes regulated by INa dysfunction (e.g. BrS) and in some countries for VT termination in patients without significant heart disease. Disopyramide is a Class Ia AAD with negative inotropic effects and can be used to suppress ventricular ectopy or in combination with β-adrenoceptor or Ca2+-channel blockers in patients with hypertrophic obstructive cardiomyopathy.33 In addition, disopyramide has significant anti-cholinergic effects, which are the primary cause of its adverse effects and have limited its use. However, these same properties make it particularly effective for certain patients with vagal AF, where a bedtime dose can be highly effective and reasonably well tolerated, provided no daytime dosing is required.
3.6.2.3. Procainamide
Procainamide is a Class Ia AAD that inhibits cardiac INa with high affinity for the open state of the Na+ channel and intermediate dissociation kinetics. It also blocks IKr. Combined, these effects reduce excitability, increase ERP, and promote dispersion by prolonging APD and augmenting post-repolarization refractoriness.34 In addition, procainamide slows conduction. Its major metabolite N-acetylprocainamide lacks INa-blocking effects but has similar APD-prolonging effects.34
Procainamide is almost completely absorbed after oral administration, with a bioavailability of 70–85% (see Supplementary material online, Table S3). Its peak plasma concentrations are typically reached within 1–2 h.35,36 Its apparent volume of distribution is 2 L/kg body weight, and about 15% is bound to plasma proteins.35 Procainamide has a half-life of 3–4 h and is eliminated 50% by hepatic metabolism and 50% via renal excretion of the unchanged drug. N-acetylprocainamide is renally excreted with a half-life of 6–10 h. Because of these relatively rapid elimination rates, procainamide is usually administered as a slow-release formulation. Given the dependency on renal clearance, dose adjustments are needed in patients with renal failure.34
Procainamide is used for the acute cardioversion of haemodynamically stable VT (Figure 5). Procainamide is also used in patients with accessory pathways and pre-excited AF, slowing conduction across the accessory pathway and lowering ventricular rate. It has recently been employed to compare electrical vs. pharmacological cardioversion of AF in emergency department settings. Finally, procainamide has been used for drug provocation testing in patients with suspected BrS, although it is less likely to provoke a Type-1 Brugada ECG pattern compared with ajmaline.
Figure 5.

Electrocardiogram (ECG) tracings of Leads II and III illustrating the termination of ventricular tachycardia (VT) after a 15 min infusion of procainamide in a patient with structural heart disease (old myocardial infarction). The tracings show VT transitioning to sinus rhythm after procainamide administration, demonstrating its anti-arrhythmic efficacy in managing VT in the presence of underlying myocardial scarring. Electrocardiogram was recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Drug-induced pro-arrhythmia is the most important adverse effect of procainamide and is directly related to the INa and IKr-blocking effects of procainamide and N-acetylprocainamide (see Supplementary material online, Table S6). N-acetylprocainamide concentrations >20μg/mL carry a higher risk of TdP, whereas procainamide concentrations >10 μg/mL appear to carry a risk of marked QRS widening and potential arrhythmia exacerbation.34
3.6.3. Class Ib
3.6.3.1. Lidocaine
In addition to its local anaesthetic effects, lidocaine is a Class Ib AAD, inhibiting cardiac INa. Lidocaine blocks Na+ channels preferentially in the inactivated state with rapid recovery from block (fast dissociation kinetics). As such, the effects of lidocaine are exacerbated in depolarized tissue (e.g. due to ischaemia) or in the presence of rapid electrical activation, when more Na+ channels are already inactivated.34 Conversely, lidocaine is less effective in the presence of hypokalaemia due to the associated RMP hyperpolarization (less Na+ channels are inactivated). Lidocaine decreases automaticity and triggered activity by reducing the slope of Phase 4 of the AP and reducing excitability. Action potential duration is either unaffected or shortened by lidocaine, with the latter due to inhibition of depolarizing INa,L.34 Nonetheless, ERP could be prolonged due to an increased post-repolarization refractoriness resulting from the INa inhibition. Of note, lidocaine is the only clinically available AAD with no relevant inhibitory effects on cardiac K+ channels.
Although lidocaine is well absorbed, it undergoes extensive first-pass hepatic metabolism, making it inappropriate for oral use (see Supplementary material online, Table S3). Accordingly, it is primarily given i.v. for the treatment of cardiac arrhythmias.34 Lidocaine is 60–80% protein bound. After i.v. administration of a bolus of lidocaine, plasma concentrations first decline rapidly (half-life of ∼8 min), which is attributed to rapid distribution of the drug from the plasma to the periphery. Thereafter, the drug is eliminated by CYP3A4-mediated hepatic metabolism, with a half-life of ∼2 h.36 Thus, steady-state plasma concentrations are reached in 8–10 h after initiation of lidocaine maintenance infusion, but these values are significantly prolonged in patients with hepatic dysfunction, e.g. in the elderly, or in the presence of HF or cardiogenic shock. Lidocaine metabolism is impaired by β-blockers, requiring dose adjustments when co-administered.
The potential use of lidocaine in the treatment of VT was already described in the 1950s and 1960s and likely results primarily from reduced myocardial excitability. Early studies in patients with acute myocardial infarction (MI) found that lidocaine suppressed premature ventricular contractions (PVCs) and non-sustained VT. However, later studies reported a higher mortality after acute MI in patients receiving lidocaine, possibly due to a higher incidence of asystole and bradyarrhythmias. As such, prophylactic lidocaine during acute MI was abandoned.34 A systematic Cochrane analysis concludes that evidence of low quality suggests that prophylactic lidocaine has very little or no effect on mortality or ventricular fibrillation (VF) in people with acute MI and that its safety profile is unclear.
3.6.3.2. Mexiletine
This lidocaine analogue inhibits both the peak and late Na+ currents (INa,P and INa,L), shortens APD and refractoriness primarily in Purkinje fibres and to a lesser extent in ventricular muscle. Additionally, it suppresses the automaticity of Purkinje fibres. However, it does not modify sinus rate, contractile force, AV nodal function, exert major haemodynamic effects, or prolong the QT. Mexiletine is advised for the treatment of VA (sustained VT),37 even in patients with recent MI, but had no favourable effect on mortality. Combined with sotalol, it has been used in patients with frequent VT recurrences who have a defibrillator. It may be appropriate as off-label add-on therapy to shorten the QT interval in LQTS Type 3 patients with a baseline corrected QT interval (QTc) >500 ms.38
3.6.3.3. Phenytoin
Phenytoin, an anti-epileptic drug with membrane-stabilizing properties, has a limited but specific role as an AAD. Its primary action is through Na+ channel blockade, which shortens the APD, particularly in ventricular myocardium. Historically, phenytoin has been used in digitalis-induced arrhythmias, especially when nodal or ventricular tachyarrhythmias occur, due to its ability to counteract the pro-arrhythmic effects of digitalis. However, its use as an AAD is rare in modern practice due to the availability of more effective and safer alternatives. Phenytoin requires careful monitoring for drug–drug interactions, as it is both a substrate and an inducer of CYP enzymes, which can complicate its PK in patients on multiple therapies.
3.6.4. Class Ic
3.6.4.1. Flecainide and propafenone
Flecainide and propafenone produce a potent frequency-dependent blockade of Na+ channels, decrease cardiac excitability (increase pacing and defibrillating thresholds) and slow conduction in fast-response cardiac tissues, with the greatest effect in the ventricular myocardium and the His-Purkinje system. They suppress ectopic automaticity, shorten the APD in Purkinje fibres but prolong the APD in the ventricular muscle, and prolong ventricular refractoriness by lengthening the reactivation of Na+ channels. Flecainide/propafenone prolong atrial APD in a frequency-dependent manner, which may facilitate the conversion of AF to SR. During orthodromic and antidromic AV re-entrant tachycardia (AVRT), flecainide/propafenone slow conduction and increase anterograde and particularly retrograde refractoriness in accessory pathways in a frequency-dependent manner. Flecainide/propafenone have minimal haemodynamic effects in patients with normal left ventricular ejection fraction (LVEF) but reduce LVEF in patients with LV dysfunction and HF.
Flecainide also blocks the INa,L channel, shortening the QT interval in patients with LQTS3 and is an open channel blocker of RyR2-Ca2+ release channels, decreasing the arrhythmogenic Ca2+ release from the sarcoplasmic reticulum in catecholaminergic polymorphic ventricular tachycardia (CPVT) patients with mutations in RYR2 and CASQ2 genes. Propafenone blocks ICa,L and RyR2 channels, being an alternative to flecainide in CPVT, and exhibits mild β-blocking properties at doses >450 mg/day.
Flecainide/propafenone are advised for the cardioversion of symptomatic new-onset AF to SR and long-term maintenance of SR following cardioversion39–42 and to enhance success of direct current (DC) cardioversion and reduce immediate/early recurrences of AF. In selected, highly symptomatic patients with rare paroxysmal AF episodes, a single self-administered oral dose of flecainide/propafenone [‘pill-in-the-pocket’ (PITP) approach] could be used to restore SR, provided that anticoagulation advice is followed and once safety has been previously established in a medical environment.43 Flecainide/propafenone can convert AF to atrial flutter (AFL) with 1:1 AV conduction and increase the ventricular rate; this can be prevented with AV blocking agents (Figure 6). Flecainide slows, but rarely terminates AFL.
Figure 6.

Single-lead (A) and 12-lead electrocardiogram (ECG) tracings (B and C) demonstrating the progression of atrial arrhythmias in a 57-year-old hypertensive male patient taking 200 mg/day of flecainide for paroxysmal atrial fibrillation (AF). (A) Baseline ECG showing AF at presentation. (B) After a few days of flecainide therapy, the patient developed atrial flutter (AFL) with 1:1 atrioventricular (AV) conduction, non-specific intraventricular conduction disturbance, and rapid ventricular response with RR intervals of 280 ms, mimicking ventricular tachycardia (VT). (C) Following the administration of 5 mg of i.v. atenolol, the AV conduction ratio changed to 3:1. This resulted in the resolution of LBBB and narrowing of the QRS complex, making the flutter waves apparent, with a cycle length of 240 ms (cycle length shortened by 40 ms after a partial washout effect of flecainide). This case illustrates flecainide-induced pro-arrhythmia with AFL, and the diagnostic clarity achieved through rate control and conduction ratio alteration. The electrocardiogram was recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Intravenous flecainide and propafenone may also be appropriate in the acute treatment of supraventricular tachycardia (SVT), including symptomatic focal atrial tachycardia (AT), pre-excited AF, and AVNRT, and orally, in the chronic treatment of focal AT and AVRT.44
It has also been used for the prophylaxis and treatment of life-threatening, sustained, haemodynamically tolerated VA, not controlled with other AADs or ablation, or when they are not tolerated or possible. The combination of flecainide/propafenone with amiodarone may be used in patients with frequent VT recurrences who have an ICD.45 Flecainide may be appropriate as add-on therapy to shorten the QT interval in LQTS3 patients with a QTc >500 ms.46,47 It is also advised in patients with CPVT who experience recurrent exercise syncope or polymorphic/bidirectional VT despite maximally tolerated β-blocker doses or when ICD implantation has risks/contraindications or is not available or accepted by the patient.48–51
Flecainide/propafenone can cause monomorphic VT and is associated with increased mortality, heart failure (HF) and cardiac arrest (CA) in patients with prior MI and impaired LV function.32,52,53 Thus, it is advised to avoid them in patients with ischaemic or SHD.54 Nevertheless, there is some controversy about the safety of flecainide use for acute arrhythmia termination or chronic prevention in patients with SHD and no prior MI or ventricular dysfunction. Their prophylactic use is potentially harmful in patients with adult congenital heart disease and asymptomatic VA.55 Flecainide and propafenone prolong the QRS duration, which can exacerbate existing conduction delays and increase the risk of complete heart block, especially in patients with pre-existing bundle branch block (BBB). In patients with BBB and no SHD, flecainide/propafenone can still be used for paroxysmal AF or SVT, but the QRS duration is advised to be closely monitored. If the QRS widens by >25% from baseline, it is advised to reduce the dose or to discontinue the drug. These drugs are not advised for patients with a baseline QRS >120 ms due to the risk of excessive conduction delay, especially in those with LBBB or bifascicular block. Flecainide and propafenone may also unmask the ECG of the BrS. Finally, flecainide may play a role in triggering takotsubo syndrome or exacerbate its occurrence and complications, and it is advised to avoid it in patients with a history of this disorder.56
3.6.4.2. Other: cibenzoline, pilsicainide, and antazoline
Cibenzoline is a Class Ic drug (also found classified as Class Ia) that also blocks L-type Ca2+ and K+ channels and exhibits antimuscarinic activity.57,58 It increases atrial and ventricular refractoriness and prolongs intracardiac conduction. Intravenous cibenzoline is advised for the cardioversion of recent-onset AF in patients with no clinically significant SHD. It is as effective as flecainide and disopyramide in haemodynamically stable patients with an accessory pathway who do not require electrical cardioversion.57–59 Orally, it can be used as a ‘PITP’ approach for paroxysmal AF.60
Pilsicainide is a Class Ic drug widely used in Japan for the cardioversion of recent-onset AF in patients with no clinically significant SHD.58,61–63 This agent does not significantly affect the Ca2+, delayed rectifier K+, inward rectifying K+, acetylcholine-induced K+ or adenosine triphosphate (ATP)–sensitive K+ currents. From these results, pilsicainide could be differentiated from other Type Ic AADs as a "pure" Na+ channel blocker. In patients with paroxysmal AF, pilsicainide prolongs refractoriness and slows conduction in the distal pulmonary veins (PVs) and left atria (LA). Pulmonary vein-LA conduction block can be observed before AF termination.62 Hybrid therapy with pilsicainide and PV isolation (by catheter ablation) appears to be an effective therapeutic approach for AF.62 There are limited data on the use of pilsicainide in VA.63 Pilsicainide can be taken as a PITP approach to terminate paroxysmal AF.59,61
Antazoline is a first-generation antihistaminic agent which blocks Na+ channel and several K+ channels and has anti-cholinergic properties.64,65 It slows intra-atrial conduction, prolongs atrial and ventricular ADP and refractoriness66 and exerts a negative inotropic effect limiting its use in patients with SHD.65 Intravenous antazoline is advised for the cardioversion of recent-onset AF in patients with preserved LV function (median time to conversion 16.0 min), being as efficacious as propafenone and amiodarone,67–70 in patients undergoing PV isolation71 or during ablation of accessory pathways.65 However, it fails to prevent AF recurrence when given orally.72
3.6.5. Class Id
3.6.5.1. Ranolazine
This anti-anginal drug selectively inhibits INa,L and IKr and prolongs atrial and ventricular APD and refractoriness, but the effect is more pronounced in the atria.73 Ranolazine does not slow intracardiac conduction velocity or modify heart rate, contractility or blood pressure. Off-label ranolazine reduces the incidence of AF post-cardiac surgery and post-electrical cardioversion, and high doses (2 g per os) may convert recent-onset AF (<48 h duration).74,75 The combination of ranolazine and low doses of dronedarone, but not each drug in monotherapy, reduced AF burden vs. placebo in patients with paroxysmal AF in one trial.76 The combination of ranolazine and amiodarone can be effective in managing refractory arrhythmias, particularly AF and VT. However, it requires careful monitoring due to the risk of QT prolongation and potential drug interactions. Additionally, ranolazine and amiodarone can increase DOAC levels, heightening the risk of bleeding through P-gp and/or CYP3A4 inhibition. In patients with unstable angina and non-ST-segment elevation MI, ranolazine significantly reduces the incidence of non-sustained VT, SVT, AF and bradycardias as compared with placebo, but not sudden cardiac death (SCD).77,78 Ranolazine may be appropriate as add-on therapy to shorten the QTc interval in LQTS3 patients with a QTc >500 ms.45,47,79 Ranolazine is approved for this purpose in the USA but not elsewhere.
3.6.6. Class III
The prototypical Class III AAD effect is prolongation of APD and, thereby, ERP, reducing the likelihood of re-entry (see Supplementary material online, Table S2).8 For most AADs, this effect is achieved through inhibition of IKr. Action potential duration prolongation by Class III AADs is most pronounced at slow rates due to the previously mentioned reverse use dependence effect. Reverse use dependence of K+ channel blockers has been attributed to the intrinsic relationship between total membrane current and APD, whereby a fixed reduction in membrane current (due to K+ channel inhibition) will have a larger impact on APD when total membrane current is small (i.e. when baseline APD is already long).80 This mechanism partly explains the potential for excessive APD prolongation by Class III anti-arrhythmic drugs in the setting of hypokalaemia or impaired repolarization reserve (e.g. due to a reduction in other repolarizing K+ currents). The primary mechanisms of drug-induced pro-arrhythmia with Class III AADs include heterogeneous APD prolongation and EAD-mediated triggered activity.81
3.6.7. Class IIIa
3.6.7.1. Amiodarone
Amiodarone inhibits a wide range of ion channels and receptors, including IKr, INa, ICa,L as well as α-adrenoceptors and β-adrenoceptors, thus exhibiting effects of all four original VW AAD classes (see Supplementary material online, Table S2).82 Consequently, amiodarone prolongs repolarization duration (primarily via IKr and IKs inhibition) and decreases conduction velocity by blocking INa. However, it has less pronounced reverse use-dependent effects than pure Class III AADs.83 Amiodarone also produces non-competitive β-adrenoceptor blockade, which can promote sinus bradycardia, and reduces ICa,L in a use-dependent manner. Of note, there are significant electrophysiological differences between i.v. amiodarone and chronically administered oral amiodarone.82,83 Intravenous application produces predominantly slowing of ventricular conduction, a smaller repolarization prolongation, little effect on sinus rate, and more potent anti-adrenergic activity.84 These differences are likely in part due to additional effects of metabolites and due to amiodarone-induced electrical remodelling during chronic treatment.82
Amiodarone has a bioavailability of 35–65% (see Supplementary material online, Table S3). The rate and extent of absorption of amiodarone increase when taken with food.85 It also has a large volume of distribution (around 60 L/kg) and is highly lipophilic, being 96% protein bound and resulting in a delayed onset of action (with the anti-arrhythmic effect plateauing after 10 weeks of therapy85 and a very long half-life (30–100 days).34,36 High oral loading doses are used to accelerate the onset of drug activity, although i.v. application has a rapid onset of action. Loading86 may be done with 600 mg per day over four weeks. Afterward, the maintenance dose is established, typically ranging between 100 and 200 mg per day. Amiodarone undergoes extensive hepatic metabolism, primarily via CYP3A4, an enzyme that it also inhibits. Consequently, amiodarone can significantly alter the metabolism of numerous other drugs, necessitating careful consideration of potential drug interactions (see Supplementary material online, Table S5).85,87 For example, amiodarone can increase simvastatin and atorvastatin concentrations through its effect on CYP3A4 and similarly affects warfarin levels, necessitating warfarin dose reductions when amiodarone is initiated. Amiodarone also inhibits P-gp transporters, e.g. increasing digoxin levels.88 The major metabolite of amiodarone is desethyl-amiodarone, which also has anti-arrhythmic properties. The metabolism of amiodarone is inhibited by grapefruit juice, leading to elevated serum levels of amiodarone. Excretion is primarily hepatic and biliary with almost no elimination via the renal route.34,36
Although originally developed as an anti-anginal agent, amiodarone is generally considered the most effective AAD available. Its efficacy and relatively low pro-arrhythmic risk (discussed below) likely result from the complex interaction between numerous molecular targets, e.g. resulting in prolongation of repolarization duration without increasing dispersion of repolarization or an increased risk of EADs.83,89 Amiodarone is approved by the Food and Drug Administration (FDA) for the treatment of VA but is also commonly used for cardioversion and rhythm control of AF. Amiodarone is first-line treatment in the setting of VF and CA.85 A randomized controlled trial compared amiodarone, lidocaine and placebo in out-of-hospital CA refractory to shock therapy. Although there was no difference in outcomes in the overall population, amiodarone demonstrated a survival benefit compared with placebo in the witnessed arrest subgroup.90 Likewise, amiodarone is commonly used in patients with recurrent VT receiving appropriate ICD shocks. In the OPTIC trial, amiodarone plus a β-blocker was associated with a significant 70% reduction in risk of appropriate ICD shocks.91 In AF patients, amiodarone is less effective than Class Ic drugs and vernakalant for acute cardioversion, likely in part due to its relatively slow onset of action.92 However, it is one of the few AADs available in HF patients. On the other hand, for long-term rhythm control in patients with AF, amiodarone is significantly more effective than dronedarone, sotalol, and propafenone, with a 1-year rate of maintaining SR of >65%.87,92,93 Amiodarone is also used for the management of peri- and post-operative AF, which is common after cardiac surgery.92 Finally, amiodarone's negative dromotropic effects can be employed for rate control when combination therapy with β-blockers and digoxin is insufficient in patients who do not qualify for non-pharmacological rate control or in the acute setting in patients with haemodynamic instability.92 Common side effects resulting from amiodarone use include nausea, vomiting, and taste disturbances (see Supplementary material online, Table S6).85
Compared with other Class III AADs, amiodarone exhibits a relatively low pro-arrhythmic risk, with an incidence of drug-induced TdP <1%, despite its QT-prolonging effects.34,87 Given its unique electrophysiological properties and lower propensity to induce TdP, amiodarone therapy may be safely maintained in patients with a QTc interval prolongation up to 550 ms, provided there are no additional risk factors such as bradycardia, electrolyte imbalances, or concomitant use of other QT-prolonging medications. It is advised to avoid the combination with other QT-prolonging drugs (notably Class Ia or Class III AADs) whenever possible and to be used with caution if deemed necessary (see Supplementary material online, Table S5). Similarly, the combination with Class II or Class IV AADs may promote sinus bradycardia or impair AV conduction. Importantly, the use of amiodarone is limited by potentially severe extra-cardiac toxicity (see the section ‘Pro-arrhythmia, toxicity, and other major adverse effects’ below). Despite its pronounced toxicity, amiodarone remains the most commonly used AAD, accounting for 48% of prescriptions in 2016 in a U.S. insurance database,94 38% in the ‘Get With the Guidelines—AF’ registry,95 and 25% in a large international AF registry.96 This predominance is likely due to the high prevalence of underlying cardiovascular disease (CVD), including ischaemic heart disease and HF, in patients at risk of atrial and VA. Under these conditions, many other AADs are contraindicated due to their increased pro-arrhythmic potential.
3.6.7.2. Dronedarone
Dronedarone is a non-iodinated derivative of amiodarone, which was designed to retain anti-arrhythmic efficacy while minimizing the potential for extra-cardiac adverse effects associated with amiodarone. Dronedarone exerts its anti-arrhythmic effects through a multifaceted mechanism, involving inhibition of multiple ion channels (see Supplementary material online, Table S2). It predominantly blocks Na+ and K+ channels, prolonging APD and refractory periods. Additionally, dronedarone possesses β-adrenoceptor blocking properties, further contributing to its anti-arrhythmic action. Notably, it lacks the iodine moiety found in amiodarone, reducing the risk of thyroid dysfunction and other extra-cardiac adverse effects. Clinical trials, including EURIDIS/ADONIS (n = 1237) and ATHENA (n = 4628), have demonstrated the efficacy of dronedarone in maintaining SR and reducing cardiovascular hospitalizations and mortality in patients with AF.97–100 While not intended for use in patients with severe HF or those recently hospitalized for HF, dronedarone has shown promise in improving outcomes in those with milder HF.98,101 Due to low bioavailability, dronedarone is advised to be taken with meals (oral absorption may increase four-fold when taken with a fatty meal) (see Supplementary material online, Tables S3 and S7). Dosing is non-complex: it is given in a fixed daily dose of 400 mg twice daily; dose adjustments do not apply. Dronedarone’s safety profile, while generally favourable, warrants careful consideration. Gastrointestinal disturbances are among the more common adverse effects. Initially, there was apprehension regarding elevations in liver enzymes, but the overall incidence of mild-to-moderate liver injuries with dronedarone is only slightly increased when compared with other AADs.102 The ANDROMEDA trial (n = 625) was conducted to evaluate the efficacy and safety of dronedarone in HF. Only 25% of patients had AF.103 Unfortunately, the trial was prematurely terminated due to safety concerns. The results indicated an increased mortality risk in patients with severe HF and a LVEF of <25%. As a consequence, dronedarone is not advised for use in this specific patient population. The PALLAS trial (n = 3236) investigated the use of dronedarone in patients with permanent AF (>6 months of continuous AF) and additional risk factors.104 However, the trial was prematurely terminated due to safety concerns. The results indicated an increased risk of cardiovascular events, including stroke, HF and death, in patients receiving dronedarone compared with the placebo group and therefore it is advised not to use dronedarone in this particular population, and it should be discontinued in patients who develop persistent AF longer than 6 months during treatment. A subanalysis of this study found a strong effect of concurrent digoxin use on the adverse effect of dronedarone on cardiovascular death, but not on the occurrence of HF.105 The elevation of digoxin concentration induced by dronedarone is attributed to its interaction with P-gp. This interaction potentially may have contributed to the less favourable outcome observed in the dronedarone arm.
In summary, its effectiveness in rhythm control, coupled with its relatively favourable adverse effect profile, positions dronedarone as a valuable option for selected patients with AF. Its use is generally advised for patients with paroxysmal or persistent AF who do not have severe left ventricular dysfunction or advanced/recently decompensated HF (NYHA III or IV). It provides an alternative to amiodarone, especially when extra-cardiac adverse effects pose significant concerns. It is advised to avoid dronedarone in patients with permanent AF (> 6 months) and in those taking digoxin.
3.6.7.3. Sotalol
Sotalol is a racemic mixture of two isomers, D- and L-sotalol. L-sotalol is a non-selective β-blocker with little Class III effects, while D-sotalol primarily blocks the IKr. At low doses (e.g. ≤160 mg/day), racemic sotalol primarily exerts β-blocking (Class II) effects, while Class III effects remain minimal. At doses starting from 160 mg/day, with a linear increase thereafter, D-L-sotalol exhibits both β-blocking and Class III anti-arrhythmic effects. Sotalol slows heart rate, prolongs cardiac APD (QT interval), and increases refractoriness throughout the heart, particularly at slower heart rates due to its reverse use dependence. In the AV node, it slows conduction and prolongs refractoriness, but it does not affect conduction velocity in fast-response tissues. In patients with reduced LVEF, sotalol can decrease the cardiac output and precipitate HF.106–108
Sotalol is less effective than amiodarone in maintaining SR after cardioversion of AF/AFL in patients with normal LV function, stable CAD, or valvular disease. However, it can enhance the success of DC cardioversion.32,109–112 To prevent AF, 80 mg twice daily may suffice, but a higher dosage may be prescribed if recurrences occur. Drug titration up to 160 mg twice daily (total 320 mg) may be performed based on arrhythmia complaints or results from ambulatory or remote rhythm monitoring. Sotalol also decreases heart rate even if AF persists,91,113,114 but it is not effective for cardioversion of AF. In patients with haemodynamically stable VT, i.v. sotalol is more effective than lidocaine for the acute termination of the arrhythmia.115 In addition, oral sotalol was significantly more effective than six Class I AADs (36 vs. 15% arrhythmia-free survival, P < 0.001) in preventing death and recurrences of VA at follow-up. This was demonstrated in 496 patients who were randomly assigned to undergo serial evaluation of drug efficacy using EP testing or Holter monitoring combined with exercise testing in the ESVEM trial.116 In patients with an ICD and ischaemic or non-ischaemic recurrent VA despite a β-blocker, sotalol reduces the recurrences of sustained VT/VF and the frequency of discharges, but it does not improve survival.32,117–119 Amiodarone plus a β-blocker is more effective for preventing ICD shocks than sotalol but has an increased risk of drug-related adverse effects.91 In survivors of acute MI, prophylactic D-sotalol therapy significantly reduces re-infarction, but not the incidence of SCD.120 Sotalol can be used in idiopathic VT from the right ventricular outflow tract (RVOT) when associated with severe symptoms or haemodynamic compromise. It may also be used in patients with a diagnosis of SQTS who present a contraindication to the ICD or refuse it, or in asymptomatic patients with a diagnosis of SQTS and a family history of SCD.121,122 Defibrillation threshold (DFT) reassessment after sotalol is not required.123
Sotalol prolongs the QT interval in a dose-dependent manner and may cause TdP (0.3–2%), especially if renal impairment and HF are present.32,107,121,124,125 Thus, sotalol is not advised in patients with less severe arrhythmias (PVCs, non-sustained VT), even if symptomatic, and may be appropriate for long-term treatment if close monitoring of QT, K+ levels, creatinine clearance (CrCl), and other pro-arrhythmia risk factors is provided.45,123,124,126
3.6.7.4. Dofetilide
Dofetilide blocks the IKr and may increase the INa,L via the inhibition of phosphoinositide-3-kinase leading to a prolongation of cardiac APD and refractoriness, without slowing intracardiac conduction.108,127,128 The APD prolongation is more prominent in the atria and at slow heart rates but diminishes as the heart rate increases (reverse use dependence).127 Dofetilide has no significant haemodynamic effects.127,129
Oral (in-hospital) dofetilide is effective for the conversion of persistent AF/AFL of >1 week duration and the maintenance of SR after cardioversion and to enhance the success of DC cardioversion in patients with SHD [HF, CAD, and hypertrophic cardiomyopathy (HCM)] or refractory to other AADs.32,92,130–135 In patients with HFrEF or recent MI, dofetilide restores and maintains the SR and reduces rehospitalizations for HF, and it does not increase all-cause or cardiac mortality.132–136 It is also effective to cardiovert macro re-entrant atrial arrhythmias to SR.44
Due to its QTc-prolonging effects and the associated risk of TdP (1–3.3% of patients), dofetilide is generally reserved for patients with symptomatic AF/AFL who are not candidates for catheter ablation and when other anti-arrhythmic drugs are ineffective or contraindicated. It is advised to avoid it in patients with risk factors for QT prolongation.132,135 It is advisable to be start dofetilide in a setting that provides continuous ECG monitoring in patients hospitalized for at least 3 days. Dosage adjustment is based on CrCl and QT interval assessment.
3.6.7.5. Ibutilide
Ibutilide is an IKr blocker that also activates INa,L, leading to the prolongation of cardiac APD and refractoriness. However, this prolongation is less pronounced at faster heart rates, demonstrating reverse use dependence.108,137,138 Ibutilide has minor haemodynamic effects or negative inotropic effects, and can be used safely in patients with SHD and prior MI.
Ibutilide is given intravenously and is advised for the rapid conversion of recent-onset AF/AFL to SR. It is more effective for the conversion of AFL137–147 and for facilitating the success of electrical cardioversion in patients with AF refractory to prior electrical cardioversion.140,142–146,148 It may also be appropriate for acute therapy of focal AT,149 macro re-entrant atrial arrhythmias,44,140,142–146 AVRT due to manifest or concealed accessory pathways,150 pre-excited AF,150 and the cardioversion of AFL and SVT during pregnancy in haemodynamically stable patients.44,92,151,152 However, it is not useful for long-term prevention of AF/AFL. Ibutilide undergoes rapid hepatic metabolism, primarily via CYP enzymes, leading to significant degradation before it can reach systemic circulation. This results in poor bioavailability, making oral administration ineffective. The half-life of ibutilide is only about 6 h, requiring frequent dosing if taken orally, which is impractical for maintaining therapeutic levels.
Ibutilide produces a dose-dependent QT interval prolongation and TdP can occur in up to 4% of patients during or within 1–4 h after drug infusion.138 Therefore, ibutilide is administered under continuous ECG monitoring, with resuscitation facilities readily available for at least 4 h, or longer in patients with hepatic or renal impairment.137,139,153
3.6.7.6. Vernakalant
Vernakalant is a multi-channel blocker with atrial specificity that prolongs atrial APD and refractoriness while slowing atrial conduction at depolarized potentials (∼−70 mV) and high heart rates, particularly during AF. It has minimal effects on ventricular or AV nodal refractoriness, heart rate, or blood pressure.154,155 Because of its fast dissociation kinetics from Na+ channels, vernakalant is not expected to cause conduction abnormalities or pro-arrhythmia once SR is restored. Intravenous vernakalant is advised for rapid cardioversion (mean time 8–14 min, > 50% conversion rate) of recent-onset AF (≤7 days for non-surgery patients and ≤3 days for post-cardiac surgery patients)92,154,156–159 and also facilitates electrical cardioversion in cardioversion-resistant AF (Figure 7).92,160 Although vernakalant produces a faster cardioversion of AF to SR and causes fewer pro-arrhythmic and extra-cardiac effects than Class Ic AADs and amiodarone, more comparative studies are needed.157,158,161–163 Drug efficacy decreases with the duration of AF, being ineffective for the conversion of AF lasting >7 days and in patients with AFL.92,158,163
Figure 7.

Electrocardiogram (ECG) tracing of Lead II demonstrating the termination of atrial fibrillation (AF) after a 7 min infusion of 350 mg of vernakalant in a patient with no structural heart disease. The transition to sinus rhythm is marked by the appearance of normal P waves (P), indicating successful restoration of organized atrial activity. This highlights the efficacy of vernakalant in achieving cardioversion in patients with AF. Electrocardiogram was recorded at a speed of 5 mm/s and a sensitivity of 10 mm/mV.
3.6.8. Class IIIb
3.6.8.1. Nicorandil
This anti-anginal drug is a nitric oxide (NO) donor and sarcolemmal and mitochondrial ATP-dependent K+ (KATP) channel opener, producing vasodilation of coronary arteries and venous capacitance vessels.164–166 During ischaemia/reperfusion, mitochondrial KATP channel opening exerts cardioprotective effects via hyperpolarization of the membrane potential that improves intracardiac conduction, shortens ventricular APD and refractoriness, prevents intra-cellular Ca2+ overload and suppresses triggered-induced arrhythmias.164–166 Nicorandil decreases ischaemia-induced VA166 and improves the no-reflow phenomenon and VA in patients undergoing percutaneous coronary angioplasty.164,167–169 In patients with LQTS1, nicorandil shortens the QT, improves repolarization abnormalities and abolishes EADs and recurrence of syncope.170,171
3.6.9. Class IIa
3.6.9.1. Bisoprolol, metoprolol, carvedilol, nadolol, and propranolol
Chronic autonomic dysfunction promotes cardiac remodelling, including hypertrophy, apoptosis and fibrosis. It also contributes to the progression of multiple CVDs, indirectly promoting a vulnerable substrate for arrhythmias. Moreover, acute autonomic imbalance is a well-accepted trigger of cardiac arrhythmias.6,172 Accordingly, β-blockers play a major role in the treatment of CVD. Here, the properties of β-blockers relevant for their anti-arrhythmic effects are briefly summarized.
Typically, β-blockers are sub-divided into three generations: first-generation β-blockers (e.g. propranolol) have similar affinity for β1- and β2-adrenoceptor sub-types, second-generation β-blockers (e.g. metoprolol, atenolol, or bisoprolol) have a higher affinity for β1-adrenoceptors, and third-generation β-blockers (e.g. carvedilol) have additional α-blocking properties.6 Most of the pro-arrhythmic effects of sympathetic stimulation have been attributed to β1-adrenoceptors and may involve Ca2+ overload due to elevated heart rates (themselves promoted by sympathetic stimulation of HCN channels) and hyperphosphorylation of Ca2+-handling proteins, thereby promoting triggered activity. In addition, sympathetic stimulation can facilitate re-entry-promoting repolarization instability when the slow delayed rectifier K+ current (IKs) is downregulated or dysfunctional due to genetic mutations (in the case of LQTS Types 1 and 5)173 or in the presence of abnormal autonomic innervation.174
All β-blockers inhibit automaticity and have negative chronotropic effects. Their inhibition of L-type Ca2+ channel phosphorylation, decreasing ICa,L, contribute to the β-blocker-induced inhibition of AV conduction (negative dromotropy). The reduction in phosphorylation of L-type Ca2+ channels and other Ca2+-handling proteins also reduces intra-cellular Ca2+ levels, explaining the potential negative inotropic effects of β-blockers in the acute setting. This reduction in Ca2+ levels also decreases the likelihood of ectopic (triggered) activity.45,173 Finally, β-blockers reduce electrophysiological heterogeneity caused by inhomogeneous autonomic innervation. With long-term use, they may help prevent pro-arrhythmic remodelling by lowering myocardial energy consumption and oxidative stress, partly due to their negative inotropic and chronotropic effects.
Due to their negative dromotropic effects and rapid onset of action, β-blockers are the first-line treatment for rate control in AF.92 In addition, β-blockers suppress PVCs, reduce the likelihood of VT, (in)appropriate ICD interventions, and SCD. Overall, they improve morbidity and mortality in a wide range of patients, including in the setting of acute coronary syndrome (ACS), MI, HF, LQTS, and CPVT.45 The prognostic benefit of β-blocker seen in HF patients with SR has not been consistently detected in patients with AF.175 Perioperative β-blocker therapy is commonly used for the prevention of post-operative AF after cardiac surgery, but must not be used in patients undergoing non-cardiac surgery.92
Although all β-blockers have anti-arrhythmic properties and detailed comparisons between different β-blockers are rare, a number of relevant clinical distinctions have to be noted.176 Non-selective β-blockers (propranolol and particularly nadolol) appear more effective than β1-adrenoceptor selective blockers in patients with LQTS or CPVT,177 possibly due to a sensitization of β2-adrenoceptors with β1-adrenoceptor selective blockers.173 In agreement, the combination of i.v. amiodarone and oral propranolol is safe, effective, and superior to amiodarone and metoprolol in the management of electrical storm (ES) in ICD patients.178 The better efficacy of nadolol compared with propranolol has been partially attributed to better compliance due to more favourable pharmacokinetic properties. In particular, propranolol is highly lipophilic, allowing it to cross the blood-brain barrier and is therefore associated with more central nervous system (CNS) side effects than nadolol.173 In addition, as mentioned earlier, short-acting β-blockers like propranolol are advised to be taken with food to improve absorption, as they are primarily metabolized in the liver and undergo significant first-pass metabolism. This hepatic processing can lead to substantial interindividual variability in plasma drug concentrations, necessitating careful dosing and monitoring. If a short-acting, hepatically metabolized β-blockers appear ineffective, switching to a renally excreted β-blocker such as nadolol may be a more effective alternative. Finally, propranolol appears to have a higher affinity for Na+ channels than other β-blockers, which may have pro- or anti-arrhythmic consequences.173 As such, nadolol is among the most commonly used β-blockers in patients with channelopathies.177 On the other hand, carvedilol stabilizes RyR2 gating and has anti-inflammatory and anti-oxidant effects. Experimental work using carvedilol analogues without β-blocking properties has suggested that direct RyR2-stabilizing effects of carvedilol may contribute to its anti-arrhythmic effects.5 Carvedilol also possesses α-blocking properties, which can promote vasodilation and potentially lead to hypotension. Finally, pharmacokinetic considerations as well as indications for specific comorbidities may direct the choice of individual β-blockers (see Supplementary material online, Table S3).176,179
3.6.9.2. Other (nebivolol, esmolol, and landiolol)
Nebivolol is a selective β1-adrenoceptor blocker with an NO-potentiating vasodilatory effect, which makes it suitable for the prevention of arrhythmias particularly in patients with CAD.180 Esmolol is a selective β1-adrenoceptor blocker with a rapid onset (1–2 min) of action but a short duration of action (∼10 min), which is used intravenously to terminate supraventricular arrhythmias.181 Landiolol, another selective β1-adrenoceptor blocker with a very rapid onset (<1 min) of action and very short half-life (undefined), was developed by modifying the chemical structure of esmolol to produce a compound with a higher β1-adrenoceptor selectivity and potency without increasing its duration of action.182 Landiolol is a pure S-enantiomer with minimal negative inotropic effects, in contrast to esmolol.
3.6.10. Class IIb
3.6.10.1. Isoprenaline (isoproterenol)
Isoprenaline, also known as isoproterenol in some countries, such as the USA, is a non-selective β-adrenoceptor agonist. Activation of both β1- and β2-adrenoceptors causes the α-sub-unit of G-protein-coupled receptors to exchange GMP for GTP, activating them, and allowing the α-sub-unit to dissociate from the β and γ-sub-units.
Dissociation of the α-sub-unit activates adenylate cyclase, converting ATP to cyclic AMP. Cyclic AMP activates protein kinase A (PKA), which phosphorylates cardiac L-type Ca2+ channels. These channels depolarize SA and AV nodal cells by inward active transport of Ca2+ ions. Activation of β₁-adrenoceptors increases contractility, enhances conduction velocity, accelerates relaxation, raises heart rate, and shortens the QT. Activation of β2-adrenoceptors leads to glycogenolysis in the liver, glucagon release from the pancreas, and activation of the renin-angiotensin-aldosterone system. Isoprenaline is metabolized by catechol O-methyltransferase and its elimination half-life following i.v. administration is 2.5–5 min.
Patients experiencing an overdose may present with tachycardia, arrhythmias, palpitations, angina, hypotension, or hypertension. In case of overdose, treatment involves stopping the administration of isoprenaline and monitoring blood pressure, pulse, respiration, and ECG.
Isoprenaline infusion is advised for ES due to bradycardia-related acquired LQTS, BrS, early repolarization syndrome (ERS) and SQTS.183 It is highly effective in acute suppression of recurrent ICD shocks in these latter settings. It may also be useful in the acute management of unstable third-degree AV block while awaiting definitive pacemaker (PM) implantation. Isoprenaline infusion is contraindicated in ACS, HCM and uncontrolled hypertension. The main side effects are sinus tachycardia, vasodilatation, tremor, sweating, and nausea.
3.6.11. Class IIc
3.6.11.1. Atropine
Atropine is a competitive antagonist of muscarinic M2 receptors located in SA and AV nodes. It reverts sinus bradycardia to normal SR, shortens AV nodal refractoriness, and enhances AV node conduction but has little effect on infranodal conduction tissues.184 It is advised for patients with symptomatic or haemodynamically unstable sinus bradycardia and second- or third-degree AV block.185–188 Additionally, it is used for symptomatic or unstable bradyarrhythmias caused by increased vagal activity in the setting of acute inferior MI and for CA associated with brady-asystole.185–190 However, atropine may worsen AV conduction block in the presence of intra-His or distal conduction disease191 and may be ineffective in heart transplant recipients due to vagal denervation.192
3.6.12. Class IId
3.6.12.1. Digoxin
Digoxin increases cardiac vagal tone via activation of IKACh in the atria, which inhibits If in the SA node and ICa,L in the AV node, and activates acetylcholine-activated potassium current (IK,Ach) in the atria. As a result, digoxin decreases SA automaticity, prolongs AV conduction and refractoriness and produces a non-uniform shortening of atrial APD and refractoriness, respectively.92,130,193 Digoxin is advised to slow the ventricular rate in patients with permanent and persistent AF. However, digoxin slows ventricular rate at rest, when vagal tone predominates, but is less effective when sympathetic activity increases (i.e. during exercise, fever, hyper-thyroidism, post-operative AF).92,130 Thus, digoxin has been replaced by β-blockers and diltiazem/verapamil that control heart rate both at rest and/or during exercise. Digoxin can be combined with these drugs when the ventricular rate remains uncontrolled, or these drugs are not tolerated or are contraindicated.92,113,130,194–196 Because of its positive inotropic effect, digoxin and/or β-blockers are appropriate for rate control in patients with HFrEF.92,130 Digoxin may be used to slow the ventricular rate in patients with AF and ACS or with acute HF when β-blockers and diltiazem/verapamil are contraindicated. In HF patients who cannot tolerate higher doses of β-blockers, low doses of digoxin can be added to reach the desired heart rate and symptom control. It is important to note, as mentioned before, that both dronedarone and amiodarone increase digoxin levels, which potentially may cause drug toxicity. Digoxin abbreviates atrial refractoriness and is ineffective in the cardioversion of AF/AFL to SR or the maintenance of SR and is advised to be avoided in pre-excited AF.92,130,196,197
3.6.13. Class IIe
3.6.13.1. Adenosine
Adenosine is a purine nucleoside that interacts with adenosine Gi-protein–coupled A1 receptors in atrial muscle, SA, and AV nodal cells.198,199 It activates IK,ACh that hyperpolarizes the membrane potential, slows SA PM activity and shortens atrial APD and refractoriness. It also inhibits the If and reduces adenylyl cyclase activity and intra-cellular cAMP levels, which indirectly inhibits the ICa,L during sympathetic stimulation. As a consequence, adenosine slows sinus rate and AV conduction (Figure 8A and B) and prolongs AV refractoriness, leading to a transient AV block responsible for AV nodal-dependent tachycardia termination, and abolishes EADs/DADs induced by catecholamines.155,198,199 Stimulation of cardiac Gs-protein–coupled A2 receptors in endothelium and vascular smooth muscle results in coronary vasodilation.
Figure 8.

Electrocardiogram (ECG) tracings illustrating the effects of adenosine on different atrial rhythms: sinus rhythm (A), atrial tachycardia (B), atrial flutter (AFL, C), and paroxysmal supraventricular tachycardia (PSVT, D). (A) Sinus rhythm at a rate of 88 b.p.m. slows significantly with PR interval prolongation following adenosine infusion. (B) Atrial tachycardia at 125 b.p.m., characterized by a non-sinus P-wave morphology (P′). Initially, conduction is 1:1 atrioventricular (AV) (left). After adenosine administration, conduction changes to 2:1 AV (right) without a significant change in atrial rate. (C) Common AFL with 2:1 AV conduction. Adenosine-induced AV block reveals prominent F waves, enhancing visualization of the flutter waves. (D) Termination of atrioventricular re-entrant tachycardia (AVRT) mediated by a left-sided concealed accessory pathway. Termination occurs after AV node conduction block, interrupting conduction of the final retrograde P′ wave and restoring sinus rhythm. All ECGs were recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Due to its rapid onset and short duration of action (10–30 s), i.v. adenosine is the drug of choice for the rapid termination of SVT when vagal manoeuvres are ineffective. It is effective in treating sinus node (SN) re-entry tachycardia, triggered focal AT, AV nodal re-entrant tachycardia (AVNRT), and AVRT due to accessory pathways (Figure 8D). Additionally, it may be useful for certain VTs and SVTs in congenital heart disease.44,198–202. Adenosine is preferable to verapamil or diltiazem, particularly in patients treated with i.v. β-blockers or with a history of HF or severe hypotension, and in children. Adenosine also slows sinus rate, may cause sinus exit block and can terminate SAN re-entry.44,203 Like digoxin, adenosine is unlikely to terminate AF or AFL because it shortens atrial refractoriness, which promotes re-entry (Figure 8C).92,130 For the same reason, it does not interrupt macro re-entrant ATs unless the circuit involves the AV node,92,204 and does not affect conduction velocity through the His-Purkinje or normal accessory pathways (Figure 9). However, conduction may be blocked in pathways with long conduction times or decremental conduction properties.44,203–205 In general, VT does not respond to adenosine, but adenosine can terminate idiopathic right outflow tract VT caused by cAMP-mediated triggered DADs and, less commonly, left fascicular idiopathic VT.
Figure 9.

Twelve-lead electrocardiograms (ECGs) of a patient with Wolff–Parkinson–White (WPW) syndrome caused by a right posteroseptal accessory pathway which becomes more prominent following adenosine infusion. (A) Pre-excitation becomes more apparent following the infusion of 12 mg of adenosine, which blocks conduction through the atrioventricular (AV) node. This is evidenced by a pronounced delta wave, indicative of increased conduction via the accessory pathway. (B) Pre-excitation diminishes as AV nodal conduction resumes after the effects of adenosine dissipate, reducing the contribution of the accessory pathway to ventricular depolarization. These findings demonstrate the dynamic inter-play between AV nodal conduction and accessory pathway activation in WPW syndrome, highlighting the diagnostic utility of adenosine in unmasking pre-excitation. The recordings were obtained at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Adenosine may cause transient new arrhythmias, such as AF, at the time of cardioversion because it heterogeneously shortens atrial APD and refractoriness and produces transient sympathetic stimulation through baroreflex activation in response to hypotension. Adenosine may lead to hyperpolarization of dormant PV myocytes, increasing their excitability and automaticity206,207 and could accelerate pre-excited atrial arrhythmias.199,208
3.6.14. Class IV
3.6.14.1. Verapamil and diltiazem
These agents block cardiac ICa,L, decrease heart rate and cardiac contractility, slow conduction, and prolong refractoriness at the AV node. They also suppress abnormal automaticity in depolarized cells and inhibit triggered activity induced by EADs.209,210 However, they do not alter excitability, conduction velocity, or refractoriness in atrial or ventricular muscle or His-Purkinje fibres that generate Na+-driven APs. Since SN rate and cardiac contractility may be suppressed, these agents are advised to be used with caution in patients with impaired left ventricular function or those receiving β-blockers. However, the impact on sinus rate depression may be less pronounced, as their vasodilatory effect induces a sympathetic reflex activation that counteracts their direct cardiac effects.
Because of their depressant effect on the AV node, diltiazem and verapamil are advised to control the ventricular rate at rest and during exercise in patients with AF/AFL. They may be used alone or in combination with β-blockers or digoxin.113,195,211–218 Intravenous verapamil and diltiazem are often used to slow ventricular heart rate in the acute setting in patients without pre-excitation.216,219–221
Verapamil and diltiazem are advised for acute ventricular rate control in haemodynamically stable patients with SVT, including focal or multi-focal AT,222–224 narrow QRS tachycardia, IAST, AFL, macro re-entrant atrial arrhythmias,219,223,225 AVNRT,220,226 and AVRT if no signs of pre-excitation are present.220,227–231 They are appropriate when vagal manoeuvres and adenosine fail. Verapamil and diltiazem are not advised for cardioversion of AF or AFL or for maintaining SR after cardioversion, as they shorten atrial refractoriness, potentially promoting re-entry.
Verapamil is advised in idiopathic LV fascicular VT, symptomatic patients with papillary muscle tachycardia and mitral and tricuspid annular VT.45,232–234 Occasionally, diltiazem and verapamil can suppress VA associated with myocardial ischaemia. In short-coupled TdP, i.v. verapamil can suppress and prevent ES or recurrent ICD interventions.235,236
Verapamil and diltiazem are contraindicated in patients with hypotension or HFrEF,200,202,237–239 haemodynamic instability or pre-excited AF as they may increase the ventricular response.240,241 They can also cause severe haemodynamic deterioration in patients with wide QRS tachycardia of unknown aetiology.233,242,243
Finally, there are differences between verapamil and diltiazem. Verapamil has stronger negative inotropic and chronotropic effects, making it more effective for arrhythmias like AF and SVT, but it is more likely to cause bradycardia, worsen HF, and lead to constipation due to its impact on gastrointestinal smooth muscle. It also has sympatholytic properties and can relieve bronchospasm. Diltiazem has a balanced action on both vascular smooth muscle and the heart, making it more suitable for hypertension and angina without significantly reducing cardiac output. It is generally better tolerated, with fewer side effects such as constipation, but it is more likely than verapamil to cause leg oedema. It is preferred for patients who require a gentler approach to rate control or blood pressure management.
3.6.14.2. Bepridil
This anti-anginal drug is a multi-channel blocker and acts intra-cellularly as a calmodulin antagonist, which reduces sarcoplasmic reticulum Ca2+ release and inhibits ischaemia-induced catecholamine release.244 Bepridil prolongs atrial and AV nodal refractoriness but has minor effects on ventricular refractoriness and reduces heart rate, peripheral vascular resistance, and blood pressure.244,245 Bepridil is effective for the conversion of persistent AF in patients without SHD and normal QT, and for blocking AV nodal conduction. It may decrease heart rate even if AF persists.59,244,246,247 The efficacy of bepridil in preventing AF recurrence is considered limited.59,248–250
4. Treatment by arrhythmia
4.1. General
Several considerations come into play when choosing an AAD, with a key determinant being whether the drug is intended for arrhythmia termination or prevention. While it is generally true that drugs effective in terminating a specific arrhythmia often exhibit preventive properties, exceptions abound. For instance, amiodarone is highly effective in preventing AF but has a weaker efficacy in terminating acute episodes. Beyond these dynamics, factors such as the route of administration and the drug's use-dependent or reverse use-dependent effects can significantly impact its effectiveness for prevention vs. termination of arrhythmias.
Additionally, AADs exert distinct effects on different regions of cardiac tissue. Class II and IV agents primarily slow conduction and prolong refractoriness in the sinus and AV nodes, while Class I and III agents predominantly affect the working myocardium at both atrial and ventricular levels (Figure 10). The presence of ventricular structural changes or scarring generally discourages the use of Class I AADs, particularly Class Ic, due to their potential to slow conduction and promote ventricular re-entry and pro-arrhythmia. Another key consideration is the degree of ventricular myocardial contractility depression and the risk of HF aggravation, which are most pronounced with Class Ic and Class IV agents but less significant with quinidine or amiodarone. Both pro-arrhythmic risks and extra-cardiac effects and toxicities—discussed in a separate section—are critical factors in the selection of an AAD. Recognizing these region-specific effects is essential for selecting the appropriate drug for each patient.
Figure 10.

Anti-arrhythmic drug (AAD) selection based on cardiac substrate and main target of action. This figure advises on the selection of AADs based on their primary target [sinus/AV nodes vs. working atrial (A) and ventricular (V) myocardium] and the presence of ventricular dysfunction, scarring, or heart failure (HF). Class 0, II, and IV agents [e.g. ivabradine, β-blocker (BB), Ca2+ channel blocker (CCB), and digoxin] are preferred for rate control by acting on the sinus and AV nodes. Class I and III agents (e.g. flecainide, propafenone, amiodarone, dronedarone, sotalol, and dofetilide) are used for rhythm control, but their choice depends on the structural integrity of the ventricles. Structural heart disease discourages Class I use, favouring Class III instead. In HF, amiodarone is the preferred option, while other AADs are generally avoided to prevent worsening of the condition. aIvabradine is primarily advised for slowing the sinus rate, with some evidence suggesting it may also influence AV nodal conduction. bBBs and CCBs also affect cardiac tissues beyond the sinus and AV nodes and may be the AADs of choice for certain disorders, such as ectopic atrial tachycardia (AT) or idiopathic fascicular ventricular tachycardia (VT), respectively. cDigoxin is less effective in sinus tachycardia compared with BBs or CCBs. However, digoxin toxicity can lead to severe bradycardia, sinus arrest, or junctional escape rhythms due to excessive vagal stimulation. dClass I and III agents also influence the sinus node and AV conduction but are not the preferred choices for this purpose. eDronedarone is not advised in patients with symptomatic HF or left ventricular ejection fraction (LVEF) <40%. fDofetilide does not worsen survival in HF with reduced ejection fraction (HFrEF) but can prolong the QT interval and cause torsades de pointes. gSotalol is not advised in patients with advanced HF or severe left ventricular dysfunction (LVEF <35%) due to the risk of worsening HF. VW, Vaughan Williams AAD classification.
Finally, it is important to keep in mind some key considerations for optimizing the safe and effective use of AADs, emphasizing patient education, risk management, and integrated care strategies (Box 2).
The following sections review the published literature on the use of specific AADs in the management of various arrhythmia disorders, both for prevention and termination. The key recommendations from the European Society of Cardiology (ESC) guidelines for selecting AADs to treat or prevent these conditions are summarized in Table 3, while Table 4 outlines the typical indications and contraindications of major AADs.
Table 3.
Advised AADs and agents for various heart rhythm disorders based on clinical practice guidelines
| First-choice AAD | Strength of advice | Second-choice AAD | Strength of advice | ESC guideline (year/topic) | |
|---|---|---|---|---|---|
| Tachycardia prevention | |||||
| Sinus tachycardiaa | Ivabradine or β-blockers | Medium | Alternative or combined | Medium | 2019 SVT |
| AT focal | β-blockers, CCBs, or Ic | Medium | alternative | 2019 SVT | |
| AFL | β-blockers or CCBsb | Medium | Amiodaronec | Low | 2019 SVT |
| AF—no SHD or HF | Ic or dronedarone | High | Alternative | 2024 AF | |
| AF—SHD or HFpEF/HFmrEF | Dronedarone | High | Alternative | 2024 AF | |
| AF—HFrEF | Amiodarone | High | 2024 AF | ||
| PSVT—non pre-excited | β-blockers or CCBs | Mediumd | Alternative | Medium | 2019 SVT |
| PVT/VF SHD or ischaemia | β-blockers and K+/Mg2+ repletion | High | Amiodarone | Medium | 2022 VA |
| PVCs/VT idiopathic from outflow tract or fascicular | β-blockers or CCBs or Ic | Mediumd | Alternative | 2022 VA | |
| PVCs/VT idiopathic from other origin | β-blockers or CCBs | High | Alternative or ablation | 2022 VA | |
| VT SHD | β-blockers | Highe | Amiodarone or sotalol | Medium | 2022 VA |
| TdP/VF non-SHD | Nadolol/propranolol (LQTS 1 and 2, CPVT) Mexiletine (LQTS 3) Quinidine (SQTS, idiopathic VF, ERS, Brugada) |
High and Medium | Flecainide (CPVT) | Medium | 2022 VA |
| Tachycardia termination control | |||||
| AT focal | Adenosine i.v. | Medium | CCBs i.v. β-blockers i.v. |
Medium | 2019 SVT |
| AFL | Ibutilide/dofetilide i.v. | High | Amiodarone | Low | 2019 SVT |
| AFL (HR control) | β-blockers or CCBsb | Medium | Amiodaronec | Low | 2019 SVT |
| AF—no SHD/HF | Vernakalant i.v.f or flecainide/propafenone i.v. or PITP |
High | Alternative Ibutilideg |
2024 AF | |
| AF—SHD/HF | Vernakalant i.v.f or amiodarone i.v. |
High | Alternative | 2024 AF | |
| AF—no SHD or HF (HR control) | β-blockers, CCBs, or digoxinh | High | Alternative | 2024 AF | |
| AF—SHD or HF (HR control) | β-bockers or digoxin | High | Alternative | 2024 AF | |
| Narrow QRS T | Adenosine i.v.i | High | CCBs i.v. β-blockers i.v. |
Medium | 2019 SVT |
| Wide QRS T | Adenosine i.v. | High | Procainamide i.v. | Medium | 2019 SVT |
| SVT—Pre-excited | Ic or ibutilide or procainamide i.v.i | Medium | Alternative | 2019 SVT | |
| AF—Pre-excited | Ibutilide or procainamide i.v. | Medium | Ic | Low | 2019 SVT |
| PVCs/VT idiopathic from outflow tract or fascicular | β-blockers (outflow tract) or CCBs (fascicular) i.v. | High | Alternative | 2022 VA | |
| VT SHD or unknown | Procainamide i.v.j | Medium | Amiodarone | Low | 2022 VA |
| TdP/VFj non-SHD | Mg2+, K+, β-blockers (congenital LQTS) Isoprenaline (acquired LQTS, idiopathic VF, ERS, Brugada) Verapamil, Quinidine (idiopathic VF) |
High Medium |
2022 VA | ||
| PVT/VFj SHD or ischaemia | β-blockers and K+/Mg2+ repletion | High | Amiodarone | Medium | 2022 VA |
Alternative, the second alternative AAD is advised to be used when two options are offered.
Abbreviations: AADs, anti-arrhythmic drugs; AF, atrial fibrillation; AFL, atrial flutter; AT, atrial tachycardia; CCB, calcium channel blockers; CPVT, catecholaminergic PVT; ERS, early repolarization syndrome; HF, heart failure; HFpEF/HFmrEF/HFrEF, HF with preserved/mildly reduced/reduced left ventricle ejection fraction; HR, heart rate; LQTS, long QT syndrome; PITP, pill-in-the-pocket; PVC, premature ventricular contraction; PVT, polymorphic VT; SHD, structural heart disease; SVT, supraventricular tachycardia; TdP, torsades de pointes; VA, ventricular arrhythmias; VF, ventricular fibrillation; VT, ventricular tachycardia.
aThe treatment of reversible causes is the first-line option.
bUsed for HR control with little effect on AFL prevention.
cSotalol and dofetilide are also recommended in the 2023 AHA/ACC/HRS AF guidelines, with dronedarone identified as another reasonable alternative to amiodarone in this consensus.
dCatheter ablation is advised as the first-line option.
eβ-Blockers are advised as a first-line option to treat HF but have low efficacy to prevent sustained episodes of VT in this setting.
fVernakalant can be given to patients with SHD but no severe aortic stenosis, recent ACS or moderate-to-severe HF.
gIn geographies with no access to vernakalant or to i.v. type Ic drugs advised by the 2023 AHA/ACC/HRS AF guidelines.
hMay have limited efficacy if high adrenergic tone.
iVagal manoeuvres are the first-line option.
jElectrical cardioversion is the first-line option.
Table 4.
Typical indications and contraindications of major AADs
| Modified VW class | AAD | Main indications | Not to be used/main contraindication |
|---|---|---|---|
| 0 | Ivabradine | Inappropriate sinus tachycardia | AF termination |
| Ia | Ajmaline | AF (pre-excitation) termination MVT (no significant SHD) termination |
BrS, HFrEF |
| Quinidine | PVT (channelopathies) prevention | Long QT | |
| Procainamide | MVT (SHD) termination | HFrEF | |
| Disopyramide | AF (vagotonic) prevention | HFrEF | |
| Ib | Lidocaine | PVT/VF (ischaemia) termination | Bradycardia |
| Mexiletine | TdP (LQTS 3) prevention | Bradycardia | |
| Phenytoin | JET (digitalis toxicity) termination | Bradycardia | |
| Ic | Flecainide/Propafenone | AF (idiopathic) prevention/termination WPW syndrome |
BrS, SHD |
| Antazoline | AF (idiopathic) termination | Bradycardia, SHD | |
| Pilsicainide | AF (idiopathic) termination | SHD | |
| Id | Ranolazine | AF prevention/termination | Long QT except LQT3 |
| IIa | β1-blockers (e.g. bisoprolol, metoprolol) | AF/AFL rate control VT/PVCs (idiopathic) |
Bradycardia |
| β1- + β2-blockers (e.g. nadolol, propranolol) |
TdP (LQTS 1 and 2) prevention CPVT |
Bradycardia | |
| IIb | Isoprenaline | TdP (acquired LQTS) PVT (BrS) |
CPVT |
| IIc | Atropine | Sinus bradycardia | Inappropriate sinus tachycardia |
| IId | Digoxin | AF/AFL rate control | Amyloidosis |
| IIe | Adenosine | PSVT termination | Asthma/COPD |
| III | Amiodarone | AF/AFL (HFrEF) prevention/termination MVT (SHD) prevention/termination |
Bradycardia |
| Dronedarone | AF/AFL (SHD) prevention | Permanent AF, HFrEF | |
| Dofetilide | AF/AFL (HFrEF) prevention | Long QT | |
| Ibutilide | AFL termination | Long QT | |
| Sotalol | MVT (SHD) prevention | CKD, hypokalaemia, hypomagnesaemia or bradycardia | |
| Vernakalant | AF (≤7 d) termination | Aortic stenosis, NYHA III/IV | |
| IV | Verapamil/ Diltiazem |
AF/AFL rate control VT (fascicular) prevention/termination |
MVT (SHD), HFrEF |
Conditions for indications and contraindications are stated in brackets.
Abbreviations: AADs, anti-arrhythmic drugs; AF, atrial fibrillation; AFL, atrial flutter; BrS, Brugada syndrome; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; CPVT, catecholaminergic polymorphic ventricular tachycardia; HFrEF, heart failure with reduced ejection fraction; JET, junctional ectopic tachycardia; LQTS, long QT syndrome; LVEF, left ventricular ejection fraction; MVT, monomorphic VT; PVC, premature ventricular contraction; PVT, polymorphic ventricular tachycardia; RVOT, right ventricular outflow tract; SHD, structural heart disease; TdP, torsades de pointes; VF, ventricular fibrillation; VT, ventricular tachycardia; WPW, Wolff–Parkinson–White.
4.2. Arrhythmia prevention
4.2.1. Atrial arrhythmias
Atrial arrhythmias, including AF and AFL, are the most common sustained cardiac rhythm disorders and are associated with an increased risk of stroke and HF. Anti-arrhythmic drugs play a key role in the management of AF, AFL, and other atrial arrhythmias. The preferred AADs for preventing AF and AFL are shown in Figure 11.
Figure 11.

Schematic representation of the preferred anti-arrhythmic drugs (AADs) for prevention of atrial arrhythmias. The figure serves as a general reference for selecting the most appropriate drug; however, the final choice—or consideration of alternative therapeutic options [e.g. catheter ablation for cavotricuspid isthmus–dependent atrial flutter (AFL)]—is based on the general patient characteristics and conditions, as outlined in the relevant sections of this document. Additionally, not all AADs are available in all regions. For secondary or alternative drug options, refer to Table 3. AAs, atrial arrhythmias; AF, atrial fibrillation; LVEF, left ventricular ejection fraction; SHD, structural heart disease.
4.2.1.1. Premature atrial contractions and focal atrial tachycardia
Independent of underlying heart disease, β-blockers are a particularly good choice for adrenergic premature atrial contractions (PACs) or focal non-sustained ATs (Box 3). If no or minimal heart disease is present, flecainide251 and propafenone252 may be used when β-blockers are ineffective. Flecainide is especially effective in vagal- or (relative) bradycardia-dependent PACs or non-sustained AT (NSAT). Note that β-blockers may be pro-arrhythmic by inducing bradycardia-dependent ectopy. If β-blockers or Class Ic drugs are ineffective, then sotalol may be used.203,253,254 Propranolol, verapamil, and procainamide have been reported to specifically suppress PACs from the PVs.255 Rate slowing with β-blockers, digoxin and verapamil/diltiazem may help suppress symptoms in NSATs. Amiodarone is advised to be used only exceptionally in patients with significant SHD.203,253,254 Flecainide and verapamil were shown effective in unspecified recurrent SVT, including ATs.256 Case reports suggest ivabradine may be useful,257,258 and amiodarone must only be used as a last resort drug therapy.259,260 Combination of sotalol with flecainide—producing an amiodarone-like Ic plus III effect—may be tried in resilient cases. Focal PACs and ATs with tachycardiomyopathy are best managed with catheter ablation.253
Box 2 Practical tips on using AADs.
-
To enhance safety of AAD use, it is advised to involve patients in AAD treatment:
Patients have to be taught about warning symptoms (progressive palpitations, unexpected dizzy spells or syncope, development of chest pain, dyspnoea, and recent-onset exercise intolerance)
Patients have to be taught about critical circumstances (to avoid concomitant QT-prolonging drugs, to report when a new drug is prescribed, the risk of developing hypokalaemia with diarrhoea and/or vomiting, excessive sweating during fever, dietary deficiencies, or the addition of diuretics)
Patients have to be taught about over-the-counter agents, including supplements and herbal remedies, and which may interact with AADs, potentially affecting their efficacy or increasing the risk of adverse effects. Patients have to promptly report any additions or discontinuations of such agents
These have to be repeated during regular follow-up visits
Integrated nurse-driven care with experienced nurses supervised by the physician can substantially improve AAD management
It is advisable to perform an exercise test on Class Ic drugs to rule out exercise-induced excessive QRS widening or ventricular tachycardia, if in doubt
Flecainide or propafenone are not contraindicated in patients with a high cardiovascular risk profile (e.g. incidental Agatston score <400) in the absence of angina pectoris or with uncomplicated mild left ventricular hypertrophy (both in the absence of left ventricular scar tissue and dysfunction)
CNS side effects of Class Ic drugs may be tackled by changing to an extended-release formulation
If dronedarone is prescribed correctly, patients may greatly benefit from its often-overlooked pleiotropic effects, including amelioration of acute coronary syndrome, reduction of stroke rate and improving of survival. Dronedarone must always be taken with food to increase its oral bioavailability
Class Ic drugs exert excess anti-arrhythmic effects during tachycardia (atrial or ventricular) and sotalol and amiodarone during bradycardia: therefore, observe ventricular Class Ic effects during infusion for tachycardia conversion or with exercise and ventricular Class III effects after cardioversion. Use dependency of dronedarone is unknown. Direct clinical manifestations of use dependency of AADs at the atrial level are not well known
Abbreviations: AAD, anti-arrhythmic drug; CNS, central nervous system.
4.2.1.2. Inappropriate sinus tachycardia
General measures, such as ruling out any cause for sinus tachycardia or treating aggravating factors, are advisable before initiating any AAD therapy for inappropriate sinus tachycardia.44 β-Blockers and ivabradine, up-titrated to the effective dose, may bring relief of symptoms, and both drugs may be combined44,261 to enhance efficacy. Hydropyridine CCBs may be pro-arrhythmic by causing rebound sinus tachycardia.44,262
4.2.1.3. Multi-focal atrial tachycardia
Management of the underlying condition, in particular lung diseases and HF, is of utmost importance for chronic prevention. Digoxin is ineffective for the treatment and may contribute to its cause. Verapamil or diltiazem (in the absence of HFrEF),222,224 or β-blockade may be helpful aiming at rate control. Class I or Class III drug therapy usually fails but sometimes the combination, looking for an amiodarone-like ‘Class Ic plus III effect’, is a rational option in resilient cases when there are no contraindications (Box 3).
Box 3 Practical tips for use of AADs for atrial arrhythmia.
Focal atrial tachycardia
Diurnal pattern may reveal adrenergic PACs or NSATs suggesting β-blocker as preferred rhythm control therapy. If a vagal pattern prevails, flecainide should be tried first and β-blockers should be avoided. Alternatively, disopyramide may be particularly effective for vagally mediated AF when the former fails or cannot be used
Rate control drugs are a rational option in NSATs
Multi-focal atrial tachycardia
Combining sotalol with flecainide, aiming to achieve an amiodarone-like effect through the synergy of Class Ic and Class III properties, could be a potential option in refractory cases for patients without significant heart disease. However, this approach has to be undertaken with extreme caution, as it requires careful and regular monitoring due to the risks of pro-arrhythmia, myocardial contractility depression, and the limited clinical evidence supporting its use
Atrial flutter
Rhythm control of AFL is rarely achieved with AADs; catheter ablation is generally preferred, especially for cavotricuspid isthmus–dependent cases (see Figure 6)
Nevertheless, using cardioversion as needed for infrequent AFL is an excellent, patient-specific treatment option
Rate control is also difficult to achieve in AFL, but when effective—using β-blockers, verapamil, diltiazem, or combinations thereof—allows long-term treatment in patients who are not suitable for catheter ablation.
Beware of inadvertent flutter elicited by Class Ic drugs during termination or prevention of AF and know how to recognise pseudo-ventricular tachycardias due to aberrant conduction (Figure 6B)
General
Atrial arrhythmias and sinus node dysfunction often co-exist. AADs with significant effect on the sinus node are discouraged when the latter is suspected
Abbreviations: AAD, anti-arrhythmic drug; AF, atrial fibrillation; AFL, atrial flutter; NSAT, non-sustained atrial tachycardia; PAC, premature atrial contraction.
4.2.1.4. Atrial flutter/macro re-entrant atrial tachycardia
Patients refusing catheter ablation of AFL or infrequent recurrences may opt for scheduled cardioversion (Box 3).263 Alternatively, rate control using β-blockers, verapamil or diltiazem may be applied if recurrences are relatively well tolerated. Rate control may be difficult to achieve, and frequently a combination of rate control drugs is needed.92,203 Therefore, it is important to emphasise that catheter ablation is advised as the first-line therapy for AFL, particularly when it is cavotricuspid isthmus-dependent. For acute termination, cardioversion may be supported by chronic AAD therapy. If no significant SHD is present, flecainide or propafenone can be used.264–266 However, dronedarone,98 sotalol,267 or amiodarone268–270 may be more effective in broader clinical scenarios (Figure 11).
Atrial flutter may occur in patients treated to prevent recurrences of AF or drug termination of AF with Class Ic drugs or amiodarone, with the classical saw-tooth flutter pattern potentially changed (hampering recognition of the classical flutter pattern) and the flutter revolution time being prolonged, typically to 240–360 ms (Figure 6B).131 This lengthened cycle may be associated with 1:1 AV conduction. The wide QRS tachycardias associated with aberrant conduction during 1:1 AV conduction may show a bizarre QRS mistaken for VT (Figure 6).271 To prevent 1:1 AV conduction, it is common to prescribe one of the negative dromotropic drugs, like β-blockers, verapamil or diltiazem, along with the prophylactic Class Ic drug (not amiodarone, which itself may provide sufficient AV block), but it is particularly useful to advise patients to avoid exercise or stress during breakthrough AF or flutter.92 Deterioration of aberrancy with pseudo-VT to true VT or VF is highly unlikely to occur if the indication for Class Ic agents was correct (absence of underlying heart disease). If AFL occurs during flecainide treatment, cavotricuspid isthmus ablation is a treatment of choice,272,273 and, in any event, the Class Ic agent is advised to be discontinued.
Spontaneous termination of a breakthrough flutter while on a Class Ic drug is very unlikely.131 If concomitant HF occurs during atrial flutter, which is not amenable to cardioversion and ablation, β-blockers are the most preferred rate control therapy.
Flutter or macro re-entrant tachycardia during the blanking period after AF ablation (about 8 weeks) may be managed by cardioversion and AADs since they often resolve spontaneously.
Box 4 Practical tips to use AAD for AF prevention.
Safety must take precedence over efficacy when selecting AADs for AF prevention
Recognition of the autonomic pattern of paroxysms may guide specific AAD treatment decisions in vagal or adrenergic AF
Despite add-on β-blocker or verapamil/diltiazem, advise patients on Class Ic drugs to avoid exercise during breakthrough episodes until AF has stopped or cardioversion has been performed. This may help to avoid side effects of these add-on drugs.
Oral amiodarone may convert 25% of persistent AF patients, thus avoiding cardioversion.
A decrease of the dose of rate control medication may be needed shortly after starting amiodarone to prevent bradycardia.
In cases of pro-arrhythmia or breakthrough AF, evaluate for potential triggers, including ischaemia, heart failure, electrolyte imbalances, thyroid dysfunction, infections, drug interactions, and abnormal plasma AAD concentrations (e.g. due to non-adherence or dosing errors).
Abbreviations: AAD, anti-arrhythmic drug; AF, atrial fibrillation.
4.2.1.5. Atrial fibrillation
Rhythm control in AF, including the application of AAD therapy, is increasing274 and can be applied safely (Box 4).275,276 Flecainide, propafenone, sotalol, dofetilide,274 dronedarone, and amiodarone are among the most frequently used drugs to maintain SR and prevent recurrences (see below ‘Anti-arrhythmic drugs for atrial fibrillation‘, Figure 11). In the past decades, flecainide use has increased, while sotalol use has declined.277 Quinidine and disopyramide are best avoided for the risk of pro-arrhythmia,121 and procainamide is hardly used to prevent AF due to complex drug application, the need for sampling plasma concentrations, and potentially severe side effects. Current ESC guidelines recommend dronedarone, flecainide, or propafenone for AF prevention in patients with no or minimal SHD; amiodarone and dronedarone in patients with CAD, valvular disease, or HF with preserved ventricular ejection fraction (HFpEF); and amiodarone in patients with HFrEF. In the USA, where dofetilide is available, it is also advised for patients with AF and HF. Sotalol is considered a second-line option for the first two patient groups.92
At follow-up, a breakthrough episode does not mean that therapy failed. Patients may report breakthrough episodes but still be perfectly content with continuing the AAD in use because of overall effectiveness and improved quality of life. To terminate breakthrough episodes, many patients apply one or more extra doses of their prescribed AAD, i.e. add-on therapy (see also the section ‘Atrial fibrillation (oral, pill-in-the-pocket)’, but this approach may be hazardous unless carefully reviewed and controlled. In case of troublesome recurrences, thyroid-stimulating hormone (TSH) (especially if on amiodarone) and any change in underlying medical conditions (HF, angina, and infection) should be checked and treated. Also, it is important to check whether the AAD dose is still right and increase or even decrease the dose depending on clinical judgement: QRS or QT duration on therapy, drug side effects, and drug efficacy parameters (see also the section ‘Follow-up and monitoring of patients on anti-arrhythmic drugs’).
4.2.1.6. Atrial fibrillation after cardiac surgery
Post-operative AF is common (20–50%) in the 3 days after cardiac surgery. Most patients present with AF although AFL is also common. Several mechanisms, such as ischaemia and inflammation may be causative, but a hyperadrenergic state is believed to be the main cause. For this reason, β-blockers are the first-line therapy in this situation and can be started 24 h before the operation and continued during the post-operative period. Amiodarone is appropriate in combination with them in resistant cases, and vernakalant may be appropriate for AF termination.
4.2.1.7. Autonomic atrial fibrillation
As with PACs and NSATs, β-blockers may prevent AF recurrences, especially if adrenergic factors play a role, e.g. after cardiac surgery, exercise-induced AF or AF occurring exclusively during daytime, stress or anxiety, or systolic HF.278 β-Blockers (including sotalol) and digoxin may, however, worsen vagally mediated AF279 with increasing attacks and progression to permanent AF.280 Sotalol and amiodarone are effective in the suppression of adrenergic AF, but it is advisable to avoid sotalol if AF-promoting conditions such as HF are present. Anti-arrhythmic drugs to treat vagal AF include disopyramide, flecainide, and amiodarone. Disopyramide is no longer a mainstream AAD, but patients with vagal AF may benefit from its marked anti-cholinergic effects, which may also cause typical side effects of dry mouth, urinary hesitancy, and constipation. Disopyramide may induce HF, AV block in susceptible patients, and TdP.
4.2.1.8. Aberrant conduction vs. ventricular pro-arrhythmia
It is important to differentiate aberrant conduction from VT when using Class I and Class III AADs. Monomorphic VT hardly ever happens, even with exercise, if flecainide or propafenone is used appropriately.281 If a wide QRS rhythm occurs during exercise on class Ic drugs, it mostly is due to aberrant conduction. Aberrantly conducted QRS complexes are bizarrely shaped due to a normal initial but very broad last part of the QRS (class Ic drugs). Hallmark features of aberrancy with Class III AADs include (i) atypical types of aberrancy, including left bundle branch block (LBBB) with extreme left axis, (ii) aberrancy onset with atypically long coupling intervals (due to prolonged refractory period in the Purkinje system), and (iii) sequential bilateral BBB.282 Electrocardiographic criteria differentiating aberrant conduction from VT do not apply due to AAD effects on QRS morphology. Aberrant conduction may be a reason to reduce the dose or stop the drug.
4.2.1.9. Anti-arrhythmic drugs for atrial fibrillation
Flecainide is used to prevent AF recurrences in patients with paroxysmal or persistent AF without SHD.275,277 Also, in lone, focal, vagal AF279,283,284 flecainide may be very effective, while in adrenergic AF—frequently associated with underlying heart disease—it is commonly either ineffective or contraindicated. Pre-treatment before cardioversion of persistent AF may help reduce immediate and subacute recurrences.263 When AV conduction is controlled, flecainide does not exert negative dromotropic effects during ongoing AF. Pre-treatment during uncontrolled heart rates may be pro-arrhythmic and reduce quality of life. Therefore, pre-treatment with flecainide is advised to be done in hospital with ECG monitoring, and patients have to be advised to refrain from exercise until after the cardioversion. Similarly, patients progressing from paroxysmal to persistent AF while on flecainide may suffer from uncontrolled high heart rates and reduced quality of life. The use of propafenone is largely the same as for flecainide.
Dronedarone has been approved to maintain normal heart rhythm in adults whose rhythm has been restored after a period of paroxysmal or persistent AF but is not advised for use in patients with permanent AF and those with left ventricular systolic dysfunction and LVEF <35% or previous episodes of severe HF.103,104,285 It does not convert persistent AF to SR. The drug is very safe in patients without SHD and in stable patients with heart disease, including CAD.286 It has a very low risk for pro-arrhythmia.97,98,276 It is a reasonable first-line alternative comparable to Class Ic drugs, and before sotalol.287 It has been shown to reduce the progression of self-terminating AF to more persistent forms.99,288 Dronedarone has vastly under appreciated pleiotropic effects, in part explaining its success in the ATHENA trial.98 It reduces vasoconstriction and blood pressure. Through vascular effects, lowering heart rate and cellular protective effects, it ameliorates ACS. Dronedarone is also associated with reduced stroke rate in AF.289
Ranolazine has shown promise in AF prevention, particularly as an adjunct therapy in combination with other AADs like dronedarone.76 While some trials suggest benefit, larger studies are needed to confirm its role. Due to its off-label status and potential QT interval prolongation, its use is advised to be carefully individualized based on patient risk factors.
The effectiveness of amiodarone to prevent recurrent AF exceeds that of other anti-arrhythmic agents. Amiodarone is advised to be reserved for second-line treatment of AF but may be given safely as a first-line agent in patients with AF and HF, in whom most AADs are discouraged. Amiodarone is the most effective AAD for AF but frequently causes significant adverse effects,196,290 and it may negatively affect time in the INR target range291 in patients using vitamin K antagonists. Amiodarone and dronedarone are also P-gp inhibitors and may increase the anticoagulant effect of direct oral anticoagulants (DOACs). During the loading phase in patients with persistent AF, conversion to normal SR may occur in up to a quarter of patients.111 Conversion is not usually associated with bradycardia, but negative chronotropic and dromotropic drugs are advised to be reduced during the loading phase (see below). Amiodarone slows AV nodal conduction and heart rate during AF and prolongs the PR interval in SR, both of which are advised to be monitored during initiation of therapy. The use of rate control drugs is advised to be adapted according to the heart rate. As a rule, β-blockers, verapamil, or diltiazem may be stopped around 6 weeks after initiation of amiodarone loading.
Sotalol is a β-blocker with reverse use dependency with stronger AP prolongation during bradycardia or after pauses. Conversely, the anti-arrhythmic effect may be reduced during tachycardias. At the atrial level, the latter means that during the fast atrial rates of AF, atrial effects are considered minimal. Nevertheless, during chronic oral treatment, sotalol may, similarly to amiodarone, convert persistent AF to SR in up to 25% of cases.111 Pre-treatment with sotalol before cardioversion of persistent AF may be applied. However, immediately after electrical cardioversion of persistent AF, it is important to measure reverse use-dependent QT interval prolongation. This latter may be excessive when a relatively high ventricular rate during AF changes to a relatively low sinus rate. For Class III drugs, safety is best ensured by measuring at the time the risk is maximal, i.e. during relative bradycardia.
4.2.2. Paroxysmal supraventricular tachycardias
Paroxysmal supraventricular tachycardia may be the result of different arrhythmia mechanisms. While catheter ablation is the preferred treatment for recurrent AVNRT and AVRT due to its high efficacy, AADs are commonly used in the interim while awaiting ablation, in cases where ablation is nor recommened or unsuccessful, or for acute termination of episodes.44,292
4.2.2.1. Atrioventricular nodal re-entrant tachycardia
AVNRT may occur as a single, sporadic episode or, more frequently, as a recurrent arrhythmia. In a series of patients presenting with AVNRT (mean age 33.5 ± 18.1 years), a substantial proportion experienced arrhythmia recurrence during long-term follow-up, even among those with initially non-severe symptoms.293 For patients with important symptoms and recurrent AVNRT, ablation is the definitive treatment. In AVNRT, catheter ablation has a success rate of 97% with a recurrence of 1.3–4% and a risk of AV block <1%.44 A randomized controlled trial was performed in patients with at least one symptomatic episode of tachycardia per month and an electrophysiological diagnosis of AVNRT. Patients were randomly assigned to catheter ablation or chronic AAD therapy (bisoprolol and/or diltiazem).254 Hospital admissions for tachycardia cardioversion were significantly lower in patients treated with ablation, and AADs were not well tolerated over the long term.254
In appropriately selected patients with infrequent, well-tolerated episodes of AVNRT, episodic treatment with an anti-arrhythmic agent (PITP approach) could be used. However, acute testing is advised in order to exclude adverse effects.294 Single oral doses of flecainide (3 mg/kg) or diltiazem (120 mg) plus propranolol (80 mg) have been used, resulting in a high conversion rate to SR within 2 h.294
The efficacy of diltiazem and verapamil has been validated for the prevention of recurrences of AVNRT,253 although adherence over the long term may be problematic and overall efficacy is in the range of 30–50%.295 Also, β-blockers have been used for prevention of recurrences of AVNRT but the indication is based on expert opinion.253
In case of documented AVNRT resistant to β-blockers or CCBs, or in case of PSVT of uncertain mechanism, prevention of recurrences can be achieved effectively by flecainide or propafenone at adequate dosages in patients without contraindications to Class Ic agents.295–298 Amiodarone and sotalol must generally be avoided for the prevention of AVNRT recurrences, as safer alternatives are usually available.
4.2.2.2. Atrioventricular re-entrant tachycardia
Atrioventricular re-entry tachycardias occur in the presence of an accessory pathway that bypasses the normal conduction system between the atria and the ventricles. This pathway can conduct the impulses retrogradely or anterogradely, leading to orthodromic and antidromic AVRT, respectively. There is a progressive decline with age in the proportion of SVT that correspond to AVRT, from 60% in the first decade of age to 9% after 70 years.299 Antidromic AVRT constitutes 5–10% of all AVRTs.300 Wolff–Parkinson–White syndrome (WPW) is the combination of accessory pathway activation, manifested on the ECG by a delta wave, expression of anterograde conduction through the accessory pathway, and episodes of SVT.301 The simple presence of a delta wave (called ‘WPW ECG pattern’) can be detected in 0.2% of the general population, but many individuals with the WPW pattern do not exhibit the tachycardias required to fulfil the definition of WPW syndrome. The risk associated with WPW is related to the possibility that an episode of AF may occur, with rapid conduction down an accessory pathway, potentially leading to VF and sudden death- an event reported in <0.1% of the patients with the WPW pattern.301,302 In the 2019 ESC Guidelines, the risk of CA/VF was estimated to be 2.4 per 1000 person-years.44
For the prevention of supraventricular tachyarrhythmias and WPW-related adverse events, the treatment of choice is ablation of the accessory pathway, which is advised for symptomatic patients and selected asymptomatic individuals, particularly athletes and younger patients at risk.303
Therapy with AADs may be used in symptomatic patients while waiting for ablation or in patients who are not suitable candidates for ablation or who refuse the procedure. In these cases, Class Ic anti-arrhythmic agents, i.e. flecainide or propafenone, can be used to prevent AVRTs. Drugs that act mainly on AV conduction, such as diltiazem, verapamil, and β-blockers, are discouraged in patients with ventricular pre-excitation because of the risk of blocking AV conduction through the AV node and favouring conduction through the accessory pathway if AF occurs. In addition, CCBs are associated with vasodilation and a secondary adrenergic response, which may further promote conduction through the accessory pathway.253 Digoxin is contraindicated, as it may also shorten refractoriness of accessory pathways.197
4.2.3. Ventricular arrhythmias
Anti-arrhythmic drugs play a crucial role in preventing VA, which can lead to SCD. By modulating ion channels and stabilizing cardiac EP, AADs help reduce arrhythmia recurrence and improve patient outcomes. The preferred AADs for preventing monomorphic VAs are shown in Figure 12.
Figure 12.

Schematic representation of the advised anti-arrhythmic drugs (AADs) for prevention of monomorphic ventricular arrhythmias. The figure serves as a general reference for selecting the most appropriate drug; however, the final choice—or consideration of alternative therapeutic options [e.g. catheter ablation for idiopathic right ventricular outflow tract (RVOT) premature ventricular contractions (PVCs)]—is advised to be based on the general patient characteristics and conditions, as outlined in the relevant sections of this document. Additionally, not all AADs are available in all regions. For secondary or alternative drug options, refer to Table 3. Adrenergic PVCs/VT are characterized by an increased burden and/or severity in response to exercise or mental stress. Ca2, calcium; LVEF, left ventricular ejection fraction; MVT, monomorphic VT; SHD, structural heart disease; VT, ventricular tachycardia.
4.2.3.1. Idiopathic premature ventricular contractions and ventricular tachycardia
Premature ventricular contractions may produce symptoms, haemodynamic deterioration, and ventricular dysfunction. However, their treatment is not clearly associated with prognostic benefits in patients either with or without SHD, especially if the latter is not present. Responses to different pharmacological agents are considered essentially the same for PVCs and VT in patients without SHD, and they will be treated as a single entity in this practical compendium.
Pharmacological preventive therapies for PVCs and VTs in patients without SHD have been mostly studied in non-randomized or small series of patients with monomorphic PVCs. Most of these studies made no distinction about the arrhythmic origin or mechanism, and their results are extrapolated for the different types of idiopathic PVCs/VTs in the ESC guidelines and this practical compendium (Table 5).304,305 A few studies specifically included just patients with PVCs/VTs originating from the RVOT or the left fascicles of the His bundle, providing more specific evidence.307,308 The information available for other forms or sources of idiopathic VT/PVCs is more limited. β-Blockers and non-dihydropyridine CCBs are among the most studied drugs, and both were shown to effectively suppress the arrhythmia in this clinical setting.304,307 There are also studies demonstrating the efficacy of Class Ic drugs to suppress PVCs in patients with no or minimal SHD.304,305 In addition, one study demonstrated that they were effective for PVC suppression and tachycardiomyopathy recovery in patients with idiopathic PVCs.309 Mexiletine has also been demonstrated to suppress PVCs in some old studies.310 However, its relative efficacy is inferior to other drugs and it is not available in many countries. Sotalol has been demonstrated effective for both PVC patients with and without SHD, and some studies have even shown better efficacy than other β-blockers.106,304 However, the risk of TdP makes its use more complex and less preferable than other drugs, especially in patients with otherwise little arrhythmic risk. Preference is advised to be given to β-blockers when there is a correlation between the number of PVCs and heart rate or they are more frequent during exercise.311 If there is no such correlation, the use of Class Ic or CCB drugs has been associated with better PVC suppression.304,311 It is also advised to select β-blockers when a focal triggered activity mechanism is suspected, the origin is not apparently in the RVOT, or the patient shows signs of ventricular function deterioration. CCBs are advised to be the drugs of choice for fascicular PVC/VTs, although this advice is primarily based on the common termination of fascicular VT by i.v. verapamil.308
Table 5.
AADs reported with positive and negative effects for the treatment of PVCs and idiopathic VT306
| β-Blockers | CCB | Ic | Sotalol | Amiodarone | Ranolazine | |
|---|---|---|---|---|---|---|
| Idiopathic RVOT PVCs/VT no SHD |
++ | ++ | ++ | + | − | ? |
| Idiopathic fascicular PVCs/VT no SHD |
+ | ++ | + | ? | − | ? |
| Idiopathic non-RVOT/fascicular PVCs/VT No SHD |
++ | ++ | + a | + | − | ? |
| PVCs/VT PVC/VT-induced cardiomyopathy |
++ | − | + b | ? | + | ? |
| PVCs SHD |
++ | − | − | + | + | + c |
++, preferred positive AAD effects; +, conditional use and/or less established positive effect; −, to be avoided; ?, not enough data.
Abbreviations: AAD, anti-arrhythmic drug; CCB, calcium channel blockers; PVC, premature ventricular contraction; RVOT, right ventricular outflow tract; SHD, structural heart disease; VT, ventricular tachycardia.
aNot to be used if unmasked SHD or malignant short-coupled PVCs are suspected.
bOnly if no heart failure or severe ventricular dysfunction are present (risk of myocardial contractility depression).
cIf ischaemic heart disease is present.
Some forms of SHD may present with PVCs or VT as their initial manifestation, mimicking an idiopathic mechanism. For this reason, β-blockers and CCBs are more appropriate than Class I drugs for non-RVOT or fascicular idiopathic PVCs/VT because of the pro-arrhythmia risk in a less defined clinical setting. In addition, a recent report found short-coupled PVCs induced during Na+ blocker infusion in some patients with structurally normal hearts and suspected or documented ventricular polymorphic arrhythmias, and therefore they are discouraged in this setting.312 Calcium channel blockers must also be avoided in patients with signs of ventricular dysfunction because they may depress myocardial contractility, similarly to Class Ic drugs. In this situation, β-blockers and amiodarone are the preferred drugs. However, amiodarone is associated with severe systemic toxicity and is advised to be only used if other drugs fail or cannot be used.313 Dronedarone, which was developed only as an anti-arrhythmic agent for the treatment of AF, is devoid of most of the amiodarone toxicity, but there are no reports of its use to treat PVCs or VTs in patients without SHD.
Children need to be treated like adults. A recent registry found that only flecainide reduced the burden of PVCs compared with no treatment, β-blockers, or verapamil.314 Verapamil is not advised as the first-line therapy in children <1 year old because it has been associated with hypotension in some case reports, although all of them had HF, overdosing of verapamil, and/or other concurrent AADs at the time this drug was given.315
4.2.3.2. Premature ventricular contractions and structural heart disease
Structural heart disease generally refers to the presence of any morphological, functional, or recognized histological abnormality in the ventricles, encompassing cardiomyopathies, HFrEF, HFpEF, significant LVH, congenital heart disease, ischaemic, valvular, or other myocardial disorders. However, preventive therapies for PVCs and VTs in patients with SHD may differ, as some drugs used to treat the underlying condition can reduce the PVC burden but are not specifically targeted or sufficiently potent to suppress VT. This section of the practical compendium only addresses those aspects of pharmacological therapy for PVCs in patients with SHD that may differ from those for VT prevention, which is considered in the following section.
A high burden of PVCs may be associated with left ventricular dysfunction, and in recent years this has led to the concepts of both a form of cardiomyopathy induced by PVCs (PVC-induced cardiomyopathy) or to the concept of a worsening of systolic function in patients with pre-existing cardiomyopathy (PVC-worsened cardiomyopathy).316 The baseline PVC burden plays a central role in the development of PVC-induced cardiomyopathy and a PVC burden higher than 24% was found to best distinguish the patients with impaired compared with those with preserved LV function among consecutive patients referred for ablation.317 In patients with a suspected cardiomyopathy induced or aggravated by PVCs, ablation is a valuable option since improvement in left ventricular function was demonstrated in patients with a tachycardia-mediated mechanism, also in patients with prior infarction.316,318 Alternatively, in patients without a prior infarction, the observational study309 mentioned above showed that flecainide and propafenone effectively suppressed PVCs in patients with a mean LVEF of 37% who were suspected of having PVC-induced cardiomyopathy. This suppression led to LVEF recovery in most of these patients. In patients with PVCs in the setting of known CAD, treatment with β-blockers is advised, while suppression of PVCs with anti-arrhythmics other than β-blockers has not demonstrated any survival benefit and was harmful, since it is associated with worsening of survival in the case of Class Ic AADs, as shown in the CAST trial.319 This advice has been extrapolated to other forms of SHD, especially when myocardial scarring is present. Some reports found a significant decrease of PVC burden using ranolazine in patients with ischaemic heart disease and this drug may be appropriate in this population. However, in general, β-blockers are advised to be the drugs of choice to reduce the burden of PVCs in patients with SHD.77
Box 5 Current AAD aims in patients with structural heart disease and ventricular tachycardia.
Improve symtoms and quality of life
-
Improve cardiac functiona if:
deteriorated because of the VT and
deteriorated by dyssynchrony (frequent PVCs and NSVT)
Prevent aggravation to malignant or intolerable VT
Prevent recurrent shocks in ICD patients
No AAD, except for β-blockers, has demonstrated reduction in all-cause mortality in patients with VT.
Abbreviations: AAD, anti-arrhythmic drug; ICD, implantable cardioverter defibrillator; NSVT, non-sustained ventricular tachycardia; PVC, premature ventricular contraction; VT, ventricular tachycardia.
aCatheter ablation is the first-line therapy for tachycardia-induced cardiomyopathy and is advised to be the preferred treatment approach.
4.2.3.3. Ventricular tachycardia and structural heart disease
Currently, AADs for malignant ventricular tachyarrhythmias in the setting of SHD predominantly serve as adjunctive therapy to the ICD to prevent VT, to prevent frequent shocks, to avoid transformation of well-tolerated arrhythmias into malignant arrhythmias, or to prevent deterioration of cardiac function because of tachycardia, irregular rhythm, or desynchrony, rather than to cure the arrhythmia itself (Box 5). Shared decision-making is important when initiating AADs, balancing the risk for pro-arrhythmia and efficacy, particularly if the indication is symptomatic therapy. It is advised to instruct patients to contact physicians when they suffer from syncope, dizziness or palpitations.
β-blockers are considered the basic medication in SHD and can be very effective in polymorphic VT (PVT) but have low efficacy in preventing monomorphic sustained VT. Recurrent polymorphic VTs (QRS morphology changing from beat to beat) are often signs of acute ischaemia or incomplete reperfusion and mandate a search for, and correction of, reversible causes (hypokalaemia, hypomagnesaemia, exacerbation of HF, and pro-arrhythmic drugs). Patients with SHD have higher risks of ventricular tachyarrhythmias and are at higher risk of pro-arrhythmia when using AADs.320 In principle, patients with HF or cardiomyopathy are not candidates for VW Class Ic or Class III AADs other than amiodarone or sotalol (see ESVEM and OPTIC trials above).320 Although sotalol can be used in patients with CAD, the drug is advised to be used with caution due to its increased risk for HF, pro-arrhythmia, and mortality.91,121 Catheter ablation is increasingly being used for the management of recurrent VT and outperforms drug escalation in patients with VTs despite baseline AAD therapy.321 Patients with LVH have increased dispersion of repolarization and higher risks of PVT,322 which may support the concern over pro-arrhythmia and the caution against using Class Ic or Class III AAD. However, observational studies reported comparable mortality rates in patients with LVH and AF treated with Class Ic and Class III agents as in those treated with amiodarone.323
Given this background, it is important to evaluate the risk for pro-arrhythmia before starting AADs and to optimize treatment of comorbidities already at baseline. It is also crucial to assess clinical status, symptoms, concomitant drugs (www.crediblemeds.org), ECG changes, left ventricular function, and objective signs of relevant changes that could provoke pro-arrhythmia on a regular basis during follow-up.324 The appropriate timing of such follow-ups depends on the disease state of the patient. Given the increased risk of pro-arrhythmia with Class III AADs in females, it is advisable to use the lowest effective doses and avoid concomitant use of any other QT-prolonging agent or pro-arrhythmia-promoting factors, e.g. hypokalaemia (Figure 13).
Figure 13.

Twelve-lead electrocardiogram (ECG) demonstrating QT interval prolongation and a 5.5 s run of non-sustained polymorphic ventricular tachycardia [torsade de pointes (TdP)] following a post-extrasystolic pause in a patient with hypomagnesaemia. This highlights the association between electrolyte imbalances, prolonged repolarization, and pro-arrhythmic events such as TdP. The typical TdP twisting pattern of QRS complexes around the isoelectric line (horizontal line) is marked with arrows of varying amplitude above lead V1. Cycle lengths and QT intervals are annotated with black and red numbers, respectively. Torsade de pointes is triggered by a pause (two-arrowhead line) that further prolongs the QT interval. The ECG was recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
4.2.3.4. Ventricular fibrillation
Polymorphic VT and VF are life-threatening ventricular tachyarrhythmias. Polymorphic VT occurring in a setting without prolongation of the QT interval has a different management when compared with TdP, which is a PVT occurring in congenital or acquired long QT syndrome.253 Prevention of VF and PVT is often required in patients implanted with an ICD in order to avoid recurrent shocks that may occur in the form of ‘storms’. Correction of myocardial ischaemia, with revascularization and avoidance of electrolyte abnormalities, and of drugs with a pro-arrhythmic potential are important preventive measures. With regard to drugs, the combination of amiodarone and a β-blocker (metoprolol, carvedilol, or bisoprolol) was more effective than a β-blocker alone in reducing ICD activation for ventricular tachyarrhythmias, while sotalol (240 mg/day) had a trend towards higher efficacy when compared with a β-blocker without Class III anti-arrhythmic activity.91
4.3. Tachycardia termination
In the termination of tachycardia, the choice between oral and i.v. administration of AAD hinges on the urgency of intervention and the patient's clinical stability. Oral administration is often considered in stable patients with well-tolerated tachycardia, allowing for gradual onset and sustained effect. Conversely, i.v. administration is preferred in acute and unstable situations, aiming for a rapid onset of action. The decision to use electrical cardioversion arises when prompt restoration of normal rhythm is imperative, especially in cases of haemodynamic compromise or severely symptomatic tachycardia. This intervention may ensure a swift and effective reset of the cardiac rhythm, offering an immediate resolution in critical scenarios.
Box 6 Anti-arrhythmic agents and dosing for ‘PITP’ treatment of AF.
Flecainidea (immediate release formulation): 300 mg (single dose and consider 200 mg for weight <70 kg)
Propafenonea (immediate release formulation): 600 mg (single dose and consider 450 mg for weight <70 kg)
Ranolazineb: 2000 mg single dose (or 1000 mg × 2 given 4 h or less apart).
Abbreviations: AF, atrial fibrillation; PITP, pill-in-the-pocket.
aConsider further reducing the dose (100 mg of flecainide or 300 mg of propafenone) in elderly patients or those with suspected sinus node dysfunction.
bRanolazine has not been approved as an anti-arrhythmic drug by the European Medicines Agency or the U.S. Food and Drug Administration except for the LQTS.
The selection among these strategies is directed by a comprehensive assessment of the patient's clinical status, the nature of tachycardia, and the urgency of intervention. A tailored approach that factors in these considerations allows a more effective and patient-centred management of tachycardia (Figure 14).
Figure 14.

Schematic representation of the advised anti-arrhythmic drugs (AADs) for termination of atrial fibrillation (AF). The figure serves as a general reference for selecting the most appropriate drug; however, the final choice is advised to be done by patient-specific characteristics and conditions, as detailed in the various sections of this document. Additionally, drug availability plays a crucial role in decision-making. For example, vernakalant is available in many European countries but not in the United States. Flecainide and propafenone i.v. are not available in many American countries. Antazoline is primarily produced and marketed in Poland, where it is registered for anti-arrhythmic use, though its availability in other European countries remains limited. Pilsicainide is primarily approved and used in Japan and Korea, but its presence in Europe is scarce. Cibenzoline is also used in Japan and has been available in certain European countries. aClass Ic AADs are contraindicated in patients with SHD, HF, or significant conduction disturbances due to the risk of pro-arrhythmia and conduction block. bOral ranolazine has been used off-label for AF conversion in patients with ischaemic heart disease. However, it is advised to be used with caution in NYHA Class III andIV HF and avoided in patients with QT prolongation due to the risk of pro-arrhythmia. AF, atrial fibrillation; HF, heart failure; NYHA, New York Heart Association functional class; i.v., intravenous; p.o., per os; SHD, structural heart disease.
4.3.1. Atrial fibrillation (oral—pill-in-the-pocket)
Pill-in-the-pocket therapy325 refers to the use of an orally administered AAD for the termination of a recent-onset arrhythmia—most commonly occasional326 AF with well-tolerated episodes.325 Agents and conditions required to use a PITP strategy are shown in Boxes 6 and 7, respectively.
Because conversion to AFL may occur (with or without subsequent reversion to SR), an AV node blocker must precede administration of flecainide or propafenone (e.g. β-blockers or CCB given 0.5–1 h prior to PITP initiation, unless taken chronically). Dofetilide can convert AF, but its time course is too slow (days) to be used as PITP. Amiodarone is much too slow, sotalol is not effective and potentially unsafe as PITP, and dronedarone has not been tested. Typical conversion rates with the Ic AADs and ranolazine are 70–80% by 8 h (about twice that of placebo) with a mean time of 3–4 h for Ic drugs and 3–6 h for ranolazine.
4.3.2. Atrial fibrillation (intravenous)
The primary mechanisms underlying the termination of AF with i.v. drugs are diverse, reflecting the complex and still incompletely understood nature of this arrhythmia. The effectiveness of i.v. drugs in terminating AF is influenced by various factors, including patient characteristics, AF duration, underlying SHD, and the presence and functional Class of HF (see Table 6).
Table 6.
AADs currently advised for AF terminationa
| AAD | No SHD and AF ≤7 daysb | No SHD and AF >7 daysc | SHD with HF NYHA I–II and AF ≤7 days | HF NYHA III–IVd |
|---|---|---|---|---|
| Vernakalant i.v. | 1 | − | 1e | No |
| Procainamide i.v. | − | 3 | No | |
| Flecainide/propafenone i.v./p.o. PITP | 2 | 1 | No | No |
| Sotalolf,g i.v. | − | − | − | No |
| Ibutilideg i.v. | 3 | 2 | 2 | 2 |
| Amiodaroneh i.v. | 3 | 2 | 2 | 1 |
‘1’, first choice; ‘2’, second choice; ‘3’, third choice; ‘−’, minimal effect/not advised; ‘No’, contraindicated.
Abbreviations: AAD, anti-arrhythmic drug; AF, atrial fibrillation; HF, heart failure; i.v., intravenous; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association functional class; PITP, pill-in-the-pocket; p.o., per os; PVC, premature ventricular contraction; SHD, structural heart disease.
aAnticoagulation and/or exclusion of left atrial thrombi must always be ensured before attempting AF termination, in accordance with the recommendations outlined in ESC AF guidelines.
bAntazoline, cibenzoline, and pilsicainide are also used in some countries, such as in Japan, Korea, and Poland, for the treatment of short-lasting AF, provided that patients do not have significant SHD, HF, or bradycardia.
cThe efficacy of conversion with each drug decreases as the duration of AF increases, with a significant decline in success for episodes lasting more than 7 days, making electrical cardioversion a more effective option in such cases.
dConsider electrical cardioversion first if there is haemodynamic instability.
eContraindicated if severe aortic stenosis or acute myocardial ischaemia.
fSotalol is not advised in patients with moderate-to-severe heart failure but still may be appropriate if AF lasts >7 days.
gNot to be given if renal insufficiency, hypokalaemia, QT prolongation risk, or LVEF ≤ 40%.
hAmiodarone has a slow and delayed effect on AF termination, with most of its benefits stemming from heart rate control rather than immediate rhythm conversion.
Box 7 Conditions required to use PITP strategy for AF termination.
Recent onset of AF (<7 days)
Properly anticoagulated if advised
No underlying sinus node, AV node dysfunction (in the absence of a pacemaker), Brugada syndrome or other contraindications to AADs, ischaemia, or haemodynamic intolerance
Established or acutely administered rate control therapy to prevent 1:1 AV conduction in case of transient conversion to AFL prior to return to sinus rhythm
Acceptance of the need to stay at rest for at least 3 h after drug administration to minimize the risk of pro-arrhythmia
Prior demonstration of tolerance to the AAD or initial PITP usage is advised to be conducted under observation to verify effectiveness and ensure no adverse effects
Abbreviations: AADs, anti-arrhythmic drugs; AF, atrial fibrillation; AFL, atrial flutter; PITP, pill-in-the-pocket.
In cases of recent-onset AF, particularly within the first 48 h, Class I and III agents have demonstrated high efficacy rates, often exceeding 70%. However, it is important to note that, in most instances, these drugs may not achieve higher conversion rates compared with placebo but may lead to earlier conversion.327 The efficacy of ibutilide and dofetilide is lower for converting AF compared with AFL. Vernakalant stands out with a favourable efficacy and safety profile, making it a valuable option for AF termination, especially in patients with recent-onset AF (within 7 days). A recent meta-analysis evaluating the efficacy of different anti-arrhythmic agents in restoring SR in paroxysmal AF identified vernakalant, amiodarone-ranolazine, flecainide, and ibutilide as the most effective medications.328
While efficacy is a critical consideration, safety is of paramount importance in the selection of i.v. drugs for AF termination. The pro-arrhythmic and ventricular contractility depression potential, particularly with Class I anti-arrhythmics, necessitates meticulous patient selection, precluding their use in individuals with SHD or significant ventricular dysfunction. Class III agents, while generally safe, require vigilant monitoring of renal function and the QT interval to mitigate potential risks.
Another important factor in AAD selection is the timing of AF termination. Vernakalant has a median time to termination of 11 min in patients with AF lasting <48 h, which is significantly shorter compared with i.v. flecainide or amiodarone (Figure 7). The rapid onset of action of vernakalant enhances the likelihood of restoring SR within 48 h, resulting in cost savings compared with alternative treatment approaches.329 With i.v. amiodarone, only 5.2% of patients converted to SR within 90 min.157
Finally, it is worth noting that β-blocker infusion is not advised for AF termination, although it may still be advised for heart rate control.
4.3.3. Atrial flutter
The AFL cycle length is determined by atrial conduction velocity and re-entrant circuit size, typically slightly longer than the APD. Modest APD prolongation, achievable with Class III agents like IV dofetilide and ibutilide, is ideal for terminating AFL. A randomized multicentre trial showed IV dofetilide terminated AFL more frequently (75%) than IV amiodarone (0%) or placebo (10%) (P < 0.001).330 While these agents might not be available in many countries, sotalol (1.5 mg/kg body weight over 5–10 min) can also be applied with careful dosing since too low drug blood levels may result in failure.
Class Ia and Ic drugs (flecainide, propafenone, and cibenzoline) and vernakalant are ineffective in terminating AFL, as they fail to sufficiently suppress conduction within the atrial re-entrant circuit. Instead, they typically prolong the atrial cycle length by ∼100 ms without interrupting tachycardia. This slower atrial rate increases the risk of 1:1 AV conduction, given the weak negative dromotropic effect of Class I AADs at the AV node. AFL with 1:1 AV conduction is often associated with aberrant conduction, producing wide, bizarre QRS complexes that mimic VT and can lead to haemodynamic instability. Due to these risks, Class I AADs are generally discouraged, and AFL termination is advised to be pursued with selective Class III AADs or electrical cardioversion.
4.3.4. Paroxysmal supraventricular tachycardia
Patients with SVT, either corresponding to AVNRT or AVRT, may respond to vagal manoeuvres, carotid massage, or adenosine i.v.253,331,332 Assessment of the exact diagnosis of the PSVT, and specifically of AVNRT vs. AVRT, is important, but in some cases the exact diagnosis may remain uncertain.331 The first and very important step in the approach to patients with PSVT, as with other re-entrant arrhythmias, is to assess haemodynamic stability. If the situation is unstable, synchronized cardioversion is recommended by the ESC guidelines of SVT.44
Adenosine is widely used in patients with tolerated PSVT because the resulting transient AV blockade is helpful both for arrhythmia termination and for differential diagnosis of other supraventricular tachyarrhythmias (e.g. AFL or AT). Adenosine is advised to be used with caution and always under ECG monitoring, since it may induce the onset of AF with a rapid ventricular response in the presence of an accessory pathway with antegrade conduction capabilities, even when previously unknown.201
Intravenous diltiazem, verapamil, metoprolol, esmolol, or other β-blockers can be useful in terminating haemodynamically stable regular SVT of uncertain type or when a diagnosis of AVNRT or AVRT is suspected. However, drugs that mainly act by slowing the conduction through the AV node (e.g. diltiazem, verapamil, and β-blockers) are discouraged in patients with known pre-excitation with antegrade conduction capabilities, in consideration of the risk of AV nodal blockade and acceleration of the ventricular rate if AF occurs.253 Also, i.v. amiodarone may precipitate a VF in case of AF with anterograde conduction over an accessory pathway.333 Assessment of the underlying mechanism of PSVT is important, as mentioned above but in some cases, ruling out participation of an accessory pathway remains uncertain.331
Procainamide, flecainide, or propafenone are advised for interrupting antidromic AVRT without haemodynamic instability, but Class Ic AADs (flecainide and propafenone) are contraindicated in the presence of left ventricular dysfunction, ischaemic heart disease, severe LVH, or conduction system disturbances.253
4.3.5. Focal atrial tachycardia
Focal AT is characterized as a tachyarrhythmia arising from a focal atrial area and may occur in many clinical conditions, including catecholamine excess, digoxin toxicity, congenital heart disease, chronic obstructive pulmonary disease, and different types of cardiomyopathy334 For the differential diagnosis between a focal AT and other supraventricular tachyarrhythmias, i.v. adenosine can be useful. Adenosine is also useful for therapeutic purposes, as an alternative to vagal manoeuvres. However, it has to be noted that focal AT is frequently terminated by adenosine, and this is not usually the case with vagal manoeuvres.
There is limited evidence on the acute treatment of focal AT. In cases of haemodynamic instability, DC cardioversion may be required, although AT is typically resistant to this. Intravenous β-blockers, diltiazem, or verapamil can be used initially. If these treatments fail, i.v. flecainide, propafenone, or amiodarone may be appropriate after an appropriate washout period to avoid the mixing of different agents in a short time frame.44
4.3.6. Junctional ectopic tachycardia
This is a tachyarrhythmia arising from the region of the AV node or AV junction, including the bundle of His, due to enhanced automaticity. It is usually observed in the post-operative settings of surgery for congenital heart disease or in children as a congenital disorder. Usually the QRS is narrow, but aberrant conduction can occur. For treating this arrhythmia, i.v. amiodarone has been successfully used, but flecainide, procainamide, propafenone, landiolol, and sotalol have also been used with some success.335,336 Also, digoxin and anti-inflammatory agents such as steroids or even colchicine have been proposed.335,336 In an open-label randomized controlled trial, oral ivabradine was not inferior to i.v. amiodarone in converting post-operative JET to SR, and no difference was found in the time taken to SR conversion between the groups, although the rate control was earlier in patients who received amiodarone.337 Therefore, according to this study and other contributions, monotherapy with ivabradine may be appropriate as an alternative to amiodarone in the management of post-operative JET, as well as an adjunct to amiodarone for refractory JETs after surgery for congenital heart disease.338
4.3.7. Ventricular tachycardia—non-structural heart disease
Patients without SHD often present with PVCs and occasional non-sustained bursts of VT. However, caution is advised when dealing with sustained VT in this population, as it could be the first manifestation of an underlying SHD. This consideration is crucial, especially when selecting an AAD for termination (see Box 8).
Box 8 Factors associated with concealed structural heart disease in patients presenting with apparent idiopathic ventricular tachycardia.
Poor haemodynamic tolerance
-
ECG:
Sustained VTa
Tachycardia cycle length <250 ms
Tachycardia QRS complex duration >140 ms
Atypical QRS complex morphology for an RVOT or fascicular-type VT
Several VT morphologies or pleomorphic VTa
Abnormal sinus rhythm ECG (Q waves, ventricular hypertrophy or low voltage QRS complex in frontal plane leads, inverted T waves beyond V2, etc.)
Abnormal cardiac findings on chest X-ray or echocardiogram
No response to adenosine
Abbreviations: ECG, electrocardiogram; RVOT, right ventricular outflow tract; VT, ventricular tachycardia.
aFascicular idiopathic VT typically presents with sustained monomorphic VT though; RVOT typically presents with multiple bursts of non-sustained monomorphic VT and frequent same-morphology PVCs.
Clinical studies have predominantly focused on VT originating from the RVOT or the left fascicles of the His bundle. Adenosine and verapamil are often advised for acute termination of outflow tract and fascicular idiopathic VT, respectively. It is worth noting that there are reports and small series supporting the use of both drugs in both VT mechanisms.
However, the use of verapamil requires caution due to its association with myocardial contractility depression and hypotension. This caution is particularly relevant when considering VT termination in patients with less established absence of SHD.
4.3.8. Ventricular tachycardia—structural heart disease
For acute termination of haemodynamically stable monomorphic VT of unknown aetiology, procainamide or amiodarone may be used339,340 with preference for procainamide341 for safer and shorter time to conversion (Figures 5 and 15). The ESC and the AHA/ACC/HRS guidelines45 recommend procainamide over amiodarone, largely based on the randomized controlled PROCAMIO trial,341 which showed that procainamide was associated with fewer major cardiac adverse events and a higher proportion of tachycardia termination within 40 min.342 However, if the patient suffers from severe HF, acute MI, or end-stage kidney disease, amiodarone is the acute treatment of choice.
Figure 15.

Twelve-lead electrocardiogram (ECG) illustrating the termination of haemodynamically tolerated monomorphic ventricular tachycardia (VT) following the infusion of amiodarone in an 81-year-old female patient with a history of anterior wall myocardial infarction. (A) Initial VT with a cycle length of 330 ms during i.v. amiodarone infusion, showing no significant change in cycle length. (B) Subsequent change in VT morphology and acceleration to a cycle length of 240 ms, followed by VT termination and resumption of sinus rhythm. This case highlights the dynamic response of VT to anti-arrhythmic therapy. The ECG was recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Amiodarone is also preferred in ES with frequent VT episodes.45,343 When underlying SHD is suspected, ajmaline, sotalol, and flecainide are not advised.304,344 If the underlying diagnosis is not clear, amiodarone may be preferred, acknowledging that initial effects occur only within hours and include mainly early β-adrenergic and calcium channel blockade.345 Initial treatment with β-blockers, preferably non-selective β-blockers like propranolol, is advised.
Intravenous lidocaine is only moderately effective but may be appropriate for the treatment of recurrent, haemodynamically stable sustained VT not responding to β-blockers or amiodarone or in the presence of contraindications to amiodarone.45 Lidocaine has been advised as an alternative to amiodarone also for acute treatment of shock-refractory VF/pulseless VT.346 Although there is no evidence of improved survival to hospital discharge associated with lidocaine, return of spontaneous circulation was higher in patients receiving lidocaine compared with placebo after CA, and survival to hospital admission was also higher compared with placebo.346
If incessant slow monomorphic VT occurs as a result of AAD treatment, catheter ablation may be needed with AAD often continued after the intervention.45
Besides AAD selection, other aspects are also important. In all cases of haemodynamically stable monomorphic VT, documentation on 12-lead ECG is important. Also, it is advised to monitor and document with a 12-lead ECG the VT cycle length and morphology as well as QRS width and QT interval of the QRS complex during VT under AAD infusion (Table 7).
Table 7.
Potential ECG changes after AAD administration for monomorphic VT termination
| ECG parameter | Electrophysiologic mechanism | VM class of AAD |
|---|---|---|
| VT cycle length prolongation |
|
|
| QRS complex morphology change | Multiple exits, which result from stopping one circuit and initiating another | May occur especially with Class Ic AAD |
| QRS complex widening during VT | Reduction of conduction velocity of the ventricular activation through the myocardium independent from the AAD effect in the re-entrant circuit; comes with the risk of acute heart failure, AV block, and sinusoidal VT | Mainly with Class Ic AADs and more marked with faster VTs (use dependency) |
| QT interval lengthening during VT | Prolongation of the action potential may associate TdP due to combination of relative bradycardia and bradycardia-dependent AAD-induced long QT after tachycardia termination | Mainly with Class III drugs. Not expected with Class Ia drugs Post-termination long QT may relate to reverse use dependency of Class III AAD |
Abbreviations: AAD, anti-arrhythmic drug; ECG, electrocardiogram; VM, Vaughan Williams; VT, ventricular tachycardia.
4.3.9. Polymorphic ventricular tachycardia and ventricular fibrillation
Ventricular fibrillation and PVT are life-threatening cardiac arrhythmias. Prompt and appropriate treatment is crucial in managing these conditions. Treatment involves immediate initiation of cardiopulmonary resuscitation and early defibrillation. In patients without SHD, the management of PVT involves addressing underlying causes, such as electrolyte imbalances, medication side effects, bradycardia or a channelopathy. Magnesium sulphate and potassium are often administered intravenously to stabilize the myocardium. It is advised to manage patients with bradycardia, or acquired or inherited LQTS, by increasing the heart rate using isoprenaline infusion or pacing at supra-normal rates. Brugada syndrome patients are advised to be managed by isoprenaline or quinidine. Patients with CPVT are advised to be treated with β-blockers and flecainide.
Polymorphic VT in SHD frequently is a marker of myocardial ischaemia, and apart from resolving ischaemia with a standard coronary intervention, i.v. β-blockers and amiodarone are considered the most suitable AADs in haemodynamically stable cases. Lidocaine347 and mexiletine may also be effective, and the latter AADs may be used as add-on therapy. Sometimes polymorphic VT/VF occur in post-infarct patients without any evidence of myocardial ischaemia and which are due to triggering from surviving Purkinje fibres. They are initiated with a relatively short-coupled PVC (350 ms) and may occur with normal QT (PVT with normal QT) or a long QT (pseudo-TdP). Storms due to these Purkinje-triggered PVTs respond well to quinidine but are refractory to β-blockers, lidocaine, mexiletine, Class Ic drugs, and amiodarone.348–350
5. Practical aspects
5.1. Initiation of anti-arrhythmic drug
The initiation of AADs requires a comprehensive and safety-driven approach to optimize outcomes while minimizing risks. This involves careful management of concomitant conditions, vigilant monitoring, and patient education. Underlying conditions such as ischaemic heart disease are advised to be addressed with revascularization and statin therapy, adequate β-blockade, and elimination of triggers like electrolyte imbalances. For patients with HF, therapy is advised to be tailored to the subtype: in HFrEF, optimization involves β-blockers, aldosterone antagonists (e.g. spironolactone or eplerenone), ACE inhibitors, sacubitril/valsartan, and sodium–glucose co-transporter-2 inhibitors (SGLT2 inhibitors), while in HFpEF, SGLT2 inhibitors play a central role. Baseline ECG, echocardiography, renal and hepatic function, along with a haematology and biochemical profile, including lipid, glucose, and electrolyte parameters, are advised to be established for future reference (Table 8). For amiodarone, baseline assessments must also include thyroid function tests, a chest X-ray, and PFTs, including diffusion capacity, and, ideally, visual assessment by a corneal slit-lamp exam and a fundoscopic evaluation. An exercise test to assess QRS widening during exercise or detect subclinical myocardial ischaemia may also be considered for patients after initiating Class Ic AADs.
Table 8.
Advisable tests at baseline and during follow-up for patients taking AADs
| Evaluation | Test/parameter | Frequency | Toxicity/interaction evaluation |
|---|---|---|---|
| AADs other than amiodarone | |||
| ECG | Rhythm, PR, QRS, QTc | Baseline, shortly after initiation or dose adjustments (1–2 days for Class Ia, sotalol, dofetilide) and periodically (e.g. every 6 months) | QT interval prolongation (for Class Ia and III drugs) QRS duration prolongation (for Class Ic drugs) Pro-arrhythmic tachycardia (e.g. type Ic AFL), bradycardia or BBB/atrioventricular block |
| Echocardiography | Ventricular function | Baseline and updated if change suspected/risk | Systolic dysfunction (contraindication for Class Ic and IV AADs) |
| Blood test and serum electrolytes | GFR, K+, Mg2+ | Baseline, periodically (e.g. every 6 months) | Reduced drug elimination, Pro-arrhythmia risk |
| Liver function | ALT, AST, and total bilirubin | Baseline, periodically (e.g. every 6 months) | Reduced drug elimination |
| Exercise test | QRS at peak exercise, myocardial ischaemia | To consider for Class Ic at follow-up | QRS widening at exercise |
| Amiodarone | |||
| ECG | Rhythm, PR, QRS, QTc | Baseline, steady state (1–3 months), annually | QT interval prolongation, pro-arrhythmic tachycardia (e.g. AFL), bradycardia, or atrioventricular block |
| Echocardiography | Ventricular function | Baseline, update if potential changes suspected | Systolic dysfunction |
| Serum electrolytes | K+, Mg2+ | Baseline, every 6 months | Pro-arrhythmia risk |
| Liver function | ALT, AST, and total bilirubin | Baseline, every 6 months | Hepatotoxicity |
| Thyroid function | TSH, free T4, and free T3 | Baseline, every 6 months | Hypo-thyroidism or hyper-thyroidism |
| Pulmonary function | Chest X-ray and pulmonary function tests (diffusion capacity) | Baseline, annually | Interstitial lung disease |
| Visual function | Corneal slit-lamp exam and fundoscopic evaluation | Baseline, annually | Corneal microdeposits and, rarely, optic neuropathy |
Abbreviations: AAD, anti-arrhythmic drug; AFL, atrial flutter; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BBB, bundle branch block; ECG, electrocardiogram; GFR, glomerular filtration rate; TSH, thyroid-stimulating hormone.
Regular blood pressure measurements are critical when initiating AADs, especially via i.v. infusion, as these drugs can cause vasodilation and hypotension. It is advised to carefully control infusion rates to ensure appropriate peak plasma concentration and to avoid hypotension, and a physician has to remain close to the patient during administration (Box 9).
Box 9 ECG parameters to monitor/observe during AAD infusion.
Atrial rate, atrial cycle length (in AFL)
Bradycardia
Enhanced AV conduction, 1:1 AV conduction
Unexpected AV block (Class Ic and III AADs)
Termination of AF or AFL
Signs of sinus node dysfunction upon AF/AFL termination
QRS prolongation and aberrant conduction (Class Ic AADs)
QT prolongation (Class Ia and III AADs)
Signs of Brugada ECG in right precordial leads (Class I AADs)
TdP and other ventricular arrhythmias
Abbreviations: AAD, anti-arrhythmic drug; AF, atrial fibrillation; AFL, atrial flutter; AV, atrioventricular; TdP, torsades de pointes.
Controversy (Box 10) exists regarding the safety of initiating AADs in outpatient settings, with advice varying depending on the drug and patient profile (Box 11; Table 9). The choice between inpatient and outpatient initiation is primarily driven by safety considerations.351 Importantly inpatient initiation is advised for high-risk patients, such as those with SHD or significant arrhythmias, as it allows for close monitoring and timely intervention. Outpatient initiation, while more convenient, requires adequate monitoring using tools like smartwatches, trans-telephonic devices, or other patient-activated ECG methods. For example, sotalol is advised for inpatient initiation in some AF patients, but some trials suggest that outpatient initiation with daily and symptomatic ECG transmissions via event recorders, smartphone apps, or smartwatches for 10 days could be a safe alternative.352 However, these were small and non-controlled and have to be taken with caution. Any abnormal findings recorded on digital devices must prompt a confirmatory 12-lead ECG.
Table 9.
Advisable agents for in-hospital/outpatient initiation of AADs

|
Abbreviations: AFL, atrial flutter; AV, atrioventricular; ECG, electrocardiogram; FDA, Food and Drug Administration; PVC, premature ventricular contraction; TdP, torsades de pointes.
aIf uncertain sinus node function or risk for AFL conversion with 1:1 AV conduction.
bIf known absence potential risk of sinus node dysfunction or AV conduction disorders.
cIf no TdP risk markers and in sinus rhythm (see Box 12). For women and patients over 65, sotalol must only be initiated in an outpatient setting with close monitoring in the absence of other risk factors. Patients have to be educated to recognize warning symptoms, avoid certain medications, and adhere to follow-up appointments. The U.S. FDA advises hospitalizing all patients being initiated or re-initiated on sotalol for at least 3 days or until steady-state drug levels are achieved in a facility that can provide cardiac resuscitation and continuous ECG monitoring.
Box 10 Advantages, disadvantages, and advice for inpatient and outpatient initiation of AADs.
In-hospital initiation
Advantages
Direct monitoring of drug effects on arrhythmia
Faster drug loading (e.g. sotalol)
Use of parenteral AADs if needed
-
Immediate response to acute adverse effects:
Sinus node/AV conduction disturbances
Conversion to AFL with 1:1 conduction
QT prolongation, TdP
Heart failure, early drug intolerance, interactions
Addresses medical–legal concerns for specific AADs
Disadvantages
Requires hospitalization (inconvenient, disruptive)
Higher costs and logistical challenges
Long half-life drugs (e.g. amiodarone, digoxin) will not reach steady state
Pro-arrhythmia risk may still occur later due to evolving conditions (e.g. electrolyte changes, new drug interactions, heart rate change)
Outpatient initiation
Advantages
Patient preference and practicality
Lower cost; avoids hospitalization for most low-risk cases
-
Safe for low-risk groups:
Class Ic AADs, dronedarone, amiodarone in non-SHD patients
Sotalol in males in sinus rhythm with normal renal function, electrolytes, and no LV hypertrophy
Predictable drug interactions can be managed
AFL with 1:1 conduction preventable with AV nodal blockers
Disadvantages
Rare but serious pro-arrhythmic events may go undetected and untreated
Abbreviations: AAD, anti-arrhythmic drug; AFL, atrial flutter; AV, atrioventricular; LV, left ventricle; TdP, torsades de pointes.
Since urgency is less critical in the outpatient setting, dosing must start low and progress gradually, with adjustments based on the longest known half-life of the drug to ensure a steady state before dose increases.
Box 11 Advice/requirements for in-hospital/outpatient initiation of AADs.
In-hospital initiation
Class Ia: required for most drugs (some exceptions).
Class III (dofetilide): must always be initiated and dose-adjusted in-hospital.
Class III (Sotalol): in-hospital if QTc ≥450 ms (500 ms if intraventricular conduction delay), HR ≤60 b.p.m., or specific risk factors (e.g. SHD and renal dysfunction). See Box 12
QT prolongation or unconfirmed sinus rhythm (risk of sick sinus syndrome or bradycardic pauses): require in-hospital initiation
Pro-arrhythmic risk: high ventricular pro-arrhythmia risk (TdP, syncope, cardiac arrest) necessitates in-hospital monitoring
Outpatient initiation
Class Ib (mexiletine): Allowed for non-tachycardic ventricular ectopy or type III long QT syndrome.
Class Ic (fleainide/propafenone): Permitted in patients without SHD with ECG checks unless normal sinus rhythm has not been previously documented.
Class Id (ranolazine): Safe for patients with or without SHD.
Class III (dronedarone and amiodarone): permitted with ECG checks in low-risk patients.
Patients with ICDs: ICDs provide protection against pro-arrhythmia, enabling outpatient initiation
Abbreviations: AADs, anti-arrhythmic drugs; ECG, electrocardiogram; HR, heart rate; ICD, implantable cardioverter defibrillator; QTc, corrected QT interval; SHD, structural heart disease.
Patient education and counselling are essential for safety and adherence. This includes defining the goals of therapy, such as symptom relief or arrhythmia prevention, and educating patients on recognizing potential adverse effects, including rapid palpitations or (pre-)syncope during exercise or at rest, which might suggest pro-arrhythmia. Patients must also be informed of potential food–drug and drug–drug interactions, particularly with QT-prolonging medications.
5.2. Follow-up and monitoring of patients on anti-arrhythmic drugs
The follow-up and monitoring of AADs necessitate a structured approach to ensure both safety and efficacy while minimizing the risks of pro-arrhythmic effects and other adverse events (Table 8). For patients in SR, obtaining a follow-up ECG shortly after initiation—typically within one week—is practical. For drugs with a prolonged loading phase, such as amiodarone, an ECG after achieving steady-state is advisable. Specifically, for Class Ia and III AADs (excluding amiodarone), a follow-up ECG is advised to be performed within 2 days of initiation to monitor for excessive QT prolongation and the associated risk of TdP.
Subsequent regular ECGs are advised every 6–12 months, tailored to the specific AAD and the patient’s clinical profile. These ECGs must monitor for QTc prolongation, QRS widening, new BBBs, bradycardia, or tachycardia.
Beyond ECG assessments, regular monitoring must include liver function tests (ALT, AST, and total bilirubin), creatinine, and serum electrolytes—particularly K⁺ and Mg2⁺—within 3–6 months of initiation to identify potential hepatic or renal impairment. If transaminase levels exceed three times the normal value, or double in a patient with elevated baseline levels, the AAD dose is advised to be reduced or discontinued. Patients on flecainide or propafenone must undergo routine checks of QRS duration and renal and hepatic function at yearly or half-yearly intervals.
Amiodarone requires additional and specific monitoring. ECG assessments of rhythm, PR, QRS, and QTc intervals must occur at steady-state (1–3 months) and annually. Given amiodarone’s low TdP risk, it may be continued despite QT prolongation, but QTc must not exceed 550 ms to prevent pro-arrhythmic complications. Biannual assessments of K⁺ and Mg2⁺ levels are necessary to mitigate pro-arrhythmia risks. Liver function tests are advised to be performed every 6 months to detect hepatotoxicity. Pulmonary evaluations, including chest X-rays and PFTs with diffusion capacity, are advised to be conducted at baseline and annually to monitor for interstitial lung disease. Visual function, including corneal slit-lamp examinations and fundoscopic evaluations, must also be assessed annually to identify potential ocular complications. Thyroid function (TSH; free thyroxine, T4; and free triiodothyronine, T3) is advised to be evaluated every 6 months to screen for hypo- or hyper-thyroidism.353
For dronedarone, a moderate, asymptomatic increase in creatinine (∼0.1 mg/dL) is commonly observed due to reduced tubular secretion, without altering glomerular filtration rate (GFR). This elevation stabilizes after 7 days and should be taken as the patient's new baseline, rather than prompting discontinuation of renin-angiotensin system inhibitors or dronedarone. Monitoring must include electrolytes, QT (ensuring QTc does not exceed 500 ms), and hepatic function. Repeat hepatic tests are advised within the first 6 months and yearly thereafter; the drug is advised to be discontinued if permanent AF develops.354–356
For patients on sotalol or dofetilide, regular monitoring of serum creatinine, potassium, and magnesium levels is essential, with dose adjustments as necessary to minimize the risk of pro-arrhythmia. The QTc interval is advised to be maintained below 500 ms, with monitoring conducted annually or semi-annually, as well as promptly following any changes in clinical conditions or the addition of medications that could interact with the drug or prolong the QT.
In addition to ECG and laboratory evaluations, echocardiography is advised to be scheduled periodically to assess LVEF, particularly in patients with SHD or HF. For intermittent arrhythmia detection, Holter monitoring, event recorders, or implantable cardiac monitors may be employed. Exercise testing can assess myocardial ischaemia, a potential contributor to arrhythmic events in patients taking Class Ic AADs.
Beyond drug-specific advice, adherence to treatment is advised to be evaluated at every visit, with risk factors for pro-arrhythmia carefully assessed. This systematic approach ensures that AAD therapy is managed effectively, balancing therapeutic goals with patient safety.
During follow-up, the clinical condition of patients may change, leading to drug accumulation or the development of an arrhythmogenic substrate, including electrolyte disturbances, ischaemia, and HF. Therefore, it is important to ensure at each visit that patients can recognize warning symptoms, including worsening palpitations, unexpected dizzy spells or syncope, development of chest pain, dyspnoea, and recent-onset exercise intolerance. It is essential to reiterate these warning symptoms during regular follow-up visits. For all drugs, it is important that patients themselves know that renal function must remain constant. For Class III drugs, patients have to be instructed to avoid QT-prolonging drugs and, with new prescriptions, refer to the treating cardiologist or arrhythmologist. Additionally, the risk of developing hypokalaemia has to be emphasized, which may occur in cases of diarrhoea, excessive sweating during fever, dietary deficiencies, or the addition of thiazides or loop diuretics, especially when unprotected by angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers. Integrated nurse-driven care with experienced nurses supervised by a cardiologist can be extremely helpful in safely applying AAD therapy AF.357
5.3. Electrocardiogram anti-arrhythmic drug effects
Initiation of AADs may induce within days alterations of the surface ECG encompassing slowing of the sinus rate, SA block, AV prolongation, higher degree AV block, QRS, and QT prolongation (see Supplementary material online, Table S8). The electrophysiological effects of AADs are different, and therefore, the impact on the surface ECG may differ between Class I and Class III AADs. The occurrence of PVCs and non-sustained VT might be the first signs of pro-arrhythmic events that could lead to fatal VT or VF. Occurrence of symptomatic electrophysiological changes (bradycardia, SN arrest, AV block or repetitive PVCs, etc.) must lead to dose reduction of the AAD or even termination of therapy. Prolongation of the QRS width >25% or prolongation of the QTc above 125% from baseline (or QTc above 500 ms) must trigger termination of the AAD therapy.
After initiation of flecainide, an increase up to 25% of baseline QRS duration on steady-state therapy (after ∼5 plasma half-lives, after 3–4 days) is a sign of drug action and underlies effective treatment (Figure 16A and B). An increase in QRS duration of >25–50% (depending on baseline QRS duration) compared with baseline represents a potential risk for pro-arrhythmia or induction of HF (Figure 16C). In that case, the dose is advised to be reduced or flecainide be discontinued. Exercise enhances use-dependent effects. Therefore, exercise testing to exclude excessive use-dependent QRS widening and show the potential for causing VA may be used after reaching steady state.358 Recently, it has been proposed to apply a test dose of 250 mg of fast-acting oral flecainide (or 200 mg if body weight is below 70 kg) to determine the appropriate flecainide starting dose and exclude treatment in potentially high-risk patients.359 The scheme includes checking of blood pressure and changes in QRS complex duration at the predicted peak plasma concentration at 2 h. After initiation of sotalol and amiodarone, an excessive increase in QT beyond 60 ms may be associated with TdP pro-arrhythmia, and discontinuation, drug reduction, or avoidance of concomitant drugs with a known potential to prolong the QT are warranted.
Figure 16.

Electrocardiogram (ECG) tracings (Lead III and monitor lead) illustrating the progression of atrial fibrillation (AF) and its response to 300 mg/day flecainide, culminating in ventricular tachycardia (VT) in a 66-year-old female with no underlying heart disease. (A) Baseline AF at a heart rate of 105 b.p.m. with a QRS duration of 90 ms. (B) After administration of 300 mg of flecainide, QRS duration prolongs to 120 ms, while the heart rate remains unchanged at 105 b.p.m. (C) Spontaneous acceleration of AF to 180 b.p.m. leads to further QRS widening to 210 ms, and regularization (atrial flutter conversion with bundle-branch blockvs VT) attributed to the use-dependent effect of flecainide. This sequence underscores the potential pro-arrhythmic effects of flecainide in AF management, if a high dosage is used. The ECG was recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
5.4. Anti-arrhythmic drug tests for electrophysiological evaluation
These pharmacological interventions are instrumental in evaluating the electrophysiological characteristics of the His-Purkinje system and AV nodal conduction, particularly in diagnosing conduction disorders and susceptibility to arrhythmias. A more comprehensive review of these interventions has been published recently.360
Anti-arrhythmic drugs are essential tools in the pharmacological assessment of patients’ electrophysiological properties, enabling clinicians to identify and evaluate various cardiac conduction abnormalities.
Class I AADs
Class I are utilized to assess His-Purkinje system conduction. By inhibiting sodium channels, these agents can unmask latent conduction defects within the His-Purkinje network, facilitating the diagnosis of conditions such as BBBs or intraventricular conduction delays. In addition, ajmaline or flecainide are employed to unmask conditions such as BrS by revealing characteristic ECG patterns.
Adenosine
Adenosine may be used for evaluation of AV nodal conduction.
Adrenaline
Adrenaline infusion is employed in the evaluation of congenital LQTS by provoking characteristic ECG changes, thereby aiding in the diagnosis of this condition. It is also used in the diagnosis of catecholaminergic polymorphic VT.
Isoprenaline, atropine, and autonomic blockade
Isoprenaline increases heart rate and enhances conduction through the AV node and His-Purkinje system, making it useful in identifying latent conduction abnormalities and assessing susceptibility to tachyarrhythmias under sympathetic stimulation. Atropine, by inhibiting parasympathetic influences, accelerates SN activity and improves AV nodal conduction, helping to differentiate between intrinsic conduction system disease and vagally mediated conduction delays in the AV node and in the SN function. The combined administration of a β-blocker and atropine achieves autonomic blockade, minimizing autonomic influences on the heart. This approach allows for the assessment of intrinsic SN function and AV conduction properties without autonomic interference.
5.5. Pro-arrhythmia
Anti-arrhythmic drugs share a narrow therapeutic window due to their association with multiple adverse effects, particularly with pro-arrhythmic effects and organ toxicity (see Supplementary material online, Table S6). Therefore, the pharmacological management of AF and other arrhythmias requires a strategy of ‘First, do no harm’ perspective.361 The full profile of potential adverse effects is advised to be taken into consideration in every patient. Knowledge of potentially dangerous pro-arrhythmia and toxic effects (see below) is therefore of paramount importance on a patient-by-patient basis.
Paradoxical worsening or new onset of arrhythmias caused by an AAD or other medications that affect cardiac EP is termed pro-arrhythmia. The pro-arrhythmic effects of AADs have been noted as early as in the 1960s (description of quinidine syncope).361 In the 1990s, two landmark trials of AAD, the CAST and the SWORD trials, demonstrated increased mortality in post-infarction patients, presumably due to the pro-arrhythmic effects of the AAD studied.362,363 Such drug-induced ventricular pro-arrhythmic effects have also been described in studies evaluating AADs in subjects with AF. Since this arrhythmia constitutes the major field of AAD use nowadays, risk stratification for and avoidance of pro-arrhythmia is critical.
The potential for pro-arrhythmic effects is shared in common by all AADs121 and may manifest as a pathological bradyarrhythmia (i.e. sinus bradycardia, AV conduction disturbances) or as tachyarrhythmias (i.e. polymorphic VT of the TdP type or incessant monomorphic VT) (Table 10). Systematic studies have revealed distinct risk factors for the occurrence of pro-arrhythmia, such as female gender, age, presence of SHD, reduced left ventricular function, impaired renal function (i.e. in case of sotalol), or concomitant polypharmacy. In addition, genetic variants may influence the metabolism of a particular AAD, which is particularly important in drugs primarily eliminated by a single affected pathway. This is the case for digoxin, propafenone, sotalol and dofetilide, which show higher plasma levels in poor metabolizers. Finally, thecombination of AADs may substantially increase the pro-arrhythmic effects. Although ranolazine mitigates the pro-arrhythmic risks of Class III AADs by blocking early EADs and TdP through INa,L current inhibition, making it a potentially safer adjunct in AAD combination therapy, it is also a moderately P-gp inhibitor, which may impair elimination of DOACs. The risk of pro-arrhythmia also has implications in terms of where to initiate therapy with AADs (i.e. in or out of hospital) since pro-arrhythmic events have the tendency to occur shortly after drug initiation (i.e. pharmacological cardioversion of AF) as discussed before. Details of pro-arrhythmic effects associated with the use of specific AADs are provided in the following sections.
Table 10.
Pro-arrhythmia risk and typical pro-arrhythmia forms of different AADs
| Class | Drug | Risk | Type of pro-arrhythmia |
|---|---|---|---|
| 0 | Ivabradine | Low | Bradycardia, AV block, AF |
| Ia | Quinidine | High | TdP |
| Procainamide | Moderate | AV block, monomorphic VTa, TdP | |
| Disopyramide | Low | Bradycardia | |
| Ib | Mexiletine/lidocaine | Low | Bradycardia, AV block |
| Ic | Flecainide | Moderate | AFLb, monomorphic VTa, bradycardiac |
| Propafenone | Moderate | AFLb, monomorphic VTa, Bradycardiac | |
| Id | Ranolazine | Low | QT prolongation |
| IIa | β-Blockers | Low | Bradycardia, AV block |
| IIb | Isoprenaline | Low | Sinus tachycardia, PACs, PVCs, VT |
| IIc | Atropine | Low | Sinus tachycardia, paradoxical AV blockd |
| IId | Digoxin/digitoxin | Moderate | AV block, junctional tachycardia, polymorphic VT, AT with AV block |
| IIe | Adenosine | Moderate | Transient sinus bradycardia and AV block, AF, PACs, PVCs |
| III | Amiodarone | Low | Bradycardia, AFLb |
| Dronedarone | Lowe | Bradycardia | |
| Dofetilide | High | TdP | |
| Ibutilide | High | TdP | |
| Sotalol | High | TdP, bradycardia | |
| Vernakalant | Low | Sinus bradycardia, NSVT | |
| IV | Verapamil | Low | Bradycardia, AV block |
| Diltiazem | Low | Bradycardia, AV block |
Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AT, atrial tachycardia; AV, atrioventricular; NSVT, non-sustained VT; PAC, premature atrial contraction; PVC, premature ventricular contraction; TdP, torsades de pointes; VT, ventricular tachycardia.
aIn patients with structural heart disease.
bIn patients with AF.
cIn patients with sinus node dysfunction or AV conduction disorders.
dWorsening of AV block on the ECG, such as a progression from second-degree AV block to AV block as atropine increases the sinus rate.
eHigh when combined with digitalis, as dronedarone reduces the renal excretion of digitalis, amplifying its associated risks. The combination may also increase the likelihood of AV block and other pro-arrhythmic effects.
5.5.1. Sinus bradycardia and arrest
All AADs can cause sinus bradyarrhythmias.364 AADs can influence SN function by exacerbating both intrinsic and extrinsic factors that contribute to SN dysfunction (SND).365 Intrinsic factors include pre-existing, either manifest or subclinical, SND, which makes the SN more susceptible to dysfunction. Extrinsic factors include metabolic or autonomic system disturbances that affect SN activity. Anti-arrhythmic drugs can exert direct effects by depressing PM currents or impairing SA conduction, leading to bradycardia or sinus arrest (Table 11). Indirectly, AADs may modulate autonomic influences on the SN, either by inhibiting sympathetic stimulation or potentiating parasympathetic tone, further suppressing SN activity. These effects underscore the need for careful assessment of baseline SN function and close monitoring of patients receiving AAD therapy.
Table 11.
Effects of AADs on sinus node function
| Modified VW class | Drug | Potential for SN depression |
|---|---|---|
| 0 | Ivabradine | Moderate |
| Ia | Quinidine | Milda |
| Procainamide | Mild | |
| Disopyramide | Milda | |
| Ib | Mexiletine/lidocaine | ∼None |
| Ic | Flecainide | Mild to moderateb |
| Propafenone | Mild to moderateb | |
| Id | Ranolazine | ∼None |
| IIa | β-Blockers | Potent |
| IIb | Isoprenaline | ∼None (reverse depression) |
| IIc | Atropine | ∼None (reverse depression) |
| IId | Digitalis | Variable (depends upon autonomic balance; mild direct effect) |
| IIe | Adenosine | Potent |
| III | Amiodarone | Potent |
| Dronedarone | Mild | |
| Dofetilide | ∼None | |
| Ibutilide | ∼None | |
| Sotalol | Potent | |
| Vernakalant | Mild to moderate | |
| IV | Verapamil | Milda |
| Diltiazem | Milda |
Abbreviations: AAD, anti-arrhythmic drug; SN, sinus node.
aAssociated mild vasodilation partially offsets SN depression.
bModerate in patients with SN dysfunction.
5.5.2. Atrioventricular block
High-grade AV block is a rare pro-arrhythmic effect of Class Ic and Class Ia anti-arrhythmic agents253 and may also be observed during treatment with β-blockers, verapamil, diltiazem, digoxin, and even with amiodarone and dronedarone. When seen, the clinical question is if AV block is mainly caused by the pharmacological agent or is the result of pharmacological effects acting on an already altered substrate that will eventually lead to AV block in the absence of the drug. It is important to distinguish between cases of drug overdosing or interactions leading to supra-therapeutic drug levels and cases with the appearance of AV block at normal levels of anti-arrhythmic agents, frequently after a long period of treatment. Beyond overdosing and drug interactions, the traditional view that anti-arrhythmic agents are the sole cause of AV block—and that PM implantation is unnecessary after drug discontinuation—has recently been challenged. Emerging data indicate that these patients often do not follow a benign course after stopping the suspected pro-arrhythmic medication, with more than 50% experiencing recurrence of AV block during follow-up despite the absence of ongoing therapy.366,367 In these series, AV block ‘truly caused by drugs’ was found in only 15% of patients who had second-nd or third-degree AV block during therapy with β-blockers, verapamil, or diltiazem, suggesting that AV block is more commonly ‘unmasked by drugs’, while rarely ‘caused by drugs’ and that in daily practice permanent pacing may be appropriate.366,367
5.5.3. New-onset, sustained, monomorphic ventricular tachycardia
The first occurrence of spontaneous monomorphic, sustained VT, soon after initiating anti-arrhythmic therapy in a patient without previous sustained VT, is considered a pro-arrhythmic response.368,369 This type of pro-arrhythmia is most likely to occur in the presence of organic heart disease, left ventricular dysfunction and Class Ic agents.370,371 Increasing the drug dose further may lead to a slower but more frequent sustained VT, and the offending drug is advised to be discontinued as soon as this pro-arrhythmic response is recognized.
5.5.4. Increased frequency of sustained ventricular tachycardia
The occurrence of an increased frequency of sustained VT in a patient with a clinical history of ventricular tachyarrhythmias is also a pro-arrhythmic response.368,369 However, this condition is often secondary to a spontaneous recurrence and the inefficacy of the AAD. Increasing the drug dose further in this situation may worsen the arrhythmia or, alternatively, may resolve it if the inefficacy was secondary to inadequate anti-arrhythmic blood levels.372 Stopping the drug that caused this arrhythmic response will improve this situation and may prevent incessant VT from developing.
5.5.5. Incessant ventricular tachycardia
Incessant VT is a pro-arrhythmic response that can occur during AAD therapy.368–371 Class Ic agents have been associated with the highest occurrence of this type of pro-arrhythmia.370,371 These drugs profoundly slow conduction with minimal effects on refractoriness; therefore, these drugs may alter the balance between refractoriness and conduction in an arrhythmogenic zone. Incessant VT can also occur with other anti-arrhythmic agents. Its occurrence is most common in patients with a history of sustained VT associated with left ventricular dysfunction306,373,374 and characterized by a wide complex, ‘sine wave’ tachycardia that has broad, undulating complexes. The rate of the tachycardia is usually slower than that of a spontaneous tachycardia. Incessant VT often cannot be terminated by pacing or even by cardioversion. It may present as sustained VT or as long runs of non-sustained VT with periodic sinus beats and quick resumption of paroxysms of VT. Adding other AADs is usually not helpful. Discontinuing the provoking AAD and cardioverting the patient after the drug's effects have passed is the best treatment. In case of haemodynamic compromise, mechanical left ventricular support devices can transiently maintain the patient's haemodynamic status.
5.5.6. Torsades de pointes
Dessertenne375 described TDP as ‘twisting around the points’ VT. However, TDP is more than a QRS changing pattern and is classically described as a pause-dependent, polymorphic VT associated with QT prolongation and U waves. Many polymorphic VTs are miss-classified as TDP that do not meet these classic criteria.376 The mechanism of this arrhythmia is secondary to prolongation of repolarization that results in activation of EADs, which may promote triggered activity.376 Re-entry, due to a dispersion of refractory periods of the different layers of the ventricle, is another potential mechanism of TDP.377–380 QT prolongation is due to blockade of one of the cardiac K+ channels expressed by the human ether-a-go-go-related gene (hERG).377–381 This results in inhibition of a major repolarizing potassium current, IKr. TDP may result from pro-arrhythmia which occurs secondary to QT-prolonging agents. Although TDP is usually secondary to an AAD overdose with marked prolongation of the QT, some episodes are idiosyncratic and may occur after only a few doses of AAD. The Class Ia AADs, quinidine, procainamide and disopyramide, and the Class III anti-arrhythmic agents, sotalol, dofetilide, ibutilide, and some psychotropic drugs which block the delayed rectifier potassium current have the highest frequency of causing this arrhythmia.153,381,382 In particular, the heart rate slowing-effect of sotalol may enhance its pro-arrhythmic effect due to reversed use dependency with stronger AP prolongation during bradycardia or after pauses. These drugs cause TDP in up to 5% of cases and are advised to be initiated under telemetry conditions since most TDP occurs early during drug initiation, as mentioned before. A list of drugs that can provoke TDP and are discouraged in patients with LQTS or previous TDP has been published (see Supplementary material online, Table S9; www.crediblemeds.org).383 Amiodarone and dronedarone, multi-channel blockers including Class III effects that also prolong the QT interval, are rarely associated with this form of pro-arrhythmia.384,385 The low incidence of amiodarone-induced TDP may be related to its lack of reverse use dependence (see above) and its lesser effect on prolonging APD in the ‘M’ cell region compared to other Class Ia and IIIa agents.385 Most amiodarone-induced episodes of TDP occur when the drug is combined with a type Ia anti-arrhythmic agent. Class lb AADs and β-blocking agents, which shorten the QT interval, are useful treatments for this syndrome. Class Ic agents have little effect on repolarization and are only rarely associated with this form of pro-arrhythmia.
Box 12 Risk factors associated with TdP.
Age >65 yearsa
Female sexa
Congenital long QT syndrome (clinical or subclinical due to incomplete penetrance, either mono- or polygenetic)
-
Personal history
History of syncope
History of TdP or significant bradycardia
Current nausea, vomiting, diarrhoea, laxative use
-
Structural heart disease
Myocardial ischaemia
Heart failure
Left ventricular hypertrophy
-
Systemic disorders
Renal or liver failure
Hypo-thyroidism
Subarachnoid haemorrhage
Hypothermia
-
Electrolyte disorders
Hypokalaemia (<3.5 mmol/L)
Hypocalcaemia (<8.5 mmol/L)
Hypomagnesaemia (≤0.7 mg/dL)
-
Drugs
QT-prolonging medications
Diuretic therapy
Drug–drug interactions
ECG signs (Box 13)
Abbreviations: ECG, electrocardiogram; TdP, torsades de pointes.
aAge and sex alone are not sufficient to contraindicate certain AADs, though they are advised for heightened monitoring and control of other risk factors
A close relationship between QT interval prolongation and the development of TDP has not been well established for Class Ia agents.376,381 With D,L-sotalol and dofetilide, QT interval prolongation and higher doses increase the risk of TDP. For D,L-sotalol, avoiding QTc interval more than 525 ms and doses more than 320 mg will decrease the incidence of TDP from 5% to <2%. In general, patients with baseline QT interval prolongation must avoid drugs that prolong the APD. Many other factors (Boxes 12 and 13)386–388 are related to the development of QT interval prolongation, including concomitant therapy with other drugs that prolong APD, the presence of congenital prolonged QT interval syndrome, hypokalaemia, hypocalcaemia, hypomagnesaemia, diuretic use, female gender, renal dysfunction, and severe bradycardia. Certain drugs involved in drug interactions are common contributors to TDP risk. QT-prolonging medications, like as clarithromycin, levofloxacin, or haloperidol, when taken concurrently with CYP inhibitors, such as fluoxetine, cimetidine, or certain foods such as grapefruit, can lead to elevated levels of medications that prolong the QT interval and increase the risk of developing TDP.383
Box 13 ECG signs indicative of risk for TdP.
Bradycardia (<60 bpm), including recent conversion from AF
QTc >500 ms
QT increase >60 ms from baseline
T-wave alternans
T- or U-wave distortion
Ventricular ectopy and non-sustained VT triggered after a pause
Abbreviations: AF, atrial fibrillation; ECG, electrocardiogram; TdP, torsades de pointes; VT, ventricular tachycardia.
5.5.7. Atrial pro-arrhythmia
Box 14 lists potential criteria for atrial pro-arrhythmia.389 The development of AT with digitalis and the increased frequency of incessant AFL exemplify atrial pro-arrhythmia. This phenomenon has been observed with all class Ia agents, amiodarone, and most notably with class Ic agents. Class Ic anti-arrhythmic drugs slow atrial conduction, which can stabilize macro re-entry circuits in predisposed anatomical regions, leading to more frequent and incessant AFL.390–392
Box 14 Criteria for supraventricular pro-arrhythmia.
Conversion of AF to incessant AFL
Atrial flutter with slower atrial rate and new-onset 1:1 AV conduction
New-onset supraventricular tachyarrhythmia
More frequent with slower rate paroxysmal supraventricular tachycardia
Incessant accessory pathway mediated tachycardia following drug-induced bundle branch block
Digitalis-induced atrial tachycardia with AV block
Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AT, atrial tachycardia; AV, atrioventricular.
Occasionally, AFL with 1:1 AV conduction may occur due to a combination of slowed atrial rates and enhanced AV nodal conduction, potentially resulting from vagolytic effects or incidental sympathetic stimulation. Adrenergic events, such as stress testing, may unmask 1:1 AV conduction.281 Due to rapid rates and wide QRS morphology secondary to rate dependence, these tachycardias may be misclassified as ventricular in origin.
Re-entrant arrhythmias may occur more frequently, although slower after anti-arrhythmic therapy.390 A classic example of this is with Na+ channel blockers used to treat patients with orthodromic SVT in the WPW syndrome. Typically, AADs will slow conduction and prolong refractoriness more in the antegrade than the retrograde direction of the pathway. Thus, a PAC is more likely to develop unidirectional block and initiate SVT, although at a slower rate. The occurrence of more frequent but slower SVTs can also be noted with drugs such as digitalis, verapamil or β-blockers, which slow conduction in the AV node.
5.5.8. Brugada mechanism
Class Ic anti-arrhythmic agents are largely used in the treatment of AF and other supraventricular tachyarrhythmias.92,253,393 In clinical practice, unselected patients treated with propafenone or flecainide at therapeutic doses may exhibit a typical type 1 Brugada pattern with a right BBB and ST-segment elevations in the right pre-cordial leads, even in the absence of symptoms such as unexplained syncope or a family history of sudden death or CA. In a series of 176 patients, serial ECGs before and after achieving steady-state concentrations (>5 half-lives) of propafenone and flecainide were performed, and a Brugada ECG pattern was found in only 2.3% of the patients, in some cases several months after therapy initiation.394 Of note, drug therapy was continued in all patients regardless of its effects on ST segments or the development of BBB, and no VA events occurred in any patient during follow-up.394 These and other data suggest the opportunity to reconsider the specificity of a Brugada pattern induced by Class Ic drugs in asymptomatic patients. Indeed, the recent ESC Guidelines on VA and SCD clearly state that BrS is diagnosed in patients without other heart disease and with a spontaneous Type 1 pattern, regardless of symptoms.45 The guidelines emphasize that a Type 1 ECG pattern induced by a Nav-blocking drug, whether as part of diagnostic testing or resulting from anti-arrhythmic treatment, should be considered less specific than previously thought, as it can be seen in 2–4% of healthy individuals without a spontaneous Type 1 pattern. In the opinion of the ESC Guidelines panel of experts, an induced Type 1 Brugada pattern therefore requires other clinical features, such as documented ventricular tachyarrhythmias, arrhythmic syncope, or relevant family history, to make a diagnosis of BrS.45 Other data confirm the good prognosis of patients who do not have symptoms typical of BrS but who develop a Brugada pattern during treatment with Class Ic drugs or other agents.395 Regardless of their good prognosis in these cases, drug discontinuation is advised,395 and therefore, it appears important in daily practice to plan an ECG check a few days after initiation of propafenone or flecainide, as suggested by Guidelines.92
5.6. Toxicity and adverse effects
All AADs have the potential for significant toxic effects encompassing different organs (Table 12). Direct organ toxicity may be encountered with specific AADs. Furthermore, some AADs like quinidine and amiodarone have substantial vasodilatory effects, and thereby, profound hypotension can be induced by the combination with other vasodilators or rapid i.v. injections. An example for a substance with significant organ toxicity is amiodarone, one of the most popular AADs. This drug may affect several organ systems, including the thyroid, lungs, skin, liver, eyes and others.396 This extra-cardiac toxicity has largely been attributed to the iodine moieties of amiodarone, its high lipophilicity, and its direct effects on thyroid function.82
Table 12.
Key side effects and toxicities of AADs aside from pro-arrhythmia
| Modified VW Class | AAD | #1 | #2 | #3 | Other significant or less frequent effects |
|---|---|---|---|---|---|
| 0 | Ivabradine | Phosphenes (2–14%) | Hypertension (8%) | Fatigue (5%), | CNS effects (5%) |
| Ia | Quinidine | Gastrointestinal (nausea, vomiting, diarrhoea, —30–50%) | Light headedness/headache (15%) | Hypotension (5–10%), | Cinchonism a , allergic reaction (5–10%), thrombocytopenia (1–2%), CNS effects (5%) |
| Procainamide | Lupus erythematosus-like syndrome (20–30%) | Hypotension (5%) | Gastrointestinal (nausea, vomiting, diarrhoea—5–10%) | Allergic reaction (5%), blood dyscrasias (agranulocytosis, thrombocytopenia—1%), CNS effects (5%) | |
| Disopyramide | Anti-cholinergic (urinary retention, constipation, dry mouth—30–40%) | Hypotension (5–10%) | Gastrointestinal (nausea, vomiting—5–10%) | Heart failure (5%), hypoglycaemia (2%), CNS effects (5%) | |
| Ib | Lidocaine | CNS effects (dizziness, tremors, tinnitus—1–10%) | Hypotension (1–5%) | Gastrointestinal (nausea, vomiting, constipation—1–5%) | Skin rash (1%) |
| Mexiletine | Gastrointestinal (nausea, vomiting, diarrhoea, —5–40%) | CNS effects (dizziness, ataxia, tremor, 5–20%) | Hypotension/fatigue/weakness (5–10%) | Skin rash, limb swelling (3%) | |
| Phenytoin | CNS effects (nystagmus, ataxia, other, 5–50%) | Gingival hyperplasia (20–40%), | Gastrointestinal (nausea, vomiting, constipation—13%) | Hypotension (<10%), thrombocytopenia (<1%) | |
| Ic | Flecainide | CNS effects (dizziness, blurred vision—15–50%) | Fatigue/weakness (5–10%) | Gastrointestinal (nausea, vomiting—5–10%) | Dyspnoea/heart failure (5%) |
| Propafenone | CNS effects (Dizziness, blurred vision—10–30%) | Metallic taste (5–10%) | Gastrointestinal (nausea, vomiting—10–15%) | Fatigue/dyspnoea/heart failure (5%) | |
| Id | Ranolazine | Dizziness (6–15%) | Gastrointestinal (nausea, vomiting, constipation—10%) | Headache (5%) | Asthenia (<5%) |
| IIa | β-Blockers | Fatigue/asthenia (10–30%) | Dizziness/hypotension (10%) | Cold extremities (10%) | Sexual dysfunction (1–28%), insomnia (1–5%), depression (1–5%), bronchospasm (1–5%) |
| IId | Digitalis | Nausea/vomiting (>10%) | CNS effects (confusion, dizziness—5–10%) | Visual disturbances (yellow vision, halos around light—5%) | Fatigue (5%), gynaecomastia (<1%) |
| III | Amiodaroneb | Corneal microdeposits (>90%), optic neuritis with risk of blindness (1%) | Photosensitivity (25–75%) and blue-grey skin discolouration (10%) | Hypo- (5–20%) and hyper-thyroidism (1–5%) | CNS effects (30%), nausea/vomiting (10–25%), liver toxicity (15–30% elevated enzymes) lung toxicity (1–10%) |
| Dronedarone | Increased serum creatinine c (10–20%) | Gastrointestinal (nausea, vomiting, diarrhoea—5–15%) | Fatigue and asthenia (5–10%) | Skin reactions (5%), mild elevation of liver enzymes (1–5%, hepatotoxicity <1%)) | |
| Dofetilide | Headache and dizziness (10%) | Chest pain (10%) | Gastrointestinal (nausea, vomiting, diarrhoea—5%) | Insomnia (<5%) | |
| Ibutilide | Headache and Dizziness (5%) | Hypo/hypertension (2%) | Gastrointestinal (nausea—5%) | Flushing (<5%) | |
| Sotalol | Fatigue (10–20%) | Dizziness (5–15%) | Hypotension (5–10%) | Gastrointestinal (nausea, vomiting, diarrhoea—5%) Worsening heart failure (1–3%) |
|
| Vernakalant | Dysgeusia (25%) | Sneezing (15%) | Hypotension and dizziness (5–10%) | Cough, nausea (5%) | |
| IV | Verapamil | Constipation (10%) | Hypotension and dizziness (5–10%) | Headache (2%) | Gingival hyperplasia, nausea, peripheral oedema, worsening heart failure (<5%) |
| Diltiazem | Peripheral oedema (10%) | Hypotension and dizziness (5–10%) | Headache (2–5%) | Gingival hyperplasia, gastrointestinal (nausea, vomiting, diarrhoea, constipation, worsening heart failure (<5%) |
Side effects and toxicities are listed in columns: #1 represents the most frequent, followed by #2 and #3 as the second most frequent. Additionally, the most characteristic side effects are highlighted in bold. A more comprehensive list of all effects can be found in the supplementary material online.
Abbreviations: AAD, anti-arrhythmic drug; CNS: central nervous system effects, including dizziness, ataxia, tremor, blurred vision, confusion, and headache; VW, Vaughan Williams.
aTinnitus, reversible hearing loss, flushing, confusion, diarrhoea, and visual disturbances, including permanent blindness in some cases.
bUp to 70% incidence of adverse effects (15% during the first year, 50% during long-term use) with an 18–37% rate of adverse-effect-driven drug discontinuation at 5 years follow-up. Fifteen per cent during the first year of amiodarone use increasing to up to 50% during long-term use.
cDue to inhibition of tubular secretion of creatinine without affecting kidney function.
5.6.1. Amiodarone-induced thyroid dysfunction
Amiodarone may influence thyroid function tests (transient elevation of TSH and decrease of T3 are commonly seen) and, in addition, it may induce thyroid dysfunction [amiodarone-induced hypothyroidism (AIH) and amiodarone-induced thyrotoxicosis (AIT)]. Amiodarone-induced hypo-thyroidism and AIT might occur in apparently normal thyroid glands. There are two types of AIT: Type 1 AIT (AIT 1), a form of iodine-induced hyper-thyroidism occurring in nodular goitres or latent Graves’ disease, and Type 2 AIT (AIT 2), due to destructive thyroiditis (Table 13).
Table 13.
Main features of the two types of amiodarone-induced thyrotoxicosisa
| AIT1 | AIT2 | |
|---|---|---|
| Geographical areas of incidence | Iodine depleted | No relation |
| Pre-existing thyroid abnormalities | Present (latent or overt Graves’ disease or nodular goitre) | Absent (normal thyroid) |
| Main mechanism | Iodine overload from amiodarone | Destructive thyroiditis (cytolysis with release of stored thyroid hormones) |
| Onset | Gradual shortly after amiodarone start (3 months) | Sudden long after amiodarone start (30 months) |
| Laboratory findings | TSH antibodies may be present | Elevated inflammatory markers (PCR) |
| Colour flow Doppler | Increased vascularity | Decreased vascularity |
| Radioiodine uptake | Normal/increased | Suppressed |
| Treatment | Anti-thyroid drugs, Potassium perchlorate (in iodine-depleted areas) Thyroidectomy or radioactive iodine ablation may be appropriate |
Oral glucocorticoids (anti-thyroid drugs are ineffective) |
| Amiodarone discontinuation | Advised | May not be needed since it often resolves with glucocorticoids |
| Outcome | No spontaneous remission High risk of recurrence |
Spontaneous remission possible |
During amiodarone treatment, it is expected that TSH serum levels may increase up to 2.7 times by 2 weeks with a fall of TSH to the upper end of the normal range after 3 months.
Abbreviations: AIT, amiodarone-induced thyrotoxicosis; PCR, protein C-reactive; TSH, thyroid-stimulating hormone.
aBoth conditions require careful differentiation for optimal management, but sometimes they may co-exist and may necessitate combined therapeutic approaches.
5.6.2. Amiodarone-induced pulmonary and other systemic toxicities
Amiodarone toxicity affects multiple organs other than the thyroid, including the lungs, liver, skin, and eyes. Pulmonary toxicity results in interstitial pneumonitis and fibrosis in 1–2% of patients (Figure 17).85,397 Pulmonary toxicity is often not recognized in patients who commonly have other reasons for respiratory failure. Symptoms include cough, dyspnoea, fever, and pleuritic chest pain. Patients may also present with fatigue, weight loss, and hypoxia. Blood tests usually show elevated markers of inflammation, such as C-reactive protein and erythrocyte sedimentation rate. Chest X-ray may show diffuse interstitial or alveolar infiltrates and computed tomography scans may show ground-glass opacities, interstitial thickening, and consolidation in both lungs. Pulmonary function tests often show a restrictive pattern with reduced diffusion capacity. Definitive diagnosis of amiodarone-induced pulmonary toxicity is achieved through bronchoalveolar lavage, which typically reveals the presence of lipid-laden (foamy) macrophages, and biopsy can confirm the diagnosis if other tests are inconclusive. Treatment includes prompt amiodarone interruption, although due to its long half-life, it does not result in quick improvement, and patients often need complementary treatment with high-dose corticosteroids (e.g. prednisone 40–60 mg/day). In addition, amiodarone can produce hepatotoxicity in 0.5–1.0% of cases and involves elevated liver enzymes, with potential for acute hepatitis or cirrhosis. Skin toxicity typically presents as photosensitivity and blue-grey discolouration, particularly in sun-exposed areas (25–75% of patients). Ocular toxicity includes corneal microdeposits in most treated patients, which are usually asymptomatic but can also cause optic neuropathy (1–2%), leading to vision loss. All these toxicities result in discontinuation in up to 20% of patients during long-term therapy.82
Figure 17.

Chest X-ray (A and B), computed tomography (CT) scan (C), and microscopic views of a lung biopsy (D and E) from a 77-year-old former smoker with a history of old myocardial infarction and preserved left ventricular ejection fraction, taking 200 mg/day of amiodarone for paroxysmal atrial fibrillation. The patient presented with cough, dyspnoea, and weight loss. Findings were suggestive of amiodarone-induced lung toxicity. (A) Chest X-ray at presentation showing a diffuse alveolar-interstitial pattern indicative of pulmonary involvement. (B) Follow-up chest X-ray after 3 months of amiodarone withdrawal and steroid therapy showing resolution of lung abnormalities. (C) Computed tomography scan confirming the diffuse alveolar-interstitial pattern at the initial presentation. (D) Optical microscopy (haematoxylin–eosin stain) of lung parenchyma showing clusters of alveolar macrophages (arrows) with foam-like cytoplasmic changes, characteristic of amiodarone toxicity. (E) Electron microscopy of the biopsy sample revealing phospholipid inclusions (red arrow) in macrophages, further confirming the diagnosis of amiodarone-induced pulmonary toxicity. This case underscores the potential for severe pulmonary adverse effects associated with amiodarone therapy and the potential reversibility of findings following drug discontinuation and appropriate treatment.
5.6.3. Quinidine systemic toxicities
The most frequent secondary effects of quinidine are nausea, vomiting, and diarrhoea, which may appear in up to one third of patients. It may also be associated with anaemia, thrombocytopenia, urticaria and, more infrequently, lupus-like reactions. It may also provoke orthostatic hypotension due to α-adrenergic block, which is potentiated by the concurrent use of vasodilators, such as ACE inhibitors. Quinidine toxicity can lead to a condition known as cinchonism, characterized by symptoms such as tinnitus, hearing loss, and blurred vision.398,399
5.6.4. Other anti-arrhythmic drug systemic toxicities
Phenytoin has been associated with gingival hyperplasia in rare patients. Disopyramide may be responsible for glaucoma, urinary retention, and hypoglycaemia. Systemic serositis has been rarely reported with AADs such as flecainide. A systemic lupus erythematosus-like syndrome, agranulocytosis, and hypersensitivity reactions may develop with procainamide treatment. Flecainide toxicity is frequently seen in patients when renal function deteriorates, due to the high renal elimination of this drug, and propafenone is advised instead in patients with impaired renal function. Propafenone overdosage may result in hypotension, bradycardia, intra-atrial and intraventricular conduction disturbances, and VA. As with flecainide, CNS adverse effects such as dizziness, nausea, unusual taste, blurred vision, and convulsions occur most frequently with higher doses or plasma concentrations. Sotalol intoxication (2–16 g) may cause death due to pro-arrhythmia (asystole, TdP, and polymorphic VT) and congestive HF. Other extra-cardiac toxicities include hypotension, bronchospasm and hypoglycaemia, which may also be seen with other β-blockers. Digitalis toxicity can manifest through various non-arrhythmic symptoms, including gastrointestinal disturbances such as nausea, vomiting, and abdominal pain. Neurological symptoms may also occur, presenting as confusion, dizziness, and visual disturbances like blurred yellow vision or seeing halos. Verapamil effects include hypotension, bradycardia, cardiac conduction defects, arrhythmias, hyperglycaemia, and decreased mental status. In addition, there have been reports of non-cardiogenic pulmonary oedema in patients taking large overdoses of verapamil (up to ∼9 g).
5.7. Pro-arrhythmia and anti-arrhythmic drug toxicity management
5.7.1. General aspects
In addition to general supportive measures, the cardiac rhythm and blood pressure have to be monitored, and if bradycardia ensues, a β-adrenergic agonist or a PM may be used. Hypotension with inadequate tissue perfusion is advised to be treated with positive inotropic and/or vasopressor agents. Acute bradycardia and bradycardia-induced polymorphic VT can be treated with temporary ventricular pacing at a fast rate (90 b.p.m.). In some cases, with predominant β-blocking effects, the use of isoprenaline might be useful. For severe hypotension, the use of i.v. catecholamines (adrenaline and noradrenaline) may be appropriate. Haemodialysis for drug removal is effective for procainamide, disopyramide, and sotalol. All other AADs cannot be removed by haemodialysis (e.g. flecainide/propafenone, verapamil, dronedarone, and amiodarone).
5.7.2. Torsades de pointes management
The first step in managing TdP is preventing its onset by targeting modifiable risk factors and preventing TdP from occurring.386 Treatment of TdP aims to restore a normal rhythm and to prevent the arrhythmia from recurring. TdP is usually non-sustained and spontaneously reverts to a normal SR. Sustained TdP requires emergency treatment, including electrical cardioversion if needed.45,400 Treatment to prevent recurrent TdP includes withdrawal of all QT-prolonging drugs, IV magnesium sulphate and correction of hypokalaemia, hypomagnesaemia, and hypocalcaemia. Isoprenaline and pacing may help prevent pause-dependent TdP as other measures are undertaken. Treatments used to prevent TdP in specific circumstances include β-blockers or mexiletine in LQTSs 1–2 and 3, respectively.369
5.7.3. Drug-specific aspects
When appropriately prescribed and monitored, flecainide and propafenone present a low risk of pro-arrhythmia or other significant side effects.275,276 However, in rare instances, patients may develop bradyarrhythmias, sinus pauses, or AV block, necessitating dose reduction or discontinuation of the drug. A notable concern is the potential conversion of AF to AFL with 1:1 AV conduction, which can be mistaken for VT due to aberrant conduction patterns (Figure 6B). In such cases, catheter ablation of the AFL circuit is advised.272,273Additionally, if HF symptoms emerge, immediate cessation of these drugs is imperative. To mitigate side effects associated with high peak plasma concentrations—such as dizziness, tremor, visual disturbances, or nausea—transitioning to a slow-release formulation may be beneficial. Monitoring plasma concentrations can also aid in optimizing dosing. Overdosage of flecainide or propafenone is potentially life-threatening; treatment is primarily supportive, by infusion of agents like dopamine and isoprenaline to stabilize rhythm and blood pressure, as no specific antidote exists (Figure 18). Interventions may include gastrointestinal decontamination and administration of hypertonic sodium bicarbonate to counteract sodium channel blockade. Due to their high protein binding and large volume of distribution, haemodialysis is ineffective in removing these drugs. Convulsions associated with propafenone toxicity have been treated with i.v. diazepam. Both medications are metabolized by the cytochrome P450 2D6 enzyme; thus, genetic variations or interactions with other drugs metabolized by this pathway can influence plasma levels and toxicity risk. Regular monitoring and appropriate dose adjustments are essential to minimize adverse effects.
Figure 18.

Single-lead and 12-lead electrocardiograms (ECGs) of a 14-year-old girl with no prior heart disease shortly after ingesting 24 flecainide tablets (2400 mg) in a suicide attempt. (A) The ECG recorded shortly after ingestion shows severe sinus bradycardia with low-amplitude P waves and an extremely wide QRS complex, indicative of significant sodium channel blockade caused by flecainide toxicity. (B) Following resuscitation efforts with sodium chloride, bicarbonate, isoproterenol, and dopamine/dobutamine, the ECG shows sinus tachycardia with less pronounced QRS broadening and repolarization changes. (C) After 9 h of treatment, the ECG demonstrates a decrease of QRS duration and resolution of repolarization abnormalities. Non-captured ventricular temporary pacemaker spikes are also visible. These findings illustrate the severe cardiotoxic effects of flecainide overdose, the dynamic ECG changes during treatment, and the efficacy of intensive medical intervention in reversing these abnormalities. The ECGs were recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Amiodarone overdose might be fatal. In addition to general supportive measures, the patient's cardiac rhythm and blood pressure are advised to be monitored, and if bradycardia ensues, a β-adrenergic agonist or a PM may be used. Neither amiodarone nor its metabolite is dialyzable. Induced AIH does not necessarily require termination of amiodarone therapy and requires hormone replacement therapy in most cases. Type 1 AIT is advised to be treated with thionamides combined with Na+ perchlorate if necessary. Type 2 AIT is advised to be managed with oral glucocorticoids. Once euthyroid status is established, patients with AIT 2 are followed without further specific treatment. Patients with AIT 1 are advised to be treated with thyroidectomy or radioiodine after euthyroid status is reached. Oral glucocorticoids might be added from the very beginning of therapy if the type of AIT is uncertain or if the response to thionamides is poor. Termination of amiodarone therapy in AIT is advised to be individualized and balanced with the anti-arrhythmic benefits of the drug. Rapidly deteriorating cardiac conditions may require emergency thyroidectomy for all forms of AIT.
Sotalol shows lack of protein binding, and haemodialysis is useful for reducing its plasma concentrations. Patients are advised to be monitored until QT interval is normalized and the heart rate returns to levels >50 b.p.m. Sotalol-induced hypotension may be associated with an initial slow drug elimination phase (half-life of 30 h) thought to be due to a temporary reduction of renal function caused by the hypotension. In case of severe bronchospasm, the use of aminophylline or aerosol β2-receptor stimulant may be appropriate.
Dronedarone overdose requires supportive therapy based on clinical symptoms. It is not known whether dronedarone and/or its metabolites can be removed by dialysis (haemodialysis, peritoneal dialysis, or haemofiltration). As with other anti-arrhythmic drugs except digoxin, there is no specific antidote available for dronedarone.
Verapamil has no specific antidote for overdosage; thus, treatment is advised to be supportive. Delayed pharmacodynamic consequences may occur with sustained-release formulations, and patients have to be observed for at least 48 h, preferably under continuous hospital care. In acute overdosage, gastrointestinal decontamination with cathartics and whole bowel irrigation may be appropriate. Calcium, inotropes (i.e. isoprenaline, dopamine, and glucagon), atropine, vasopressors (i.e. noradrenaline and adrenaline), and cardiac pacing have been used with variable results to reverse hypotension and myocardial depression. Overdose with CCBs that was initially refractory to atropine may become responsive when large doses (close to 1 g/h for more than 24 h) of calcium chloride are administered. Calcium chloride is preferred to calcium gluconate since it provides three times more calcium per volume. Verapamil cannot be removed by haemodialysis.
Digoxin toxicity, when mild, can often be managed by simply discontinuing digoxin and monitoring the patient, as symptoms may resolve with time. In cases where hypokalaemia is present, potassium supplements may be administered to restore normal levels, as low potassium can exacerbate digoxin's effects. For arrhythmias such as ectopic junctional and VTs resulting from digitalis toxicity, anti-arrhythmic agents like phenytoin or lidocaine may be effective. In severe cases, characterized by life-threatening arrhythmias or significant hyperkalaemia, the administration of digoxin-specific antibody fragments (digoxin immune Fab) is advised to neutralize the drug.
5.8. Contraindications and precautions
5.8.1. Flecainide
As a rule, flecainide is not advised for patients with a baseline QRS >120 ms, particularly those with LBBB or bifascicular block. It is not advised in patients with CAD (including an Agatston score >400), HF, cardiogenic shock, or reduced LVEF. An incidental finding of an Agatston score <400 in the absence of angina pectoris and uncomplicated LVH, both in the absence of left ventricular scar tissue, is not a contraindication for flecainide. Also, in patients with a GFR <35 mL/min, flecainide is discouraged or should be reduced due to the significant elimination of the drug by the kidneys. It is not allowed in patients with BrS. Unless pacing rescue is available, flecainide is not advised in patients with SND, atrial conduction defects, second-degree or greater AV block, or BBB. Flecainide may be combined with β-blockers, verapamil or diltiazem in patients with AF to prevent fast rates during arrhythmia recurrences or if conversion to type Ic AFL occurs. Renal function is advised to be carefully considered when combining flecainide with β-blockers like atenolol, as both drugs have significant elimination through the kidneys. Impaired renal clearance may result in drug toxicity and severe bradycardia. It is also important to advise patients to avoid exercise during breakthrough episodes until AF has stopped or active cardioversion is performed. Flecainide is contraindicated in case of hypersensitivity to the drug.
5.8.2. Propafenone
Contraindications of propafenone are like those of flecainide. However, due to its mild β-blocking effect, combination with AV-negative dromotropic agents may not be needed to prevent high ventricular rates during AF or type Ic AFL conversion. On the other hand, bronchospastic disorders or severe obstructive pulmonary disease are relative contraindications for the use of propafenone. Dose adjustments may be needed with hepatic disease but not with renal dysfunction.
5.8.3. Amiodarone
Contraindications include cardiogenic shock, sick sinus syndrome, second- or third-degree AV block, and bradycardia leading to syncope without a functioning PM. Known hypersensitivity to the drug or to any of its components, including iodine, hyper-thyroidism, and LQTS, is also considered a contraindication.
5.8.4. Dronedarone
Contraindications include New York Heart Association (NYHA) Class IV HF or NYHA Class II–III HF with a recent decompensation requiring hospitalization or referral to a specialized HF clinic. Furthermore, second- or third-degree AV block or sick sinus syndrome (except when used in conjunction with a functioning PM), significant bradycardia, and severe hepatic impairment are considered contraindications.
5.8.5. Sotalol and dofetilide
Sotalol and dofetilide are discouraged in LQTS, bradyarrhythmias/AV block (<50 b.p.m. during walking), uncontrolled HF, hypokalaemia (<4.0 mmol/L), and CrCl below 50 mL/min. Caution is advised to be exercised in low-weight females with LVH. As previously mentioned, careful monitoring of the QTc interval is advised during both the initiation and follow-up.
5.8.6. Verapamil and diltiazem
Contraindications to verapamil include severe left ventricular dysfunction, hypotension (systolic pressure <90 mmHg), and cardiogenic shock. It is also contraindicated in patients with severe sick sinus syndrome or second- or third-degree AV block, except in those with a functioning ventricular PM. Additionally, CCBs are discouraged in patients with atrial arrhythmias and an accessory bypass tract (e.g. WPW syndrome) due to the risk of VF. Last, their use is contraindicated in individuals with a known hypersensitivity to the drug. Intravenous verapamil is discouraged after doses of i.v. β-blocker.
A more comprehensive review of contraindications and cautions associated with AADs is provided in Supplementary material online, Table S10.
5.9. Anti-arrhythmic drug plasma concentration
The PK of anti-arrhythmics exhibit significant variability among patients, influenced by factors such as age, renal or hepatic function, and drug interactions. In addition, therapeutic ranges for most AADs remain poorly defined. Appropriate drug plasma concentrations have not been carefully derived and are often extrapolated from limited patient samples, hindering precision. Finally, the lack of standardized therapeutic ranges and the limited integration of plasma concentration data into clinical studies contribute to the challenge of tailoring anti-arrhythmic therapy effectively for individual patients. Consequently, the current application of optimizing individual anti-arrhythmic therapy is highly limited and is mostly reserved for suspected cases of drug toxicity.
Flecainide plasma levels are usually measured as trough levels, e.g. in the morning prior to intake of the morning tablet, and the normal values are between 200 and 400 ng/mL. In propafenone, unlike flecainide, differences in speed of metabolism and saturable oxidative elimination between patients make plasma concentrations even less predictable. The normal therapeutic range for propafenone is between 400 and 1100 ng/mL. The main electrophysiological effects of amiodarone are mediated through intra-cellular metabolites such as desethyl-amiodarone. When using plasma concentration monitoring, N-monodesethylamiodarone is advised to be followed together with amiodarone. Finally, levels of digoxin (therapeutic range 0.8–2 ng/dL, though levels >1.2 ng/mL may increase toxicity risk without additional benefit) are nowadays rarely determined and mostly reserved for intoxication suspicion.
5.10. Drug–drug interactions
5.10.1. Anti-arrhythmic drug–drug interactions
Drug–drug interactions represent 3% of preventable in-hospital adverse drug events and contribute to hospital admissions and emergency room visits.401,402 Patients with arrhythmias are vulnerable to these interactions due to the narrow therapeutic index of anti-arrhythmic agents and the frequent use of multiple cardiovascular drugs in patients with arrhythmias.403 Pharmacodynamic interactions, such as the added AV nodal blockade of digoxin used in combination with a β-blocker or calcium blocking agent, can be a desirable effect or an unintentional adverse effect.403 Pharmacokinetic interactions relate to changes in the absorption, distribution, metabolism, and elimination of either the substrate or precipitant drug (Figure 3).
The most common pharmacokinetic drug–drug interactions involve the CYP mono-oxygenation system and the P-gp (permeability glycoprotein).404–406 Several AADs undergo biotransformation by hepatic oxidative metabolism through the CYP system. Co-administered drugs that inhibit these pathways will result in a lower metabolism of the AAD and thus a higher plasma concentration that can cause adverse drug reactions. A precipitant drug that induces these enzymes can decrease the plasma level of anti-arrhythmic agent, which can lead to an ineffective anti-arrhythmic effect. Some drugs, like ritonavir, are strong CYP3A4 and P-gp inhibitors and significantly increase the blood concentration of multiple anti-arrhythmic agents and DOACs. Because of these interactions, potent inhibitors and inducers are best avoided in combination with anti-arrhythmics. Predisposing factors that can aggravate these drug–drug interactions include advanced age, female sex, weight, racial differences, inherited enzyme systems, HF, renal and liver dysfunction, and polypharmacy.407–409 The frequency of CYP genetic polymorphisms varies across ethnicities, with 5–10% of whites (≈1% of Asians; up to 19% in blacks) being poor metabolizers of CYP2D6.409,410
CYP2D6 is the major enzyme for biotransformation of metoprolol, propranolol, flecainide, and propafenone. ‘Poor metabolizers’ (5–10% of the caucasian and black populations) have reduced amounts of CYP2D6.405 Low doses of quinidine can inhibit CYP2D6, thus increasing the peak and steady-state plasma concentrations and prolonging the half-life of parent compounds such as propafenone. CYP3A4 is responsible for the metabolism of amiodarone, bisoprolol, diltiazem, disopyramide, dronedarone, ivabradine, quinidine, ranolazine, or verapamil. CYP3A4 is inhibited by clarithromycin, erythromycin, itraconazole, or ritonavir.
Amiodarone, cimetidine, diltiazem, ketoconazole, procainamide, propranolol, and verapamil increase quinidine plasma levels.406 Quinidine is a potent CYP2D6 and P-gp inhibitor resulting in increased plasma levels of substrates of these enzyme systems. Because quinidine decreases digoxin clearance, one must reduce digoxin doses by 50% when used in combination. Class I and III anti-arrhythmics prolong the QT and are best avoided in patients treated with other QT-prolonging drugs.
β-blockers, cimetidine, and halothane increase lidocaine plasma levels.406 Mexiletine increases plasma levels of theophylline, and amiodarone increases mexiletine levels.
Fluoxetine, duloxetine, and paroxetine are potent CYP2D6 inhibitors, and co-administration increases plasma flecainide levels.410,411 Concomitant administration of flecainide with amiodarone has also been shown to increase plasma flecainide concentrations by 50%. Co-administration of flecainide with digoxin increased digoxin trough serum concentrations by an average of 24%, but usually this is not enough to make a dose adjustment.
Propafenone undergoes extensive first-pass metabolism, mainly by CYP2D6, with two active metabolites (5-hydroxypropafenone and N-depropylpropafenone), which are renally excreted.406 Because of active metabolites, poor or extensive metabolizer status does not affect the dosing of the drug. Propafenone can increase the plasma levels of digoxin, metoprolol, propranolol, and warfarin, but these interactions are minimal and usually do not require any dose adjustments.406,412
Sotalol is not hepatically metabolized, is excreted renally, and is not subject to drug interactions involving the hepatic CYP enzyme system.
Dofetilide is also mainly excreted renally with no significant CYP interactions.127 However, plasma dofetilide concentrations are significantly elevated when co-administered with verapamil, cimetidine, trimethoprim, ketoconazole, prochlorperazine, megestrol, dolutegravir, and hydrochlorothiazide, which compete with the active tubular secretion (via the cation transport system), and their concomitant use is an absolute contraindication.
Amiodarone inhibits P-gp, CYP1A2, CYP2C9, CYP2D6, and CYP3A4 and has the potential to increase plasma levels of drugs metabolized by these isoenzymes or substrates of P-gp. When used in combination with amiodarone, lower doses for digoxin, flecainide, and warfarin are often required, and one must monitor digoxin levels and the INR.413–415 Cholestyramine decreases the absorption of amiodarone. Co-administration of amiodarone with digoxin, β-blockers, verapamil, or diltiazem increases the risk of bradycardia and AV block. Severe bradycardia has been reported when amiodarone is co-administered with hepatitis C antiviral drugs (daclatasvir, ledipasvir, and sofosbuvir).416 Amiodarone can also inhibit cyclosporin metabolism and cause higher blood levels requiring a dose reduction of cyclosporin.417 Statins, especially simvastatin levels, can increase when used concomitantly with amiodarone.418
Dronedarone is highly metabolized by CYP3A4 and is a moderate inhibitor of CYP3A4 and a weak inhibitor of CYP2D6.419 Dronedarone is discouraged from being administered at the same time with potent CYP3A4 inhibitors. Dronedarone may be co-administered with moderate CYP3A4 inhibitors such as verapamil and diltiazem. Dronedarone is a P-gp inhibitor and can increase the level of digoxin and dabigatran if co-administered together.419–421 It is discouraged to be co-administered with them.
Diltiazem and verapamil are moderate inhibitors of CYP3A4 and P-gp; thus, doses of CYP3A4 and P-gp substrates are advised to be adjusted as appropriate. Concomitant QT-prolonging drugs (https://crediblemeds.org) and strong CYP3A inhibitors (discontinue before initiation) are best avoided.
As mentioned above, multiple drugs interact with the concomitant use of digoxin, but the most significant are quinidine, amiodarone, and dronedarone.412,414,420,422,423
The co-administration of adenosine with β-blockers, digoxin, diltiazem, or verapamil increases the risk of bradycardia and AV block. Dipyridamole inhibits the uptake of adenosine potentiating its effects; theophylline blocks adenosine receptors and decreases the effects of adenosine.406 These latter two drugs need to be avoided with adenosine stress testing.
Table 14 summarizes the substrates, inhibitors, and inducers of CYP3A4, 2D6, and 1A2 and P-gp, and Table 15 summarizes key drugs with potential interactions involving AADs, excluding AAD combinations (see Table 16) and anticoagulants (see section ‘Anticoagulation’). More comprehensive reviews of substrates, inducers, and inhibitors of CYP and P-gp transporters, as well as drug–drug interactions of AADs, are provided in Supplementary material online, Tables S4 and S5, respectively.
Table 14.
Cytochrome P450 complex enzymes and P-glycoprotein involved in the metabolism or affected by AADs and other substances (a more comprehensive table is provided in Supplementary material online, Table S4)
| CYP or P-gp | Inhibition, induction, or substrate | AADs | Other drugs or substances |
|---|---|---|---|
| CYP 3A4 | Inhibitors |
|
|
| Inducers | Phenytoin | Alcohol chronic exposure, carbamazepine, glucocorticoids, rifampicin, St. John's wort | |
| Substrates | Ivabradine Class I (quinidine, lidocaine, ranolazine) Class II (carvedilol, metoprolol, nebivolol, propranolol) Class III (amiodarone, dronedarone) Class IV (diltiazem, verapamil) |
Benzodiazepines, bosentan, clopidogrel, colchicine, antiXa DOACs, eplerenone, HCV protease inhibitors, HIV protease inhibitors, immunosuppressants (cyclosporine, tacrolimus), macrolides (clarithromycin, erythromycin, etc.), omeprazole, ondansetron, PDE5 inhibitors, statinsa, ticagrelor Many anticancer drugs |
|
| CYP 2D6 | Inhibitors | Class I (quinidine, propafenone) Class III (amiodarone, dronedarone) Class IV (diltiazem, verapamil) |
|
| Inducers | Phenytoin | Carbamazepine, dexamethasone, phenobarbital, rifampicin | |
| Substrates | Class I (mexiletine, flecainide, propafenone ranolazine) Class II (bisoprolol, carvedilol, metoprolol, nebivolol, propranolol) Class III (vernakalant) |
Most anti-depressants, anti-psychotics, ondansetron, opioids, tamsulosin, trazodone, tropisetron | |
| CYP 1A2 | Inhibitors | Propranolol amiodarone verapamil |
Allopurinol, cimetidine, ciprofloxacin, famotidine, fluoxetine, fluvoxamine |
| Inducers | Phenytoin | Tobacco, carbamazepine, rifampicin, ritonavir | |
| Substrates | Class I (lidocaine, mexiletine, propafenone) verapamil |
Clopidogrel, clozapine, olanzapine, tamoxifen, theophylline, tizanidine, warfarin | |
| P-gp | Inhibitors | Class I (quinidine, propafenone) Class II (bisoprolol, carvedilol, propranolol) Class III (amiodarone, dronedarone) Class IV (diltiazem, verapamil) |
Azole anti-fungals (itraconazole, etc.), conivaptan, HCV/HIV protease inhibitors, cyclosporine, proton pump inhibitors, macrolides (clarithromycin, erythromycin, etc.), tamoxifen, ticagrelor, tolvaptan |
| Inducers | None significant | Carbamazepine, phenytoin, rifampicin, St. John’s wort | |
| Substrate | Class I (quinidine, ranolazine) Class IIa (carvedilol, metoprolol, nadolol, nebivolol, propranolol) Class IIb (digoxin, digitoxin) Class III (amiodarone, dronedarone) Class IV (diltiazem, verapamil) |
Ambrisentan, cimetidine, clopidogrel, colchicine, dipyridamole, DOACs, erythromycin, fexofenadine, immunosuppressants (cyclosporine, tacrolimus), ondansetron, opioids, riociguat, statins |
Cytochrome P450 enzymes (CYP) are responsible for the metabolism of many drugs. Inhibition or induction of these enzymes can lead to significant drug interactions with agents, which are substrates of these enzymes. P-glycoprotein (P-gp) is a transporter protein expressed in various tissues (intestine, liver, and kidneys) that affects the absorption and elimination of various drugs. Inhibitors can increase drug levels, while inducers can decrease them. Anti-arrhythmic drugs often interact with these enzymes and transporters, affecting the metabolism of other drugs and vice versa. Non-AADs/substances can also inhibit or induce these enzymes and transporters, leading to potential interactions when combined with AADs.
Abbreviations: AADs, anti-arrhythmic drugs; DOACs, direct oral anticoagulants; HCV, hepatitis C virus; HIV, human immunodeficiency virus; PDE, phosphodiesterase; SSRI, selective serotonin reuptake inhibitor.
aAtorvastatin, simvastatin, and lovastatin are metabolized by CYP3A4, while rosuvastatin, pravastatin, pitavastatin, and fluvastatin are not significantly metabolized by it.
Table 15.
Key drugs with potential interactions involving AADs, excluding anticoagulants (covered in Table 21) and AAD combinations (discussed in Table 16)
| Modified VW class | AAD | Drug #1 | Advice for concurrent use with drug #1 | Drug #2 | Advice for concurrent use with drug #2 |
|---|---|---|---|---|---|
| 0 | Ivabradine | Strong CYP3A4 inhibitorsa (+ivabradine) | Avoid combination | CYP3A4 inducersb (−ivabradine) | Avoid combination |
| Ia | Quinidine | Strong CYP3A4 inhibitorsa (+quinidine) | Caution, monitor levels | Other QT-prolonging drugs | Avoid combination |
| Procainamide | Cimetidine, propranolol, verapamil (+procainamide) | Replace (e.g. cimetidine by PPIs), monitor procainamide levels | Amiodarone (+procainamide, summative effect) | Monitor ECG | |
| Disopyramide | Anti-cholinergic (H1-anti-histaminics, anti-spasmodics, tricyclic anti-depressants) (summative) | Avoid combination | β-Blockers, CCB (additive negative inotropic effect) | Monitor cardiac function | |
| Ib | Lidocaine | Amiodarone, β-blockers, cimetidine (+lidocaine) | Replace (e.g. PPIs, renal excreted β-blockers), reduce lidocaine dose or monitor levels | β-Blockers (+lidocaine) | Reduce lidocaine dose |
| Mexiletine | Theophylline (+theophylline) | Reduce and monitor theophylline levels | Phenytoin (mutual reduction) | Adjust dose, monitor levels | |
| Phenytoin | CYP3A4 inhibitorsa (+phenytoin) | Reduce phenytoin dose, monitor phenytoin levels | Oral contraceptives (reduces contraceptive efficacy) and corticosteroid (reduces efficacy) | Avoid or increase corticosteroid dose | |
| Ic | Flecainide | Digoxin (+digoxin) | Reduce digoxin dose (25%), monitor digoxin levels | Amiodarone, Fluoxetine/paroxetine (+flecainide) | Replace (e.g. escitalopram or sertraline), reduce (30%) flecainide dose, monitor levels and ECG |
| Propafenone | Digoxin (+digoxin) | Reduce digoxin dose (25%), monitor digoxin levels | Fluoxetine/paroxetine (+propafenone) | Replace (e.g. escitalopram or sertraline), reduce (30%) flecainide dose, monitor levels and ECG | |
| Id | Ranolazine | Strong CYP3A4 inhibitorsa (+ranolazine) | Avoid combination | Statins (potentiates myopathy) | Limit statin dose or use non-CYP3A4 statins (pitavastatin, pravastatin, rosuvastatin) |
| IIa | β-Blockers | Anti-diabetic drugs (mask hypoglycaemic symptoms) | Counsel patients, avoid non-selective β-blockers (use carvedilol or nebivolol) | Clonidine (hypertension if abrupt discontinuation) | Avoid abrupt clonidine discontinuation |
| IId | Digitalis | Amiodarone/dronedarone/flecainide/propafenone/quinidine/ranolazine/verapamil (+digoxin) | Reduce (50%) or avoid digoxin, monitor digoxin levels | Macrolides (+digoxin) | Monitor digoxin levels |
| IIe | Adenosine | Dipyridamole (potentiates adenosine) | Reduce adenosine dose | Theophylline/caffeine (antagonizes adenosine) | Increase adenosine dose |
| III | Amiodarone | Simvastatin, lovastatin, atorvastatin (potentiates myopathy) | Reduce statin dose or use non-CYP3A4 statins (pravastatin, rosuvastatin) | β-Blockers (summative) | Adjust dose, Monitor the ECG |
| Clopidogrel (decreases the active metabolite | Replace with prasugrel or ticagrelor | Hepatitis C antiviral drugs (potentiate bradycardia) | Monitor heart rate during the first 48 h | ||
| Dronedarone | Simvastatin, lovastatin, atorvastatin (potentiates myopathy) | Reduce statin dose or use non-CYP3A4 statins (pravastatin, rosuvastatin) | Potent CYP3A4 inhibitors (increase dronedarone) | Avoid combination | |
| Dofetilide | Cimetidine, trimethoprim, dolutegravir (reduce dofetilide OCT2 renal elimination) | Avoid combination | CYP3A4 inhibitors (+dofetilide) | Avoid combination | |
| Ibutilide/dofetilide/sotalol | Drugs producing hypokalaemia/hypomagnesaemia | Increase the risk of QT prolongation and TdP. Monitor ionic levels | Other QT-prolonging drugs | Avoid combination | |
| Vernakalant | CYP3A4 inhibitorsa (+vernakalant) | Caution | Strong CYP2D6 inhibitorsc (+vernakalant) | Caution | |
| IV | Verapamil/diltiazem | CYP3A4 substrates (+substrate) | Replace or adjust substrate dose | P-gp substrates (+substrate) | Replace or adjust substrate dose |
Supplementary material online, Table S5 gives a more comprehensive description.
+, increases levels of the AAD; −, decreases levels of the AAD; OCT2: organic cation transporter 2; AAD, anti-arrhythmic drug; CCB, calcium channel blocker; PPI, proton pump inhibitor; SSRI, selective serotonin reuptake inhibitors; TdP, torsades de pointes.
aVerapamil, grapefruit juice, azole anti-fungals, macrolides, and others (Table 14).
bPhenytoin, rifampicin and others (see Table 14).
cBupropion, SSRI (fluoxetine, fluvoxamine, paroxetine), ritonavir; others (see Table 14).
Table 16.
Main potential AAD combinations
| Potential AAD combinations424 (Supported by some evidencea) | ||||||
|---|---|---|---|---|---|---|
| Class #1 | AAD #1 | Class #2 | AAD #2 | Rationale | Objective | Study, reference |
| 0 | Ivabradine | II | β-blockers | Summative complementary effects | Inappropriate sinus tachycardia | 425 |
| Ia | Quinidine | Ia | Disopyramide | Combined reduced doses to minimize side effects | Decrease gastrointestinal intolerance (constipation for disopyramide, diarrhoea for quinidine)b | 426,427 |
| Ib | Mexiletine |
|
VAs | 428–430 | ||
| IV | Verapamil | Verapamil may prevent:
|
AAsb and VAs |
431
, PAFAC432, SOPAT433 |
||
| Ic | Flecainide | Ib | Mexiletine | Summative effects | VAs | 434 |
| Flecainide, propafenone | II, IV | β-blockers, CCBs |
Complementary effects on myocardium and AV nodec | Rate control of AF/AFL/ type Ic AFL and SVT prevention/terminationc | 92 | |
| III | Amiodarone | Ia/Ib | Quinidine/Mexiletine | Complementary effects on ventricular conduction and refractoriness | VAs | 435–437 |
| Ic/Id | Flecainide/propafenone/ranolazine | Complementary effects on conduction and refractoriness | AAs and VAS | 438–440 | ||
| II | β-blockers | Complementary effects on myocardium and adrenergic tone | SHD VAs | OPTIC91 | ||
| Dronedarone | Id | Ranolazine | Combined reduced doses to potentiate efficacy and minimize side effects (constipation for ranolazine, diarrhoea for dronedarone) | AF | HARMONY76 | |
| Sotalol | Ib/Ic | Mexiletine/flecainided | Complementary effects on conduction and refractoriness | VAs in ARVC | 441,442, | |
| IIa | β-blockers | 0, I, IIb, IId, III | Blocks sympathetic stimulation enhancing the efficacy and/or safety of other AADs | AAs and VAs | 443 | |
| IV | CCBs | IId | Digoxine | Summative effects | Rate control of AF | 444 |
| Uncertain AAD combinations (limited data on efficacy and safety) | ||||||
|---|---|---|---|---|---|---|
| Class #1 | AAD #1 | Class #2 | AAD #2 | Rationale | Objective | |
| III | Dronedarone | IV | Verapamil, diltiazemf | Additional rate control to that of dronedarone | AAs | Limited data |
| III | Sotalol dofetilide |
Id | Ranolazine | Ranolazine may mitigate EADs caused by Class III drugs, reducing TdP risk | AAs and VAs | Limited data445,446 |
| Potentially hazardous AAD combinations (To avoid or to be used at reduced doses) | ||||||
|---|---|---|---|---|---|---|
| Class #1 | AAD #1 | Class #2 | AAD #2 | Rationale | Risk | |
| Ia | Disopyramide | IV | CCBsg | Both reduce cardiac contractility | Heart failure and shock | 447 |
| Id | Ranolazine | Ia | Quinidine | Both are metabolized primarily by CYP3A4 | TdP | |
| Disopyramide | Both may produce constipation | Constipation | ||||
| Ib Ic |
Mexiletine Flecainide |
Both have CNS effects | Tremor | |||
| II | β-blockers | IV | CCBs | Both depress the SN, AV conduction and cardiac contractility | AV block | |
| III | Sotalol | II IV |
β-Blockers CCB |
Both depress the SN, AV conduction, and cardiac contractility | Bradycardia | |
| Sotalol/dofetilide | Ia | Quinidine | Both prolong the QT interval | TdP | 448,449 | |
| Dronedarone | Id | Digoxin | Dronedarone decreases renal clearance of digoxin | Digitalis toxicity | 105 | |
| Dofetilide | IV | CCBs | CCBs increase dofetilide levels | TdP | ||
Column #1 lists AADs commonly used as the first-choice drug, while Column #2 includes AADs typically added as complementary therapy when the primary drug fails to control the arrhythmia or may result in pro-arrhythmia or other adverse effects. This order may be reversed depending on specific circumstances.
Abbreviations: AA, atrial arrhythmias; ARVC, arrhythmogenic right ventricular dysplasia; AF, atrial fibrillation; AFL, atrial flutter; AV, atrioventricular; CCB, calcium channel blockers; EADs, early after depolarizations; SN, sinus node; SHD, structural heart disease; SVT, supraventricular tachycardia; VA, ventricular arrhythmias; TdP, torsades de pointes; VA, ventricular arrhythmias; VF, ventricular fibrillation; VM, Vaughan Williams; VT, ventricular tachycardia.
aCombining AADs increases risks and necessitates careful evaluation of alternatives and patient conditions, along with close dose adjustments and ECG monitoring to mitigate myocardial depression and pro-arrhythmia. Most evidence comes from small, non-controlled studies.
bIn general, ablation is advised before quinidine for AF treatment.
cContraindicated in patients with structural heart disease due to the risk of myocardial contraction depression and heart failure.
dFlecainide may potentiate the myocardial contraction depression effect of sotalol.
eConsider reducing the dose of digoxin and monitoring serum levels closely due to the risk of toxicity. CCB can increase digoxin levels by 50–75% through inhibition of P-glycoprotein activity, which decreases renal tubular elimination of digoxin.
fDiltiazem, verapamil, and dronedarone are substrates and inhibitors of CYP3A4, and their concurrent use can increase plasma concentrations of each drug, potentially amplifying their pharmacological effects and side effects. When rate control is required, combining dronedarone with a β-blocker is generally preferred over CCBs. Additionally, both dronedarone and CCBs can depress AV conduction, increasing the risk of bradycardia or heart block. Therefore, the combination of dronedarone with CCBs must only be used with caution and under close clinical and ECG monitoring.
gWith caution to improve symptoms in hypertrophic cardiomyopathy.
5.10.2. Drug–herb and drug–food interactions involving anti-arrhythmic drugs
St. John's wort (Hypericum perforatum) is a potent inducer of CYP2C9, CYP2C19, and CYP3A4 and can decrease verapamil and dronedarone plasma concentrations.450,451 St. John's wort has no effect on CYP1A2 or CYP2D6. St. John's wort can induce P-gp transport and lower plasma levels of digoxin.
Concomitant consumption of green tea (catechins) with nadolol and digoxin can significantly reduce plasma concentrations.452
Grapefruit juice is a moderate inhibitor of CYP3A4, and its effects may last from 4 to 24 h.453,454 Co-administration of grapefruit juice with amiodarone and dronedarone can cause major increases in peak plasma concentrations of these anti-arrhythmic agents.455 In addition, grapefruit juice inhibits the metabolic breakdown of amiodarone to its active metabolite n-desethyl-amiodarone. Grapefruit juice can increase plasma concentrations of other AADs, such as quinidine and verapamil, that undergo CYP3A4 metabolism, as listed in Table 14.
Food may affect the bioavailability of AADs.456 Both amiodarone and dronedarone have a food effect on absorption; peak plasma concentrations and area under the curve are increased when these drugs are taken with a meal.419,457 Taking amiodarone with a meal can increase plasma levels and may be used instead of increasing the dose in patients taking the drug on an empty stomach. The oral bioavailability of dronedarone increases when administered with a high-fat meal. Dronedarone has low absolute bioavailability (∼4%) when taken in the fasted state, but co-administration with food, particularly high-fat meals, signifcantly increases its absorption (by up to two- to four-fold).457 For this reason, it is advised to be taken with meals to enhance its bioavailability and therapeutic effect.457 All clinical studies with dronedarone were conducted with patients taking the medication with meals, and this is how the drug is encouraged to be used in clinical practice.419
5.11. Anti-arrhythmic drug switch and combinations
When an AAD is ineffective or not tolerated, changing it (drug switch) or adding another (drug combination)458 can be tried. Reasons for switching or combining are inefficacy (initial or lost over time), adverse effects and the development of a contraindication (e.g. new disorder, new potential drug interaction) (Box 15).
Box 15 Considerations when switching or combining AADs.
Switching AADs
-
Pharmacokinetics:
For AADs with similar half-lives, start the new AAD at its usual dose/interval when the next dose of the prior AAD is due
For AADs with long washout periods (e.g. amiodarone), up-titrate the new AAD gradually
Monitoring: Use serum drug levels and/or ECG markers to direct washout and initiation.
Combining AADs
Reasons
-
Additive efficacy
Combining drugs (e.g. Class Ic + III or Ia + Ib) may enhance effectiveness under close monitoring. β-Blockers: usually enhance efficacy of most AADs when added
One AAD may enhance the binding of another (e.g. Ia or III lengthens plateau phase allowing increased time for Ib binding)
-
Improved tolerance
Lower combined doses may reduce side effects
Mechanism of one AAD may decrease pro-arrhythmic profile of another (e.g. ranolazine can block EADs due to Class III AADs)
Considerations
Experience with prior AADs: tailor combinations based on previous response
Do not combine full doses of Classes Ia and III (increased risk of TdP)
Abbreviations: AAD, anti-arrhythmic drug.
The combination of flecainide or propafenone with sotalol441,459,460 may create a pseudo-amiodarone effect, but there is limited clinical experience in AF.426,461 Flecainide has been combined with amiodarone in children.462,463 but more guideline-relevant AAD studies are needed.
The main potential AAD combinations, those with uncertain safety, and those to be avoided are summarized in Table 16.
6. Anti-arrhythmic drug in special situations
6.1. Pregnancy
Anti-arrhythmic drugs are best avoided, if possible, during the first trimester of pregnancy because of the risk of teratogenic effects. Later in pregnancy, they may cause adverse effects on foetal growth and development, affect uterine contractility, or produce pro-arrhythmic events.464,465 The main characteristics of AADs during pregnancy and lactation are summarized in Table 17.
Table 17.
AADs during pregnancy and breastfeeding464
| Drug | Former FDA category | Placental transfer | Present in human milk | Safety in lactation | Adverse effects | Used for foetal arrhythmias |
|---|---|---|---|---|---|---|
| Adenosine | C | No | No | LD | No foetal adverse effects reported (limited human data) | Yes |
| Amiodarone | D | Yes | Yes | Contraindicated | Goitre, hypo- (9%), hyper-thyroidism, neurodevelopmental abnormalities, premature birth, growth retardation; bradycardia and QT prolongation in newborns | (Yes, last choice) |
| Atenolol | D | Yes | Yes | Produces IUGR Avoid its use |
Significant IUGR | |
| Atropine | C | LD | Yes | Yes | LD | |
| Bepridil | C | LD | Yes | Avoid its use | LD | |
| Bisoprolol, carvedilol, metoprolol, propranolol | C | Yes | Yes | LD | Foetal bradycardia and hypoglycaemia, IUGR and immature and preterm birth | Yes (metoprolol) |
| Cibenzoline | C | LD | In animals | LD | LD | |
| Digoxin | C | Yes | Yes | Considered safe | Serum levels unreliable, safe | Yes |
| Diltiazem | C | Yes | Yes | Avoid its use | Diltiazem is teratogenic in animals | |
| Disopyramide | C | Yes | Yes | Avoid its use | Uterine contractions, placental abruption, prolonged QT | |
| Dofetilide | C | In animals | LD | Avoid its use | Adverse effects in animals | |
| Dronedarone | X | Unknown | Contraindicated | It may cause foetal harm when administered to a pregnant woman Dronedarone is teratogenic in rats | ||
| Esmolol | C/Da | LD | Unknown | Avoid its use | See β-blockers | |
| Flecainide | C | Yes | Yes | Plasma levels in nursing infants are 5–10 times lower than therapeutic plasma levels | LD | Yes |
| Ibutilide | − | Unknown | Yes | Contraindicated | Teratogenic (abnormalities included adactyly, interventricular septal defects, and scoliosis) in rats | |
| Ivabradine b | LD | Yes (in rats) | Yes | Avoid its use | Animal reproduction studies have shown adverse effects | |
| Lidocaine | B | Yes | Yes | Considered safe | Foetal bradycardia/tachycardia, neonatal bradycardia, hypotonia or respiratory depression | Yes |
| Mexiletine | C | Yes | Yes | Caution | Limited data, but probably safe | |
| Nicorandil | B | LD | Yes in animals | LD | No harmful effects in animals | |
| Pilsicainide | C | LD | Yes in animals | LD | ||
| Procainamide | C | Yes | Yes | Avoid its use | Lupus-like syndrome, prolonged QT | |
| Propafenone | C | Yes | Unknown | LD | Limited data. Probably safe | Yes |
| Quinidine | C | Yes | Yes | Yes | Foetal/neonatal thrombocytopenia, uterine contraction, prolonged QT. | Yes |
| Ranolazine | LD | LD | Unknown | Avoid its use | Foetal bradycardia, hypoglycaemia, reduced birth-weight, QT prolongation | |
| Sotalol | B | Yes | Yes | Avoid its use | Maternal QTc prolongation | Yes |
| Verapamil | C | Yes | Yes | LD | Pre-maturity, IUGR, foetal bradycardia, impaired uterine contraction | |
| Vernakalant | Unknown | LD | Unknown | Unknown | LD |
AADs in bold are contraindicated or discouraged for use during pregnancy.
- A: Controlled studies show no risk to the foetus.
- B: May be acceptable. Either animal studies showed no risk but human studies are not available, or animal studies showed minor risks and human studies showed no risk.
- C: Use with caution if benefits outweigh risks. Animal studies show risk and human studies were not available or neither animal nor human studies were done.
- D: Use in life-threatening emergencies when no safer drug is available. Positive evidence of human foetal risk.
- X: Do not use in pregnancy. Risks involved outweigh potential benefits. Safer alternatives exist.
Abbreviations: IUGR, intrauterine growth retardation; FDA, Food and Drug Administration; LD, limited human data.
aD in the second and third trimesters.
bWomen of child-bearing potential must use appropriate contraceptive measures during treatment.
During pregnancy symptomatic PACs and PVCs rarely require treatment, although β-blockers may be used. Treatment of SVT, the most common sustained arrhythmia in pregnancy, is advised when symptomatic or causing haemodynamic compromise. In the absence of pre-excitation, vagal manoeuvres should be attempted first, followed by adenosine, β1-selective blockers (except atenolol) and verapamil (diltiazem is teratogenic in animals) or a combination of β-blockers464–468 and verapamil are advised.464,466–469 Adenosine is the drug of choice for acute conversion of SVT, AT, and orthodromic AVRT.44,198,199,203,464,470 β-Blockers are considered safe in pregnancy for the treatment of cardiac arrhythmias and other CVDs464,467–469,471,472 although they are associated with lower birth weights. Flecainide and propafenone are advised as second-line agents in patients without ischaemic or SHD and in maternal and foetal SVT with pre-excitation.464,473,474 Amiodarone and dronedarone produce foetal harm and are advised only when other measures have failed.464,466
Rhythm control is the preferred strategy for AF during pregnancy. i.v. ibutilide or flecainide may be appropriate for termination of AF/AFL in haemodynamically stable patients without SHD.92,151 Electrical cardioversion preceded by anticoagulation is advised in cases of haemodynamic instability or considerable risk to the mother or foetus.44,203,464 Foetal heart rate is encouraged to be routinely controlled after cardioversion.475 Intravenous β1-selective blockers (metoprolol, bisoprolol, but not atenolol) are advised for acute rate control; if they fail, digoxin and verapamil are appropriate in the absence of pre-excited AF.464,466 Oral flecainide, propafenone, or sotalol are appropriate to prevent AF if AV nodal-blocking drugs fail.92
Ventricular arrhythmias in the absence of SHD are usually sensitive to β-blockers.45,464,476 Sotalol or Class Ic drugs may be appropriate if β-blockers are ineffective, and catheter ablation is an option if drug treatment fails.45,464,477,478 Sotalol or procainamide IV are appropriate for acute conversion of haemodynamically stable monomorphic sustained VT, while oral metoprolol, propranolol, or verapamil are advised for long-term management of idiopathic sustained VT.45 β-blockers are advised during pregnancy and post-partum in patients with LQTS or CPVT.45,479 Finally, verapamil and diltiazem are discouraged during late pregnancy, breast feeding, and in children <1 year old because they have been associated with hypotension in some case reports. However, all reported cases had HF, overdosing and/or other concurrent AADs at the time the drug was given, and therefore some controversy exists.
6.2. Children
As a general rule, prescription of AADs in children requires a clear diagnosis with ECG documentation of the arrhythmia.135 Paediatric population present specific characteristics, including few or poorly described symptoms, differences in drug PK, lack of specific drug formulations, and immaturity of the specialized cardiac conduction tissue.480 In newborns, milk can substantially modify the absorption of some drugs (i.e. flecainide), and erratic feeding schedules and vomiting can affect AAD availability. Additionally, certain arrhythmic substrates are more common in this age group—such as permanent junctional re-entrant tachycardia (PJRT or Coumel's tachycardia) and both congenital and post-operative JET—and may require combination AAD therapy for adequate control. Although catheter ablation is possible for children of almost any size, recent European registries show that AAD therapy is generally preferred in those weighing <15 kg.481
Anti-arrhythmic drugs commonly used for arrhythmias in infants and children are summarized in Table 18 482 and Supplementary material online, Table S11. For patients without SHD, adenosine, β-blockers (metoprolol and propranolol), digoxin, flecainide, propafenone, and sotalol can be safely used. Class I AADs are discouraged in the presence of SHD and/or systolic ventricular dysfunction because of their negative inotropic effect and the risk of pro-arrhythmia. Careful dose adjustment based on renal dysfunction is needed for digoxin, flecainide, propafenone, and sotalol. As discussed before, i.v. verapamil is discouraged when possible in VA in infants <1 year of age. Nonetheless, verapamil remains the treatment of choice for some arrhythmias (e.g. posterior fascicular VT, even at this age) and can be used safely in acute settings.483 Ivabradine is emerging as an option for the prevention and treatment of JET in both post-operative and congenital presentations.337,484 Amiodarone is encouraged to be used when other AADs fail or are contraindicated.45,315,482
Table 18.
Pharmacological therapy for arrhythmias in infants and children482
| Drug | Dose | Arrhythmia | Comments |
|---|---|---|---|
| Intravenous | |||
| Lidocaine | 1 mg/kg (up to 3 doses in 10 min) and then 20–50 mg/kg/min | ||
| Flecainidea | 1.5–2 mg/kg over 5 min2–4 mg/kg/day | Avoid in patients with structural heart diseaseb. Milk reduces flecainide absorption | |
| Propafenonea | Loading: 2 mg/kg over 2 h. Maintenance: 4–7 mg/kg/min | ||
| Amiodarone | Loading: 5–10 mg/kg over 60 min. Maintenance dose: 5–15 mg/kg/min | It may take hours until successful conversion to SR occurs. The safety and efficacy of amiodarone in children have not been established | |
| Adenosine | Rapid i.v. bolus: (i) for infants: 0.15 mg/kg. For >1 year of age: 0.1 mg/kgIncreasing dosage up to 0.3 mg/kg | ||
| Esmolol | Bolus 100–500 μg/kg; then 25–100 μg/kg/min | ||
| Propranolol | 1 mg/kg/day | ||
| Verapamila | 0.1 mg/kg slowly over 2 min | Avoid in infants <1 year of age | |
| Oral | |||
| Digoxin | Neonates 5–8 µg/kg/day, infants 10–15 µg/kg/day, children (2–10 years) 8–10 µg/kg/day, children (>10 years) 3–5 µg/kg/day | SVT/VTSNRT | Bradycardiac. Children require proportionally larger doses than adults based on body weight/surface area. Avoid in WPW patientsb |
| Propranolol | 1–3 mg/kg three times daily | SVT/VT | Bradycardiac, asthmab |
| Atenolol | 0.3–1.3 mg/kg three times daily | SVT/VT | Bradycardiac, asthmab |
| Verapamil | 4–8 mg/kg three times daily | SVT/VT | Bradycardiac, reduced LV functionb Avoid in infants <1 year of agebAvoid in WPW patients |
| Flecainide | 2–7 mg/kg twice daily ‘PITP’: 3 mg/kg | WPW and SVT | QRS duration 25% above baselinec, CrCl <50 mg/mLb, reduced LVEFb. Caution if conduction system diseasebFlecainide is not approved for use in children below the age of 12 years |
| Propafenone | 200–600 mg/m2 or 10–15 mg/kg in 3× daily | WPW and SVT | QT interval >500 msc, conduction system diseaseb and renal impairmentb. Contraindicated if reduced LVEFb |
| Sotalol | 1–2 mg/kg/day twice daily for neonates and children <6 years; 1.5–3 mg/kg/day twice daily for infants and children >6 years | WPW and SVT | Contraindicatedb: significant LVH, systolic HF, pre-existing QT prolongation, hypokalaemia, CrCl <50 mg/mL and asthma. QT interval >500 ms. Dose adjustment based on renal functionc |
| Amiodarone | Loading: 10 mg/kg for 10 days. Maintenance: 5 mg/kg/day | WPW and SVT, JET, PJRT | QT interval >500 msc. Caution with QT-prolonging drugsb Reduce the dose of vitamin K and digoxinb |
Abbreviations: CrCL, creatinine clearance; HF, heart failure; JET, junctional ectopic tachycardia; LV, left ventricle; LVEF, LV ejection fraction; LVH, LV hypertrophy; PJRT, permanent junctional ectopic tachycardia; SN, sinus node; SNRT, sinus node re-entrant tachycardia; SVT, supraventricular tachycardia; VT, ventricular tachycardia, WPW, Wolff–Parkinson–White syndrome.
aMyocardial depressant effect.
bMain contraindications and precautions.
cFeatures prompting a lower dose or discontinuation.
Very few studies, all of limited scope, have compared different AADs for the prophylactic treatment of SVTs.485,486 No evidence demonstrates clear superiority of one agent over another,486,487 and combination therapy may be necessary in some cases.488 Factors influencing drug choice include local availability and safety profiles.487 The optimal duration of treatment is also under debate, with some clinicians endorsing shorter treatment courses (four to six months instead of extending up to the first year of life).489
6.3. Foetal arrhythmias
Sustained foetal tachyarrhythmias (>180 b.p.m.) develop in up to 2% of pregnancies,490–492 and can lead to foetal nonimmune hydrops, cardiac dysfunction, preterm delivery, and higher perinatal morbidity and mortality.492–494 However, data are limited regarding optimal treatment, route of administration, and drug dosages. Thus, AAD selection is encouraged to be based on the severity (presence of maternal haemodynamic instability or hydrops foetalis), associated congenital abnormalities, and maternal preferences.495 Digoxin, flecainide, or sotalol, alone or in combination, depending on the type of tachycardia, are useful in terminating foetal tachyarrhythmias,482,490,496 but flecainide is more effective than digoxin in terminating foetal SVT in patients with and without hydrops foetalis.496,497 During treatment, adverse effects of these AADs can appear in both the foetus and the mother and maternal intolerance may be a limiting factor in the appropriate treatment of foetal arrhythmias; thus, close follow-up of both mother and foetus is needed.460,473,482,491
6.4. Elderly
Normal ageing is associated with changes in body composition, cardiac electrophysiological and structural alterations, and homeostatic mechanisms that increase susceptibility to developing CVDs (see Supplementary material online, Table S12).498–501 Additionally, patients ≥80 years often present several comorbidities that markedly affect the PD (effects of the drug on the body) and PK (absorption, distribution, metabolism, and excretion) of AADs and are treated with polypharmacy, increasing the risk of adverse events and drug–drug and drug–disease interactions (Tables 12 and 15).501–505 However, very old people with arrhythmias and comorbidities are under-represented/excluded from clinical trials; therefore, the benefit–risk balance of AADs to direct effective and safe treatment of arrhythmias in this population is unknown and is extrapolated from the results obtained in younger populations.506–508
Age-related cardiac changes include the loss of SA and AV nodal cells, leading to decreased heart rate and slowed AV conduction; changes in the expression/function of cardiac ion channels, leading to prolongation of the APD (QT interval); the presence of CVD leading to ventricular hypertrophy, amyloidosis, cardiac valvular degenerative changes, and annular calcification; and fibrous infiltration of the conduction system. These structural changes render the ageing heart more susceptible to the development of cardiac arrhythmias (pro-arrhythmia).498–501
Pharmacodynamic changes. β-Blockers are probably the most used and safest AADs in older people, with once daily drugs being preferred. Hydrophilic drugs (atenolol and nadolol) produce fewer CNS side effects, but are best avoided in patients with renal dysfunction. Co-administration of β-blockers with verapamil/diltiazem and/or digoxin increases the risk of severe bradycardia or different degrees of AV block in the elderly. In older sedentary people and HF patients who cannot tolerate higher doses of β-blockers, low doses of digoxin (to maintain serum digoxin levels <1 ng/mL) can be added to reach the desired heart rate and symptom control. Therefore, it is particularly important in elderly patients to obtain 24 h Holter monitoring to ensure that there is not excessive bradycardia or pauses during night when vagal tone is highest.506 It is advised to carefully manage Class I AADs in the elderly because they often have SND or SHD; if used, the ECG is adivsed to be closely monitored. Class Ia, dofetilide and sotalol are AADs particularly prone to inducing QT-related ventricular pro-arrhythmia in this population with frequent SHD, comorbidities, renal function impairment, and ionic imbalance.370 Amiodarone is often given to the very elderly because it is more effective than other AADs, can be administered to patients with ischaemic or SHD and does not need dose adjustment for renal or hepatic function.45,92 The risk of cardiovascular events and mortality with dronedarone appears to be reduced in elderly patients with non-permanent AF.98 Although the incidence of SCD increases with age, the proportion of deaths that are sudden compared with total mortality declines markedly in the elderly.509 In a meta-analysis of randomized trials, AADs significantly reduced recurrent VT without improving mortality, and the benefit was mainly driven by amiodarone.510
Older patients may have different responses to AADs and are more susceptible to some adverse effects than younger patients.501,511–514 Furthermore, several drugs with non-cardiovascular indications prolong the QTc interval and increase the risk of developing TdP and are advised to be avoided. The Beers criteria advise to avoid the following in older adults:515 (i) amiodarone as first-line therapy for AF unless the patient has HF or LVH, if rhythm control is preferred over rate control, because safer drugs are available; (ii) disopyramide, because of its potent negative inotropic effects and anti-cholinergic properties; (iii) dronedarone in patients with permanent AF or severe (Class IV) or recently decompensated HF, because of increased risk of death; and (iv) digoxin as first-line therapy for AF or HF in doses >0.125 mg/day for any indication, because more effective and safer alternatives exist.
Pharmacokinetic changes. Oral drug absorption may be delayed in the older individuals, but full drug absorption can be achieved because most drugs are absorbed by passive diffusion.512,516–518 The most important changes refer to drug distribution, biotransformation and excretion of AADs (see Supplementary material online, Table S12). In older people, body fat mass increases, while total body water and peripheral (hepatic and renal) blood flow and lean body mass decrease.511,512,517,518 Thus, the volume of distribution (Vd) and half-life of lipophilic drugs may increase, while the Vd of hydrophilic drugs decreases, leading to a more rapid increase in plasma concentrations. Hepatic biotransformation of some AADs depends on plasma protein binding, hepatic blood flow (which decreases with age, hepatic impairment, HF, shock, or β-blockers), and expression/activity of drug metabolizing enzymes.519 Hepatic metabolism via CYP-mediated Phase I reactions (oxidization, reduction, and hydrolysis) leading to active metabolites decreases, while Phase II conjugation reactions leading to inactive metabolites remain unaltered.516,517,520,521 Thus, hepatic impairment may reduce the clearance and increase the half-lives of AADs metabolized by the liver, and, therefore, dose adjustments according to age may be required to minimize the risk of adverse effects.517 Furthermore, older people are especially prone to drug interactions, which frequently occur at the level of drug metabolism.501,512,521
Older people present a reduction in renal blood flow, estimated GFR, and tubular secretion/reabsorption, along with an increased prevalence in renal diseases that impair renal function. These changes reduce the clearance and increase the exposure, the half-lives, and the risk of adverse events of renally cleared drugs.501,511,512,517,519
As a rule, lower starting doses (for patients ≥65 years) or a 50% dose reduction (for those ≥75 years) of AADs are advised in elderly patients. Doses are advised to be gradually up-titrated based on regular monitoring of symptoms, ECG findings, plasma drug levels, relevant laboratory parameters, and overall patient tolerance to ensure safety and efficacy. The selection of AADs in the elderly is mainly determined by the treatment target, patient tolerance, potential drug interactions, comorbidities, and renal and liver function (see Table 19 of renal and hepatic failure excretion and Box 16).513
Table 19.
Main pharmacokinetic characteristics of AADs with advice for their use in patients with renal or liver impairment (a more comprehensive review is provided in Supplementary material online, Table S3)
| AAD class | AAD | Tmax (h) | t 1/2 (h) | Excretion renal/hepatic (%) | Advice in renal impairment | Advice in hepatic impairment |
|---|---|---|---|---|---|---|
| 0 | Ivabradine | 1 | 2 (11a) | 10/90 | Avoid | Avoid |
| Ia | Quinidine | 2–4 | 4–10 | 20/80b | DR (75%) | DR |
| Procainamide | 3 | 3.5–5 | 60/40 | DR | DR | |
| Disopyramide | 1–2.5 4–7c |
6.5 (4–10) | 55/45 | DR | DR | |
| Ib | Lidocaine | 45–90 s | 1.5–2 | 5/95 | Close monitoring | |
| Mexiletine | 2–4 | 10–14 | 10/90b | DR | DR | |
| Ic | Flecainide | 2–4 | 20 (7–22) | 20/80b | DR | |
| Propafenone | 2–3.5 | 2–10 10–32d |
1/99 | DR | ||
| Id | Ranolazine | 2–6 | 7 | 70/- | Avoid | Avoid |
| III | Amiodarone | 3–8 | 25–100 days | <5/95 | DR | |
| Dronedarone | 3–6 | 25–30 | 16/84 | Avoid | ||
| Sotalol | 2.5–4 | 10–20 | 90/10 | Avoid | ||
| Dofetilide | 2–4 | 7–13 | 80/20 | Avoid | ||
| Ibutilide | 1.5 | 6 (2–12) | 18/82 | Caution | Caution | |
| Vernakalant | 1–5 min | 1.5–3.5 | 7/93 | |||
| IIa | Acebutolol | 1.3–3 | 3–4 | 40/60 | DR if CrCl <50 mL/min | |
| Bisoprolol | 2–4 | 9–12 | 50/50 | DR if CrCl <50 mL/min | ||
| Nadolol | 2–4 | 20–24 | 75/25 | DR if CrCl <50 mL/min | ||
| Atenolol | 2–4 | 6–9 | 90/10 | DR | ||
| Propranolol | 2 (p.o.) 2–10 min (i.v.) |
4–6.5 8–10c |
10/90e | DR | ||
| Metoprolol | 1–2; 3–3.5c | 3–5 (2.8 IR, 7.5 ER); 24c | 5/95 | DR | ||
| Carvedilol | 1–3 | 7–10 | 2/98 | Avoid | ||
| Esmolol | 2–10 min | 9 min | – | |||
| IIb | Isoprenaline | 80/20 | ||||
| IIc | Atropine | 2–4 min | 2–4 | 60/40 | ||
| IId | Digoxin | 3–6 (1–3 min i.v.) | 35 (30–48) | 75/25 | DR done by CrCl and serum levels | |
| IIe | Adenosine | 10–30 s | <20 seg | – | ||
| IV | Diltiazem | 30–60 min p.o. 3 min i.v. |
iv: 2–5 IR: 4.5–12 ER: 12 |
10/90 | Caution | DR |
| Verapamil | 1–2,5–11c | 4–7 | 15/85 | Caution | DR |
Abbreviations: AAD, anti-arrhythmic drug; CrCl, creatinine clearance; DR, dose reduction; H, hepatic; i.v.; intravenous; p.o., oral administration; R, renal; Tmax, time to peak plasma levels; t1/2, drug half-life.
aEffective half-life.
bDrugs that alkalinize urine decrease renal excretion of the AAD.
cSlow/extended release.
dLess than 10% people are poor metabolizers of the AAD.
eShort-acting β-blockers can undergo significant first-pass liver metabolism. As a result, their serum levels may vary substantially for the same dose. These β-blockers are advised to be taken with food to improve absorption.
Box 16 Key advice for AAD use in the elderly.
β-Blockers are first line; atenolol/nadolol have fewer CNS effects but require caution in renal dysfunction
Avoid combining β-blockers with verapamil, diltiazem, or digoxin to prevent severe bradycardia and AV block. Low-dose digoxin (<1 ng/mL) may be used in HF or sedentary patients
Avoid Class Ia AADs, dofetilide, and sotalol due to a high risk of pro-arrhythmia. Use Class Ic cautiously due to potential sinus node depression
Amiodarone is often preferred in elderly patients with structural heart disease due to its efficacy and tolerance
Dronedarone may reduce cardiovascular events but is not advised in patients with permanent AF or severe HF due to increased mortality risk
Lipophilic drugs (e.g. amiodarone, propranolol) have prolonged half-lives and a increased risk of accumulation, while hydrophilic drugs (e.g. sotalol, atenolol) may reach higher plasma concentrations due to age-related changes
Declining renal and hepatic functions increase toxicity risk; frequent monitoring of renal function and plasma drug levels is essential
-
Reduce doses in older adults
Start with lower doses in patients ≥65 years
Reduce by 50% in patients ≥75 years and titrate gradually with ECG and lab monitoring
Abbreviations: AAD, anti-arrhythmic drug; AF, atrial fibrillation; CNS, central nervous system; ECG, electrocardiogram; HF, heart failure.
6.5. Athletes
Anti-arrhythmic drug therapy in athletes presents unique challenges due to their young age, hypervagotonic state, and reluctance to take medications that may impact physical performance, such as β-blockers. β-blockers and CCBs can exacerbate bradycardia, increasing the risk of fatigue, dizziness, and syncope. Class Ic drugs are advised to be used with caution due to their use-dependent effects, which can increase the risk of exercise-induced pro-arrhythmia. Conversely, drugs with reverse use dependence, such as sotalol, may heighten the risk of TdP in athletes, particularly in those with frequent bradycardia. Additionally, some drugs, including metoprolol and sotalol, are classified as prohibited substances by the World Anti-Doping Agency in precision-based sports such as archery, automobile racing, billiards, darts, golf, shooting, ski jumping, snowboarding (with jumping), and underwater apnoea sports.522 However, other AADs, including amiodarone, Class Ic agents, and CCBs, are not subject to these restrictions. Given these complexities, AAD selection must carefully balance efficacy, safety, and regulatory compliance, ensuring optimal arrhythmia control while preserving athletic performance and minimizing drug-related impairments.
6.6. Heart failure
6.6.1. Reduced ejection fraction
In patients with HFrEF, both AF and VA are more likely to occur. Anti-arrhythmic drugs are used for symptomatic atrial and ventricular tachyarrhythmias, after the use of guideline-directed medical therapies (GDMTs), all of which have a role in improving LVEF, overall survival, and reducing arrhythmias.523 β-Blockers and aldosterone receptor blockers also reduce SCD.523 In HF patients, avoiding hypokalaemia, hypomagnesaemia, and digitalis toxicity is required to minimize pro-arrhythmic events.
Rate and rhythm control strategies in AF patients with HFrEF are important to minimize the contribution of tachycardia-induced cardiomyopathy to LVEF depression.92,130,523 In addition, HF increases thromboembolic risk, and therapeutic anticoagulation is a necessary part of the treatment strategy based on risk/benefit ratio. Worldwide, amiodarone is the main AAD used for rhythm control in AF patients with HFrEF based on safety from the GESICA and CHF-STAT studies,524,525 higher efficacy rates than other drugs,526 and guideline recommendations.92,130,396 However, caution is warranted, as amiodarone increased mortality in Class III HF patients in the SCD-HeFT trial [hazard ratio (HR) 1.44; confidence interval (CI) 1.05–1.97, P = 0.01 compared with control].527 In North America, dofetilide is recommended by the AHA/ACC/HRS guidelines based on the drug's safety in the DIAMOND-HF study.528 The use of dofetilide in HFrEF patients should be restricted due to its high renal excretion rate, risk of TdP, and poor renal function in these patients.529 If a rhythm control strategy is chosen and amiodarone is ineffective, rhythm control with catheter ablation has to be considered based on the CASTLE-AF and CABANA-AF HF sub-studies530,531 and the ESC and other societies guideline recommendations.92,130 In these studies, LV function and quality of life improved although overall efficacy rates of catheter ablations were less than in patients without HFrEF.532
In patients with HFrEF who develop recurrent symptomatic AF, a rhythm control strategy with AADs is not superior to a rate control strategy.533 However, a recent study in end-stage HF patients showed that AF ablation improved survival vs. optimal medical therapy.534 Ablation is also encouraged as first-line therapy when a component of tachycardia-mediated cardiomyopathy is involved (e.g. lack of late gadolinium enhancement on cardiac magnetic resonance).535 For acute and chronic rate control of AF, β-blockers are first-line therapy followed by digoxin.92,130,523 The DIG trial, which predated current GDMT, primarily enrolled patients with NYHA Class II–III HF and showed that treatment with digoxin for 2–5 years had no effect on mortality but modestly reduced the combined risk of death and hospitalization.536 For chronic rate control, non-dihydropyridine CCBs are advised to be used with caution, and dronedarone is discouraged in patients with decompensated HF.103 Oral amiodarone can be used as added rate control prior to considering AV junction ablation for uncontrolled ventricular rates.92,130,523
For acute termination of AF in HFrEF, DC cardioversion is the safest and most effective option, along side rate control and the possibilty of spontaneous conversion.537 Intravenous amiodarone is safe, but conversion rates are acutely limited and delayed.157,537,538 Intravenous ibutilide can be used with caution for conversion and is more effective in converting AFL than AF.137,537 Intravenous vernakalant is discouraged in Class III/IV HF patients and in those with hypotension.163 Class Ic agents are best avoided for AF conversion in this subset of patients.52,92,130
For acute rate control of AF with a rapid ventricular response, i.v. β-blockers and digoxin are appropriate firs-line agents, while i.v. non-dihydropyridine CCBs are not advised if LVEF is ≤40%.92,130,523 Intravenous amiodarone is an effective short-term rate control agent in the acute management.92,130,538 Ivabradine in combination with ranolazine is being studied to determine efficacy in controlling the ventricular response in HFrEF patients.539
Because of the safety profile and effectiveness in treating VA and reducing the risk of sudden death, β-blockers are often used as first-line anti-arrhythmic therapy.477 Amiodarone is the next AAD of choice for patients with VT or VF who are not otherwise candidates for an ICD.396,477,527 This is based on the efficacy, minimal negative inotropic effects, and low pro-arrhythmic potential. Amiodarone, combined with a β-blocker, reduced frequent ICD shocks in the OPTIC trial.91 Sotalol has some efficacy in suppressing VA, but it has significant pro-arrhythmic effects and has not been shown to improve survival. Sotalol appears to reduce the DFT and decrease ICD therapies.119 Sotalol can lead to HF decompensation and is discouraged in patients with an LVEF <20%.477
6.6.2. Preserved ejection fraction
Heart failure with preserved ejection fraction and AF often co-exist, each facilitating the occurrence and aggravating the prognosis of each other.175,540,541 In symptomatic patients, it seems reasonable to start with rate control to optimize ventricular filling time and improve symptoms. The ESC guidelines recommend β-blockers, diltiazem, verapamil, and digoxin for rate control (<100–110 b.p.m.) in patients with HFpEF; amiodarone may be appropriate only in the acute setting.92 Digoxin is associated with a neutral effect on mortality and a lower rate of hospital admissions. Although the benefit of β-blocker therapy in reducing mortality in AF patients with HFrEF has been questioned,175 some real-world studies support an improved prognosis.540 Pharmacological cardioversion using i.v. amiodarone may be attempted if haemodynamic instability or worsening of HF.
A rhythm control strategy is challenging in patients with HFpEF, who are often of advanced age and have comorbidities that may influence the success of treatment and the risk of adverse events. Drugs of choice are amiodarone, dofetilide, dronedarone and sotalol.92 In a post-hoc analysis of the RACE 3 trial, recruiting patients with early persistent AF and mild-to-moderate stable HFpEF and HFrEF, AAD treatment was effective in nearly half of the patients at 1 year.541 Maintenance of SR was significantly better with amiodarone (58%) compared with flecainide (32%) and sotalol/dronedarone (23%). In an observational study, rhythm control (mainly amiodarone) was associated with lower 1-year all-cause death rate compared to rate control in older patients (≥65 years) with HFpEF.542 In a retrospective study, maintenance of SR was associated with a lower risk of the composite outcome of cardiovascular death or hospitalization for HF in patients with HFpEF and AF.543 Furthermore, in a recent systematic review comparing rhythm and rate control treatment strategies in patients with HFpEF and AF, rhythm control was associated with 15% lower mortality compared to rate control. However, no differences were found between rhythm and rate controls regarding HF admission rates, stroke/transient ischaemic attack, and cardiovascular mortality.544 Additionally, in a pooled analysis of AFFIRM and AF-CHF trials, amiodarone's efficacy in maintaining SR and reducing the burden of AF was similar in the presence or absence of severe LV dysfunction.545 In the 2024 ESC AF guidelines, dronedarone is recommended for long-term rhythm control in AF patients with HFpEF or mildly reduced but stable LV function.92 Therefore, when carefully instituted, rhythm control is a viable and relatively safe option in patients with HFpEF and AF.541 Although mineralocorticoid receptor antagonists are advised for patients with HFpEF,523 spironolactone does not reduce the risk of new-onset AF or AF recurrence in patients with HFpEF.546
Amiodarone is effective in suppressing VA and improving LV function but does not reduce the incidence of sudden death or prolong survival among patients with HFrEF, except for a trend towards reduced mortality among those with non-ischaemic cardiomyopathy. However, its effects in patients with HFpEF remain uncertain.525
Recently, the EMPEROR-Preserved trial showed that empagliflozin reduced the combined risk of cardiovascular death or hospitalization for HF in patients with HFpEF, regardless of the presence or absence of Type 2 diabetes mellitus (T2DM). In a meta-analysis, SGLT2 inhibitors were associated with significantly reduced risks of incident atrial arrhythmias and SCD in patients with T2DM with or without HF,547 but not with an overall lower risk of SCD or VAs in patients with T2DM and/or HF and/or chronic kidney disease.548 Prospective trials are warranted to confirm the anti-arrhythmic effect of SGLT2i inhibitors and whether this is a Class or drug-specific effect.
Although it seems clear that weight loss is associated with a reduced risk of AF recurrence, and the pleiotropic effects (such as anti-inflammatory effects) of the metabolic weight loss reducing agents (such as GLP-1 receptor agonists), are potentially advantageous, their value for AF rhythm control has yet to be established, though it appears promising.
6.7. Cardiomyopathies
Limited data exist regarding the use of AADs in cardiomyopathies other than HCM and arrhythmogenic right ventricular cardiomyopathy (ARVC). Therefore, the principles for AAD use in these cases must align with those for other forms of SHD. Class I AADs are generally to be avoided, and β-blockers, sotalol, and amiodarone are typically the preferred choices.45
6.7.1. Hypertrophic cardiomyopathy
β-blockers are commonly used as first-line therapy in HCM, providing dual benefits of symptom reduction and decreased risk of VA.447
Disopyramide (Class I AAD) and CCBs may be appropriate in specific situations due to its potential to reduce left ventricular outflow tract obstruction and alleviate symptoms. However, no demonstrated prognostic benefits exist, and its use requires close monitoring.
Amiodarone is reserved for refractory cases or when other medications are not well tolerated. Long-term use is not convincingly associated with sudden death reduction and is carefully weighed against potential side effects.
6.7.2. Arrhythmogenic right ventricular cardiomyopathy
β-blockers are the first-line therapy for VA in ARVC.45 Other AADs, such as flecainide, sotalol, and amiodarone, demonstrated VT suppression in small observational studies, but their ability to prevent VT recurrences at follow-up is either minimal or not established.549,550 A small study showed some benefit from combining flecainide with β-blockers or sotalol.441
6.8. Renal and liver failure
Most AADs are biotransformed in the liver via CYP enzymes, mainly CYP3A4 and CYP2D6, and their exposure and half-life increase/decrease when co-administered with CYP inhibitors/inducers, respectively, leading to important drug–drug interactions as discussed above153,551,552 (Tables 14 and 15). Additionally, some AADs (see Supplementary material online, Table S3) are biotransformed in the liver into active metabolites with similar or different electrophysiological effects from those of the parent compound (N-acetylprocainamide is a Class III drug; 5-hydroxypropafenone lacks β-adrenergic blocking effects).
Hepatic drug clearance depends on the expression/activity of drug metabolizing enzymes, hepatic blood flow and drug protein binding.519,520,553 Drugs with high hepatic clearance (diltiazem, lidocaine, metoprolol, propranolol, and verapamil) are rapidly metabolized, and the rate of drug loss is determined by the hepatic blood flow. In patients with hepatic impairment or decreased hepatic blood flow (elderly, cirrhosis, HF, shock, MI, or treated with β-blockers) drug biotransformation is inhibited, so that exposure and half-life of the parent compound significantly increase, while the formation of active metabolites decreases.
Patients vary in their responses to drug therapy, and some of that variability is genetically determined.253,520,554 CYP2D6 metabolism is under genetic control, and plasma levels, half-life and the risk of adverse effects of CYP2C6 substrates increase in poor metabolizers (∼7% of Caucasians), while a decrease in drug efficacy can be observed in ultra-rapid metabolizers. Slow acetylators of procainamide develop more often and earlier drug-induced lupus syndrome than rapid acetylators. Thus, doses are advised to be reduced in carriers of poor/slow phenotypes.
Renal failure decreases the clearance and increases the exposure and the risks of adverse effects of renally cleared drugs (digoxin, disopyramide, N-acetylprocainamide, dofetilide, flecainide, ibutilide, procainamide, and mainly sotalol). Thus, doses, clinical response and plasma levels of these drugs are advised to be carefully titrated in patients with kidney impairment. Some AADs (amiodarone, dronedarone, and quinidine) inhibit the P-gp required for renal excretion of digoxin, thereby increasing its plasma levels.552 Therefore, in patients with impaired hepatic and/or renal function, both loading and maintenance doses of AADs are advised to be reduced, and ECG monitoring is advised to minimize the risk of adverse events, mainly pro-arrhythmia (Figure 3, Table 18).
6.9. Congenital heart disease
Arrhythmias are common and poorly tolerated in patients with congenital heart disease. Various factors (Box 17), including single ventricle or systemic morphologic right ventricle, cyanosis, residual post-surgical obstructive lesions and scars, ventricular dysfunction, and pulmonary hypertension, contribute to the complexity of managing arrhythmias in this population.555,556 Despite advancements in invasive therapies, AADs remain crucial for their management. However, there is limited evidence supporting AAD selection for this specific population, and advice is largely extrapolated from those for the general arrhythmia population. Nevertheless, there are specific considerations to keep in mind when managing individuals with congenital heart disease.
Box 17 Specific factors for AAD selection in patients with congenital heart disease.
-
Frequent sinus node dysfunction and AV conduction abnormalities
Examples: Post-atriotomy, D-transposition of the great arteries
-
Accelerated AV conduction potentially leading to sudden death
Patients with atrial arrhythmias, especially in D-transposition of the great arteries535
-
Frequent post-surgical or spontaneous myocardial scars and ventricular dysfunction
Risk of pro-arrhythmia or heart failure
Particularly in Fallot or univentricular patients
-
Higher systemic venous pressures
May alter hepatic metabolism, potentially leading to AAD toxicity (e.g. amiodarone)536
Relevant for Fontan patients and those with cyanotic conditions
-
Young age factors
Low body mass
Child-bearing potential
Need for long-duration AAD treatment
Abbreviations: AAD, anti-arrhythmic drug; AV, atrioventricular.
6.10. Channelopathies
6.10.1. Long QT and short QT syndromes
Long QT syndrome is characterized by a prolonged QT interval, T-wave changes, syncope, polymorphic VT, SCA, and SCD, mainly triggered by adrenergic activation in a structurally normal heart. The annual rate of SCD in asymptomatic patients with untreated LQTS has been estimated to be <0.5%, but may increase to around 5% in high-risk patients depending on ECG, symptoms, and specific mutations. β-blockers are advised for all LQTS patients but may be omitted in asymptomatic low-risk individuals experiencing side effects. The non-selective β-blockers nadolol (oral dose 40–120 mg/day) and propranolol (oral dose 80–320 mg/day, slow-release preferred) have been shown in observation studies to have higher efficacy in preventing VA.557 Mexiletine (oral dose 5–10 mg/kg/day) may be used in addition to β-blockers in LQTS3 and some LQTS2 patients, highlighting the importance of genetic testing to direct pharmacological treatment. In LQTS2 and LQTS3, mexiletine reduces the length of the QT interval and the number of arrhythmic events. Not all mutations in sodium channel protein Type 5 subunit alpha (SCN5A) or hERG, the genes responsible for LQTS3 and LQTS2, respond to mexiletine; therefore, it is advised to perform oral testing to document that the QTc shortens by 40 ms or more in the outpatient clinic or hospital department before prescribing chronic treatment.558 Lifestyle advice includes avoidance of drugs (Figure 19) that prolong the QT interval (see Supplementary material online, Tables S8 and S9; also see www.crediblemeds.org/).
Figure 19.

Twelve-lead electrocardiograms (ECGs) of a 22-year-old woman with no prior heart disease presenting with ventricular fibrillation and pleomorphic ventricular arrhythmias. The patient was initially treated with oral quinidine and was later diagnosed with Type II long QT syndrome. The ECG demonstrates inverted T waves (T) across several leads and an extremely prolonged QT interval. This case underscores the pro-arrhythmic potential of quinidine in patients with underlying repolarization disorders. The recordings were obtained at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
Short QT syndrome is characterized by a very short QT, AF, and SCA in a structurally normal heart. Quinidine (oral dose 600–1600 mg/day; loading dose: 200 mg every 3 h until effect) is the advised AAD but is advised to be monitored for excessive QT interval prolongation and other side effects, and isoprenaline infusion (0.5–10 μg/min) is the drug of choice in ES.559 Lifestyle advice includes avoidance of drugs that shorten QT (e.g. nicorandil).
6.10.2. Brugada syndrome
The Type 1 Brugada ECG pattern is characterized by ST segment elevation and T-wave inversion in at least one right pre-cordial ECG lead. The ECG changes may be spontaneous or induced by exposure to fever or Nav-blocking agents (Figure 20).45,560 Anti-arrhythmic drug strategy depends on whether the BrS patient is asymptomatic or symptomatic. To lower the risk of SCA, lifestyle advice includes the avoidance of drugs that are known to block the Na+ current (see Supplementary material online, Table S13; also see www.Brugadadrugs.org). Quinidine (oral dose 600–1600 mg/day; loading dose: 200 mg every 3 h until effect) is the drug of choice in the prevention of VA and in the treatment of ICD shocks and ES.561 In a Canadian report of IVF, BrS, and ERS cases, ICD shocks were reduced from 7.47 in 34 months to 0.86 in 44 months after quinidine initiation. In the case of ES, isoprenaline infusion (0.5–10 μg/min) is recommended by ESC guidelines.562
Figure 20.

Electrocardiogram (ECG) tracings of leads V1–V6 (A) and V1–V3 (B) illustrating the dynamic changes in two patients with Brugada syndrome at baseline, during isoproterenol infusion, and following i.v. administration of flecainide (2 mg/kg). (A) Baseline ECG shows mild ST-segment elevation (1.5 mm) in V2, measured at 80 ms from the J point. Following flecainide infusion, ST-segment elevation increases significantly to 4.5 mm. (B) In another patient, baseline ECG reveals ST elevation and T-wave inversion in V2. These abnormalities normalize during isoproterenol infusion but are markedly exaggerated following flecainide administration. The ECG was recorded at a speed of 25 mm/s and a sensitivity of 10 mm/mV.
6.10.3. Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia is a rare inherited heart disease characterized by catecholamine-induced bidirectional VT or PVT in a structurally normal heart and in the absence of ischaemia or digitalis. Catecholaminergic polymorphic ventricular tachycardia patients often have a normal resting ECG, but an exercise stress test reveals the VA. Pharmacologic treatment is always initiated with β-blockers and non-selective β-blockers, such as nadolol (oral dose 40–120 mg/day) and propranolol (oral dose 80–320 mg/day, preferably slow-release) are preferred. As noted in the individual AAD descriptions, short-acting β-blockers like propranolol, which are metabolized by the liver, can exhibit significant variability in serum levels due to first-pass metabolism, with up to a 10-fold variation reported. If propranolol proves ineffective, switching to nadolol—a renally excreted β-blocker—may provide more consistent therapeutic effects. The effect of β-blockers is advised to be evaluated by a repeated exercise stress test assesing the number of PVCs, appearance of VAs, and maximum heart rate. Data convincingly suggest that flecainide (oral dose per day 50–200 mg) significantly reduces the VA burden in CPVT patients and is often desirable in addition to β-blockers when control of arrhythmias is incomplete.50 In a multicentre study, 22 of 29 patients treated with β-blockers and flecainide had either partial (n = 8) or complete (n = 14) suppression of exercise-induced VA with flecainide. In selected patients who show intolerance to β-blocker therapy, pharmacological therapy with flecainide alone is an option.563
6.10.4. Early repolarization syndrome
Early repolarization syndrome is diagnosed in SCA patients with documented PVT or VF, a structurally normal heart, an early repolarization pattern, and J-point elevation ≥1 mm in two or more adjacent lateral and/or inferior ECG leads. Isoprenaline infusion (0.5–10 μg/min) is effective in treating ES or recurrent ICD discharges, and also attenuates J-wave amplitude.564 Anti-arrhythmic drug that blocks the Ito seems to prevent VA. A multicentre study found a decline in recurrent VF after initiation of quinidine (oral dose 600–1600 mg/day; loading dose: 200 mg every 3 h until effect) but not with other AADs.565 Cilostazol and milrinone have been shown in an experimental models to reduce the recurrence of VF.183
The advised AADs for prevention of polymorphic VAs in patients with channelopathies are summarized in Figure 21.
Figure 21.

Schematic representation of the advised first-line anti-arrhythmic drugs (AADs) for prevention of polymorphic ventricular arrhythmias. The figure provides a general reference for selecting the most appropriate drug; however, the final choice is advised to be based on additional patient characteristics and conditions, as detailed in the various sections of this document. BrS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; ERS, early repolarization syndrome; LQTS, long QT syndrome; PVT, polymorphic ventricular tachycardia; SQTS, short QT syndrome.
6.11. Anticoagulation
Anticoagulants and AADs are commonly prescribed concurrently for the same patient. Potential interactions within these medication classes involve both pharmacokinetic and pharmacodynamic aspects, potentially resulting in an intensified anticoagulation effect (Table 20).566
Table 20.
Main interactions of AADs with anticoagulantsa
| Class | AAD | Dabigatran | Apixaban | Edoxaban567 | Rivaroxaban | Warfarin |
|---|---|---|---|---|---|---|
| I | Quinidine (inhibits CYP2D6 and P-gp) | Caution Avoid co-administration if CrCl <50 mL/min |
Caution | Caution | Caution | Reduce the warfarin dose by 10–20% Monitor INR |
| Propafenoneb (inhibits CYP2C9 and CYP3A4) | Safe | Safe | Safe | Safe | Reduce warfarin dose Monitor INR |
|
| III | Amiodarone (inhibits CYP3A4 and P-gp) | Caution if CrCl 30–50 mL/min | Safe | Caution if CrCl 15–50 mL/min | Caution if CrCl 15–50 mL/min | Reduce the warfarin dose by 40, 35, 30, and 25% if the amiodarone dose is 400, 300, 200 or 100 mg/day, respectively |
| Dronedarone (inhibits CYP2C9, CYP3A4, and P-gp) | Avoid | Caution | Reduce the edoxaban dose by 50% (to 30 mg/12 h) | Avoid | Monitor INR | |
| IIb | Digoxin (potential displacement of warfarin from plasma protein-binding sites) | Safe | Safe | Safe | Safe | Monitor INR |
| IV | Verapamil (inhibits P-gp) | Reduction the dabigatran dose (110 mg/12 h) | Caution | Caution | Caution | Safe |
‘Avoid’ is advised according to the drug official label, while ‘caution’ is advised with potential dose reduction if additional risk factors are present. Agents with some interaction with anticoagulants are shown in bold.
Abbreviations: AAD, anti-arrhythmic drug; CrCL, creatinine clearance; LMWH, low-molecular-weight heparin; P-gp, P-glycoprotein; UFH, unfractionated heparin.
aDOACs are substrates of P-gp, and P-gp inhibitors can increase their plasma concentrations, leading to a heightened bleeding risk. Warfarin is metabolized primarily by CYP2C9, along with other cytochrome P450 enzymes. Inhibitors of these enzymes can significantly increase INR and the risk of bleeding. Unfractionated heparin and LMWHs are not metabolized by CYP enzymes or P-gp. As a result, they exhibit minimal pharmacokinetic interactions with AADs.
Certain AADs hinder the degradation of warfarin through the CYP pathway, while specific DOACs experience reduced elimination via P-gp due to direct competition with some AADs (amiodarone, dronedarone, quinidine, verapamil, diltiazem, and digoxin).568,569 However, dronedarone plays a role in influencing both the degradation of warfarin and DOACs via the CYP pathway and the elimination of these anticoagulants via P-gp. In addition, verapamil and diltiazem not only contribute to decreased clearance of DOACs through competitive interactions with P-gp elimination but also exert a mild inhibitory effect on the CYP pathway, impacting the degradation of these drugs. Apixaban and rivaroxaban are most affected due to CYP3A4 metabolism, while dabigatran and edoxaban are primarily affected via P-gp inhibition.
It is noteworthy that CYP-mediated drug interactions are minimally affected by the timing of drug administration, while P-gp-mediated interactions can be mitigated by spacing the intake of the two drugs by at least 2 h. Careful consideration of these factors is crucial for optimizing therapeutic outcomes and minimizing potential complications in patients receiving both anticoagulants and AADs.
6.12. Anti-arrhythmic drug and non-pharmacological anti-arrhythmic therapies
6.12.1. Anti-arrhythmic drugs and pacemakers
Pacemakers are usually used in patients with symptomatic bradyarrhythmias, including sick sinus syndrome and AV block. Pacemakers are often implanted when an effective AAD causes significant negative chronotropic or dromotropic side effects. Anti-arrhythmic medications may be advised in patients with PM when atrial or ventricular tachyarrhythmias need to be treated in patients who are not ablation candidates. Anti-arrhythmic drugs blocking Na+ channels may increase pacing thresholds, especially at higher doses, and lead to loss of capture (Table 21).570–572 The increase in pacing thresholds is usually minimized by safety margins programmed into atrial and ventricular PMs. Drugs that slow the sinus heart rate may cause a PM to pace more frequently, and the provocation of AV block may increase the frequency of ventricular pacing.
Table 21.
Influence of AAD on pacing, ventricular defibrillation threshold, and atrial cardioversion failure
| AAD class | AAD | Pacing threshold | Ventricular defibrillation threshold | Atrial defibrillation thresholda |
|---|---|---|---|---|
| Ia | Procainamide Quinidine |
+ | + | − |
| Ib | Lidocaine Mexiletine |
0 | + | |
| Ic | Flecainide | + | + | + |
| IIa | β-Blockers | 0 | 0/− | 0 |
| IIb | Isoprenaline | − | ? | |
| III | Amiodarone | + | + | − |
| Sotalol | 0 | − | − | |
| Ibutilide | − | |||
| IV | Verapamil Diltiazem |
0 | + | 0 |
+, increases threshold; −, decreases threshold or risk of cardioversion failure. Blue: clinical positive effect.
Abbreviation: AAD, anti-arrhythmic drug.
aInferred from atrial arrhythmia cardioversion failure and/or cardioversion energy requirements.
Box 18 ICD–AAD interactions.
AADs may increase ICD pacing thresholds (see Table 21)
AADs may alter DFT (see Table 21)
AADs may aggravate bradycardia/AV block requiring more antibradycardia pacing
AADs may slow AFL leading to 1:1 conduction or pacing
AADs may slow VT rate and increase cycle length above the ICD tachycardia detection interval
AADs may alter VT sensing by slowing the dV/dT and increasing the QRS duration
AADs may cause pro-arrhythmia or incessant VT, potentially increasing the need for ICD interventions or rendering ICD therapy ineffective
Abbreviations: AAD, anti-arrhythmic drug; AFL, atrial flutter; AV, atrioventricular; DFT, defibrillation treshold; ICD, implantable cardioverter defibrillator; VT, ventricular tachycardia.
6.12.2. Anti-arrhythmic drugs in patients with implantable cardioverter defibrillators
Anti-arrhythmic drugs are commonly used in ICD patients to decrease delivered therapies, such as anti-tachycardia pacing, cardioversion, and defibrillation.91,571–574 Amiodarone combined with β-blockers is effective in reducing ICD therapy, though amiodarone adverse effects need to be considered.91 Sotalol is also effective, but less than amiodarone combined with a β-blocker.574 However, in one placebo-controlled trial, sotalol reduced ICD therapies and was effective in reducing DFT, similar to other Class III agents.119 In a small series, dofetilide also reduced ICD therapies.575 In placebo-controlled trials, azimilide (not commercially available) demonstrated the ability to decrease total all-cause shocks and VT episodes terminated by anti-tachycardia pacing.576 Ranolazine did not reduce the incidence of first VT, VF, or death but showed a 30% reduction (P = 0.028) in ICD therapies for recurrent VT or VF.577 Although no ICD interaction studies exist with dronedarone, a sister compound, celivarone, showed no benefit at the doses tested in reducing ICD therapies compared with placebo.578 Most AADs influence the DFT (Table 20), and since ICD defibrillation failure due to drug-induced high DFTs may result in SCD, it is important to check DFTs when AADs are used in patients with an ICD. Table 21 and Box 18 list the main influences of AADs on pacing threshold, ventricular DFT, and atrial cardioversion failure.579
6.12.3. Anti-arrhythmic drugs following ablation therapy
The number of randomized trials evaluating AADs after AF ablation is limited. One study, after a 12-month follow-up, showed no significant difference in the rates of AF recurrence in patients with either paroxysmal or persistent AF, but AAD increased the proportion of patients with asymptomatic AF episodes.580 The 5A study demonstrated that paroxysmal AF patients treated with AAD for 6 weeks after ablation had about a 50% reduction in AF recurrences compared to those treated with AV nodal-blocking agents.581,582 Further reports from the 5A study showed no benefit of an early rhythm suppression strategy with AADs in persistent AF after catheter ablation to decrease arrhythmia recurrence after the blanking period.583 Initiation of AADs at discharge after catheter ablation has been associated with a significant reduction in readmission within 90 days (11.6 vs. 16.2%).584 In unadjusted time-to-event analysis, amiodarone was associated with the greatest reduction in readmission, whereas dronedarone, Class II agents, and Class Ic agents had no statistically significant effect on readmission. The POWDER-AF study suggested that a longer-lasting treatment with AAD may be a strategy to reduce AF recurrences during long-term follow-up.585
Not all studies demonstrated a benefit of AAD therapy in patients who underwent catheter ablation. A retrospective, non-randomized study of 274 ablation patients demonstrated no difference in the rates of early AF recurrence among those treated with an AAD or an AV nodal-blocking agent alone.586 In a recent large retrospective German study,587 the rates of AF recurrences, cardiovascular events, and mortality did not differ between patients discharged with or without AADs after AF catheter ablation. Therefore, expert consensus statements on catheter and surgical ablation of AF588 give a moderate level of advice and state that administration of AADs following AF catheter ablation is reasonable in selected patients to prevent early post-ablation AF recurrence.1 Studies of specific AADs post-AF ablation are lacking, and drug choice is based on AF guideline, comorbidities, and prior efficacy and safety in each patient.589
Finally, a recent study examined rhythm control strategies following index catheter ablation for AF in a large patient cohort (n = 23 323).1 Over a median follow-up of 1165 days, AAD use post-ablation was prevalent (46.9%), with a notable increase among patients requiring repeat ablations (62.8–92.3%). These findings underscore the widespread clinical practice of combining catheter ablation and AAD therapy to enhance rhythm control in AF patients.589
6.12.4. Anti-arrhythmic drugs: effects on direct current cardioversion and defibrillation
Anti-arrhythmic drugs may alter the energy required for cardioversion of atrial and ventricular tachyarrhythmias, as well as the DFT (Table 21). A study involving 57 patients with persistent AF assessed the energy levels required for successful electrical cardioversion among those receiving different classes of AADs. The findings showed that patients on Class Ia or Class III anti-arrhythmic drugs had a median cardioversion energy requirement of 100 joules, whereas those on Class Ic drugs required a median of 200 joules (P = 0.03).590 Importantly, the frequency of unsuccessful cardioversions did not differ significantly between these groups.
Atrial fibrillation cannot always be converted to SR by transthoracic electrical cardioversion, although this is less likely with biphasic shock waveforms.537 Ibutilide, which lowers cardioversion energy requirements, has been used in refractory DC cardioversions of AF. One study showed that the cardioversion success rate increased from 72 to 100% after i.v. ibutilide (P < 0.001). However, ibutilide caused sustained polymorphic VT in 2 of the 64 patients in this trial, both of whom had an LVEF of 20% or less.148 Ibutilide decreased cardioversion energy requirements from 228 ± 93 to 166 ± 80 J, P < 0.001. Anti-arrhythmic drugs have the additional benefit following cardioversion of reducing immediate and early recurrence of AF.537
7. Anti-arrhythmic drugs under development
After a hiatus in recent years, several new AADs have reached the stage of Phase II or Phase III pre-approval studies, and it is possible that one or more may become available within the next few years. These constitute only a minority of the novel molecules with anti-arrhythmic potential.591 More of these may emerge from pre-clinical development in the near future.592 Only the histone deacetylase 6 (HDAC6) inhibitor (see below) has recently emerged with a fundamentally new mechanism of action.
Seven new drugs/formulations deserve mention. Two exploit new methods of drug delivery to facilitate patient self-administration in the out-of-hospital setting. Two of these drugs predominantly inhibit ion channels which have not previously been considered AAD targets. Several of these drugs are multiple ion channel blockers, although the balance of ion channel inhibition or the PK/PD properties of the formulations are sufficiently different to offer new therapeutic opportunities.
7.1.1. Etripamil
Etripamil is a novel L-type CCB, which has a rapid onset of action (Tmax ≤ 7 min) and is short lasting being inactivated by blood esterases. Etripamil is administered using a nasal spray. It has been developed for patient self-administration for the termination of PSVT where the AV node is a critical component of the re-entry circuit, i.e. AVRT and AVNRT. It prolongs AV nodal conduction and decreases the Wenckebach rate. The drug has been studied when given by medical staff in the EP laboratory for the termination of induced PSVT. It was shown that etripamil 70 mg was the most appropriate dose, terminating 90% of tachycardias without inducing significant hypotension.593 Subsequently, several large studies with substantial extensions were undertaken with patient self-administrations outside the medical setting.594–596 In the most recent of these studies—RAPID, named for the fast-acting nature of intranasal etripamil—the 30-minute conversion rate was 64% (63/99) with etripamil, compared to 31% (26/85) with placebo (HR: 2.62; 95% CI: 1.66–4.15; P < 0.001). Adverse events were few, mostly limited to local nasal irritation and similar mild effects.
Etripamil has also been evaluated for the treatment of AF presenting with a rapid ventricular rate >110 b.p.m. In the ReVeRa study, where the drug was given by medical staff to patients presenting at the emergency department with fast heart rates, the ventricular rate fell on average by 30 b.p.m. (placebo adjusted). Overall, the effect lasted as long as 150 min and was associated with patient satisfaction and relief of symptoms.597 This suggests that etripamil may be useful as a therapy to provide symptomatic relief quickly, allowing transport to medical facilities for cardioversion or giving time for other patient self-administered therapies, such as PITP with oral AADs or AV nodal blockade, to be effective.
7.1.2. Inhaled flecainide
Flecainide is an effective anti-arrhythmic agent used orally to prevent recurrences of AF or to terminate the arrhythmia (PITP). Atrial fibrillation termination following oral flecainide administration takes between 2 and 4 h on average, whereas with the highest dose of inhaled flecainide, 48% of AF episodes were terminated with a median time to conversion of 8 min. A small number of patients had post-conversion pauses, bradycardia, or AFL with 1:1 AV conduction.598 The conversion rate of recent-onset AF produced by orally inhaled flecainide acetate was 42.6% with a median time to conversion of 14.6 min in some studies.599 A recent randomized clinical trial (RESTORE-1)600 demonstrated that inhaled flecainide was significantly more effective than placebo in converting AF to SR in 90 minutes (30.8 vs. 0.0%; P = 0.04). The median time to conversion was 12.8 min. Safety data revealed no serious adverse events. Further studies are needed to optimize drug formulation and inhalation delivery to achieve higher plasma concentrations and improved AF conversion rates while maintaining a favourable safety profile.
7.1.3. Small-conductance calcium-activated potassium channel inhibitors
AP30663 is a small-conductance Ca2+-activated K+ channel (KCa2) inhibitor with mild off-target inhibition of IKr. KCa2 channels are up-regulated in patients with AF and show increased Ca2+ sensitivity, which increases their open probability. AP30663 decreases the Ca2+ sensitivity of KCa2 channels and markedly prolongs atrial refractoriness with only a mild increase of the QT interval. It seems unlikely to provoke VA since even in severely hypokalaemic guinea pig hearts, unlike dofetilide, it failed to induce VA.601 In experimental models, it terminates vernakalant-resistant pacing-induced AF.602 However, in healthy human volunteers, placebo corrected dose-dependent QTc interval prolongation of up to 18 ms was found.603 A Phase 2 trial of AP30663 involved 63 patients with an episode of ongoing AF who were randomized to AP30663 or placebo. Conversion to SR at 90 min occurred much more frequently in those treated with the active agent rather than placebo.604 There were no adverse safety signals except for QT interval prolongation. For this reason, plans are underway to commence a placebo-controlled randomized study with a second-generation molecule with greater specificity for the KCa2 channel and no off-target IKr inhibition.
7.1.4. Sulcardine (HBI-3000)
This multiple ion channel blocker (INa,P, INa,L, ICa,L, and IKr), like ranolazine, is being developed for the treatment of ventricular tachyarrhythmias and AF. Sulcardine induces dose-dependent increases in all cardiac ECG intervals except the J-point to T-wave peak interval,which it shortens.605 The drug suppresses dofetilide-induced EADs and is not expected to cause TdP.606 Sulcardine is being evaluated in a dose-finding study for the termination of AF by i.v. infusion.
7.1.5. Doxapram
Doxapram is a TWIK-related acid-sensitive K+ channel 1 (TASK1) inhibitor, which is already approved as a ventilatory stimulant. TWIK-related acid-sensitive K+ channel 1 expression is increased in AF and contributes to the shortening of the atrial AP.607 TWIK-related acid-sensitive K+ channel 1 inhibitors increase the atrial refractory period and reduce AF burden in experimental animal models.608 Since TASK-1 channels are found in atrial but not in ventricular tissue, ventricular pro-arrhythmia is not expected. The DOCTOS (Doxapram Conversion TO Sinus rhythm) trial is currently underway, testing the value of i.v. doxapram for cardioversion of AF.609
7.1.6. Bucindolol
Bucindolol is a non-specific β-blocker, which also inhibits α1-adrenoceptors, potentially causing vasodilation. In a sub-study of the BEST trial, new-onset AF was reduced by 75% in patients with the β1 389 arginine homozygotes (Arg/Arg), but there was no reduction in patients with the β1 389 Arg/Gly genotype, who constituted ∼50% of the substudy population.610
In the GENETIC AF (Genotype-Directed Comparative Effectiveness Trial of Bucindolol and Toprol-XL for the Prevention of Symptomatic AF/AFL in Patients With Heart Failure) trial, HFrEF patients with the ADR β1 Arg/Arg genotype were randomized to bucindolol or metoprolol. Bucindolol did not increase the time to recurrence of AF/AFL or all-cause mortality, but trends were seen in subgroups with AF and HF diagnoses occurring <12 years previously and AF onset that did not precede HF by >2 years.611 In a subgroup of patients implanted with a loop recorder, AF burden was significantly reduced by 33% in those assigned to bucindolol, as were AF interventions, plasma levels of noradrenaline, and N-terminal pro B-type natriuretic peptide.612 Bradycardia occurred less often in bucindolol than in metoprolol-treated patients.613
7.1.7. Budiodarone
Budiodarone is a multiple ion channel blocker, similar to amiodarone but with a short elimination half-life. It was shown to be effective at reducing AF in patients with implanted PM who suffered from paroxysmal AF (PASCAL trial).614 Recently, further interest has been shown in this molecule, and more clinical studies are planned.
7.1.8. Histone deacetylase 6 inhibitors
PKN605 is a potent and selective HDAC6 inhibitor being developed as an oral therapy for AF. In AF, cardiomyocyte refractoriness, measured by APD at 90% repolarization (APD90), is shortened, promoting re-entry circuits and facilitating AF initiation and maintenance. Histone deacetylase 6 inhibition is expected to normalize APD90, reducing re-entry substrates and restoring SR. As a cytosolic enzyme, HDAC6 regulates protein acetylation, affecting key cellular functions such as microtubule stability, intra-cellular transport, and protein degradation. One of its primary targets, α-tubulin, plays a crucial role in maintaining stable microtubules. Studies have shown that tubastatin A, a selective HDAC6 inhibitor, increased acetylated α-tubulin levels in atrial cardiomyocytes and reduced AF in a beagle dog model. Pre-clinical studies of PKN605 demonstrated its ability to restore shortened APD90 in rabbit and human atrial tissue and reduce AF duration in a canine model.615 In healthy volunteers, PKN605 was well tolerated and led to increased circulating acetylated α-tubulin, confirming its pharmacodynamic activity. These findings suggest that PKN605 has strong potential as an anti-arrhythmic agent, offering a novel mechanism-based approach for maintaining SR in AF patients by targeting electrophysiological and structural remodelling processes.
8. Areas of uncertainty and gaps of knowledge
After the unanticipated adverse results of the CAST, SWORD, and ANDROMEDA, physicians became more reliant on interventional therapies such as catheter ablation and ICDs, and pharmaceutical companies lost interest in AAD development. Paradoxically, AAD use increased, but the hybrid value of an intervention plus AAD therapy has not been sufficiently researched.
There has almost been no recent development in AAD therapy for VA. This is urgently needed because interventional therapies, such ablation, have limitations, and device-based treatments like anti-tachycardia pacing or cardioverter shock, are uncomfortable and often highly symptomatic. This latter therapy repeatedly reminds the patients of their dependence on the treatment and the fragility of their health.
Anti-arrhythmic drug targets have been single or multiple transmembrane ion channels and/or autonomic nervous system receptors. Novel drug targets are emerging (inflammatory, anti-fibrotic, electrophysiological, and genetic), but progress has been slow in evaluating the anti-arrhythmic effect of modulating these targets.
Many arrhythmia mechanisms have a genetic element. Genotyping is often employed clinically in monogenetic diseases and may facilitate effective anti-arrhythmic therapy. However, this is far from being well developed for polygenic diseases. Such advances are needed.
There are many theoretical ‘drug-based’ genetic approaches, such as gene delivery, micro ribonucleic acid regulation, modulation of non-coding RNA, etc., that are increasingly well understood at a pre-clinical level and meanwhile applied to small patient populations with genetic forms of diseases.
Precision medical approaches to match appropriate AAD therapy to the underlying cause have not been well developed at an experimental or clinical level. The development of machine learning and augmented intelligence may support progress in this area.
In the coming years, precision medicine will become an indispensable component of clinical practice, and this will extend to AAD therapy. For example, combining clinical, genetic, imaging, and ECG analysis with the cellular response of tissue derived from the patient to specific therapies may allow the most effective and safe therapy to be prescribed. Research into this so-called tailored therapy is ongoing.
Most arrhythmias occur because of substrates and/or triggers resulting from ‘underlying comorbidities. Effective treatment of the comorbidities may prevent or reduce the likelihood of arrhythmias. This has often been studied, but accurate phenotyping of the underlying condition and adequate documentation of any resulting arrhythmia have usually been poor.
When an arrhythmia first presents in the clinical domain, the pathophysiological substrate that supports the arrhythmia is usually already well developed. However, the use of lay ECG devices and artificial intelligence analysis of ECGs during SR, genetic profiles, biomarkers, etc., are now available such that potential arrhythmias may be detected at a much earlier stage. Patients with atrial cardiomyopathy and early-onset arrhythmias may respond differently and better to AAD therapy. This is a new opportunity to manage arrhythmia effectively.
Anti-arrhythmic drug choices are often made based on the underlying cardiac pathology. These choices have often been based on safety rather than efficacy. Concerns stemming from the CAST study have been widely extrapolated to restrict the use of Class Ic drugs when any form of SHD is present. Other drugs are advised against based on purely theoretical considerations, for example, Class III drugs for patients with LVH. These restrictions have limited research on AAD therapy for many forms of heart disease, such as valvular heart disease and HFpEF. New research has to address these underdeveloped areas.
Some arrhythmias are clinically ‘silent’ and may not have serious clinical consequences until many years after their onset. Conventional clinical trials, lasting only several months or years, do not capture late adverse outcomes. Alternative designs, for example, based on clinical registries, are needed to properly address this unmet need.
Anti-arrhythmic drug therapy is advised to be compared with alternative interventional strategies. The EP community has not found these trials easy to perform because of the reluctance of patients to be randomized away from ‘popular’ interventional therapy or concern about possible hazards related to an interventional approach. For similar reasons, crossovers between assigned groups are also frequent. Physician bias towards a particular therapy may also play a negative role. One solution is to perform such comparisons earlier in the life cycle of the interventional therapy, but this is countered by the ongoing improvement of the intervention and the learning curve required to implement such therapy. A needed solution is to improve the trial discipline to ensure this information is more easily and quickly acquired.
Few studies have evaluated AAD combinations, though some, like amiodarone or dronedarone with ranolazine, show promise. Further research is needed on other potential combinations.
A poly-modular approach may be needed for effective management of arrhythmia. Examples include autonomic modulation, AAD therapy, and ablation. Systematic studies are needed to ascertain the value of combinations of these therapies.
Initially, conversion of cardiac arrhythmias and suppression or delay of arrhythmia recurrence were accepted as suitable clinical and regulatory outcomes by which to evaluate AAD therapy. Regulatory agencies now consider these outcomes to be merely ‘surrogate’ endpoints, favouring more clinically significant measures such as mortality, stroke, myocardial infarction, hospitalizations, and quality of life impairments. Adequate assessment of these outcomes requires large and generally expensive clinical trials based on appropriate clinical models; in many instances, they are not yet developed and difficult to fund.
Guidelines provide recommendations on anti-arrhythmic therapy, which have changed little over the past decades. Nevertheless, AAD therapy that is prescribed is often not adherent to guideline recommendations. Future AAD use is likely to become more complicated, and major educational efforts or the implementation of automated prescription aids will be essential.
Anti-arrhythmic drugs have been classified for many years by various iterations of a scheme introduced by VW. The alternative classification known as the Sicilian Gambit was too complicated and was never used. As we begin to practice in the era of precision medicine, many elements of the Sicilian Gambit will be valuable for prescribing better and safer therapy. The use of the traditional VW classification will decline because of its simplicity and relative imprecision.
Surprisingly, despite their proven efficacy, not all AADs are available in every country, even within the European Union, due to national and international regulatory constraints and corporate marketing strategies. For example, vernakalant, cibenzoline, and dofetilide have EMA approval, but the pharmaceutical companies choose not to supply the drugs to all countries. Moricizine/ethmozine was widely approved, but the drug company chose to discontinue its production. Ibutilide is a nationally approved therapy and is not widely used. Some drugs, such as antazoline and ranolazine, are not generally approved as anti-arrhythmic agents but have been re-purposed as anti-arrhythmics. Some drugs, such as bepridil, pilsicainide, and cibenzoline, are infrequently used but do have approval in some areas. Low-sale volumes led to the withdrawal of some drugs such as quinidine, which is valuable for BrS in adult and paediatric patients, and mexiletine, which can be used for the management of LQTS3, but professional complaints led to limited supplies being made available. This is a rather chaotic situation, adding to the complexity of choosing and gaining access to anti-arrhythmic therapy, which needs resolution.
9. Conclusions
In the face of waning attention on AADs due to the emergence of alternative therapies, the persistently high prevalence of cardiac arrhythmias, the synergistic benefits of AADs alongside other treatments, and their indispensability in addressing acute episodes underscore their continued importance. In this regard, AADs still fulfil an ABC approach serving as Appropriate therapy, Backup therapy, and Complementary therapy—in the management of cardiac arrhythmias.
This significance is further underscored by the ongoing development of new and promising AADs, despite the rigorous regulatory requirements that contribute to a protracted, intricate, and costly development process. These developments may, in turn, drive new or marginally used therapeutic approaches, such as individual self-administration for arrhythmia termination. Understanding how these drugs work is essential for proper selection, but potential hazardous interactions with other medications or patient conditions have to be taken into consideration.
This practical compendium offers a comprehensive review of the knowledge necessary for prescribing these agents—tools that are not only useful and potent but also carry the potential for severe adverse effects. Navigating this balance is paramount for healthcare professionals aiming to optimize the management of cardiac arrhythmias.
10. Tables of advice
Table of Advice 1.
Definitions of supporting and strength of evidence
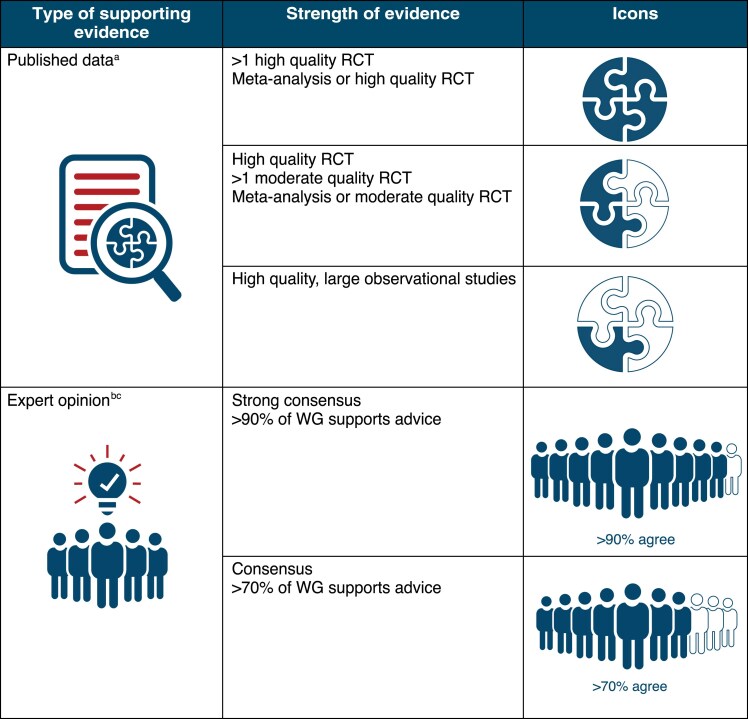
|
aThe reference and trials for the published data that fulfil the criteria are indicated in the table of advice, if applicable.
bExpert opinion also considers: Randomized, non-randomized, observational, or registry studies with limitations of design or execution, case series, meta-analyses of such studies, physiological, or mechanistic studies in human subjects.
cFor areas of uncertainty, strong consensus/consensus that the topic is relevant and important to be addressed by future trials.
Table of Advice 2.
Main advice on AAD treatment
| AAD SELECTION | Strength | Trials and references |
|---|---|---|
| Advice TO DO | ||
| For rhythm control of AFL, selective flutter ablation is generally preferred, particularly for cavotricuspid isthmus–dependent flutter, but if not possible or contraindicated, amiodarone or dronedarone has to be attempted |

|
ATHENA trial98 |
| Vernakalant is the AAD of choice for AF termination lasting less than 7 days, provided the patient has no NYHA Class III/IV heart failure or other contraindications |
|
ACT and AVRO trials157,616–619 |
| Type Ic are the AADs of choice for AF termination lasting more than 7 days, provided the patient has no SHD, heart failure, or other contraindications |
|
620 |
| May be appropriate TO DO | ||
| Flecainide could be used as the first-line treatment, while β-blockers are advised to be avoided in AT with a vagal pattern |

|
|
| Disopyramide may be particularly effective for vagally mediated AF and could be used as an alternative when other treatments fail or are contraindicated |
|
|
| Flecainide or propafenone is not contraindicated in patients with a high cardiovascular risk profile (e.g. accidental Agatston score <400) in the absence of angina pectoris or with uncomplicated mild left ventricular hypertrophy (both in the absence of left ventricular scar tissue) |
|
|
| Dronedarone can provide significant benefits beyond rhythm control, including pleiotropic effects such as mitigating acute coronary syndrome and reducing stroke risk |
|
|
| Flecainide, propafenone, or ranolazine can be used for AF termination with a PITP strategy in patients without underlying SND or other contraindications to AADs, provided there is prior demonstration of tolerance to the AAD or that the initial PITP usage is conducted under observation to verify effectiveness and ensure no adverse effects |

|
621 |
| Sotalol is an alternative when β-blockers fail for controlling PVCs in patients with SHD, while ranolazine serves as another viable option specifically for patients with ischaemic heart disease |
|
|
| Advice NOT TO DO | ||
| Dronedarone is advised to be avoided in patients taking dabigatran |
|
|
| Ivabradine, ranolazine, sotalol, and dofetilide are advised to be avoided in patients with severe renal impairment |
|
|
| Ivabradine, ranolazine, dronedarone, and carvedilol are advised to be avoided in patients with severe hepatic impairment |
|
|
| Amiodarone and dronedarone are advised to be avoided during the pregnancy to avoid foetal harm |
|
|
| AAD INITIATION AND FOLLOW-UP | ||
| Advice TO DO | ||
| Patient education and counselling during the initiation and follow-up of AAD therapy is advised to cover the goals of treatment, recognition of symptoms and signs of potential adverse effects, and awareness of possible drug interactions |
|
|
| When initiating an AAD, it is crucial to optimize the management of concomitant diseases and assess baseline parameters, including ECG, echocardiography, haematology, renal and hepatic function, and electrolyte status |
|
|
| When initiating amiodarone therapy, it is essential to assess baseline thyroid, pulmonary, and visual function, in addition to the standard tests required for AADs |
|
|
| All i.v. AADs are advised to be monitored with continuous ECG |
|
|
| Patients prescribed Class Ia AADs or the Class III agents of dofetilide and sotalol, as well as those at high risk for pro-arrhythmia, are advised to be closely monitored in the hospital during the initiation of therapy |
|
|
| After initiating an oral Class IA, sotalol, or dofetilide AAD, it is advised to perform an ECG within 2 days of initiation to evaluate its effects on heart rhythm and the RR, PR, QRS, and QTc intervals |
|
87,622 |
| After initiating an oral AAD, excluding Class Ia agents and the Class III agents of dofetilide and, in certain cases, sotalol, it is advised to perform an ECG shortly after initiation or dose adjustments (e.g. within 7 days) or at steady state (e.g. 1–3 months for amiodarone) to evaluate its effects on heart rhythm and the RR, PR, QRS, and QTc intervals |
|
|
| Adequate and regular follow-up, typically every 6–12 months, is advised to be scheduled in patients taking AADs to assess adherence to AAD therapy, to monitor for potential risk factors for pro-arrhythmia, and to evaluate ECG, haematology, renal and hepatic function, and electrolyte status |
|
|
| Adequate and regular follow-up is advised to be scheduled to evaluate thyroid (6 months), hepatic (12 months), pulmonary (12 months), and visual function (12 months), in patients taking amiodarone addition to the standard tests required for AADs |
|
|
| May be appropriate TO DO | ||
| Integrated nurse-driven care with experienced nurses supervised by the physician may substantially improve AAD management |
|
|
| An exercise test may be performed to rule out exercise-induced excessive QRS widening or VT in selected patients on Class Ic drugs |
|
|
| AADs, excluding Class Ia agents and the Class III agents of dofetilide and sotalol, may generally be initiated in the outpatient setting with appropriate ECG monitoring, unless the patient has not previously been documented in SR, in which case underlying or associated sick sinus syndrome cannot be ruled out |
|
352 |
| Class III sotalol may be initiated out-hospital unless specific conditions are present and if titration is slow with frequent ECG checks looking for QTc prolongation (≥500 ms) or HR (≤50 b.p.m.) depression |
|
623 |
| Patients with an ICD may be protected from the pro-arrhythmic effects of AADs, allowing for the initiation of AAD therapy in an outpatient setting |
|
|
| PRO-ARRHYTHMIA/TOXICITY | ||
| Advice TO DO | ||
| To enhance the safety of AAD use, patients are advised to be educated about warning symptoms and critical circumstances related to their treatment |
|
|
| AADs such as acebutolol, atenolol, nadolol, flecainide, quinidine, and digoxin are advised to be carefully monitored in patients with severe renal impairment to avoid toxicity and dose reductions may be appropriate |
|
|
| AADs such as metoprolol, propranolol, CCBs, propafenone, amiodarone, and lidocaine are advised to be carefully monitored in patients with severe hepatic impairment to avoid toxicity, and dose reductions may be appropriate |
|
|
| When pro-arrhythmia occurs, potential triggers such as ischaemia, heart failure, electrolyte disturbances, thyroid dysfunction, infection, drug interactions, and high or low plasma AAD concentrations are advised to be evaluated |
|
|
| AADs with significant effect on the SN are advised to be avoided when sinus node dysfunction is suspected |
|
|
| May be appropriate TO DO | ||
| Patients on Class Ic drugs, despite add-on β-blocker or verapamil/diltiazem, may avoid exercise during breakthrough episodes until AF has resolved or cardioversion has been performed |
|
624 |
| Ventricular use-dependent effects may be monitored during the infusion of Class Ic for tachycardia conversion or during exercise, while reverse use dependence is characteristic of Class III AADs, particularly after cardioversion |
|
|
| Regular rate control drugs may be discontinued shortly after initiating sotalol or amiodarone to prevent bradycardia in the event of conversion to SR |
|
|
| Greater caution is advised in women regarding use of sotalol, as women are at increased risk of developing TdP during administration of this drug |
|
625 |
| CNS side effects of Class Ic drugs may be tackled by changing to a slow-release preparation |
|
|
| AAD COMBINATION OR SWICTHING | ||
| Advice TO DO | ||
| Class Ic AADs are advised to be combined with β-blockers or CCBs to enhance efficacy and complement their effects on the AV node, providing a treatment option for resistant cases of atrial arrhythmias and SVT that do not respond to monotherapy or other therapies |
|
|
| May be appropriate TO DO | ||
| Serum drug concentrations and/or ECG markers are advised to be used to decide the washout process and initiation of a new AAD |
|
|
| AADs with a similar half-life may be switched by starting the new AAD at usual doses/dosing intervals when a dose of the prior AAD was due |
|
|
| The long-lasting washout of some AADs (e.g. amiodarone) is so slow that the new AAD may have dosing up-titrated over time |
|
|
| Ivabradine and β-blockers may be combined to manage resistant cases of inappropriate sinus tachycardia that do not respond to monotherapy or other treatments |
|
|
| Quinidine may be combined with Class IA, IB, or IV AADs to increase efficacy and tolerance to manage resistant cases of atrial and VA that do not respond to monotherapy or other treatments |
|
|
| Flecainide and mexiletine may be combined to enhance efficacy, providing a treatment option for resistant cases of VA that do not respond to monotherapy or other therapies |
|
|
| Combining sotalol with flecainide to achieve an amiodarone-like ‘Class Ic plus Class III effect’ can be a rational approach in refractory cases with MAT or VA and right ventricular arrhythmogenic cardiomyopathy, provided there are no other significant SHDs |
|
626 |
| Amiodarone may be combined with Class I AADs or β-blockers to enhance efficacy and complement their effects, providing a treatment option for resistant cases of atrial or VA that do not respond to monotherapy or other therapies |
|
|
| Dronedarone and ranolazine may be combined to enhance efficacy and minimize side effects in patients with AF that do not respond to monotherapy or other therapies |
|
HARMONY trial76 |
| Beta-blockers may be combined with AADs other than sotalol to enhance their efficacy |
|
|
| CCB and digoxin may be combined to complement their effects on the AV node and achieve rate control in patients with arial arrhythmias that do not respond to monotherapy or other therapies |
|
|
| Advice NOT TO DO | ||
| A combination of Class Ia and III AADs is advised to be avoided at their conventional doses due to the increased TdP risk |
|
|
| A combination of dofetilide and CCBs is advised to be avoided due to the increased risk of TdP |
|
|
| CCBs are advised to not be combined at their usual dose with β-blockers or disopyramide due to potential depression of the SN, AV conduction, and cardiac contractility |
|
|
| Ranolazine is advised to not be combined with other Class I AADs due to the potential for central nervous system effects, constipation, or an increased risk of TdP |
|
|
| A combination of dronedarone and digoxin is advised to be avoided due to the increased risk of digitalis toxicity and death |
|
PALLAS trial104 |
| AREAS OF UNCERTAINTY | ||
| The extent to which the findings of the CAST study is advised to be applied to restrict the use of Class Ic drugs in patients with mild or non-ischaemic SHD remains uncertain, posing a challenge in clinical decision-making |
|
|
| The advice against using Class III drugs such as sotalol and dofetilide in patients with LVH is based on theoretical considerations rather than robust clinical evidence, leaving some uncertainty in decision-making for this population |
|
|
| The clinical significance of drug-induced QT prolongation in the absence of TdP remains unclear, complicating decisions about whether to discontinue or adjust therapy |
|
|
| The safety and efficacy of amiodarone for long-term rhythm control in younger patients are advised to be weighed against its potential for cumulative toxicity, with no clear consensus on the best alternative |
|
|
| While dronedarone is contraindicated in patients with HFrEF, its safety in those with mildly reduced LVEF remains uncertain due to limited data |
|
|
| The effectiveness of upstream therapies, such as renin-angiotensin system inhibitors, in reducing the need for AADs in AF management remains an area of ongoing investigation |
|
|
Abbreviations: AAD, anti-arrhythmic drug; AF, atrial fibrillation; AFL, atrial flutter; AT, atrial tachycardia; AV, atrioventricular; CCB, calcium channel blocker; HF, heart failure; HFpEF/HFmrEF/HFrEF, HF with preserved/mildly reduced/reduced left ventricle ejection fraction; ICD, implantable cardioverter defibrillator; LV, left ventricle; LVEF, LV ejection fraction; LVH, LV hypertrophy; NYHA, New York Heart Association functional class; PITP, pill-in-the-pocket; SHD, structural heart disease; SN, sinus node; SND, SN dysfunction; SNRT, sinus node re-entrant tachycardia; SVT, supraventricular tachycardia; VT, ventricular tachycardia.
Supplementary Material
Contributor Information
Jose L Merino, Arrhythmia and Robotic Electrophysiology Unit, Cardiology Department, La Paz University Hospital, IdiPaz, Universidad Autonóma, P. Castellana, 261, Madrid 28046, Spain; University Hospital Quiron Ruber Juan Bravo, Madrid, Spain; University Hospital Viamed Santa Elena, Madrid, Spain.
Juan Tamargo, Department of Pharmacology and Toxicology, School of Medicine, Universidad Complutense, Madrid, Spain.
Carina Blomström-Lundqvist, School of Medical Sciences, Faculty of Medicine and Health, Örebro University, Örebro, Sweden; Department of Medical Science, Uppsala University, Uppsala, Sweden.
Giuseppe Boriani, Cardiology Division, Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Policlinico di Modena, Modena, Italy.
Harry J G M Crijns, Department of Cardiology, Cardiovascular Research Institute Maastricht (CARIM), Maastricht University, Maastricht, The Netherlands.
Dobromir Dobrev, Institute of Pharmacology, University Duisburg-Essen, Essen, Germany; Montréal Heart Institute, Université de Montréal, Montréal, Québec, Canada; Department of Integrative Physiology, Baylor College of Medicine, Houston, TX, USA.
Andreas Goette, Department of Cardiology and Intensive Care Medicine, St. Vincenz-Hospital, Paderborn, Germany; MAESTRIA Consortium at AFNET, Münster, Germany; Medical Faculty, Otto-von-Guericke University, Magdeburg, Germany.
Stefan H Hohnloser, Department of Cardiology Division of Clinical Electrophysiology J. W. Goethe University, Theodor-Stern-Kai 7, Frankfurt D 60590, Germany.
Gerald V Naccarelli, Heart and Vascular Institute, Penn State University College of Medicine, Hershey, PA, USA.
James A Reiffel, Division of Cardiology, Department of Medicine, Columbia University, New York, NY, USA.
Jacob Tfelt-Hansen, Department of Cardiology, The Heart Centre, Copenhagen University Hospital, Rigshospitalet, Blegdamsvej 9, 2100, Copenhagen, Denmark.
Marcel Martínez-Cossiani, Arrhythmia and Robotic Electrophysiology Unit, Cardiology Department, La Paz University Hospital, Idipaz, Madrid, Spain.
A John Camm, Clinical Cardiology, City St. George’s, University of London, Cranmer Terrace, London SW17 0RE, UK.
Jesus M Almendral Garrote, Arrhythmia Unit, Cardiology Department (CIEC), Hospital HM Monteprincipe, Madrid, Spain.
Beata Średniawa, Department of Cardiology and Electrotherapy Medical University of Silesia, DMS in Zabrze, Katowice, Poland; Department of Cardiology, Silesian Center of Heart Diseases, Zabrze, Poland.
Piotr Kułakowski, Department of Cardiology, Medical Centre of Postgraduate Medical Education, Grochowski Hospital, Warsaw, Poland.
Irina Savelieva, Cardiovascular Clinical Academic Group, City St. George’s University of London, London, UK.
Tatjana Potpara, Medical Faculty, University of Belgrade, Belgrade, Serbia; Cardiology Clinic, University Clinical Centre of Serbia, Belgrade, Serbia.
Bulent Gorenek, Cardiology Department, Eskisehir Osmangazi University, Eskisehir, Turkey.
Jose L Zamorano, Cardiology Department, University Hospital Ramon y Cajal, CiberCV, Madrid, Spain.
Supplementary material
Supplementary material is available at Europace online.
Funding
No funding was received for the preparation of this document.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
References
- 1. Saksena S, Ken-Opurum J, McKindley DS, Preblick R, Rashkin J, Aldaas OM et al. Arrhythmia recurrence and rhythm control strategies after catheter ablation of newly diagnosed atrial fibrillation (ARRC-AF study). JACC Clin Electrophysiol 2025:S2405-500X(24)01027-2. [Google Scholar]
- 2. Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 2014;114:1483–99. [DOI] [PubMed] [Google Scholar]
- 3. Landstrom AP, Dobrev D, Wehrens XHT. Calcium signaling and cardiac arrhythmias. Circ Res 2017;120:1969–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nattel S, Heijman J, Zhou L, Dobrev D. Molecular basis of atrial fibrillation pathophysiology and therapy: a translational perspective. Circ Res 2020;127:51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heijman J, Ghezelbash S, Dobrev D. Investigational antiarrhythmic agents: promising drugs in early clinical development. Expert Opin Investig Drugs 2017;26:897–907. [Google Scholar]
- 6. Grandi E, Ripplinger CM. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol Res 2019;146:104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nattel S. The molecular and ionic specificity of antiarrhythmic drug actions. J Cardiovasc Electrophysiol 1999;10:272–82. [DOI] [PubMed] [Google Scholar]
- 8. Dorian P, Newman D. Rate dependence of the effect of antiarrhythmic drugs delaying cardiac repolarization: an overview. Europace 2000;2:277–85. [DOI] [PubMed] [Google Scholar]
- 9. Hamer AW, Arkles LB, Johns JA. Beneficial effects of low dose amiodarone in patients with congestive cardiac failure: a placebo-controlled trial. J Am Coll Cardiol 1989;14:1768–74. [DOI] [PubMed] [Google Scholar]
- 10. Zhu W, Mazzanti A, Voelker TL, Hou P, Moreno JD, Angsutararux P et al. Predicting patient response to the antiarrhythmic mexiletine based on genetic variation. Circ Res 2019;124:539–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lemoine MD, Fabritz L. Improving antiarrhythmic therapy for patients with atrial fibrillation using common genetic variants. Heart 2025;111:145–6. [DOI] [PubMed] [Google Scholar]
- 12. Vaughan Williams EM. Classification of antidysrhythmic drugs. Pharmacol Ther B 1975;1:115–38. [DOI] [PubMed] [Google Scholar]
- 13. Karagueuzian HS, Pezhouman A, Angelini M, Olcese R. Enhanced late Na and Ca currents as effective antiarrhythmic drug targets. Front Pharmacol 2017;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nattel S. Antiarrhythmic drug classifications. A critical appraisal of their history, present status, and clinical relevance. Drugs 1991;41:672–701. [DOI] [PubMed] [Google Scholar]
- 15. Nattel S, Talajic M. Recent advances in understanding the pharmacology of amiodarone. Drugs 1988;36:121–31. [DOI] [PubMed] [Google Scholar]
- 16. Heijman J, Heusch G, Dobrev D. Pleiotropic effects of antiarrhythmic agents: dronedarone in the treatment of atrial fibrillation. Clin Med Insights Cardiol 2013;7:127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tzivoni D, Banai S, Schuger C, Benhorin J, Keren A, Gottlieb S et al. Treatment of torsade de pointes with magnesium sulfate. Circulation 1988;77:392–7. [DOI] [PubMed] [Google Scholar]
- 18. Chakraborty P, Rose RA, Nair K, Downar E, Nanthakumar K. The rationale for repurposing funny current inhibition for management of ventricular arrhythmia. Heart Rhythm 2021;18:130–7. [DOI] [PubMed] [Google Scholar]
- 19. Thomas Bigger J Jr, Breithardt G, Brown AM, John Camm A, Carmeliet E, Fozzard HA et al. The Sicilian gambit. A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Task force of the working group on arrhythmias of the European Society of Cardiology. Circulation 1991;84:1831–51. [DOI] [PubMed] [Google Scholar]
- 20. Rosen MR. Consequences of the Sicilian gambit. Eur Heart J 1995;16 Suppl G:32–6. [Google Scholar]
- 21. Lei M, Wu L, Terrar DA, Huang CL-H. Modernized classification of cardiac antiarrhythmic drugs. Circulation 2018;138:1879–96. [DOI] [PubMed] [Google Scholar]
- 22. Fontenla A, Tamargo J, Salgado R, López-Gil M, Mejía E, Matía R et al. Ivabradine for controlling heart rate in permanent atrial fibrillation: a translational clinical trial. Heart Rhythm 2023;20:822–30. [DOI] [PubMed] [Google Scholar]
- 23. Di Marco GM, De Nigris A, Pepe A, Pagano A, Di Nardo G, Tipo V. Ivabradine-flecainide as breakthrough drug combination for congenital junctional ectopic tachycardia: a case report and literature review. Pediatr Rep 2021;13:624–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vitali Serdoz L, Rittger H, Furlanello F, Bastian D. Quinidine-A legacy within the modern era of antiarrhythmic therapy. Pharmacol Res 2019;144:257–63. [DOI] [PubMed] [Google Scholar]
- 25. Crumb WJ Jr, Vicente J, Johannesen L, Strauss DG. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J Pharmacol Toxicol Methods 2016;81:251–62. [DOI] [PubMed] [Google Scholar]
- 26. Sutanto H, Laudy L, Clerx M, Dobrev D, Crijns HJGM, Heijman J. Maastricht Antiarrhythmic Drug Evaluator (MANTA): a computational tool for better understanding of antiarrhythmic drugs. Pharmacol Res 2019;148:104444. [DOI] [PubMed] [Google Scholar]
- 27. Leahey EB Jr, Reiffel JA, Drusin RE, Heissenbuttel RH, Lovejoy WP, Bigger JT Jr. Interaction between quinidine and digoxin. JAMA 1978;240:533–4. [PubMed] [Google Scholar]
- 28. Mazzanti A, Tenuta E, Marino M, Pagan E, Morini M, Memmi M et al. Efficacy and limitations of quinidine in patients with Brugada syndrome. Circ Arrhythm Electrophysiol 2019;12:e007143. [Google Scholar]
- 29. Belhassen B, Glick A, Viskin S. Excellent long-term reproducibility of the electrophysiologic efficacy of quinidine in patients with idiopathic ventricular fibrillation and Brugada syndrome. Pacing Clin Electrophysiol 2009;32:294–301. [DOI] [PubMed] [Google Scholar]
- 30. Viskin S, Wilde AAM, Guevara-Valdivia ME, Daoulah A, Krahn AD, Zipes DP et al. Quinidine, a life-saving medication for Brugada syndrome, is inaccessible in many countries. J Am Coll Cardiol 2013;61:2383–7. [DOI] [PubMed] [Google Scholar]
- 31. Selzer A, Wray HW. Quinidine syncope. Paroxysmal ventricular fibrillation occurring during treatment of chronic atrial arrhythmias. Circulation 1964;30:17–26.. [DOI] [PubMed] [Google Scholar]
- 32. Lafuente-Lafuente C, Valembois L, Bergmann J-F, Belmin J. Antiarrhythmics for maintaining sinus rhythm after cardioversion of atrial fibrillation. Cochrane Database Syst Rev 2015:CD005049. [DOI] [PubMed] [Google Scholar]
- 33. Saikawa T, Nakagawa M, Takahashi N, Ishida S, Fujino T, Ito M et al. Mexiletine and disopyramide suppress ventricular premature contractions (VPC) irrespective of the relationship between the VPC and the underlying heart rate. Jpn Heart J 1992;33:665–78. [DOI] [PubMed] [Google Scholar]
- 34. Darbar D. Standard antiarrhythmic drugs. In: Zipes DP, Jalife J (eds.), Cardiac Electrophysiology: From Cell to Bedside. 6th ed. Philadelphia: W.B. Saunders; 2013. p1095–110. [Google Scholar]
- 35. Karlsson E. Clinical pharmacokinetics of procainamide. Clin Pharmacokinet 1978;3:97–107. [DOI] [PubMed] [Google Scholar]
- 36. Kowey PR. Pharmacological effects of antiarrhythmic drugs. Review and update. Arch Intern Med 1998;158:325–32. [DOI] [PubMed] [Google Scholar]
- 37. Könemann H, Dagres N, Merino JL, Sticherling C, Zeppenfeld K, Tfelt-Hansen J et al. Spotlight on the 2022 ESC guideline management of ventricular arrhythmias and prevention of sudden cardiac death: 10 novel key aspects. Europace 2023;25:euad091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nasilli G, Yiangou L, Palandri C, Cerbai E, Davis RP, Verkerk AO et al. Beneficial effects of chronic mexiletine treatment in a human model of SCN5A overlap syndrome. Europace 2023;25:euad154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung S-C, Lai A, Lip GYH, Lambiase PD, Providencia R. Impact of anti-arrhythmic drugs and catheter ablation on the survival of patients with atrial fibrillation: a population study based on 199 433 new-onset atrial fibrillation patients in the UK. Europace 2023;25:351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rillig A, Eckardt L, Borof K, Camm AJ, Crijns HJGM, Goette A et al. Safety and efficacy of long-term sodium channel blocker therapy for early rhythm control: the EAST-AFNET 4 trial. Europace 2024;26:euae121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guerra JM, Moreno Weidmann Z, Perrotta L, Sultan A, Anic A, Metzner A et al. Current management of atrial fibrillation in routine practice according to the last ESC guidelines: an EHRA physician survey-how are we dealing with controversial approaches? Europace 2024;26:euae012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rienstra M, Tzeis S, Bunting KV, Caso V, Crijns HJGM, De Potter TJR et al. Spotlight on the 2024 ESC/EACTS management of atrial fibrillation guidelines: 10 novel key aspects. Europace 2024;26:euae298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reiffel JA, Blomström-Lundqvist C, Boriani G, Goette A, Kowey PR, Merino JL et al. Real-world utilization of the pill-in-the-pocket method for terminating episodes of atrial fibrillation: data from the multinational Antiarrhythmic Interventions for Managing Atrial Fibrillation (AIM-AF) survey. Europace 2023;25:euad162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brugada J, Katritsis DG, Arbelo E, Arribas F, Bax JJ, Blomström-Lundqvist C et al. 2019 ESC guidelines for the management of patients with supraventricular tachycardia. The task force for the management of patients with supraventricular tachycardia of the European Society of Cardiology (ESC). Eur Heart J 2020;41:655–720. [DOI] [PubMed] [Google Scholar]
- 45. Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA et al. 2022 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2022;43:3997–4126. [DOI] [PubMed] [Google Scholar]
- 46. Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S et al. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol 2005;10:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chorin E, Hu D, Antzelevitch C, Hochstadt A, Belardinelli L, Zeltser D et al. Ranolazine for congenital long-QT syndrome type III: experimental and long-term clinical data. Circ Arrhythm Electrophysiol 2016;9:e004370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 2009;15:380–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2011;4:128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 2011;57:2244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kannankeril PJ, Moore JP, Cerrone M, Priori SG, Kertesz NJ, Ro PS et al. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol 2017;2:759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991;324:781–8. [DOI] [PubMed] [Google Scholar]
- 53. Siebels J, Cappato R, Rüppel R, Schneider MA, Kuck KH. Preliminary results of the Cardiac Arrest Study Hamburg (CASH). CASH Investigators. Am J Cardiol 1993;72:109F–13F. [Google Scholar]
- 54. Valentino MA, Panakos A, Ragupathi L, Williams J, Pavri BB. Flecainide toxicity: a case report and systematic review of its electrocardiographic patterns and management. Cardiovasc Toxicol 2017;17:260–6. [DOI] [PubMed] [Google Scholar]
- 55. Fish FA, Gillette PC, Benson DW Jr. Proarrhythmia, cardiac arrest and death in young patients receiving encainide and flecainide. The Pediatric Electrophysiology Group. J Am Coll Cardiol 1991;18:356–65. [DOI] [PubMed] [Google Scholar]
- 56. Moretti A, Polselli M, Carbone I, Pannarale G, Acconcia MC, Torromeo C et al. Takotsubo cardiomyopathy and flecainide toxicity: a case report and brief literature review. Eur Rev Med Pharmacol Sci 2021;25:4069–73. [DOI] [PubMed] [Google Scholar]
- 57. Harron DW, Brogden RN, Faulds D, Fitton A. Cibenzoline. A review of its pharmacological properties and therapeutic potential in arrhythmias. Drugs 1992;43:734–59. [DOI] [PubMed] [Google Scholar]
- 58. Kodama I, Ogawa S, Inoue H, Kasanuki H, Kato T, Mitamura H et al. Profiles of aprindine, cibenzoline, pilsicainide and pirmenol in the framework of the Sicilian Gambit. The Guideline Committee for Clinical Use of Antiarrhythmic Drugs in Japan (working group of arrhythmias of the Japanese Society of Electrocardiology). Jpn Circ J 1999;63:1–12. [DOI] [PubMed] [Google Scholar]
- 59. JCS Joint Working Group . Guidelines for pharmacotherapy of atrial fibrillation (JCS 2013). Circ J 2014;78:1997–2021. [DOI] [PubMed] [Google Scholar]
- 60. Shimada M, Yokozuka H, Inoue S, Koyama T, Kodama H, Suzuki Y et al. Pill-in-the-pocket approach for paroxysmal atrial fibrillation by cibenzoline succinate. Jpn J Electrocardiol 2006;26:710–9. [Google Scholar]
- 61. Atarashi H, Inoue H, Hiejima K, Hayakawa H. Conversion of recent-onset atrial fibrillation by a single oral dose of pilsicainide (Pilsicainide Suppression Trial on atrial fibrillation). The PSTAF Investigators. Am J Cardiol 1996;78:694–7. [DOI] [PubMed] [Google Scholar]
- 62. Kumagai K, Nakashima H, Tojo H, Yasuda T, Noguchi H, Matsumoto N et al. Pilsicainide for atrial fibrillation. Drugs 2006;66:2067–73. [DOI] [PubMed] [Google Scholar]
- 63. Plosker GL. Pilsicainide. Drugs 2010;70:455–67. [DOI] [PubMed] [Google Scholar]
- 64. Bińkowski BJ, Makowski M, Kubiński P, Lubiński A. Effect of antazoline on electrophysiological properties of atrial muscle and conduction system of the heart. Cardiovasc Drugs Ther 2018;32:169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Piotrowski R, Giebułtowicz J, Baran J, Sikorska A, Gralak-Łachowska D, Soszyńska M et al. Antazoline-insights into drug-induced electrocardiographic and hemodynamic effects: results of the ELEPHANT II substudy. Ann Noninvasive Electrocardiol 2017;22:e12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kline SR, Dreifus LS, Watanabe Y, McGarry TF, Likoff W. Evaluation of the antiarrhythmic properties of antazoline. A preliminary study. Am J Cardiol 1962;9:564–7. [DOI] [PubMed] [Google Scholar]
- 67. Wybraniec MT, Wróbel W, Wilkosz K, Wrona K, Bula K, Mizia-Stec K. Pharmacological cardioversion with antazoline in atrial fibrillation: the results of the CANT Study. J Am Heart Assoc 2018;7:e010153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Farkowski MM, Maciąg A, Żurawska M, Pytkowski M, Kowalik I, Woźniak J et al. Comparative effectiveness and safety of antazoline-based and propafenone-based strategies for pharmacological cardioversion of short-duration atrial fibrillation in the emergency department. Pol Arch Med Wewn 2016;126:381–7. [DOI] [PubMed] [Google Scholar]
- 69. Farkowski MM, Maciag A, Kowalik I, Konka M, Szwed H, Pytkowski M. Intravenous antazoline, a first-generation antihistaminic drug with antiarrhythmic properties, is a suitable agent for pharmacological cardioversion of atrial fibrillation induced during pulmonary vein isolation due to the lack of influence on atrio-venous conduction and high clinical effectiveness (AntaEP Study). Br J Clin Pharmacol 2019;85:1552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maciag A, Farkowski MM, Chwyczko T, Beckowski M, Syska P, Kowalik I et al. Efficacy and safety of antazoline in the rapid cardioversion of paroxysmal atrial fibrillation (the AnPAF Study). Europace 2017;19:1637–42. [DOI] [PubMed] [Google Scholar]
- 71. Balsam P, Koźluk E, Peller M, Piątkowska A, Lodziński P, Kiliszek M et al. Antazoline for termination of atrial fibrillation during the procedure of pulmonary veins isolation. Adv Med Sci 2015;60:231–5. [DOI] [PubMed] [Google Scholar]
- 72. Reynolds EW Jr, Baird WM, Clifford ME. A clinical trial of antazoline in the treatment of arrhythmias. Am J Cardiol 1964;14:513–21. [DOI] [PubMed] [Google Scholar]
- 73. Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 2011;8:1281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gong M, Zhang Z, Fragakis N, Korantzopoulos P, Letsas KP, Li G et al. Role of ranolazine in the prevention and treatment of atrial fibrillation: a meta-analysis of randomized clinical trials. Heart Rhythm 2017;14:3–11. [DOI] [PubMed] [Google Scholar]
- 75. Guerra F, Romandini A, Barbarossa A, Belardinelli L, Capucci A. Ranolazine for rhythm control in atrial fibrillation: a systematic review and meta-analysis. Int J Cardiol 2017;227:284–91. [DOI] [PubMed] [Google Scholar]
- 76. Reiffel JA, Camm AJ, Belardinelli L, Zeng D, Karwatowska-Prokopczuk E, Olmsted A et al. The HARMONY trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ Arrhythm Electrophysiol 2015;8:1048–56. [DOI] [PubMed] [Google Scholar]
- 77. Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation 2007;116:1647–52. [DOI] [PubMed] [Google Scholar]
- 78. Scirica BM, Belardinelli L, Chaitman BR, Waks JW, Volo S, Karwatowska-Prokopczuk E et al. Effect of ranolazine on atrial fibrillation in patients with non-ST elevation acute coronary syndromes: observations from the MERLIN-TIMI 36 trial. Europace 2015;17:32–7. [DOI] [PubMed] [Google Scholar]
- 79. Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol 2008;19:1289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bányász T, Horváth B, Virág L, Bárándi L, Szentandrássy N, Harmati G et al. Reverse rate dependency is an intrinsic property of canine cardiac preparations. Cardiovasc Res 2009;84:237–44. [DOI] [PubMed] [Google Scholar]
- 81. Heist EK, Ruskin JN. Drug-induced arrhythmia. Circulation 2010;122:1426–35. [DOI] [PubMed] [Google Scholar]
- 82. Mujović N, Dobrev D, Marinković M, Russo V, Potpara TS. The role of amiodarone in contemporary management of complex cardiac arrhythmias. Pharmacol Res 2020;151:104521. [DOI] [PubMed] [Google Scholar]
- 83. Singh BN. Amiodarone: a multifaceted antiarrhythmic drug. Curr Cardiol Rep 2006;8:349–55. [DOI] [PubMed] [Google Scholar]
- 84. Kułakowski P, Karczmarewicz S, Karpiński G, Soszyńska M, Ceremuzyński L. Effects of intravenous amiodarone on ventricular refractoriness, intraventricular conduction, and ventricular tachycardia induction. Europace 2000;2:207–15. [DOI] [PubMed] [Google Scholar]
- 85. Hamilton D Sr, Nandkeolyar S, Lan H, Desai P, Evans J, Hauschild C et al. Amiodarone: a comprehensive guide for clinicians. Am J Cardiovasc Drugs 2020;20:549–58. [DOI] [PubMed] [Google Scholar]
- 86. Julian DG, Camm AJ, Frangin G, Janse MJ, Munoz A, Schwartz PJ et al. Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. European Myocardial Infarct Amiodarone Trial Investigators. Lancet 1997;349:667–74. [DOI] [PubMed] [Google Scholar]
- 87. Zimetbaum P. Antiarrhythmic drug therapy for atrial fibrillation. Circulation 2012;125:381–9. [DOI] [PubMed] [Google Scholar]
- 88. Nademanee K, Kannan R, Hendrickson J, Ookhtens M, Kay I, Singh BN. Amiodarone-digoxin interaction: clinical significance, time course of development, potential pharmacokinetic mechanisms and therapeutic implications. J Am Coll Cardiol 1984;4:111–6. [DOI] [PubMed] [Google Scholar]
- 89. Árpádffy-Lovas T, Husti Z, Baczkó I, Varró A, Virág L. Different effects of amiodarone and dofetilide on the dispersion of repolarization between well-coupled ventricular and Purkinje fibers 1. Can J Physiol Pharmacol 2021;99:48–55. [DOI] [PubMed] [Google Scholar]
- 90. Kudenchuk PJ, Brown SP, Daya M, Rea T, Nichol G, Morrison LJ et al. Amiodarone, lidocaine, or placebo in out-of-hospital cardiac arrest. N Engl J Med 2016;374:1711–22. [DOI] [PubMed] [Google Scholar]
- 91. Connolly SJ, Dorian P, Roberts RS, Gent M, Bailin S, Fain ES et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: the OPTIC study: a randomized trial. JAMA 2006;295:165–71. [DOI] [PubMed] [Google Scholar]
- 92. Van Gelder IC, Rienstra M, Bunting KV, Casado-Arroyo R, Caso V, Crijns HJGM et al. 2024 ESC guidelines for the management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J 2024;45:3314–414. [DOI] [PubMed] [Google Scholar]
- 93. Dasí A, Nagel C, Pope MTB, Wijesurendra RS, Betts TR, Sachetto R et al. In silico trials guide optimal stratification of atrial fibrillation patients to catheter ablation and pharmacological medication: the i-STRATIFICATION study. Europace 2024;26:euae150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Markman TM, Geng Z, Epstein AE, Nazarian S, Deo R, Marchlinski FE et al. Trends in antiarrhythmic drug use among patients in the United States between 2004 and 2016. Circulation 2020;141:937–9. [DOI] [PubMed] [Google Scholar]
- 95. Field ME, Holmes DN, Page RL, Fonarow GC, Matsouaka RA, Turakhia MP et al. Guideline-concordant antiarrhythmic drug use in the get with the guidelines-atrial fibrillation registry. Circ Arrhythm Electrophysiol 2021;14:e008961. [DOI] [PubMed] [Google Scholar]
- 96. Chiang C-E, Goethals M, O’Neill JO, Naditch-Brûlé L, Brette S, Gamra H et al. Inappropriate use of antiarrhythmic drugs in paroxysmal and persistent atrial fibrillation in a large contemporary international survey: insights from RealiseAF. Europace 2013;15:1733–40.. [DOI] [PubMed] [Google Scholar]
- 97. Singh BN, Connolly SJ, Crijns HJGM, Roy D, Kowey PR, Capucci A et al. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007;357:987–99. [DOI] [PubMed] [Google Scholar]
- 98. Hohnloser SH, Crijns HJGM, van Eickels M, Gaudin C, Page RL, Torp-Pedersen C et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009;360:668–78. [DOI] [PubMed] [Google Scholar]
- 99. Blomström-Lundqvist C, Naccarelli GV, McKindley DS, Bigot G, Wieloch M, Hohnloser SH. Effect of dronedarone vs. placebo on atrial fibrillation progression: a post hoc analysis from ATHENA trial. Europace 2023;25:845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Curtis AB, Zeitler EP, Malik A, Bogard A, Bhattacharyya N, Stewart J et al. Efficacy and safety of dronedarone across age and sex subgroups: a post hoc analysis of the ATHENA study among patients with non-permanent atrial fibrillation/flutter. Europace 2022;24:1754–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vaduganathan M, Piccini JP, Camm AJ, Crijns HJGM, Anker SD, Butler J et al. Dronedarone for the treatment of atrial fibrillation with concomitant heart failure with preserved and mildly reduced ejection fraction: a post-hoc analysis of the ATHENA trial. Eur J Heart Fail 2022;24:1094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Khachatryan A, Merino JL, de Abajo FJ, Botto GL, Kirchhof P, Breithardt G et al. International cohort study on the effectiveness of dronedarone and other antiarrhythmic drugs for atrial fibrillation in real-world practice (EFFECT-AF). Europace 2022;24:899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Køber L, Torp-Pedersen C, McMurray JJV, Gøtzsche O, Lévy S, Crijns H et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008;358:2678–87. [DOI] [PubMed] [Google Scholar]
- 104. Connolly SJ, Camm AJ, Halperin JL, Joyner C, Alings M, Amerena J et al. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011;365:2268–76. [DOI] [PubMed] [Google Scholar]
- 105. Hohnloser SH, Halperin JL, Camm AJ, Gao P, Radzik D, Connolly SJ et al. Interaction between digoxin and dronedarone in the PALLAS trial. Circ Arrhythm Electrophysiol 2014;7:1019–25. [DOI] [PubMed] [Google Scholar]
- 106. Hohnloser SH, Meinertz T, Stubbs P, Crijns HJ, Blanc JJ, Rizzon P et al. Efficacy and safety of d-sotalol, a pure class III antiarrhythmic compound, in patients with symptomatic complex ventricular ectopy. Results of a multicenter, randomized, double-blind, placebo-controlled dose-finding study. The d-Sotalol PVC Study Group. Circulation 1995;92:1517–25. [DOI] [PubMed] [Google Scholar]
- 107. Kpaeyeh JA Jr, Wharton JM. Sotalol. Card Electrophysiol Clin 2016;8:437–52. [DOI] [PubMed] [Google Scholar]
- 108. Tamargo J, Caballero R, Delpón E. Class III antiarrhythmic drugs. In: Martínez-Rubio A, Tamargo J, Dan GA (eds.), Antiarrhythmic Drugs. 1st ed. Cham: Springer; 2020. p107–80. [Google Scholar]
- 109. Reimold SC, Cantillon CO, Friedman PL, Antman EM. Propafenone versus sotalol for suppression of recurrent symptomatic atrial fibrillation. Am J Cardiol 1993;71:558–63. [DOI] [PubMed] [Google Scholar]
- 110. Wanless RS, Anderson K, Joy M, Joseph SP. Multicenter comparative study of the efficacy and safety of sotalol in the prophylactic treatment of patients with paroxysmal supraventricular tachyarrhythmias. Am Heart J 1997;133:441–6. [DOI] [PubMed] [Google Scholar]
- 111. Singh BN, Singh SN, Reda DJ, Tang XC, Lopez B, Harris CL et al. Amiodarone versus sotalol for atrial fibrillation. N Engl J Med 2005;352:1861–72. [DOI] [PubMed] [Google Scholar]
- 112. Greenberg JW, Lancaster TS, Schuessler RB, Melby SJ. Postoperative atrial fibrillation following cardiac surgery: a persistent complication. Eur J Cardiothorac Surg 2017;52:665–72. [DOI] [PubMed] [Google Scholar]
- 113. Tamariz LJ, Bass EB. Pharmacological rate control of atrial fibrillation. Cardiol Clin 2004;22:35–45. [DOI] [PubMed] [Google Scholar]
- 114. Singh S, Saini RK, DiMarco J, Kluger J, Gold R, Chen YW. Efficacy and safety of sotalol in digitalized patients with chronic atrial fibrillation. The Sotalol Study Group. Am J Cardiol 1991;68:1227–30. [DOI] [PubMed] [Google Scholar]
- 115. Ho DS, Zecchin RP, Richards DA, Uther JB, Ross DL. Double-blind trial of lignocaine versus sotalol for acute termination of spontaneous sustained ventricular tachycardia. Lancet 1994;344:18–23. [DOI] [PubMed] [Google Scholar]
- 116. Mason JW. A comparison of seven antiarrhythmic drugs in patients with ventricular tachyarrhythmias. Electrophysiologic Study versus Electrocardiographic Monitoring Investigators. N Engl J Med 1993;329:452–8. [DOI] [PubMed] [Google Scholar]
- 117. Kettering K, Mewis C, Dörnberger V, Vonthein R, Bosch RF, Kühlkamp V. Efficacy of metoprolol and sotalol in the prevention of recurrences of sustained ventricular tachyarrhythmias in patients with an implantable cardioverter defibrillator. Pacing Clin Electrophysiol 2002;25:1571–6. [DOI] [PubMed] [Google Scholar]
- 118. Kühlkamp V, Mewis C, Mermi J, Bosch RF, Seipel L. Suppression of sustained ventricular tachyarrhythmias: a comparison of d,l-sotalol with no antiarrhythmic drug treatment. J Am Coll Cardiol 1999;33:46–52. [DOI] [PubMed] [Google Scholar]
- 119. Pacifico A, Hohnloser SH, Williams JH, Tao B, Saksena S, Henry PD et al. Prevention of implantable-defibrillator shocks by treatment with sotalol. d,l-Sotalol Implantable Cardioverter-Defibrillator Study Group. N Engl J Med 1999;340:1855–62. [DOI] [PubMed] [Google Scholar]
- 120. Julian DG, Prescott RJ, Jackson FS, Szekely P. Controlled trial of sotalol for one year after myocardial infarction. Lancet 1982;1:1142–7. [DOI] [PubMed] [Google Scholar]
- 121. Valembois L, Audureau E, Takeda A, Jarzebowski W, Belmin J, Lafuente-Lafuente C. Antiarrhythmics for maintaining sinus rhythm after cardioversion of atrial fibrillation. Cochrane Database Syst Rev 2019;9:CD005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011;58:587–95. [DOI] [PubMed] [Google Scholar]
- 123. Hohnloser SH, Dorian P, Roberts R, Gent M, Israel CW, Fain E et al. Effect of amiodarone and sotalol on ventricular defibrillation threshold: the optimal pharmacological therapy in cardioverter defibrillator patients (OPTIC) trial. Circulation 2006;114:104–9. [DOI] [PubMed] [Google Scholar]
- 124. Agusala K, Oesterle A, Kulkarni C, Caprio T, Subacius H, Passman R. Risk prediction for adverse events during initiation of sotalol and dofetilide for the treatment of atrial fibrillation. Pacing Clin Electrophysiol 2015;38:490–8. [DOI] [PubMed] [Google Scholar]
- 125. Lin C-Y, Lin Y-J, Lo L-W, Chen Y-Y, Chong E, Chang S-L et al. Factors predisposing to ventricular proarrhythmia during antiarrhythmic drug therapy for atrial fibrillation in patients with structurally normal heart. Heart Rhythm 2015;12:1490–500. [DOI] [PubMed] [Google Scholar]
- 126. Marcus FI. Risks of initiating therapy with sotalol for treatment of atrial fibrillation. J Am Coll Cardiol 1998;32:177–80. [DOI] [PubMed] [Google Scholar]
- 127. Mounsey JP, DiMarco JP. Cardiovascular drugs. Dofetilide. Circulation 2000;102:2665–70. [DOI] [PubMed] [Google Scholar]
- 128. Yang T, Meoli DF, Moslehi J, Roden DM. Inhibition of the α-subunit of phosphoinositide 3-kinase in heart increases late sodium current and is arrhythmogenic. J Pharmacol Exp Ther 2018;365:460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Roukoz H, Saliba W. Dofetilide: a new class III antiarrhythmic agent. Expert Rev Cardiovasc Ther 2007;5:9–19. [DOI] [PubMed] [Google Scholar]
- 130. Joglar JA, Chung MK, Armbruster AL, Benjamin EJ, Chyou JY, Cronin EM et al. 2023 ACC/AHA/ACCP/HRS guideline for the diagnosis and management of atrial fibrillation: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation 2024;149:e1–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Crijns HJ, Van Gelder IC, Kingma JH, Dunselman PH, Gosselink AT, Lie KI. Atrial flutter can be terminated by a class III antiarrhythmic drug but not by a class IC drug. Eur Heart J 1994;15:1403–8. [DOI] [PubMed] [Google Scholar]
- 132. Pedersen OD, Bagger H, Keller N, Marchant B, Køber L, Torp-Pedersen C. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: a Danish investigations of arrhythmia and mortality on dofetilide (diamond) substudy. Circulation 2001;104:292–6. [DOI] [PubMed] [Google Scholar]
- 133. Falk RH, Pollak A, Singh SN, Friedrich T. Intravenous dofetilide, a class III antiarrhythmic agent, for the termination of sustained atrial fibrillation or flutter. Intravenous Dofetilide Investigators. J Am Coll Cardiol 1997;29:385–90. [DOI] [PubMed] [Google Scholar]
- 134. Shamiss Y, Khaykin Y, Oosthuizen R, Tunney D, Sarak B, Beardsall M et al. Dofetilide is safe and effective in preventing atrial fibrillation recurrences in patients accepted for catheter ablation. Europace 2009;11:1448–55. [DOI] [PubMed] [Google Scholar]
- 135. Singh S, Zoble RG, Yellen L, Brodsky MA, Feld GK, Berk M et al. Efficacy and safety of oral dofetilide in converting to and maintaining sinus rhythm in patients with chronic atrial fibrillation or atrial flutter: the symptomatic atrial fibrillation investigative research on dofetilide (SAFIRE-D) study. Circulation 2000;102:2385–90. [DOI] [PubMed] [Google Scholar]
- 136. Køber L, Bloch Thomsen PE, Møller M, Torp-Pedersen C, Carlsen J, Sandøe E et al. Effect of dofetilide in patients with recent myocardial infarction and left-ventricular dysfunction: a randomised trial. Lancet 2000;356:2052–8. [DOI] [PubMed] [Google Scholar]
- 137. Murray KT. Ibutilide. Circulation 1998;97:493–7. [DOI] [PubMed] [Google Scholar]
- 138. Nair M, George LK, Koshy SKG. Safety and efficacy of ibutilide in cardioversion of atrial flutter and fibrillation. J Am Board Fam Med 2011;24:86–92. [DOI] [PubMed] [Google Scholar]
- 139. Naccarelli GV, Wolbrette DL, Khan M, Bhatta L, Hynes J, Samii S et al. Old and new antiarrhythmic drugs for converting and maintaining sinus rhythm in atrial fibrillation: comparative efficacy and results of trials. Am J Cardiol 2003;91:15D–26D. [Google Scholar]
- 140. Volgman AS, Carberry PA, Stambler B, Lewis WR, Dunn GH, Perry KT et al. Conversion efficacy and safety of intravenous ibutilide compared with intravenous procainamide in patients with atrial flutter or fibrillation. J Am Coll Cardiol 1998;31:1414–9. [DOI] [PubMed] [Google Scholar]
- 141. Bernard EO, Schmid ER, Schmidlin D, Scharf C, Candinas R, Germann R. Ibutilide versus amiodarone in atrial fibrillation: a double-blinded, randomized study. Crit Care Med 2003;31:1031–4. [DOI] [PubMed] [Google Scholar]
- 142. Stambler BS, Wood MA, Ellenbogen KA, Perry KT, Wakefield LK, VanderLugt JT. Efficacy and safety of repeated intravenous doses of ibutilide for rapid conversion of atrial flutter or fibrillation. Ibutilide Repeat Dose Study Investigators. Circulation 1996;94:1613–21. [DOI] [PubMed] [Google Scholar]
- 143. Stambler BS, Wood MA, Ellenbogen KA. Antiarrhythmic actions of intravenous ibutilide compared with procainamide during human atrial flutter and fibrillation: electrophysiological determinants of enhanced conversion efficacy. Circulation 1997;96:4298–306. [DOI] [PubMed] [Google Scholar]
- 144. Ellenbogen KA, Stambler BS, Wood MA, Sager PT, Wesley RC Jr, Meissner MC et al. Efficacy of intravenous ibutilide for rapid termination of atrial fibrillation and atrial flutter: a dose-response study. J Am Coll Cardiol 1996;28:130–6. [DOI] [PubMed] [Google Scholar]
- 145. Ellenbogen KA, Clemo HF, Stambler BS, Wood MA, VanderLugt JT. Efficacy of ibutilide for termination of atrial fibrillation and flutter. Am J Cardiol 1996;78:42–5. [Google Scholar]
- 146. Vos MA, Golitsyn SR, Stangl K, Ruda MY, Van Wijk LV, Harry JD et al. Superiority of ibutilide (a new class III agent) over DL-sotalol in converting atrial flutter and atrial fibrillation. The Ibutilide/Sotalol Comparator Study Group. Heart 1998;79:568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. VanderLugt JT, Mattioni T, Denker S, Torchiana D, Ahern T, Wakefield LK et al. Efficacy and safety of ibutilide fumarate for the conversion of atrial arrhythmias after cardiac surgery. Circulation 1999;100:369–75. [DOI] [PubMed] [Google Scholar]
- 148. Oral H, Souza JJ, Michaud GF, Knight BP, Goyal R, Strickberger SA et al. Facilitating transthoracic cardioversion of atrial fibrillation with ibutilide pretreatment. N Engl J Med 1999;340:1849–54. [DOI] [PubMed] [Google Scholar]
- 149. Eidher U, Freihoff F, Kaltenbrunner W, Steinbach K. Efficacy and safety of ibutilide for the conversion of monomorphic atrial tachycardia. Pacing Clin Electrophysiol 2006;29:358–62. [DOI] [PubMed] [Google Scholar]
- 150. Glatter KA, Dorostkar PC, Yang Y, Lee RJ, Van Hare GF, Keung E et al. Electrophysiological effects of ibutilide in patients with accessory pathways. Circulation 2001;104:1933–9. [DOI] [PubMed] [Google Scholar]
- 151. Kockova R, Kocka V, Kiernan T, Fahy GJ. Ibutilide-induced cardioversion of atrial fibrillation during pregnancy. J Cardiovasc Electrophysiol 2007;18:545–7. [DOI] [PubMed] [Google Scholar]
- 152. Burkart TA, Kron J, Miles WM, Conti JB, Gonzalez MD. Successful termination of atrial flutter by ibutilide during pregnancy. Pacing Clin Electrophysiol 2007;30:283–6. [DOI] [PubMed] [Google Scholar]
- 153. Kowey PR, VanderLugt JT, Luderer JR. Safety and risk/benefit analysis of ibutilide for acute conversion of atrial fibrillation/flutter. Am J Cardiol 1996;78:46–52. [DOI] [PubMed] [Google Scholar]
- 154. Ritchie LA, Qin S, Penson PE, Henney NC, Lip GY. Vernakalant hydrochloride for the treatment of atrial fibrillation: evaluation of its place in clinical practice. Future Cardiol 2020;16:585–95. [DOI] [PubMed] [Google Scholar]
- 155. Tamargo J. Other antiarrhythmic drugs. In: Martínez-Rubio A, Tamargo J, Dan G-A (eds.), Antiarrhythmic Drugs. Cham: Springer International Publishing; 2020. p265–306. [Google Scholar]
- 156. Savelieva I, Graydon R, Camm AJ. Pharmacological cardioversion of atrial fibrillation with vernakalant: evidence in support of the ESC Guidelines. Europace 2014;16:162–73. [DOI] [PubMed] [Google Scholar]
- 157. Camm AJ, Capucci A, Hohnloser SH, Torp-Pedersen C, Van Gelder IC, Mangal B et al. A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fibrillation. J Am Coll Cardiol 2011;57:313–21. [DOI] [PubMed] [Google Scholar]
- 158. Akel T, Lafferty J. Efficacy and safety of intravenous vernakalant for the rapid conversion of recent-onset atrial fibrillation: a meta-analysis. Ann Noninvasive Electrocardiol 2018;23:e12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Arsenault KA, Yusuf AM, Crystal E, Healey JS, Morillo CA, Nair GM et al. Interventions for preventing post-operative atrial fibrillation in patients undergoing heart surgery. Cochrane Database Syst Rev 2013;2013:CD003611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Müssigbrodt A, John S, Kosiuk J, Richter S, Hindricks G, Bollmann A. Vernakalant-facilitated electrical cardioversion: comparison of intravenous vernakalant and amiodarone for drug-enhanced electrical cardioversion of atrial fibrillation after failed electrical cardioversion. Europace 2016;18:51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Conde D, Costabel JP, Caro M, Ferro A, Lambardi F, Corrales Barboza A et al. Flecainide versus vernakalant for conversion of recent-onset atrial fibrillation. Int J Cardiol 2013;168:2423–5. [DOI] [PubMed] [Google Scholar]
- 162. Pohjantähti-Maaroos H, Hyppölä H, Lekkala M, Sinisalo E, Heikkola A, Hartikainen J. Intravenous vernakalant in comparison with intravenous flecainide in the cardioversion of recent-onset atrial fibrillation. Eur Heart J Acute Cardiovasc Care 2019;8:114–20. [DOI] [PubMed] [Google Scholar]
- 163. Hall AJ, Mitchell AR. Introducing vernakalant into clinical practice. Arrhythm Electrophysiol Rev 2019;8:70–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Simpson D, Wellington K. Nicorandil: a review of its use in the management of stable angina pectoris, including high-risk patients. Drugs 2004;64:1941–55. [DOI] [PubMed] [Google Scholar]
- 165. Sato T, Sasaki N, O’Rourke B, Marbán E. Nicorandil, a potent cardioprotective agent, acts by opening mitochondrial ATP-dependent potassium channels. J Am Coll Cardiol 2000;35:514–8. [DOI] [PubMed] [Google Scholar]
- 166. Foster MN, Coetzee WA. KATP channels in the cardiovascular system. Physiol Rev 2016;96:177–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ueda H, Nakayama Y, Tsumura K, Yoshimaru K, Hayashi T, Yoshikawa J. Intravenous nicorandil can reduce the occurrence of ventricular fibrillation and QT dispersion in patients with successful coronary angioplasty in acute myocardial infarction. Can J Cardiol 2004;20:625–9. [PubMed] [Google Scholar]
- 168. Geng N, Ren L, Xu L, Zou D, Pang W. Clinical outcomes of nicorandil administration in patients with acute ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention: a systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc Disord 2021;21:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kato T, Kamiyama T, Maruyama Y, Tanaka S, Yoshimoto N. Nicorandil, a potent cardioprotective agent, reduces QT dispersion during coronary angioplasty. Am Heart J 2001;141:940–3. [DOI] [PubMed] [Google Scholar]
- 170. Shimizu W, Kurita T, Matsuo K, Suyama K, Aihara N, Kamakura S et al. Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long-QT syndrome. Circulation 1998;97:1581–8. [DOI] [PubMed] [Google Scholar]
- 171. Sato T, Hata Y, Yamamoto M, Morita H, Mizuo K, Yamanari H et al. Early afterdepolarization abolished by potassium channel opener in a patient with idiopathic long QT syndrome. J Cardiovasc Electrophysiol 1995;6:279–82. [DOI] [PubMed] [Google Scholar]
- 172. Heijman J, Linz D, Schotten U. Dynamics of atrial fibrillation mechanisms and comorbidities. Annu Rev Physiol 2021;83:83–106. [DOI] [PubMed] [Google Scholar]
- 173. Saadeh K, Shivkumar K, Jeevaratnam K. Targeting the β-adrenergic receptor in the clinical management of congenital long QT syndrome. Ann N Y Acad Sci 2020;1474:27–46. [DOI] [PubMed] [Google Scholar]
- 174. Tapa S, Wang L, Francis Stuart SD, Wang Z, Jiang Y, Habecker BA et al. Adrenergic supersensitivity and impaired neural control of cardiac electrophysiology following regional cardiac sympathetic nerve loss. Sci Rep 2020;10:18801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kotecha D, Holmes J, Krum H, Altman DG, Manzano L, Cleland JGF et al. Efficacy of β blockers in patients with heart failure plus atrial fibrillation: an individual-patient data meta-analysis. Lancet 2014;384:2235–43. [DOI] [PubMed] [Google Scholar]
- 176. Roston TM, Chua D, Lum E, Krahn AD. Switching between β-blockers: an empiric tool for the cardiovascular practitioner. Can J Cardiol 2019;35:539–43. [DOI] [PubMed] [Google Scholar]
- 177. Ackerman MJ, Priori SG, Dubin AM, Kowey P, Linker NJ, Slotwiner D et al. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: are all beta-blockers equivalent? Heart Rhythm 2017;14:e41–4. [DOI] [PubMed] [Google Scholar]
- 178. Chatzidou S, Kontogiannis C, Tsilimigras DI, Georgiopoulos G, Kosmopoulos M, Papadopoulou E et al. Propranolol versus metoprolol for treatment of electrical storm in patients with implantable cardioverter-defibrillator. J Am Coll Cardiol 2018;71:1897–906. [DOI] [PubMed] [Google Scholar]
- 179. Denardo SJ, Gong Y, Cooper-DeHoff RM, Farsang C, Keltai M, Szirmai L et al. Effects of verapamil SR and atenolol on 24-hour blood pressure and heart rate in hypertension patients with coronary artery disease: an international verapamil SR-trandolapril ambulatory monitoring substudy. PLoS One 2015;10:e0122726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Fongemie J, Felix-Getzik E. A review of nebivolol pharmacology and clinical evidence. Drugs 2015;75:1349–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Garnock-Jones KP. Esmolol: a review of its use in the short-term treatment of tachyarrhythmias and the short-term control of tachycardia and hypertension. Drugs 2012;72:109–32. [DOI] [PubMed] [Google Scholar]
- 182. Syed YY. Landiolol: a review in tachyarrhythmias. Drugs 2018;78:377–88. [DOI] [PubMed] [Google Scholar]
- 183. Patocskai B, Barajas-Martinez H, Hu D, Gurabi Z, Koncz I, Antzelevitch C. Cellular and ionic mechanisms underlying the effects of cilostazol, milrinone, and isoproterenol to suppress arrhythmogenesis in an experimental model of early repolarization syndrome. Heart Rhythm 2016;13:1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Mangiardi LM, Bonamini R, Conte M, Gaita F, Orzan F, Presbitero P et al. Bedside evaluation of atrioventricular block with narrow QRS complexes: usefulness of carotid sinus massage and atropine administration. Am J Cardiol 1982;49:1136–45. [DOI] [PubMed] [Google Scholar]
- 185. Brady WJ, Swart G, DeBehnke DJ, Ma OJ, Aufderheide TP. The efficacy of atropine in the treatment of hemodynamically unstable bradycardia and atrioventricular block: prehospital and emergency department considerations. Resuscitation 1999;41:47–55. [DOI] [PubMed] [Google Scholar]
- 186. Swart G, Brady WJ Jr, DeBehnke DJ, Ma OJ, Aufderheide TP. Acute myocardial infarction complicated by hemodynamically unstable bradyarrhythmia: prehospital and ED treatment with atropine. Am J Emerg Med 1999;17:647–52. [DOI] [PubMed] [Google Scholar]
- 187. Feigl D, Ashkenazy J, Kishon Y. Early and late atrioventricular block in acute inferior myocardial infarction. J Am Coll Cardiol 1984;4:35–8. [DOI] [PubMed] [Google Scholar]
- 188. Sodeck GH, Domanovits H, Meron G, Rauscha F, Losert H, Thalmann M et al. Compromising bradycardia: management in the emergency department. Resuscitation 2007;73:96–102. [DOI] [PubMed] [Google Scholar]
- 189. Kusumoto FM, Schoenfeld MH, Barrett C, Edgerton JR, Ellenbogen KA, Gold MR et al. 2018 ACC/AHA/HRS guideline on the evaluation and management of patients with bradycardia and cardiac conduction delay: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Rhythm Society. Circulation 2019;140:e382–482. [DOI] [PubMed] [Google Scholar]
- 190. Scheinman MM, Thorburn D, Abbott JA. Use of atropine in patients with acute myocardial infarction and sinus bradycardia. Circulation 1975;52:627–33. [DOI] [PubMed] [Google Scholar]
- 191. Katritsis DG, Josephson ME. Electrophysiological testing for the investigation of bradycardias. Arrhythm Electrophysiol Rev 2017;6:24–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Bernheim A, Fatio R, Kiowski W, Weilenmann D, Rickli H, Brunner-La Rocca HP. Atropine often results in complete atrioventricular block or sinus arrest after cardiac transplantation: an unpredictable and dose-independent phenomenon. Transplantation 2004;77:1181–5. [DOI] [PubMed] [Google Scholar]
- 193. Tamargo J, Delpón E, Caballero R. The safety of digoxin as a pharmacological treatment of atrial fibrillation. Expert Opin Drug Saf 2006;5:453–67. [DOI] [PubMed] [Google Scholar]
- 194. Khand AU, Rankin AC, Martin W, Taylor J, Gemmell I, Cleland JGF. Carvedilol alone or in combination with digoxin for the management of atrial fibrillation in patients with heart failure? J Am Coll Cardiol 2003;42:1944–51. [DOI] [PubMed] [Google Scholar]
- 195. Farshi R, Kistner D, Sarma JS, Longmate JA, Singh BN. Ventricular rate control in chronic atrial fibrillation during daily activity and programmed exercise: a crossover open-label study of five drug regimens. J Am Coll Cardiol 1999;33:304–10. [DOI] [PubMed] [Google Scholar]
- 196. McNamara RL, Tamariz LJ, Segal JB, Bass EB. Management of atrial fibrillation: review of the evidence for the role of pharmacologic therapy, electrical cardioversion, and echocardiography. Ann Intern Med 2003;139:1018–33. [DOI] [PubMed] [Google Scholar]
- 197. Sellers TD Jr, Bashore TM, Gallagher JJ. Digitalis in the pre-excitation syndrome. Analysis during atrial fibrillation. Circulation 1977;56:260–7. [DOI] [PubMed] [Google Scholar]
- 198. Layland J, Carrick D, Lee M, Oldroyd K, Berry C. Adenosine: physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv 2014;7:581–91. [DOI] [PubMed] [Google Scholar]
- 199. Lerman BB, Markowitz SM, Cheung JW, Liu CF, Thomas G, Ip JE. Supraventricular tachycardia: mechanistic insights deduced from adenosine. Circ Arrhythm Electrophysiol 2018;11:e006953. [DOI] [PubMed] [Google Scholar]
- 200. Brady WJ Jr, DeBehnke DJ, Wickman LL, Lindbeck G. Treatment of out-of-hospital supraventricular tachycardia: adenosine vs verapamil. Acad Emerg Med 1996;3:574–85. [DOI] [PubMed] [Google Scholar]
- 201. Glatter KA, Cheng J, Dorostkar P, Modin G, Talwar S, Al-Nimri M et al. Electrophysiologic effects of adenosine in patients with supraventricular tachycardia. Circulation 1999;99:1034–40. [DOI] [PubMed] [Google Scholar]
- 202. Delaney B, Loy J, Kelly A-M. The relative efficacy of adenosine versus verapamil for the treatment of stable paroxysmal supraventricular tachycardia in adults: a meta-analysis. Eur J Emerg Med 2011;18:148–52. [DOI] [PubMed] [Google Scholar]
- 203. Page RL, Joglar JA, Caldwell MA, Calkins H, Conti JB, Deal BJ et al. 2015 ACC/AHA/HRS guideline for the management of adult patients with supraventricular tachycardia: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2016;67:e27–115. [DOI] [PubMed] [Google Scholar]
- 204. Kall JG, Kopp D, Olshansky B, Kinder C, O’Connor M, Cadman CS et al. Adenosine-sensitive atrial tachycardia. Pacing Clin Electrophysiol 1995;18:300–6. [DOI] [PubMed] [Google Scholar]
- 205. Markowitz SM, Stein KM, Mittal S, Slotwiner DJ, Lerman BB. Differential effects of adenosine on focal and macroreentrant atrial tachycardia. J Cardiovasc Electrophysiol 1999;10:489–502. [DOI] [PubMed] [Google Scholar]
- 206. Ip JE, Cheung JW, Chung JH, Liu CF, Thomas G, Markowitz SM et al. Adenosine-induced atrial fibrillation: insights into mechanism. Circ Arrhythm Electrophysiol 2013;6:e34–37. [DOI] [PubMed] [Google Scholar]
- 207. Li N, Csepe TA, Hansen BJ, Sul LV, Kalyanasundaram A, Zakharkin SO et al. Adenosine-induced atrial fibrillation: localized reentrant drivers in lateral right atria due to heterogeneous expression of adenosine A1 receptors and GIRK4 subunits in the Human Heart. Circulation 2016;134:486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Garratt CJ, Griffith MJ, O’Nunain S, Ward DE, Camm AJ. Effects of intravenous adenosine on antegrade refractoriness of accessory atrioventricular connections. Circulation 1991;84:1962–8. [DOI] [PubMed] [Google Scholar]
- 209. Godfraind T. Calcium channel blockers in cardiovascular pharmacotherapy. J Cardiovasc Pharmacol Ther 2014;19:501–15. [DOI] [PubMed] [Google Scholar]
- 210. Abernethy DR, Schwartz JB. Calcium-antagonist drugs. N Engl J Med 1999;341:1447–57. [DOI] [PubMed] [Google Scholar]
- 211. Hilleman DE, Hunter CB, Mohiuddin SM, Maciejewski S. Pharmacological management of atrial fibrillation following cardiac surgery. Am J Cardiovasc Drugs 2005;5:361–9. [DOI] [PubMed] [Google Scholar]
- 212. Dougherty AH, Jackman WM, Naccarelli GV, Friday KJ, Dias VC. Acute conversion of paroxysmal supraventricular tachycardia with intravenous diltiazem. IV Diltiazem Study Group. Am J Cardiol 1992;70:587–92. [DOI] [PubMed] [Google Scholar]
- 213. Scheuermeyer FX, Grafstein E, Stenstrom R, Christenson J, Heslop C, Heilbron B et al. Safety and efficiency of calcium channel blockers versus beta-blockers for rate control in patients with atrial fibrillation and no acute underlying medical illness. Acad Emerg Med 2013;20:222–30. [DOI] [PubMed] [Google Scholar]
- 214. Schreck DM, Rivera AR, Tricarico VJ. Emergency management of atrial fibrillation and flutter: intravenous diltiazem versus intravenous digoxin. Ann Emerg Med 1997;29:135–40. [DOI] [PubMed] [Google Scholar]
- 215. Segal JB, McNamara RL, Miller MR, Kim N, Goodman SN, Powe NR et al. The evidence regarding the drugs used for ventricular rate control. J Fam Pract 2000;49:47–59. [PubMed] [Google Scholar]
- 216. Siu C-W, Lau C-P, Lee W-L, Lam K-F, Tse H-F. Intravenous diltiazem is superior to intravenous amiodarone or digoxin for achieving ventricular rate control in patients with acute uncomplicated atrial fibrillation. Crit Care Med 2009;37:2174–9; quiz 2180. [DOI] [PubMed] [Google Scholar]
- 217. Tisdale JE, Padhi ID, Goldberg AD, Silverman NA, Webb CR, Higgins RS et al. A randomized, double-blind comparison of intravenous diltiazem and digoxin for atrial fibrillation after coronary artery bypass surgery. Am Heart J 1998;135:739–47. [DOI] [PubMed] [Google Scholar]
- 218. Ulimoen SR, Enger S, Pripp AH, Abdelnoor M, Arnesen H, Gjesdal K et al. Calcium channel blockers improve exercise capacity and reduce N-terminal pro-B-type natriuretic peptide levels compared with beta-blockers in patients with permanent atrial fibrillation. Eur Heart J 2014;35:517–24. [DOI] [PubMed] [Google Scholar]
- 219. Ellenbogen KA, Dias VC, Plumb VJ, Heywood JT, Mirvis DM. A placebo-controlled trial of continuous intravenous diltiazem infusion for 24-hour heart rate control during atrial fibrillation and atrial flutter: a multicenter study. J Am Coll Cardiol 1991;18:891–7. [DOI] [PubMed] [Google Scholar]
- 220. Platia EV, Michelson EL, Porterfield JK, Das G. Esmolol versus verapamil in the acute treatment of atrial fibrillation or atrial flutter. Am J Cardiol 1989;63:925–9. [DOI] [PubMed] [Google Scholar]
- 221. Salerno DM, Dias VC, Kleiger RE, Tschida VH, Sung RJ, Sami M et al. Efficacy and safety of intravenous diltiazem for treatment of atrial fibrillation and atrial flutter. The Diltiazem-Atrial Fibrillation/Flutter Study Group. Am J Cardiol 1989;63:1046–51. [DOI] [PubMed] [Google Scholar]
- 222. Hazard PB, Burnett CR. Verapamil in multifocal atrial tachycardia. Hemodynamic and respiratory changes. Chest 1987;91:68–70. [DOI] [PubMed] [Google Scholar]
- 223. Salerno DM, Anderson B, Sharkey PJ, Iber C. Intravenous verapamil for treatment of multifocal atrial tachycardia with and without calcium pretreatment. Ann Intern Med 1987;107:623–8. [DOI] [PubMed] [Google Scholar]
- 224. Levine JH, Michael JR, Guarnieri T. Treatment of multifocal atrial tachycardia with verapamil. N Engl J Med 1985;312:21–5. [DOI] [PubMed] [Google Scholar]
- 225. Ellenbogen KA, Dias VC, Cardello FP, Strauss WE, Simonton CA, Pollak SJ et al. Safety and efficacy of intravenous diltiazem in atrial fibrillation or atrial flutter. Am J Cardiol 1995;75:45–9. [DOI] [PubMed] [Google Scholar]
- 226. Blackshear JL, Stambler BS, Strauss WE, Roy D, Dias VC, Beach CL et al. Control of heart rate during transition from intravenous to oral diltiazem in atrial fibrillation or flutter. Am J Cardiol 1996;78:1246–50. [DOI] [PubMed] [Google Scholar]
- 227. Rinkenberger RL, Prystowsky EN, Heger JJ, Troup PJ, Jackman WM, Zipes DP. Effects of intravenous and chronic oral verapamil administration in patients with supraventricular tachyarrhythmias. Circulation 1980;62:996–1010. [DOI] [PubMed] [Google Scholar]
- 228. Winniford MD, Fulton KL, Hillis LD. Long-term therapy of paroxysmal supraventricular tachycardia: a randomized, double-blind comparison of digoxin, propranolol and verapamil. Am J Cardiol 1984;54:1138–9. [DOI] [PubMed] [Google Scholar]
- 229. Mauritson DR, Winniford MD, Walker WS, Rude RE, Cary JR, Hillis LD. Oral verapamil for paroxysmal supraventricular tachycardia: a long-term, double-blind randomized trial. Ann Intern Med 1982;96:409–12. [DOI] [PubMed] [Google Scholar]
- 230. Sakurai M, Yasuda H, Kato N, Nomura A, Fujita M, Nishino T et al. Acute and chronic effects of verapamil in patients with paroxysmal supraventricular tachycardia. Am Heart J 1983;105:619–28. [DOI] [PubMed] [Google Scholar]
- 231. Yeh SJ, Lin FC, Chou YY, Hung JS, Wu D. Termination of paroxysmal supraventricular tachycardia with a single oral dose of diltiazem and propranolol. Circulation 1985;71:104–9. [DOI] [PubMed] [Google Scholar]
- 232. Nogami A, Naito S, Tada H, Taniguchi K, Okamoto Y, Nishimura S et al. Demonstration of diastolic and presystolic Purkinje potentials as critical potentials in a macroreentry circuit of verapamil-sensitive idiopathic left ventricular tachycardia. J Am Coll Cardiol 2000;36:811–23. [DOI] [PubMed] [Google Scholar]
- 233. Belhassen B, Rotmensch HH, Laniado S. Response of recurrent sustained ventricular tachycardia to verapamil. Br Heart J 1981;46:679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Tsuchiya T, Okumura K, Honda T, Iwasa A, Ashikaga K. Effects of verapamil and lidocaine on two components of the re-entry circuit of verapamil-senstitive idiopathic left ventricular tachycardia. J Am Coll Cardiol 2001;37:1415–21. [DOI] [PubMed] [Google Scholar]
- 235. Leenhardt A, Glaser E, Burguera M, Nürnberg M, Maison-Blanche P, Coumel P. Short-coupled variant of torsade de pointes. A new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation 1994;89:206–15. [DOI] [PubMed] [Google Scholar]
- 236. Eisenberg SJ, Scheinman MM, Dullet NK, Finkbeiner WE, Griffin JC, Eldar M et al. Sudden cardiac death and polymorphous ventricular tachycardia in patients with normal QT intervals and normal systolic cardiac function. Am J Cardiol 1995;75:687–92. [DOI] [PubMed] [Google Scholar]
- 237. Hood MA, Smith WM. Adenosine versus verapamil in the treatment of supraventricular tachycardia: a randomized double-crossover trial. Am Heart J 1992;123:1543–9. [DOI] [PubMed] [Google Scholar]
- 238. Lim SH, Anantharaman V, Teo WS, Chan YH. Slow infusion of calcium channel blockers compared with intravenous adenosine in the emergency treatment of supraventricular tachycardia. Resuscitation 2009;80:523–8. [DOI] [PubMed] [Google Scholar]
- 239. Brubaker S, Long B, Koyfman A. Alternative treatment options for atrioventricular-nodal-reentry tachycardia: an emergency medicine review. J Emerg Med 2018;54:198–206. [DOI] [PubMed] [Google Scholar]
- 240. Gulamhusein S, Ko P, Carruthers SG, Klein GJ. Acceleration of the ventricular response during atrial fibrillation in the Wolff-Parkinson-White syndrome after verapamil. Circulation 1982;65:348–54. [DOI] [PubMed] [Google Scholar]
- 241. Kim RJ, Gerling BR, Kono AT, Greenberg ML. Precipitation of ventricular fibrillation by intravenous diltiazem and metoprolol in a young patient with occult Wolff-Parkinson-White syndrome. Pacing Clin Electrophysiol 2008;31:776–9. [DOI] [PubMed] [Google Scholar]
- 242. Buxton AE, Marchlinski FE, Doherty JU, Flores B, Josephson ME. Hazards of intravenous verapamil for sustained ventricular tachycardia. Am J Cardiol 1987;59:1107–10. [DOI] [PubMed] [Google Scholar]
- 243. Dancy M, Camm AJ, Ward D. Misdiagnosis of chronic recurrent ventricular tachycardia. Lancet 1985;2:320–3. [DOI] [PubMed] [Google Scholar]
- 244. Gill A, Flaim SF, Damiano BP, Sit SP, Brannan MD. Pharmacology of bepridil. Am J Cardiol 1992;69:11D–16D. [Google Scholar]
- 245. Flammang D, Waynberger M, Jansen FH, Paillet R, Coumel P. Electrophysiological profile of bepridil, a new anti-anginal drug with calcium blocking properties. Eur Heart J 1983;4:657–654. [PubMed] [Google Scholar]
- 246. Fujiki A, Tsuneda T, Sugao M, Mizumaki K, Inoue H. Usefulness and safety of bepridil in converting persistent atrial fibrillation to sinus rhythm. Am J Cardiol 2003;92:472–5. [DOI] [PubMed] [Google Scholar]
- 247. Nakazato Y, Yasuda M, Sasaki A, Iida Y, Kawano Y, Nakazato K et al. Conversion and maintenance of sinus rhythm by bepridil in patients with persistent atrial fibrillation. Circ J 2005;69:44–8. [DOI] [PubMed] [Google Scholar]
- 248. Yamase M, Nakazato Y, Daida H. Effectiveness of amiodarone versus bepridil in achieving conversion to sinus rhythm in patients with persistent atrial fibrillation: a randomised trial. Heart 2012;98:1067–71. [DOI] [PubMed] [Google Scholar]
- 249. Shiga T, Suzuki A, Naganuma M, Hosaka F, Shoda M, Hagiwara N. Clinical outcome in patients with paroxysmal or persistent atrial fibrillation receiving bepridil. Circ J 2011;75:1334–42. [DOI] [PubMed] [Google Scholar]
- 250. Kondo T, Miake J, Kato M, Ogura K, Iitsuka K, Yamamoto K. Impact of postprocedural antiarrhythmic drug therapy with bepridil on maintaining sinus rhythm after catheter ablation for persistent atrial fibrillation. J Cardiol 2016;68:229–35. [DOI] [PubMed] [Google Scholar]
- 251. Alhede C, Lauridsen TK, Johannessen A, Dixen U, Jensen JS, Raatikainen P et al. Antiarrhythmic medication is superior to catheter ablation in suppressing supraventricular ectopic complexes in patients with atrial fibrillation. Int J Cardiol 2017;244:186–91. [DOI] [PubMed] [Google Scholar]
- 252. Beck OA, Ihle C, Hochrein H. [Propafenon and propranolol in the management of cardiac extrasystoles (author’s transl)]. Dtsch Med Wochenschr 1982;107:579–83. [DOI] [PubMed] [Google Scholar]
- 253. Dan G-A, Martinez-Rubio A, Agewall S, Boriani G, Borggrefe M, Gaita F et al. Antiarrhythmic drugs-clinical use and clinical decision making: a consensus document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology (ESC) working group on cardiovascular pharmacology, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and International Society of Cardiovascular Pharmacotherapy (ISCP). Europace 2018;20:731–732an. [DOI] [PubMed] [Google Scholar]
- 254. Katritsis DG, Zografos T, Katritsis GD, Giazitzoglou E, Vachliotis V, Paxinos G et al. Catheter ablation vs. antiarrhythmic drug therapy in patients with symptomatic atrioventricular nodal re-entrant tachycardia: a randomized, controlled trial. Europace 2017;19:602–6. [DOI] [PubMed] [Google Scholar]
- 255. Chen SA, Hsieh MH, Tai CT, Tsai CF, Prakash VS, Yu WC et al. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation 1999;100:1879–86. [DOI] [PubMed] [Google Scholar]
- 256. Dorian P, Naccarelli GV, Coumel P, Hohnloser SH, Maser MJ. A randomized comparison of flecainide versus verapamil in paroxysmal supraventricular tachycardia. The Flecainide Multicenter Investigators Group. Am J Cardiol 1996;77:89A–95A. [Google Scholar]
- 257. Bohora S, Lokhandwala Y, Parekh P, Vasavda A. Reversal of tachycardiomyopathy due to left atrial tachycardia by ivabradine. J Cardiovasc Electrophysiol 2011;22:340–2. [DOI] [PubMed] [Google Scholar]
- 258. Meles E, Carbone C, Maggiolini S, Moretti P, DE Carlini CC, Gentile G et al. A case of atrial tachycardia treated with ivabradine as bridge to ablation. J Cardiovasc Electrophysiol 2015;26:565–8. [DOI] [PubMed] [Google Scholar]
- 259. Guccione P, Paul T, Garson A Jr. Long-term follow-up of amiodarone therapy in the young: continued efficacy, unimpaired growth, moderate side effects. J Am Coll Cardiol 1990;15:1118–24. [DOI] [PubMed] [Google Scholar]
- 260. von Bernuth G, Engelhardt W, Kramer HH, Singer H, Schneider P, Ulmer H et al. Atrial automatic tachycardia in infancy and childhood. Eur Heart J 1992;13:1410–5. [DOI] [PubMed] [Google Scholar]
- 261. Ptaszynski P, Kaczmarek K, Ruta J, Klingenheben T, Cygankiewicz I, Wranicz JK. Ivabradine in combination with metoprolol succinate in the treatment of inappropriate sinus tachycardia. J Cardiovasc Pharmacol Ther 2013;18:338–44. [DOI] [PubMed] [Google Scholar]
- 262. Olshansky B, Sullivan RM. Inappropriate sinus tachycardia. Europace 2019;21:194–207. [DOI] [PubMed] [Google Scholar]
- 263. Brandes A, Crijns HJGM, Rienstra M, Kirchhof P, Grove EL, Pedersen KB et al. Cardioversion of atrial fibrillation and atrial flutter revisited: current evidence and practical guidance for a common procedure. Europace 2020;22:1149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Aliot E, Denjoy I. Comparison of the safety and efficacy of flecainide versus propafenone in hospital out-patients with symptomatic paroxysmal atrial fibrillation/flutter. The Flecainide AF French Study Group. Am J Cardiol 1996;77:66A–71A. [Google Scholar]
- 265. Van Gelder IC, Crijns HJ, Van Gilst WH, Verwer R, Lie KI. Prediction of uneventful cardioversion and maintenance of sinus rhythm from direct-current electrical cardioversion of chronic atrial fibrillation and flutter. Am J Cardiol 1991;68:41–6. [DOI] [PubMed] [Google Scholar]
- 266. Pietersen AH, Hellemann H. Usefulness of flecainide for prevention of paroxysmal atrial fibrillation and flutter. Danish-Norwegian Flecainide Multicenter Study Group. Am J Cardiol 1991;67:713–7. [DOI] [PubMed] [Google Scholar]
- 267. Benditt DG, Williams JH, Jin J, Deering TF, Zucker R, Browne K et al. Maintenance of sinus rhythm with oral d,l-sotalol therapy in patients with symptomatic atrial fibrillation and/or atrial flutter. d,l-Sotalol Atrial Fibrillation/Flutter Study Group. Am J Cardiol 1999;84:270–7. [DOI] [PubMed] [Google Scholar]
- 268. Crijns HJ, Van Gelder IC, Tieleman RG, Brügemann J, De Kam PJ, Gosselink AT et al. Long-term outcome of electrical cardioversion in patients with chronic atrial flutter. Heart 1997;77:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Da Costa A, Thévenin J, Roche F, Romeyer-Bouchard C, Abdellaoui L, Messier M et al. Results from the Loire-Ardèche-Drôme-Isère-Puy-de-Dôme (LADIP) trial on atrial flutter, a multicentric prospective randomized study comparing amiodarone and radiofrequency ablation after the first episode of symptomatic atrial flutter. Circulation 2006;114:1676–81. [DOI] [PubMed] [Google Scholar]
- 270. Katritsis DG, Boriani G, Cosio FG, Hindricks G, Jaïs P, Josephson ME et al. European Heart Rhythm Association (EHRA) consensus document on the management of supraventricular arrhythmias, endorsed by Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS), and Sociedad Latinoamericana de Estimulación Cardiaca y Electrofisiologia (SOLAECE). Europace 2017;19:465–511. [DOI] [PubMed] [Google Scholar]
- 271. Crijns HJ, van Gelder IC, Lie KI. Supraventricular tachycardia mimicking ventricular tachycardia during flecainide treatment. Am J Cardiol 1988;62:1303–6. [DOI] [PubMed] [Google Scholar]
- 272. Nabar A, Rodriguez LM, Timmermans C, van Mechelen R, Wellens HJ. Class IC antiarrhythmic drug induced atrial flutter: electrocardiographic and electrophysiological findings and their importance for long term outcome after right atrial isthmus ablation. Heart 2001;85:424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of ‘class IC atrial flutter’ in patients with resistant atrial fibrillation. Am J Cardiol 1999;83:785–7, A10. [DOI] [PubMed] [Google Scholar]
- 274. Camm AJ, Naccarelli GV, Mittal S, Crijns HJGM, Hohnloser SH, Ma C-S et al. The increasing role of rhythm control in patients with atrial fibrillation: JACC state-of-the-art review. J Am Coll Cardiol 2022;79:1932–48. [DOI] [PubMed] [Google Scholar]
- 275. Andersen SS, Hansen ML, Gislason GH, Schramm TK, Folke F, Fosbøl E et al. Antiarrhythmic therapy and risk of death in patients with atrial fibrillation: a nationwide study. Europace 2009;11:886–91. [DOI] [PubMed] [Google Scholar]
- 276. Kirchhof P, Camm AJ, Goette A, Brandes A, Eckardt L, Elvan A et al. Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med 2020;383:1305–16. [DOI] [PubMed] [Google Scholar]
- 277. Aliot E, Capucci A, Crijns HJ, Goette A, Tamargo J. Twenty-five years in the making: flecainide is safe and effective for the management of atrial fibrillation. Europace 2011;13:161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Nasr IA, Bouzamondo A, Hulot J-S, Dubourg O, Le Heuzey J-Y, Lechat P. Prevention of atrial fibrillation onset by beta-blocker treatment in heart failure: a meta-analysis. Eur Heart J 2007;28:457–62. [DOI] [PubMed] [Google Scholar]
- 279. Carpenter A, Frontera A, Bond R, Duncan E, Thomas G. Vagal atrial fibrillation: what is it and should we treat it? Int J Cardiol 2015;201:415–21. [DOI] [PubMed] [Google Scholar]
- 280. de Vos CB, Nieuwlaat R, Crijns HJGM, Camm AJ, LeHeuzey J-Y, Kirchhof CJ et al. Autonomic trigger patterns and anti-arrhythmic treatment of paroxysmal atrial fibrillation: data from the Euro Heart Survey. Eur Heart J 2008;29:632–9. [DOI] [PubMed] [Google Scholar]
- 281. Anastasiou-Nana MI, Anderson JL, Stewart JR, Crevey BJ, Yanowitz FG, Lutz JR et al. Occurrence of exercise-induced and spontaneous wide complex tachycardia during therapy with flecainide for complex ventricular arrhythmias: a probable proarrhythmic effect. Am Heart J 1987;113:1071–7. [DOI] [PubMed] [Google Scholar]
- 282. Crijns HJGM. Changes of intracardiac conduction induced by antiarrhythmic drugs. Importance of use- and reverse use-dependence. Thesis. Groningen: University of Groningen, 1993.
- 283. Zimmermann M, Kalusche D. Fluctuation in autonomic tone is a major determinant of sustained atrial arrhythmias in patients with focal ectopy originating from the pulmonary veins. J Cardiovasc Electrophysiol 2001;12:285–91. [DOI] [PubMed] [Google Scholar]
- 284. Rattanawong P, Kewcharoen J, S Srivathsan K, Shen W-K. Drug therapy for vagally-mediated atrial fibrillation and sympatho-vagal balance in the genesis of atrial fibrillation: a review of the current literature. J Atr Fibrillation 2020;13:2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Connolly SJ, Crijns HJGM, Torp-Pedersen C, van Eickels M, Gaudin C, Page RL et al. Analysis of stroke in ATHENA: a placebo-controlled, double-blind, parallel-arm trial to assess the efficacy of dronedarone 400 mg BID for the prevention of cardiovascular hospitalization or death from any cause in patients with atrial fibrillation/atrial flutter. Circulation 2009;120:1174–80. [DOI] [PubMed] [Google Scholar]
- 286. Pisters R, Hohnloser SH, Connolly SJ, Torp-Pedersen C, Naditch-Brûlé L, Page RL et al. Effect of dronedarone on clinical end points in patients with atrial fibrillation and coronary heart disease: insights from the ATHENA trial. Europace 2014;16:174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Singh JP, Blomström-Lundqvist C, Turakhia MP, Camm AJ, Fazeli MS, Kreidieh B et al. Dronedarone versus sotalol in patients with atrial fibrillation: a systematic literature review and network meta-analysis. Clin Cardiol 2023;46:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Page RL, Connolly SJ, Crijns HJGM, van Eickels M, Gaudin C, Torp-Pedersen C et al. Rhythm- and rate-controlling effects of dronedarone in patients with atrial fibrillation (from the ATHENA trial). Am J Cardiol 2011;107:1019–22. [DOI] [PubMed] [Google Scholar]
- 289. Kirchhof P, Camm J, Crijns HJGM, Piccini JP, Torp-Pedersen C, McKindley DS et al. Dronedarone provides effective early rhythm control: post-hoc analysis of the ATHENA trial using EAST-AFNET 4 criteria. Europace. In press. [Google Scholar]
- 290. Lafuente-Lafuente C, Mouly S, Longás-Tejero MA, Mahé I, Bergmann J-F. Antiarrhythmic drugs for maintaining sinus rhythm after cardioversion of atrial fibrillation: a systematic review of randomized controlled trials. Arch Intern Med 2006;166:719–28. [DOI] [PubMed] [Google Scholar]
- 291. Flaker G, Lopes RD, Hylek E, Wojdyla DM, Thomas L, Al-Khatib SM et al. Amiodarone, anticoagulation, and clinical events in patients with atrial fibrillation: insights from the ARISTOTLE trial. J Am Coll Cardiol 2014;64:1541–50. [DOI] [PubMed] [Google Scholar]
- 292. Doldi F, Geßler N, Anwar O, Kahle A-K, Scherschel K, Rath B et al. In-hospital mortality and major complications related to radiofrequency catheter ablations of over 10 000 supraventricular arrhythmias from 2005 to 2020: individualized case analysis of multicentric administrative data. Europace 2023;25:130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. D’Este D, Zoppo F, Bertaglia E, Zerbo F, Picciolo A, Scarabeo V et al. Long-term outcome of patients with atrioventricular node reentrant tachycardia. Int J Cardiol 2007;115:350–3. [DOI] [PubMed] [Google Scholar]
- 294. Alboni P, Tomasi C, Menozzi C, Bottoni N, Paparella N, Fucà G et al. Efficacy and safety of out-of-hospital self-administered single-dose oral drug treatment in the management of infrequent, well-tolerated paroxysmal supraventricular tachycardia. J Am Coll Cardiol 2001;37:548–53. [DOI] [PubMed] [Google Scholar]
- 295. Blomström-Lundqvist C. Drug treatment of supraventricular tachycardia. Heart 2009;95:1803–7. [DOI] [PubMed] [Google Scholar]
- 296. Hoff PI, Tronstad A, Oie B, Ohm OJ. Electrophysiologic and clinical effects of flecainide for recurrent paroxysmal supraventricular tachycardia. Am J Cardiol 1988;62:585–9. [DOI] [PubMed] [Google Scholar]
- 297. Garcia-Civera R, Sanjuan R, Morell S, Ferrero JA, Miralles L, Llavador J et al. Effects of propafenone on induction and maintenance of atrioventricular nodal reentrant tachycardia. Pacing Clin Electrophysiol 1984;7:649–55. [DOI] [PubMed] [Google Scholar]
- 298. Anderson JL, Platt ML, Guarnieri T, Fox TL, Maser MJ, Pritchett EL. Flecainide acetate for paroxysmal supraventricular tachyarrhythmias. The Flecainide Supraventricular Tachycardia Study Group. Am J Cardiol 1994;74:578–84. [DOI] [PubMed] [Google Scholar]
- 299. Porter MJ, Morton JB, Denman R, Lin AC, Tierney S, Santucci PA et al. Influence of age and gender on the mechanism of supraventricular tachycardia. Heart Rhythm 2004;1:393–6. [DOI] [PubMed] [Google Scholar]
- 300. Bardy GH, Packer DL, German LD, Gallagher JJ. Preexcited reciprocating tachycardia in patients with Wolff-Parkinson-White syndrome: incidence and mechanisms. Circulation 1984;70:377–91. [DOI] [PubMed] [Google Scholar]
- 301. Kulig J, Koplan BA. Cardiology patient page. Wolff-Parkinson-White syndrome and accessory pathways. Circulation 2010;122:e480–483. [DOI] [PubMed] [Google Scholar]
- 302. Delise P, Sciarra L. Sudden cardiac death in patients with ventricular preexcitation. Card Electrophysiol Clin 2020;12:519–25. [DOI] [PubMed] [Google Scholar]
- 303. Scheinman MM, Huang S. The 1998 NASPE prospective catheter ablation registry. Pacing Clin Electrophysiol 2000;23:1020–8. [DOI] [PubMed] [Google Scholar]
- 304. Gill JS, Mehta D, Ward DE, Camm AJ. Efficacy of flecainide, sotalol, and verapamil in the treatment of right ventricular tachycardia in patients without overt cardiac abnormality. Br Heart J 1992;68:392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Kjekshus J, Bathen J, Orning OM, Storstein L. A double-blind, crossover comparison of flecainide acetate and disopyramide phosphate in the treatment of ventricular premature complexes. Am J Cardiol 1984;53:72B–78B. [Google Scholar]
- 306. Podrid PJ, Lampert S, Graboys TB, Blatt CM, Lown B. Aggravation of arrhythmia by antiarrhythmic drugs--incidence and predictors. Am J Cardiol 1987;59:38E–44E. [DOI] [PubMed] [Google Scholar]
- 307. Krittayaphong R, Bhuripanyo K, Punlee K, Kangkagate C, Chaithiraphan S. Effect of atenolol on symptomatic ventricular arrhythmia without structural heart disease: a randomized placebo-controlled study. Am Heart J 2002;144:e10. [Google Scholar]
- 308. Ward DE, Nathan AW, Camm AJ. Fascicular tachycardia sensitive to calcium antagonists. Eur Heart J 1984;5:896–905. [DOI] [PubMed] [Google Scholar]
- 309. Hyman MC, Mustin D, Supple G, Schaller RD, Santangeli P, Arkles J et al. Class IC antiarrhythmic drugs for suspected premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 2018;15:159–63. [DOI] [PubMed] [Google Scholar]
- 310. Capucci A, Di Pasquale G, Boriani G, Carini G, Balducelli M, Frabetti L et al. A double-blind crossover comparison of flecainide and slow-release mexiletine in the treatment of stable premature ventricular complexes. Int J Clin Pharmacol Res 1991;11:23–33. [PubMed] [Google Scholar]
- 311. Hamon D, Swid MA, Rajendran PS, Liu A, Boyle NG, Shivkumar K et al. Premature ventricular contraction diurnal profiles predict distinct clinical characteristics and beta-blocker responses. J Cardiovasc Electrophysiol 2019;30:836–43. [DOI] [PubMed] [Google Scholar]
- 312. Escande W, Gourraud J-B, Haissaguerre M, Gandjbakhch E, Lavergne T, Martins R et al. Malignant Purkinje ectopy induced by sodium channel blockers. Heart Rhythm 2022;19:1595–1603. [DOI] [PubMed] [Google Scholar]
- 313. Primeau R, Agha A, Giorgi C, Shenasa M, Nadeau R. Long term efficacy and toxicity of amiodarone in the treatment of refractory cardiac arrhythmias. Can J Cardiol 1989;5:98–104. [PubMed] [Google Scholar]
- 314. Bertels RA, Kammeraad JAE, Zeelenberg AM, Filippini LH, Knobbe I, Kuipers IM et al. The efficacy of anti-arrhythmic drugs in children with idiopathic frequent symptomatic or asymptomatic premature ventricular complexes with or without asymptomatic ventricular tachycardia: a retrospective multi-center study. Pediatr Cardiol 2021;42:883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Lapage MJ, Bradley DJ, Dick M II. Verapamil in infants: an exaggerated fear? Pediatr Cardiol 2013;34:1532–4. [DOI] [PubMed] [Google Scholar]
- 316. Cojocaru C, Penela D, Berruezo A, Vatasescu R. Mechanisms, time course and predictability of premature ventricular contractions cardiomyopathy-an update on its development and resolution. Heart Fail Rev 2022;27:1639–51. [DOI] [PubMed] [Google Scholar]
- 317. Baman TS, Lange DC, Ilg KJ, Gupta SK, Liu T-Y, Alguire C et al. Relationship between burden of premature ventricular complexes and left ventricular function. Heart Rhythm 2010;7:865–9. [DOI] [PubMed] [Google Scholar]
- 318. Penela D, Teres C, Fernández-Armenta J, Aguinaga L, Tercedor L, Soto-Iglesias D et al. Premature ventricular complex site of origin and ablation outcomes in patients with prior myocardial infarction. Heart Rhythm 2021;18:27–33. [DOI] [PubMed] [Google Scholar]
- 319. Cairns JA, Connolly SJ, Roberts R, Gent M. Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT. Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet 1997;349:675–82. [DOI] [PubMed] [Google Scholar]
- 320. Cardiac Arrhythmia Suppression Trial (CAST) Investigators . Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989;321:406–12. [DOI] [PubMed] [Google Scholar]
- 321. Sapp JL, Wells GA, Parkash R, Stevenson WG, Blier L, Sarrazin J-F et al. Ventricular tachycardia ablation versus escalation of antiarrhythmic drugs. N Engl J Med 2016;375:111–21. [DOI] [PubMed] [Google Scholar]
- 322. Gillis AM, Mathison HJ, Kulisz E, Lester WM. Dispersion of ventricular repolarization in left ventricular hypertrophy: influence of afterload and dofetilide. J Cardiovasc Electrophysiol 1998;9:988–97. [DOI] [PubMed] [Google Scholar]
- 323. Chung R, Houghtaling PL, Tchou M, Niebauer MJ, Lindsay BD, Tchou PJ et al. Left ventricular hypertrophy and antiarrhythmic drugs in atrial fibrillation: impact on mortality. Pacing Clin Electrophysiol 2014;37:1338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Frommeyer G, Eckardt L. Drug-induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol 2016;13:36–47. [DOI] [PubMed] [Google Scholar]
- 325. Reiffel JA, Capucci A. “Pill in the pocket” antiarrhythmic drugs for orally administered pharmacologic cardioversion of atrial fibrillation. Am J Cardiol 2021;140:55–61. [DOI] [PubMed] [Google Scholar]
- 326. Reiffel JA, Blomstrom-Lundqvist C, Boriani G, Goette A, Kowey PR, Merino JL et al. Abstract 10986: use of the “pill in the pocket” approach for atrial fibrillation termination as determined from the Antiarrhythmic Medication for Atrial Fibrillation (AIM-AF) study: a physician survey on the prescription of antiarrhythmic drugs in the USA and Europe. Circulation 2021;144:A10986–A10986. [Google Scholar]
- 327. Azpitarte J, Alvarez M, Baún O, García R, Moreno E, Martín F et al. Value of single oral loading dose of propafenone in converting recent-onset atrial fibrillation. Results of a randomized, double-blind, controlled study. Eur Heart J 1997;18:1649–54. [DOI] [PubMed] [Google Scholar]
- 328. Orso D, Santangelo S, Guglielmo N, Bove T, Cilenti F, Cristiani L et al. Bayesian network meta-analysis of randomized controlled trials on the efficacy of antiarrhythmics in the pharmacological cardioversion of paroxysmal atrial fibrillation. Am J Cardiovasc Drugs 2023;23:355–77. [DOI] [PubMed] [Google Scholar]
- 329. Lamotte M, Gerlier L, Caekelbergh K, Lalji K, Polifka J, Lee E. Impact of a pharmacological cardioversion with vernakalant on the management cost of recent atrial fibrillation in Belgium. Value Health 2014;17:A490. [Google Scholar]
- 330. Bianconi L, Castro A, Dinelli M, Alboni P, Pappalardo A, Richiardi E et al. Comparison of intravenously administered dofetilide versus amiodarone in the acute termination of atrial fibrillation and flutter. A multicentre, randomized, double-blind, placebo-controlled study. Eur Heart J 2000;21:1265–73. [DOI] [PubMed] [Google Scholar]
- 331. Al-Khatib SM, Page RL. Acute treatment of patients with supraventricular tachycardia. JAMA Cardiol 2016;1:483–5. [DOI] [PubMed] [Google Scholar]
- 332. Helton MR. Diagnosis and management of common types of supraventricular tachycardia. Am Fam Physician 2015;92:793–800. [PubMed] [Google Scholar]
- 333. Boriani G, Biffi M, Frabetti L, Azzolini U, Sabbatani P, Bronzetti G et al. Ventricular fibrillation after intravenous amiodarone in Wolff-Parkinson-White syndrome with atrial fibrillation. Am Heart J 1996;131:1214–6. [DOI] [PubMed] [Google Scholar]
- 334. Leonelli F, Bagliani G, Boriani G, Padeletti L. Arrhythmias originating in the atria. Card Electrophysiol Clin 2017;9:383–409. [DOI] [PubMed] [Google Scholar]
- 335. Kylat RI, Samson RA. Junctional ectopic tachycardia in infants and children. J Arrhythm 2020;36:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Alasti M, Mirzaee S, Machado C, Healy S, Bittinger L, Adam D et al. Junctional ectopic tachycardia (JET). J Arrhythm 2020;36:837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Arvind B, Kothari SS, Juneja R, Saxena A, Ramakrishnan S, Gupta SK et al. Ivabradine versus amiodarone in the management of postoperative junctional ectopic tachycardia: a randomized, open-label, noninferiority study. JACC Clin Electrophysiol 2021;7:1052–60. [DOI] [PubMed] [Google Scholar]
- 338. Kumar V, Kumar G, Tiwari N, Joshi S, Sharma V, Ramamurthy R. Ivabradine as an adjunct for refractory junctional ectopic tachycardia following pediatric cardiac surgery: a preliminary study. World J Pediatr Congenit Heart Surg 2019;10:709–14. [DOI] [PubMed] [Google Scholar]
- 339. Gorgels AP, van den Dool A, Hofs A, Mulleneers R, Smeets JL, Vos MA et al. Comparison of procainamide and lidocaine in terminating sustained monomorphic ventricular tachycardia. Am J Cardiol 1996;78:43–6. [DOI] [PubMed] [Google Scholar]
- 340. deSouza IS, Martindale JL, Sinert R. Antidysrhythmic drug therapy for the termination of stable, monomorphic ventricular tachycardia: a systematic review. Emerg Med J 2015;32:161–7. [DOI] [PubMed] [Google Scholar]
- 341. Ortiz M, Martín A, Arribas F, Coll-Vinent B, Del Arco C, Peinado R et al. Randomized comparison of intravenous procainamide vs. intravenous amiodarone for the acute treatment of tolerated wide QRS tachycardia: the PROCAMIO study. Eur Heart J 2017;38:1329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Kelson K, deSouza I. Procainamide versus amiodarone for stable ventricular tachycardia. Acad Emerg Med 2019;26:1099–101. [DOI] [PubMed] [Google Scholar]
- 343. Baldi E, Conte G, Zeppenfeld K, Lenarczyk R, Guerra JM, Farkowski MM et al. Contemporary management of ventricular electrical storm in Europe: results of a European Heart Rhythm Association Survey. Europace 2023;25:1277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Manz M, Mletzko R, Jung W, Lüderitz B. Electrophysiological and haemodynamic effects of lidocaine and ajmaline in the management of sustained ventricular tachycardia. Eur Heart J 1992;13:1123–8. [DOI] [PubMed] [Google Scholar]
- 345. Schwartz A, Shen E, Morady F, Gillespie K, Scheinman M, Chatterjee K. Hemodynamic effects of intravenous amiodarone in patients with depressed left ventricular function and recurrent ventricular tachycardia. Am Heart J 1983;106:848–56. [DOI] [PubMed] [Google Scholar]
- 346. Panchal AR, Berg KM, Kudenchuk PJ, Del Rios M, Hirsch KG, Link MS et al. 2018 American heart association focused update on advanced cardiovascular life support use of antiarrhythmic drugs during and immediately after cardiac arrest: an update to the american heart association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2018;138:e740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Martí-Carvajal AJ, Simancas-Racines D, Anand V, Bangdiwala S. Prophylactic lidocaine for myocardial infarction. Cochrane Database Syst Rev 2015;2015:CD008553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Rosso R, Hochstadt A, Viskin D, Chorin E, Schwartz AL, Tovia-Brodie O et al. Polymorphic ventricular tachycardia, ischaemic ventricular fibrillation, and torsade de pointes: importance of the QT and the coupling interval in the differential diagnosis. Eur Heart J 2021;42:3965–75. [DOI] [PubMed] [Google Scholar]
- 349. Viskin S, Chorin E, Viskin D, Hochstadt A, Schwartz AL, Rosso R. Polymorphic ventricular tachycardia: terminology, mechanism, diagnosis, and emergency therapy. Circulation 2021;144:823–39. [DOI] [PubMed] [Google Scholar]
- 350. Viskin S, Chorin E, Viskin D, Hochstadt A, Halkin A, Tovia-Brodie O et al. Quinidine-responsive polymorphic ventricular tachycardia in patients with coronary heart disease. Circulation 2019;139:2304–14. [DOI] [PubMed] [Google Scholar]
- 351. Reiffel JA. Inpatient versus outpatient antiarrhythmic drug initiation: safety and cost-effectiveness issues. Curr Opin Cardiol 2000;15:7–11. [DOI] [PubMed] [Google Scholar]
- 352. LaBreck ME, Chopra N, Robinson A, Billakanty SR, Fu EY, Nemer DM et al. Home sotalol initiation for the management of atrial and ventricular arrhythmias using remote electrocardiographic monitoring. JACC Clin Electrophysiol 2024;11:386–396. [DOI] [PubMed] [Google Scholar]
- 353. Ali SA, Ersbøll M, Vinding NE, Butt JH, Rørth R, Selmer C et al. Incidence of thyroid dysfunction following initiation of amiodarone treatment in patients with and without heart failure: a nationwide cohort study. Europace 2023;25:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Knapp E, Watson K. Medication management of atrial fibrillation: emerging therapies for rhythm control and stroke prevention. P T 2011;36:518–28. [PMC free article] [PubMed] [Google Scholar]
- 355. Gonzalez JE, Sauer WH, Krantz MJ. Ventricular ectopy and QTc-interval prolongation associated with dronedarone therapy. Pharmacotherapy 2013;33:e179–181. [DOI] [PubMed] [Google Scholar]
- 356. Kao DP, Hiatt WR, Krantz MJ. Proarrhythmic potential of dronedarone: emerging evidence from spontaneous adverse event reporting. Pharmacotherapy 2012;32:767–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Wijtvliet EPJP, Tieleman RG, van Gelder IC, Pluymaekers NAHA, Rienstra M, Folkeringa RJ et al. Nurse-led vs. usual-care for atrial fibrillation. Eur Heart J 2020;41:634–41. [DOI] [PubMed] [Google Scholar]
- 358. Ranger S, Talajic M, Lemery R, Roy D, Nattel S. Amplification of flecainide-induced ventricular conduction slowing by exercise. A potentially significant clinical consequence of use-dependent sodium channel blockade. Circulation 1989;79:1000–6. [DOI] [PubMed] [Google Scholar]
- 359. Lavalle C, Magnocavallo M, Straito M, Santini L, Forleo GB, Grimaldi M et al. Flecainide how and when: a practical guide in supraventricular arrhythmias. J Clin Med 2021;10:1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Behr ER, Winkel BG, Ensam B, Alfie A, Arbelo E, Berry C et al. The diagnostic role of pharmacological provocation testing in cardiac electrophysiology: a clinical consensus statement of the European Heart Rhythm Association and the European Association of Percutaneous Cardiovascular Interventions (EAPCI) of the ESC, the ESC Working Group on Cardiovascular Pharmacotherapy, the Association of European Paediatric and Congenital Cardiology (AEPC), the Paediatric & Congenital Electrophysiology Society (PACES), the Heart Rhythm Society (HRS), the Asia Pacific Heart Rhythm Society (APHRS), and the Latin American Heart Rhythm Society (LAHRS). Europace 2025;27:euaf067. 10.1093/europace/euaf067. [DOI] [Google Scholar]
- 361. Heijman J, Hohnloser SH, Camm AJ. Antiarrhythmic drugs for atrial fibrillation: lessons from the past and opportunities for the future. Europace 2021;23:ii14–22. [DOI] [PubMed] [Google Scholar]
- 362. Remme CA, Heijman J, Gomez AM, Zaza A, Odening KE. 25 years of basic and translational science in EP Europace: novel insights into arrhythmia mechanisms and therapeutic strategies. Europace 2023;25:euad210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Schuijt E, Scherr D, Plank G, Schotten U, Heijman J. Evolution in electrophysiology 100 years after Einthoven: translational and computational innovations in rhythm control of atrial fibrillation. Europace 2024;27:euae304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Bigger JT Jr, Sahar DI. Clinical types of proarrhythmic response to antiarrhythmic drugs. Am J Cardiol 1987;59:2E–9E. [Google Scholar]
- 365. Choudhury M, Boyett MR, Morris GM. Biology of the sinus node and its disease. Arrhythm Electrophysiol Rev 2015;4:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Zeltser D, Justo D, Halkin A, Rosso R, Ish-Shalom M, Hochenberg M et al. Drug-induced atrioventricular block: prognosis after discontinuation of the culprit drug. J Am Coll Cardiol 2004;44:105–8. [DOI] [PubMed] [Google Scholar]
- 367. Osmonov D, Erdinler I, Ozcan KS, Altay S, Turkkan C, Yildirim E et al. Management of patients with drug-induced atrioventricular block. Pacing Clin Electrophysiol 2012;35:804–10. [DOI] [PubMed] [Google Scholar]
- 368. Friedman PL, Stevenson WG. Proarrhythmia. Am J Cardiol 1998;82:50N–58N. [DOI] [PubMed] [Google Scholar]
- 369. Naccarelli GV, Wolbrette DL, Luck JC. Proarrhythmia. Med Clin North Am 2001;85:503–26, xii. [DOI] [PubMed] [Google Scholar]
- 370. Stanton MS, Prystowsky EN, Fineberg NS, Miles WM, Zipes DP, Heger JJ. Arrhythmogenic effects of antiarrhythmic drugs: a study of 506 patients treated for ventricular tachycardia or fibrillation. J Am Coll Cardiol 1989;14:209–15; discussion 216–7. [DOI] [PubMed] [Google Scholar]
- 371. Herre JM, Titus C, Oeff M, Eldar M, Franz MR, Griffin JC et al. Inefficacy and proarrhythmic effects of flecainide and encainide for sustained ventricular tachycardia and ventricular fibrillation. Ann Intern Med 1990;113:671–6. [DOI] [PubMed] [Google Scholar]
- 372. Ávila P, Berruezo A, Jiménez-Candil J, Tercedor L, Calvo D, Arribas F et al. Bayesian analysis of the substrate ablation vs. antiarrhythmic drug therapy for symptomatic ventricular tachycardia trial. Europace 2023;25:euad181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Morganroth J. Risk factors for the development of proarrhythmic events. Am J Cardiol 1987;59:32E–37E. [Google Scholar]
- 374. Slater W, Lampert S, Podrid PJ, Lown B. Clinical predictors of arrhythmia worsening by antiarrhythmic drugs. Am J Cardiol 1988;61:349–53. [DOI] [PubMed] [Google Scholar]
- 375. Dessertenne F. [Ventricular tachycardia with 2 variable opposing foci]. Arch Mal Coeur Vaiss 1966;59:263–72. [PubMed] [Google Scholar]
- 376. Jackman WM, Friday KJ, Anderson JL, Aliot EM, Clark M, Lazzara R. The long QT syndromes: a critical review, new clinical observations and a unifying hypothesis. Prog Cardiovasc Dis 1988;31:115–72. [DOI] [PubMed] [Google Scholar]
- 377. Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol 2002;17:43–51. [DOI] [PubMed] [Google Scholar]
- 378. Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV et al. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 1999;10:1124–52. [DOI] [PubMed] [Google Scholar]
- 379. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart 2003;89:1363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Napolitano C, Priori SG, Schwartz PJ. Torsade de pointes. Mechanisms and management. Drugs 1994;47:51–65. [DOI] [PubMed] [Google Scholar]
- 381. Roden DM, Woosley RL, Primm RK. Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am Heart J 1986;111:1088–1093. [DOI] [PubMed] [Google Scholar]
- 382. Kuck KH, Kunze KP, Roewer N, Bleifeld W. Sotalol-induced torsade de pointes. Am Heart J 1984;107:179–80. [DOI] [PubMed] [Google Scholar]
- 383. Li M, Ramos LG. Drug-induced QT prolongation and torsades de pointes. P T 2017;42:473–7. [PMC free article] [PubMed] [Google Scholar]
- 384. Brown MA, Smith WM, Lubbe WF, Norris RM. Amiodarone-induced torsades de pointes. Eur Heart J 1986;7:234–9. [DOI] [PubMed] [Google Scholar]
- 385. Sicouri S, Moro S, Litovsky S, Elizari MV, Antzelevitch C. Chronic amiodarone reduces transmural dispersion of repolarization in the canine heart. J Cardiovasc Electrophysiol 1997;8:1269–79. [DOI] [PubMed] [Google Scholar]
- 386. Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V et al. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. J Am Coll Cardiol 2010;55:934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Schwartz PJ, Woosley RL. Predicting the unpredictable: drug-induced QT prolongation and torsades de pointes. J Am Coll Cardiol 2016;67:1639–50. [DOI] [PubMed] [Google Scholar]
- 388. Usuda K, Hayashi K, Nakajima T, Kurata Y, Cui S, Kusayama T et al. Mechanisms of fever-induced QT prolongation and torsades de pointes in patients with KCNH2 mutation. Europace 2023;25:euad161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Berns E, Rinkenberger RL, Jeang MK, Dougherty AH, Jenkins M, Naccarelli GV. Efficacy and safety of flecainide acetate for atrial tachycardia or fibrillation. Am J Cardiol 1987;59:1337–41. [DOI] [PubMed] [Google Scholar]
- 390. Tisdale JE, Chung MK, Campbell KB, Hammadah M, Joglar JA, Leclerc J et al. Drug-induced arrhythmias: a scientific statement from the American Heart Association. Circulation 2020;142:e214–33. [DOI] [PubMed] [Google Scholar]
- 391. Falk RH. Flecainide-induced ventricular tachycardia and fibrillation in patients treated for atrial fibrillation. Ann Intern Med 1989;111:107–11. [DOI] [PubMed] [Google Scholar]
- 392. Echt DS, Ruskin JN. Use of flecainide for the treatment of atrial fibrillation. Am J Cardiol 2020;125:1123–33. [DOI] [PubMed] [Google Scholar]
- 393. Boriani G, Martignani C, Biffi M, Capucci A, Branzi A. Oral loading with propafenone for conversion of recent-onset atrial fibrillation: a review on in-hospital treatment. Drugs 2002;62:415–23. [DOI] [PubMed] [Google Scholar]
- 394. Beldner S, Lin D, Marchlinski FE. Flecainide and propafenone induced ST-segment elevation in patients with atrial fibrillation: clue to specificity of Brugada-type electrocardiographic changes. Am J Cardiol 2004;94:1184–5. [DOI] [PubMed] [Google Scholar]
- 395. Yap YG, Behr ER, Camm AJ. Drug-induced Brugada syndrome. Europace 2009;11:989–94. [DOI] [PubMed] [Google Scholar]
- 396. Epstein AE, Olshansky B, Naccarelli GV, Kennedy JI Jr, Murphy EJ, Goldschlager N. Practical management guide for clinicians who treat patients with amiodarone. Am J Med 2016;129:468–75. [DOI] [PubMed] [Google Scholar]
- 397. Tsaban G, Ostrovsky D, Alnsasra H, Burrack N, Gordon M, Babayev AS et al. Amiodarone and pulmonary toxicity in atrial fibrillation: a nationwide Israeli study. Eur Heart J 2024;45:379–88. [DOI] [PubMed] [Google Scholar]
- 398. Grace AA, Camm AJ. Quinidine. N Engl J Med 1998;338:35–45. [DOI] [PubMed] [Google Scholar]
- 399. Cosín-Aguilar J, Hernandiz-Martinez A. The clinical usefulness of the antiarrhythmic drug quinidine. Eur Heart J 1987;8 Suppl A:1–9. [Google Scholar]
- 400. Thomas SHL, Behr ER. Pharmacological treatment of acquired QT prolongation and torsades de pointes. Br J Clin Pharmacol 2016;81:420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Leape LL, Bates DW, Cullen DJ, Cooper J, Demonaco HJ, Gallivan T et al. Systems analysis of adverse drug events. ADE Prevention Study Group. JAMA 1995;274:35–43. [PubMed] [Google Scholar]
- 402. Raschetti R, Morgutti M, Menniti-Ippolito F, Belisari A, Rossignoli A, Longhini P et al. Suspected adverse drug events requiring emergency department visits or hospital admissions. Eur J Clin Pharmacol 1999;54:959–63. [DOI] [PubMed] [Google Scholar]
- 403. Proietti M, Raparelli V, Olshansky B, Lip GYH. Polypharmacy and major adverse events in atrial fibrillation: observations from the AFFIRM trial. Clin Res Cardiol 2016;105:412–20. [DOI] [PubMed] [Google Scholar]
- 404. Trujillo TC, Nolan PE. Antiarrhythmic agents: drug interactions of clinical significance. Drug Saf 2000;23:509–32. [DOI] [PubMed] [Google Scholar]
- 405. Lund M, Petersen TS, Dalhoff KP. Clinical implications of P-glycoprotein modulation in drug-drug interactions. Drugs 2017;77:859–83. [DOI] [PubMed] [Google Scholar]
- 406. Mar PL, Horbal P, Chung MK, Dukes JW, Ezekowitz M, Lakkireddy D et al. Drug interactions affecting antiarrhythmic drug use. Circ Arrhythm Electrophysiol 2022;15:e007955. [DOI] [PubMed] [Google Scholar]
- 407. Davies EA, O’Mahony MS. Adverse drug reactions in special populations—the elderly. Br J Clin Pharmacol 2015;80:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Belle DJ, Singh H. Genetic factors in drug metabolism. Am Fam Physician 2008;77:1553–60. [PubMed] [Google Scholar]
- 409. Soldin OP, Mattison DR. Sex differences in pharmacokinetics and pharmacodynamics. Clin Pharmacokinet 2009;48:143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Zhou S-F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet 2009;48:689–723. [DOI] [PubMed] [Google Scholar]
- 411. Lim KS, Cho J-Y, Jang I-J, Kim B-H, Kim J, Jeon J-Y et al. Pharmacokinetic interaction of flecainide and paroxetine in relation to the CYP2D6*10 allele in healthy Korean subjects. Br J Clin Pharmacol 2008;66:660–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Calvo MV, Martin-Suarez A, Martin Luengo C, Avila C, Cascon M, Domínguez-Gil Hurlé A. Interaction between digoxin and propafenone. Ther Drug Monit 1989;11:10–5. [DOI] [PubMed] [Google Scholar]
- 413. Naccarelli GV, Rinkenberger RL, Dougherty AH, Fitzgerald DM. Adverse effects of amiodarone. Pathogenesis, incidence and management. Med Toxicol Adverse Drug Exp 1989;4:246–53. [DOI] [PubMed] [Google Scholar]
- 414. Robinson K, Johnston A, Walker S, Mulrow JP, McKenna WJ, Holt DW. The digoxin-amiodarone interaction. Cardiovasc Drugs Ther 1989;3:25–8. doi: 10.1007/BF01881526 [DOI] [PubMed] [Google Scholar]
- 415. Holm J, Lindh JD, Andersson ML, Mannheimer B. The effect of amiodarone on warfarin anticoagulation: a register-based nationwide cohort study involving the Swedish population. J Thromb Haemost 2017;15:446–53. [DOI] [PubMed] [Google Scholar]
- 416. Back DJ, Burger DM. Interaction between amiodarone and sofosbuvir-based treatment for hepatitis C virus infection: potential mechanisms and lessons to be learned. Gastroenterology 2015;149:1315–7. [DOI] [PubMed] [Google Scholar]
- 417. Nicolau DP, Uber WE, Crumbley AJ III, Strange C. Amiodarone-cyclosporine interaction in a heart transplant patient. J Heart Lung Transplant 1992;11:564–8. [PubMed] [Google Scholar]
- 418. Prom R, Umscheid CA, Kasbekar N, Spinler SA. Effect of simvastatin-amiodarone drug interaction alert on appropriate prescribing. Am J Health Syst Pharm 2013;70:1878–9. [DOI] [PubMed] [Google Scholar]
- 419. Naccarelli GV, Wolbrette DL, Levin V, Samii S, Banchs JE, Penny-Peterson E et al. Safety and efficacy of dronedarone in the treatment of atrial fibrillation/flutter. Clin Med Insights Cardiol 2011;5:103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Vallakati A, Chandra PA, Pednekar M, Frankel R, Shani J. Dronedarone-induced digoxin toxicity: new drug, new interactions. Am J Ther 2013;20:e717–719. [DOI] [PubMed] [Google Scholar]
- 421. Mochalina N, Juhlin T, Platonov PG, Svensson PJ, Wieloch M. Concomitant use of dronedarone with dabigatran in patients with atrial fibrillation in clinical practice. Thromb Res 2015;135:1070–4. [DOI] [PubMed] [Google Scholar]
- 422. Brown DD, Spector R, Juhl RP. Drug interactions with digoxin. Drugs 1980;20:198–206. [DOI] [PubMed] [Google Scholar]
- 423. Rodin SM, Johnson BF. Pharmacokinetic interactions with digoxin. Clin Pharmacokinet 1988;15:227–44. [DOI] [PubMed] [Google Scholar]
- 424. Reiffel JA, Robinson VM, Kowey PR. Perspective on antiarrhythmic drug combinations. Am J Cardiol 2023;192:116–23. [DOI] [PubMed] [Google Scholar]
- 425. Nedoshivin A, Petrova PTS, Karpov Y. Efficacy and safety of ivabradine in combination with beta-blockers in patients with stable angina pectoris: a systematic review and meta-analysis. Adv Ther 2022;39:4189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Reiffel JA. Reduced-dose antiarrhythmic drugs: valuable or valueless? J Innov Card Rhythm Manag 2020;11:4063–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Baker BJ, Gammill J, Massengill J, Schubert E, Karin A, Doherty JE. Concurrent use of quinidine and disopyramide: evaluation of serum concentrations and electrocardiographic effects. Am Heart J 1983;105:12–5. [DOI] [PubMed] [Google Scholar]
- 428. Bonavita GJ, Pires LA, Wagshal AB, Cuello C, Mittleman RS, Greene TO et al. Usefulness of oral quinidine-mexiletine combination therapy for sustained ventricular tachyarrhythmias as assessed by programmed electrical stimulation when quinidine monotherapy has failed. Am Heart J 1994;127:847–51. [PubMed] [Google Scholar]
- 429. Giardina EG, Wechsler ME. Low dose quinidine-mexiletine combination therapy versus quinidine monotherapy for treatment of ventricular arrhythmias. J Am Coll Cardiol 1990;15:1138–45. [DOI] [PubMed] [Google Scholar]
- 430. Whitford EG, McGovern B, Schoenfeld MH, Garan H, Newell JB, McElroy M et al. Long-term efficacy of mexiletine alone and in combination with class Ia antiarrhythmic drugs for refractory ventricular arrhythmias. Am Heart J 1988;115:360–6. [DOI] [PubMed] [Google Scholar]
- 431. Shimizu W, Ohe T, Kurita T, Kawade M, Arakaki Y, Aihara N et al. Effects of verapamil and propranolol on early afterdepolarizations and ventricular arrhythmias induced by epinephrine in congenital long QT syndrome. J Am Coll Cardiol 1995;26:1299–309. [DOI] [PubMed] [Google Scholar]
- 432. Fetsch T, Bauer P, Engberding R, Koch HP, Lukl J, Meinertz T et al. Prevention of atrial fibrillation after cardioversion: results of the PAFAC trial. Eur Heart J 2004;25:1385–94. [DOI] [PubMed] [Google Scholar]
- 433. Patten M, Maas R, Bauer P, Lüderitz B, Sonntag F, Dluzniewski M et al. Suppression of paroxysmal atrial tachyarrhythmias--results of the SOPAT trial. Eur Heart J 2004;25:1395–404. [DOI] [PubMed] [Google Scholar]
- 434. Mendes L, Podrid PJ, Fuchs T, Franklin S. Role of combination drug therapy with a class IC antiarrhythmic agent and mexiletine for ventricular tachycardia. J Am Coll Cardiol 1991;17:1396–402. [DOI] [PubMed] [Google Scholar]
- 435. Toivonen L, Kadish A, Morady F. A prospective comparison of class IA, B, and C antiarrhythmic agents in combination with amiodarone in patients with inducible, sustained ventricular tachycardia. Circulation 1991;84:101–8. [DOI] [PubMed] [Google Scholar]
- 436. Marcus FI. Drug combinations and interactions with class III agents. J Cardiovasc Pharmacol 1992;20 Suppl 2:S70–74. [Google Scholar]
- 437. Madadi S, Nemati M, Fazelifar A, Kamali F, Haghjoo M. Clinical results of combined amiodarone and mexiletine therapy in refractory ventricular tachycardias. Res Cardiovasc Med 2019;8:11–3. [Google Scholar]
- 438. Lüderitz B, Mletzko R, Jung W, Manz M. Combination of antiarrhythmic drugs. J Cardiovasc Pharmacol 1991;17 Suppl 6:S48–52. [Google Scholar]
- 439. Kagal DR, Crystal E, Lashevsky I, Tiong I, Lau C, Vitali AC et al. Amiodarone plus Flecainide combination therapy in patients with amiodarone refractory paroxysmal atrial fibrillation. Int J Cardiol 2013;168:4262–3. [DOI] [PubMed] [Google Scholar]
- 440. Simopoulos V, Tagarakis GI, Daskalopoulou SS, Daskalopoulos ME, Lenos A, Chryssagis K et al. Ranolazine enhances the antiarrhythmic activity of amiodarone by accelerating conversion of new-onset atrial fibrillation after cardiac surgery. Angiology 2014;65:294–7. [DOI] [PubMed] [Google Scholar]
- 441. Ermakov S, Gerstenfeld EP, Svetlichnaya Y, Scheinman MM. Use of flecainide in combination antiarrhythmic therapy in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2017;14:564–9. [DOI] [PubMed] [Google Scholar]
- 442. Ermakov S, Hoffmayer KS, Gerstenfeld EP, Scheinman MM. Combination drug therapy for patients with intractable ventricular tachycardia associated with right ventricular cardiomyopathy. Pacing Clin Electrophysiol 2014;37:90–4. [DOI] [PubMed] [Google Scholar]
- 443. Capucci A, Piangerelli L, Ricciotti J, Gabrielli D, Guerra F. Flecainide-metoprolol combination reduces atrial fibrillation clinical recurrences and improves tolerability at 1-year follow-up in persistent symptomatic atrial fibrillation. Europace 2016;18:1698–704. [DOI] [PubMed] [Google Scholar]
- 444. Wattanasuwan N, Khan IA, Mehta NJ, Arora P, Singh N, Vasavada BC et al. Acute ventricular rate control in atrial fibrillation: IV combination of diltiazem and digoxin vs. IV diltiazem alone. Chest 2001;119:502–6. [DOI] [PubMed] [Google Scholar]
- 445. Frommeyer G, Kaiser D, Uphaus T, Kaese S, Osada N, Rajamani S et al. Effect of ranolazine on ventricular repolarization in class III antiarrhythmic drug-treated rabbits. Heart Rhythm 2012;9:2051–8. [DOI] [PubMed] [Google Scholar]
- 446. Shah SA, Kluger J, White CM. Monotherapy versus combination therapy with class III antiarrhythmic agents to attenuate transmural dispersion of repolarization: a potential risk factor for torsade de pointes. Pharmacotherapy 2007;27:1297–305. [DOI] [PubMed] [Google Scholar]
- 447. Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C et al. 2023 ESC guidelines for the management of cardiomyopathies. Eur Heart J 2023;44:3503–626. [DOI] [PubMed] [Google Scholar]
- 448. Dorian P, Newman D, Berman N, Hardy J, Mitchell J. Sotalol and type IA drugs in combination prevent recurrence of sustained ventricular tachycardia. J Am Coll Cardiol 1993;22:106–13. [DOI] [PubMed] [Google Scholar]
- 449. Lee SD, Newman D, Ham M, Dorian P. Electrophysiologic mechanisms of antiarrhythmic efficacy of a sotalol and class Ia drug combination: elimination of reverse use dependence. J Am Coll Cardiol 1997;29:100–5. [DOI] [PubMed] [Google Scholar]
- 450. Borrelli F, Izzo AA. Herb-drug interactions with St John’s wort (Hypericum perforatum): an update on clinical observations. AAPS J 2009;11:710–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Tannergren C, Engman H, Knutson L, Hedeland M, Bondesson U, Lennernäs H. St John’s wort decreases the bioavailability of R- and S-verapamil through induction of the first-pass metabolism. Clin Pharmacol Ther 2004;75:298–309. [DOI] [PubMed] [Google Scholar]
- 452. Kim T-E, Shin K-H, Park J-E, Kim M-G, Yun Y-M, Choi D-H et al. Effect of green tea catechins on the pharmacokinetics of digoxin in humans. Drug Des Devel Ther 2018;12:2139–47. [Google Scholar]
- 453. Bailey DG, Dresser G, Arnold JMO. Grapefruit-medication interactions: forbidden fruit or avoidable consequences? CMAJ 2013;185:309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Mazi-Kotwal N, Seshadri M. Drug interactions with grapefruit juice. Br J Med Pract 2012;5:31–4. [Google Scholar]
- 455. Libersa CC, Brique SA, Motte KB, Caron JF, Guédon-Moreau LM, Humbert L et al. Dramatic inhibition of amiodarone metabolism induced by grapefruit juice. Br J Clin Pharmacol 2000;49:373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Jáuregui-Garrido B, Jáuregui-Lobera I. Interactions between antiarrhythmic drugs and food. Nutr Hosp 2012;27:1399–407. [DOI] [PubMed] [Google Scholar]
- 457. Meng X, Mojaverian P, Doedée M, Lin E, Weinryb I, Chiang ST et al. Bioavailability of amiodarone tablets administered with and without food in healthy subjects. Am J Cardiol 2001;87:432–5. [DOI] [PubMed] [Google Scholar]
- 458. Lévy S. Combination therapy for cardiac arrhythmias. Am J Cardiol 1988;61:A95–100. [Google Scholar]
- 459. Narayan G, Akhtar M, Sra J. Combined use of 1C and III agents for highly symptomatic, refractory atrial fibrillation. J Interv Card Electrophysiol 2006;15:175–8. [DOI] [PubMed] [Google Scholar]
- 460. Price JF, Kertesz NJ, Snyder CS, Friedman RA, Fenrich AL. Flecainide and sotalol: a new combination therapy for refractory supraventricular tachycardia in children <1 year of age. J Am Coll Cardiol 2002;39:517–20. [DOI] [PubMed] [Google Scholar]
- 461. Singh BN. Augmenting maintenance of sinus rhythm in the control of atrial fibrillation by antiarrhythmic drug combinations. J Cardiovasc Pharmacol Ther 2010;15:31S–5S. [DOI] [PubMed] [Google Scholar]
- 462. Purcaro A, Capestro F, Ciampani N, Fratadocchi GB, Mazzanti M, Costantini C et al. [Treatment of recurrent supraventricular tachyarrhythmias with flecainide or a flecainide and amiodarone combination]. G Ital Cardiol 1987;17:690–8. [PubMed] [Google Scholar]
- 463. Fenrich AL Jr, Perry JC, Friedman RA. Flecainide and amiodarone: combined therapy for refractory tachyarrhythmias in infancy. J Am Coll Cardiol 1995;25:1195–8. [DOI] [PubMed] [Google Scholar]
- 464. Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, Blomström-Lundqvist C, Cífková R, De Bonis M et al. 2018 ESC guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J 2018;39:3165–241. [DOI] [PubMed] [Google Scholar]
- 465. Tamirisa KP, Elkayam U, Briller JE, Mason PK, Pillarisetti J, Merchant FM et al. Arrhythmias in pregnancy. JACC Clin Electrophysiol 2022;8:120–35. [DOI] [PubMed] [Google Scholar]
- 466. Halpern DG, Weinberg CR, Pinnelas R, Mehta-Lee S, Economy KE, Valente AM. Use of medication for cardiovascular disease during pregnancy: JACC state-of-the-art review. J Am Coll Cardiol 2019;73:457–76. [DOI] [PubMed] [Google Scholar]
- 467. Lee SH, Chen SA, Wu TJ, Chiang CE, Cheng CC, Tai CT et al. Effects of pregnancy on first onset and symptoms of paroxysmal supraventricular tachycardia. Am J Cardiol 1995;76:675–8. [DOI] [PubMed] [Google Scholar]
- 468. Tanaka K, Tanaka H, Kamiya C, Katsuragi S, Sawada M, Tsuritani M et al. Beta-blockers and fetal growth restriction in pregnant women with cardiovascular disease. Circ J 2016;80:2221–6. [DOI] [PubMed] [Google Scholar]
- 469. Bateman BT, Heide-Jørgensen U, Einarsdóttir K, Engeland A, Furu K, Gissler M et al. β-blocker use in pregnancy and the risk for congenital malformations: an international cohort study. Ann Intern Med 2018;169:665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Ghosh N, Luk A, Derzko C, Dorian P, Chow C-M. The acute treatment of maternal supraventricular tachycardias during pregnancy: a review of the literature. J Obstet Gynaecol Can 2011;33:17–23. [DOI] [PubMed] [Google Scholar]
- 471. Enriquez AD, Economy KE, Tedrow UB. Contemporary management of arrhythmias during pregnancy. Circ Arrhythm Electrophysiol 2014;7:961–7. [DOI] [PubMed] [Google Scholar]
- 472. Ersbøll AS, Hedegaard M, Søndergaard L, Ersbøll M, Johansen M. Treatment with oral beta-blockers during pregnancy complicated by maternal heart disease increases the risk of fetal growth restriction. BJOG 2014;121:618–26. [DOI] [PubMed] [Google Scholar]
- 473. Jaeggi ET, Carvalho JS, De Groot E, Api O, Clur S-AB, Rammeloo L et al. Comparison of transplacental treatment of fetal supraventricular tachyarrhythmias with digoxin, flecainide, and sotalol: results of a nonrandomized multicenter study. Circulation 2011;124:1747–54. [DOI] [PubMed] [Google Scholar]
- 474. Ebenroth ES, Cordes TM, Darragh RK. Second-line treatment of fetal supraventricular tachycardia using flecainide acetate. Pediatr Cardiol 2001;22:483–7. [DOI] [PubMed] [Google Scholar]
- 475. Barnes EJ, Eben F, Patterson D. Direct current cardioversion during pregnancy should be performed with facilities available for fetal monitoring and emergency caesarean section. BJOG 2002;109:1406–7. [DOI] [PubMed] [Google Scholar]
- 476. Joglar JA, Page RL. Treatment of cardiac arrhythmias during pregnancy: safety considerations. Drug Saf 1999;20:85–94. [DOI] [PubMed] [Google Scholar]
- 477. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Rhythm Society. Circulation 2018;138:e272–391. [DOI] [PubMed] [Google Scholar]
- 478. Jeejeebhoy FM, Zelop CM, Lipman S, Carvalho B, Joglar J, Mhyre JM et al. Cardiac arrest in pregnancy: a scientific statement from the American Heart Association. Circulation 2015;132:1747–73. [DOI] [PubMed] [Google Scholar]
- 479. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M et al. Long QT syndrome and pregnancy. J Am Coll Cardiol 2007;49:1092–8. [DOI] [PubMed] [Google Scholar]
- 480. Richardson C, Silver ES. Management of supraventricular tachycardia in infants. Paediatr Drugs 2017;19:539–51. [DOI] [PubMed] [Google Scholar]
- 481. Krause U, Paul T, Bella PD, Gulletta S, Gebauer RA, Paech C et al. Pediatric catheter ablation at the beginning of the 21st century: results from the European Multicenter Pediatric Catheter Ablation Registry ‘EUROPA’. Europace 2021;23:431–40. [DOI] [PubMed] [Google Scholar]
- 482. Brugada J, Blom N, Sarquella-Brugada G, Blomstrom-Lundqvist C, Deanfield J, Janousek J et al. Pharmacological and non-pharmacological therapy for arrhythmias in the pediatric population: EHRA and AEPC-Arrhythmia Working Group joint consensus statement. Europace 2013;15:1337–82. [DOI] [PubMed] [Google Scholar]
- 483. Kehr J, Binfield A, Maxwell F, Hornung T, Skinner JR. Fascicular tachycardia in infancy and the use of verapamil: a case series and literature review. Arch Dis Child 2019;104:789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Devaprasath S, Buddhavarapu S, Mariam S, Krishna MR. Ivabradine monotherapy in congenital junctional ectopic tachycardia. Ann Pediatr Cardiol 2022;15:61–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Sanatani S, Potts JE, Reed JH, Saul JP, Stephenson EA, Gibbs KA et al. The study of antiarrhythmic medications in infancy (SAMIS): a multicenter, randomized controlled trial comparing the efficacy and safety of digoxin versus propranolol for prophylaxis of supraventricular tachycardia in infants. Circ Arrhythm Electrophysiol 2012;5:984–91. [DOI] [PubMed] [Google Scholar]
- 486. Wei N, Lamba A, Franciosi S, Law IH, Ochoa LA, Johnsrude CL et al. Medical management of infants with supraventricular tachycardia: results from a registry and review of the literature. CJC Pediatr Congenit Heart Dis 2022;1:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Mojahedi A, Mirshekari A. Evaluating antiarrhythmic drugs for managing infants with supraventricular tachycardia; a review. Am J Cardiovasc Dis 2024;14:144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Lim YT, Kim YH, Kwon JE. Effective control of supraventricular tachycardia in neonates may requires combination pharmacologic therapy. J Clin Med 2022;11:3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Aljohani OA, Herrick NL, Borquez AA, Shepard S, Wieler ME, Perry JC et al. Antiarrhythmic treatment duration and tachycardia recurrence in infants with supraventricular tachycardia. Pediatr Cardiol 2021;42:716–20. [DOI] [PubMed] [Google Scholar]
- 490. Oudijk MA, Ruskamp JM, Ambachtsheer BE, Ververs TFF, Stoutenbeek P, Visser GHA et al. Drug treatment of fetal tachycardias. Paediatr Drugs 2002;4:49–63. [DOI] [PubMed] [Google Scholar]
- 491. Jaeggi E, Öhman A. Fetal and neonatal arrhythmias. Clin Perinatol 2016;43:99–112. [DOI] [PubMed] [Google Scholar]
- 492. Bravo-Valenzuela NJ, Rocha LA, Machado Nardozza LM, Araujo Júnior E. Fetal cardiac arrhythmias: current evidence. Ann Pediatr Cardiol 2018;11:148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Yaksh A, van der Does LJ, Lanters EA, de Groot NM. Pharmacological therapy of tachyarrhythmias during pregnancy. Arrhythm Electrophysiol Rev 2016;5:41–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Kleinman CS, Nehgme RA. Cardiac arrhythmias in the human fetus. Pediatr Cardiol 2004;25:234–51. [DOI] [PubMed] [Google Scholar]
- 495. Munoz JL, Lewis AL, Song J, Ramsey PS. Fetal intervention for refractory supraventricular tachycardia complicated by hydrops fetalis. Case Rep Obstet Gynecol 2022;2022:5148250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Sridharan S, Sullivan I, Tomek V, Wolfenden J, Škovránek J, Yates R et al. Flecainide versus digoxin for fetal supraventricular tachycardia: comparison of two drug treatment protocols. Heart Rhythm 2016;13:1913–9. [DOI] [PubMed] [Google Scholar]
- 497. Alsaied T, Baskar S, Fares M, Alahdab F, Czosek RJ, Murad MH et al. First-line antiarrhythmic transplacental treatment for fetal tachyarrhythmia: a systematic review and meta-analysis. J Am Heart Assoc 2017;6:e007164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Paneni F, Diaz Cañestro C, Libby P, Lüscher TF, Camici GG. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol 2017;69:1952–67. [DOI] [PubMed] [Google Scholar]
- 499. Lakatta EG. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev 2002;7:29–49. [DOI] [PubMed] [Google Scholar]
- 500. Damluji AA, Forman DE, van Diepen S, Alexander KP, Page RL II, Hummel SL et al. Older adults in the cardiac intensive care unit: factoring geriatric syndromes in the management, prognosis, and process of care: a scientific statement from the American Heart Association. Circulation 2020;141:e6–32. [DOI] [PubMed] [Google Scholar]
- 501. Tamargo J, Kjeldsen KP, Delpón E, Semb AG, Cerbai E, Dobrev D et al. Facing the challenge of polypharmacy when prescribing for older people with cardiovascular disease. A review by the European Society of Cardiology Working Group on Cardiovascular Pharmacotherapy. Eur Heart J Cardiovasc Pharmacother 2022;8:406–19. [DOI] [PubMed] [Google Scholar]
- 502. Guthrie B, Makubate B, Hernandez-Santiago V, Dreischulte T. The rising tide of polypharmacy and drug-drug interactions: population database analysis 1995–2010. BMC Med 2015;13:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Dunlay SM, Chamberlain AM. Multimorbidity in older patients with cardiovascular disease. Curr Cardiovasc Risk Rep 2016;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Kuzuya M. Era of geriatric medical challenges: multimorbidity among older patients. Geriatr Gerontol Int 2019;19:699–704. [DOI] [PubMed] [Google Scholar]
- 505. Schwartz JB, Schmader KE, Hanlon JT, Abernethy DR, Gray S, Dunbar-Jacob J et al. Pharmacotherapy in older adults with cardiovascular disease: report from an american college of cardiology, american geriatrics society, and national institute on aging workshop. J Am Geriatr Soc 2019;67:371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Curtis AB, Karki R, Hattoum A, Sharma UC. Arrhythmias in patients ≥80 years of age: pathophysiology, management, and outcomes. J Am Coll Cardiol 2018;71:2041–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Salmela B, Jaakkola J, Kalatsova K, Inkovaara J, Aro AL, Teppo K et al. Sex- and age-specific differences in the use of antiarrhythmic therapies among atrial fibrillation patients: a nationwide cohort study. Europace 2024;26:euae264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Savelieva I, Fumagalli S, Kenny RA, Anker S, Benetos A, Boriani G et al. EHRA expert consensus document on the management of arrhythmias in frailty syndrome, endorsed by the Heart Rhythm Society (HRS), Asia Pacific Heart Rhythm Society (APHRS), Latin America Heart Rhythm Society (LAHRS), and Cardiac Arrhythmia Society of Southern Africa (CASSA). Europace 2023;25:1249–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Krahn AD, Connolly SJ, Roberts RS, Gent M; ATMA Investigators . Diminishing proportional risk of sudden death with advancing age: implications for prevention of sudden death. Am Heart J 2004;147:837–40. [DOI] [PubMed] [Google Scholar]
- 510. Santangeli P, Muser D, Maeda S, Filtz A, Zado ES, Frankel DS et al. Comparative effectiveness of antiarrhythmic drugs and catheter ablation for the prevention of recurrent ventricular tachycardia in patients with implantable cardioverter-defibrillators: a systematic review and meta-analysis of randomized controlled trials. Heart Rhythm 2016;13:1552–9. [DOI] [PubMed] [Google Scholar]
- 511. Aymanns C, Keller F, Maus S, Hartmann B, Czock D. Review on pharmacokinetics and pharmacodynamics and the aging kidney. Clin J Am Soc Nephrol 2010;5:314–27. [DOI] [PubMed] [Google Scholar]
- 512. Corsonello A, Pedone C, Incalzi RA. Age-related pharmacokinetic and pharmacodynamic changes and related risk of adverse drug reactions. Curr Med Chem 2010;17:571–84. [DOI] [PubMed] [Google Scholar]
- 513. Deneer VHM, van Hemel NM. Is antiarrhythmic treatment in the elderly different? A review of the specific changes. Drugs Aging 2011;28:617–33. [DOI] [PubMed] [Google Scholar]
- 514. Frishman WH, Aronow WS. Pharmacology of antiarrhythmic drugs in elderly patients. Clin Geriatr Med 2012;28:575–615. [DOI] [PubMed] [Google Scholar]
- 515. By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel . American Geriatrics Society 2023 updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2023;71:2052–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Tan JL, Eastment JG, Poudel A, Hubbard RE. Age-related changes in hepatic function: an update on implications for drug therapy. Drugs Aging 2015;32:999–1008. [DOI] [PubMed] [Google Scholar]
- 517. Shi S, Klotz U. Age-related changes in pharmacokinetics. Curr Drug Metab 2011;12:601–10. [DOI] [PubMed] [Google Scholar]
- 518. Drenth-van Maanen AC, Wilting I, Jansen PAF. Prescribing medicines to older people-how to consider the impact of ageing on human organ and body functions. Br J Clin Pharmacol 2020;86:1921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004;56:163–84. [DOI] [PubMed] [Google Scholar]
- 520. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013;138:103–41. [DOI] [PubMed] [Google Scholar]
- 521. Cheng JWM, Frishman WH, Aronow WS. Updates on cytochrome p450-mediated cardiovascular drug interactions. Dis Mon 2010;56:163–79. [DOI] [PubMed] [Google Scholar]
- 522. Zorzi A, Cipriani A, Corrado D. Anti-arrhythmic therapy in athletes. Pharmacol Res 2019;144:306–14. [DOI] [PubMed] [Google Scholar]
- 523. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association joint committee on clinical practice guidelines. J Am Coll Cardiol 2022;79:e263–421. [DOI] [PubMed] [Google Scholar]
- 524. Doval HC, Nul DR, Grancelli HO, Perrone SV, Bortman GR, Curiel R. Randomised trial of low-dose amiodarone in severe congestive heart failure. Grupo de Estudio de la Sobrevida en la Insuficiencia Cardiaca en Argentina (GESICA). Lancet 1994;344:493–8. [DOI] [PubMed] [Google Scholar]
- 525. Singh SN, Fletcher RD, Fisher SG, Singh BN, Lewis HD, Deedwania PC et al. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival trial of antiarrhythmic therapy in congestive heart failure. N Engl J Med 1995;333:77–82. [DOI] [PubMed] [Google Scholar]
- 526. Roy D, Talajic M, Dorian P, Connolly S, Eisenberg MJ, Green M et al. Amiodarone to prevent recurrence of atrial fibrillation. Canadian trial of atrial fibrillation investigators. N Engl J Med 2000;342:913–20. [DOI] [PubMed] [Google Scholar]
- 527. Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R et al. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med 2005;352:225–37. [DOI] [PubMed] [Google Scholar]
- 528. Torp-Pedersen C, Møller M, Bloch-Thomsen PE, Køber L, Sandøe E, Egstrup K et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish investigations of arrhythmia and mortality on Dofetilide Study Group. N Engl J Med 1999;341:857–65. [DOI] [PubMed] [Google Scholar]
- 529. Wolbrette DL, Hussain S, Maraj I, Naccarelli GV. A quarter of a century later: what is dofetilide’s clinical role today? J Cardiovasc Pharmacol Ther 2019;24:3–10. [DOI] [PubMed] [Google Scholar]
- 530. Marrouche NF, Brachmann J, Andresen D, Siebels J, Boersma L, Jordaens L et al. Catheter ablation for atrial fibrillation with heart failure. N Engl J Med 2018;378:417–27. [DOI] [PubMed] [Google Scholar]
- 531. Packer DL, Piccini JP, Monahan KH, Al-Khalidi HR, Silverstein AP, Noseworthy PA et al. Ablation versus drug therapy for atrial fibrillation in heart failure: results from the CABANA trial. Circulation 2021;143:1377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Prabhu S, Taylor AJ, Costello BT, Kaye DM, McLellan AJA, Voskoboinik A et al. Catheter ablation versus medical rate control in atrial fibrillation and systolic dysfunction: the CAMERA-MRI study. J Am Coll Cardiol 2017;70:1949–61. [DOI] [PubMed] [Google Scholar]
- 533. Roy D, Talajic M, Nattel S, Wyse DG, Dorian P, Lee KL et al. Rhythm control versus rate control for atrial fibrillation and heart failure. N Engl J Med 2008;358:2667–77. [DOI] [PubMed] [Google Scholar]
- 534. Sohns C, Fox H, Marrouche NF, Crijns HJGM, Costard-Jaeckle A, Bergau L et al. Catheter ablation in end-stage heart failure with atrial fibrillation. N Engl J Med 2023;389:1380–9. [DOI] [PubMed] [Google Scholar]
- 535. Serban T, Badertscher P, du Fay de Lavallaz J, Providencia R, Migliore F, Mugnai G et al. Definition and management of arrhythmia-induced cardiomyopathy: findings from the European Heart Rhythm Association survey. Europace 2024;26:euae112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Digitalis Investigation Group . The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 1997;336:525–33. [DOI] [PubMed] [Google Scholar]
- 537. Naccarelli GV, Dell’Orfano JT, Wolbrette DL, Patel HM, Luck JC. Cost-effective management of acute atrial fibrillation: role of rate control, spontaneous conversion, medical and direct current cardioversion, transesophageal echocardiography, and antiembolic therapy. Am J Cardiol 2000;85:36D–45D. [Google Scholar]
- 538. Galve E, Rius T, Ballester R, Artaza MA, Arnau JM, García-Dorado D et al. Intravenous amiodarone in treatment of recent-onset atrial fibrillation: results of a randomized, controlled study. J Am Coll Cardiol 1996;27:1079–82. [DOI] [PubMed] [Google Scholar]
- 539. Verrier RL, Silva AFG, Bonatti R, Batatinha JAP, Nearing BD, Liu G et al. Combined actions of ivabradine and ranolazine reduce ventricular rate during atrial fibrillation. J Cardiovasc Electrophysiol 2015;26:329–35. [DOI] [PubMed] [Google Scholar]
- 540. Filippatos G, Farmakis D. How to use beta-blockers in heart failure with reduced ejection fraction and atrial fibrillation. J Am Coll Cardiol 2017;69:2897–900. [DOI] [PubMed] [Google Scholar]
- 541. Al-Jazairi MIH, Nguyen B-O, De With RR, Smit MD, Weijs B, Hobbelt AH et al. Antiarrhythmic drugs in patients with early persistent atrial fibrillation and heart failure: results of the RACE 3 study. Europace 2021;23:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Kelly JP, DeVore AD, Wu J, Hammill BG, Sharma A, Cooper LB et al. Rhythm control versus rate control in patients with atrial fibrillation and heart failure with preserved ejection fraction: insights from get with the guidelines-heart failure. J Am Heart Assoc 2019;8:e011560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Machino-Ohtsuka T, Seo Y, Ishizu T, Yamamoto M, Hamada-Harimura Y, Machino T et al. Relationships between maintenance of sinus rhythm and clinical outcomes in patients with heart failure with preserved ejection fraction and atrial fibrillation. J Cardiol 2019;74:235–44. [DOI] [PubMed] [Google Scholar]
- 544. Sartipy U, Dahlström U, Fu M, Lund LH. Atrial fibrillation in heart failure with preserved, mid-range, and reduced ejection fraction. JACC Heart Fail 2017;5:565–74. [DOI] [PubMed] [Google Scholar]
- 545. Cadrin-Tourigny J, Wyse DG, Roy D, Blondeau L, Levesque S, Talajic M et al. Efficacy of amiodarone in patients with atrial fibrillation with and without left ventricular dysfunction: a pooled analysis of AFFIRM and AF-CHF trials. J Cardiovasc Electrophysiol 2014;25:1306–13. [DOI] [PubMed] [Google Scholar]
- 546. Neefs J, van den Berg NWE, Krul SPJ, Boekholdt SM, de Groot JR. Effect of spironolactone on atrial fibrillation in patients with heart failure with preserved ejection fraction: post-hoc analysis of the randomized, placebo-controlled TOPCAT trial. Am J Cardiovasc Drugs 2020;20:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Fernandes GC, Fernandes A, Cardoso R, Penalver J, Knijnik L, Mitrani RD et al. Association of SGLT2 inhibitors with arrhythmias and sudden cardiac death in patients with type 2 diabetes or heart failure: a meta-analysis of 34 randomized controlled trials. Heart Rhythm 2021;18:1098–105. [DOI] [PubMed] [Google Scholar]
- 548. Sfairopoulos D, Zhang N, Wang Y, Chen Z, Letsas KP, Tse G et al. Association between sodium-glucose cotransporter-2 inhibitors and risk of sudden cardiac death or ventricular arrhythmias: a meta-analysis of randomized controlled trials. Europace 2022;24:20–30. [DOI] [PubMed] [Google Scholar]
- 549. Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation 1992;86:29–37. [DOI] [PubMed] [Google Scholar]
- 550. Mahida S, Venlet J, Saguner AM, Kumar S, Baldinger SH, AbdelWahab A et al. Ablation compared with drug therapy for recurrent ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy: results from a multicenter study. Heart Rhythm 2019;16:536–43. [DOI] [PubMed] [Google Scholar]
- 551. Preston CL (ed.), Stockley’s Drug Interactions. 12th ed. London: Pharmaceutical Press; 2019. [Google Scholar]
- 552. Tamargo J, Caballero R, Delpón E. Chapter 8.1 cardiovascular drugs—from A to Z. In: Kaski JC, Kjeldsen KP (eds.), The ESC Handbook on Cardiovascular Pharmacotherapy. Oxford: Oxford University Press; 2019. p413–809. [Google Scholar]
- 553. Klotz U. Antiarrhythmics: elimination and dosage considerations in hepatic impairment. Clin Pharmacokinet 2007;46:985–96. [DOI] [PubMed] [Google Scholar]
- 554. Darbar D, Roden DM. Pharmacogenetics of antiarrhythmic therapy. Expert Opin Pharmacother 2006;7:1583–90. [DOI] [PubMed] [Google Scholar]
- 555. Khairy P, Harris L, Landzberg MJ, Fernandes SM, Barlow A, Mercier L-A et al. Sudden death and defibrillators in transposition of the great arteries with intra-atrial baffles: a multicenter study. Circ Arrhythm Electrophysiol 2008;1:250–7. [DOI] [PubMed] [Google Scholar]
- 556. Thorne SA, Barnes I, Cullinan P, Somerville J. Amiodarone-associated thyroid dysfunction: risk factors in adults with congenital heart disease. Circulation 1999;100:149–54. [DOI] [PubMed] [Google Scholar]
- 557. Mazzanti A, Maragna R, Vacanti G, Monteforte N, Bloise R, Marino M et al. Interplay between genetic substrate, QTc duration, and arrhythmia risk in patients with long QT syndrome. J Am Coll Cardiol 2018;71:1663–71. [DOI] [PubMed] [Google Scholar]
- 558. Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol 2016;67:1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. El-Battrawy I, Besler J, Li X, Lan H, Zhao Z, Liebe V et al. Impact of antiarrhythmic drugs on the outcome of short QT syndrome. Front Pharmacol 2019;10:771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Crotti L, Brugada P, Calkins H, Chevalier P, Conte G, Finocchiaro G et al. From gene-discovery to gene-tailored clinical management: 25 years of research in channelopathies and cardiomyopathies. Europace 2023;25:euad180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Malhi N, Cheung CC, Deif B, Roberts JD, Gula LJ, Green MS et al. Challenge and impact of quinidine access in sudden death syndromes: a national experience. JACC Clin Electrophysiol 2019;5:376–82. [DOI] [PubMed] [Google Scholar]
- 562. Ohgo T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T et al. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm 2007;4:695–700. [DOI] [PubMed] [Google Scholar]
- 563. Padfield GJ, AlAhmari L, Lieve KVV, AlAhmari T, Roston TM, Wilde AA et al. Flecainide monotherapy is an option for selected patients with catecholaminergic polymorphic ventricular tachycardia intolerant of β-blockade. Heart Rhythm 2016;13:609–13. [DOI] [PubMed] [Google Scholar]
- 564. Aizawa Y, Chinushi M, Hasegawa K, Naiki N, Horie M, Kaneko Y et al. Electrical storm in idiopathic ventricular fibrillation is associated with early repolarization. J Am Coll Cardiol 2013;62:1015–9. [DOI] [PubMed] [Google Scholar]
- 565. Haïssaguerre M, Sacher F, Nogami A, Komiya N, Bernard A, Probst V et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J Am Coll Cardiol 2009;53:612–9. [DOI] [PubMed] [Google Scholar]
- 566. Ferri N, Colombo E, Tenconi M, Baldessin L, Corsini A. Drug-drug interactions of direct oral anticoagulants (DOACs): from pharmacological to clinical practice. Pharmaceutics 2022;14:1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S et al. Drug-drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs 2013;13:331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Konieczny KM, Dorian P. Clinically important drug-drug interactions between antiarrhythmic drugs and anticoagulants. J Innov Card Rhythm Manag 2019;10:3552–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569. Wiggins BS, Dixon DL, Neyens RR, Page RL II, Gluckman TJ. Select drug-drug interactions with direct oral anticoagulants: JACC review topic of the week. J Am Coll Cardiol 2020;75:1341–50. [DOI] [PubMed] [Google Scholar]
- 570. Goldschlager N, Epstein A, Friedman P, Gang E, Krol R, Olshansky B et al. Environmental and drug effects on patients with pacemakers and implantable cardioverter/defibrillators: a practical guide to patient treatment. Arch Intern Med 2001;161:649–55. [DOI] [PubMed] [Google Scholar]
- 571. Hook BG, Perlman RL, Callans DJ, Hanna MS, Kleiman RB, Flores BT et al. Acute and chronic cycle length dependent increase in ventricular pacing threshold. Pacing Clin Electrophysiol 1992;15:1437–44. [DOI] [PubMed] [Google Scholar]
- 572. Brode SE, Schwartzman D, Callans DJ, Gottlieb CD, Marchlinski FE. ICD-antiarrhythmic drug and ICD-pacemaker interactions. J Cardiovasc Electrophysiol 1997;8:830–42. [DOI] [PubMed] [Google Scholar]
- 573. Rajawat YS, Patel VV, Gerstenfeld EP, Nayak H, Marchlinski FE. Advantages and pitfalls of combining device-based and pharmacologic therapies for the treatment of ventricular arrhythmias: observations from a tertiary referral center. Pacing Clin Electrophysiol 2004;27:1670–81. [DOI] [PubMed] [Google Scholar]
- 574. Van Herendael H, Pinter A, Ahmad K, Korley V, Mangat I, Dorian P. Role of antiarrhythmic drugs in patients with implantable cardioverter defibrillators. Europace 2010;12:618–25. [DOI] [PubMed] [Google Scholar]
- 575. Baquero GA, Banchs JE, Depalma S, Young SK, Penny-Peterson ED, Samii SM et al. Dofetilide reduces the frequency of ventricular arrhythmias and implantable cardioverter defibrillator therapies. J Cardiovasc Electrophysiol 2012;23:296–301. [DOI] [PubMed] [Google Scholar]
- 576. Dorian P, Borggrefe M, Al-Khalidi HR, Hohnloser SH, Brum JM, Tatla DS et al. Placebo-controlled, randomized clinical trial of azimilide for prevention of ventricular tachyarrhythmias in patients with an implantable cardioverter defibrillator. Circulation 2004;110:3646–54. [DOI] [PubMed] [Google Scholar]
- 577. Zareba W, Daubert JP, Beck CA, Huang DT, Alexis JD, Brown MW et al. Ranolazine in high-risk patients with implanted cardioverter-defibrillators: the RAID trial. J Am Coll Cardiol 2018;72:636–45. [DOI] [PubMed] [Google Scholar]
- 578. Kowey PR, Crijns HJGM, Aliot EM, Capucci A, Kulakowski P, Radzik D et al. Efficacy and safety of celivarone, with amiodarone as calibrator, in patients with an implantable cardioverter-defibrillator for prevention of implantable cardioverter-defibrillator interventions or death: the ALPHEE study. Circulation 2011;124:2649–60. [DOI] [PubMed] [Google Scholar]
- 579. Zipes DP, Jalife J. Antiarrhythmic drug-implantable cardioverter/defibrillator interactions. In: Zipes DP, Jalife J (eds.), Cardiac Electrophysiology: From Cell to Bedside. 2nd ed. Philadelphia: W. B. Saunders Co.; 1995. p1426–33. [Google Scholar]
- 580. Turco P, De Simone A, La Rocca V, Iuliano A, Capuano V, Astarita C et al. Antiarrhythmic drug therapy after radiofrequency catheter ablation in patients with atrial fibrillation. Pacing Clin Electrophysiol 2007;30 Suppl 1:S112–115. [DOI] [PubMed] [Google Scholar]
- 581. Roux J-F, Zado E, Callans DJ, Garcia F, Lin D, Marchlinski FE et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study). Circulation 2009;120:1036–40. [DOI] [PubMed] [Google Scholar]
- 582. Leong-Sit P, Roux J-F, Zado E, Callans DJ, Garcia F, Lin D et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study): six-month follow-up study. Circ Arrhythm Electrophysiol 2011;4:11–4. [DOI] [PubMed] [Google Scholar]
- 583. Gu J, Liu X, Tan H, Zhou L, Gu J, Jiang W et al. Extensive antiarrhythmic drugs after catheter ablation of persistent atrial fibrillation. Acta Cardiol 2012;67:407–14. [DOI] [PubMed] [Google Scholar]
- 584. Noseworthy PA, Van Houten HK, Sangaralingham LR, Deshmukh AJ, Kapa S, Mulpuru SK et al. Effect of antiarrhythmic drug initiation on readmission after catheter ablation for atrial fibrillation. JACC Clin Electrophysiol 2015;1:238–44. [DOI] [PubMed] [Google Scholar]
- 585. Duytschaever M, Demolder A, Phlips T, Sarkozy A, El Haddad M, Taghji P et al. Pulmonary vein isolation with vs. without continued antiarrhythmic drug treatment in subjects with recurrent atrial fibrillation (POWDER AF): results from a multicentre randomized trial. Eur Heart J 2018;39:1429–37. [DOI] [PubMed] [Google Scholar]
- 586. Sohns C, von Gruben V, Sossalla S, Bergau L, Seegers J, Lüthje L et al. Antiarrhythmic drug therapy for maintaining sinus rhythm early after pulmonary vein ablation in patients with symptomatic atrial fibrillation. Cardiovasc Ther 2014;32:7–12. [DOI] [PubMed] [Google Scholar]
- 587. Schleberger R, Metzner A, Kuck K-H, Andresen D, Willems S, Hoffmann E et al. Antiarrhythmic drug therapy after catheter ablation for atrial fibrillation-insights from the German Ablation Registry. Pharmacol Res Perspect 2021;9:e00880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Tzeis S, Gerstenfeld EP, Kalman J, Saad EB, Sepehri Shamloo A, Andrade JG et al. 2024 European Heart Rhythm Association/Heart Rhythm Society/Asia Pacific Heart Rhythm Society/Latin American Heart Rhythm Society expert consensus statement on catheter and surgical ablation of atrial fibrillation. Europace 2024;26:euae043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Reiffel JA. Atrial fibrillation: why are we hiding reality? Circulation 2024;149:979–80. [DOI] [PubMed] [Google Scholar]
- 590. Guarnieri T, Tomaselli G, Griffith LS, Brinker J. The interaction of antiarrhythmic drugs and the energy for cardioversion of chronic atrial fibrillation. Pacing Clin Electrophysiol 1991;14:1007–12. [DOI] [PubMed] [Google Scholar]
- 591. Peyronnet R, Ravens U. Atria-selective antiarrhythmic drugs in need of alliance partners. Pharmacol Res 2019;145:104262. [DOI] [PubMed] [Google Scholar]
- 592. Saljic A, Heijman J, Dobrev D. Recent advances in antiarrhythmic drug therapy. Drugs 2023;83:1147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Stambler BS, Dorian P, Sager PT, Wight D, Douville P, Potvin D et al. Etripamil nasal spray for rapid conversion of supraventricular tachycardia to sinus rhythm. J Am Coll Cardiol 2018;72:489–97. [DOI] [PubMed] [Google Scholar]
- 594. Stambler BS, Plat F, Sager PT, Shardonofsky S, Wight D, Potvin D et al. First Randomized, multicenter, placebo-controlled study of self-administered intranasal etripamil for acute conversion of spontaneous paroxysmal supraventricular tachycardia (NODE-301). Circ Arrhythm Electrophysiol 2022;15:e010915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Ip JE, Coutu B, Bennett MT, Pandey AS, Stambler BS, Sager P et al. Etripamil nasal spray for conversion of repeated spontaneous episodes of paroxysmal supraventricular tachycardia during long-term follow-up: results from the NODE-302 study. J Am Heart Assoc 2023;12:e028227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Stambler BS, Camm AJ, Alings M, Dorian P, Heidbuchel H, Houtgraaf J et al. Self-administered intranasal etripamil using a symptom-prompted, repeat-dose regimen for atrioventricular-nodal-dependent supraventricular tachycardia (RAPID): a multicentre, randomised trial. Lancet 2023;402:118–28. [DOI] [PubMed] [Google Scholar]
- 597. Camm AJ, Piccini JP, Alings M, Dorian P, Gosselin G, Guertin M-C et al. Multicenter, phase 2, randomized controlled study of the efficacy and safety of etripamil nasal spray for the acute reduction of rapid ventricular rate in patients with symptomatic atrial fibrillation (ReVeRA-201). Circ Arrhythm Electrophysiol 2023;16:639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Crijns HJGM, Elvan A, Al-Windy N, Tuininga YS, Badings E, Aksoy I et al. Open-label, multicenter study of flecainide acetate oral inhalation solution for acute conversion of recent-onset, symptomatic atrial fibrillation to sinus rhythm. Circ Arrhythm Electrophysiol 2022;15:e010204. [DOI] [PubMed] [Google Scholar]
- 599. Ruskin JN, Camm AJ, Dufton C, Woite-Silva AC, Tuininga Y, Badings E et al. Orally inhaled flecainide for conversion of atrial fibrillation to sinus rhythm: INSTANT phase 2 trial. JACC Clin Electrophysiol 2024;10:1021–33. [DOI] [PubMed] [Google Scholar]
- 600. Rienstra M, Kuijper A, Eijsbouts S, Kraaier K, Janota T, Van Ofwegen C et al. Flecainide acetate inhalation solution for cardioversion of recent-onset, symptomatic atrial fibrillation: results of the phase 3 RESTORE-1 trial. Eur Heart J 2024;45:ehae666.519. [Google Scholar]
- 601. Diness JG, Abildgaard L, Bomholtz SH, Skarsfeldt MA, Edvardsson N, Sørensen US et al. Inhibition of KCa2 channels decreased the risk of ventricular arrhythmia in the guinea pig heart during induced hypokalemia. Front Pharmacol 2020;11:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Diness JG, Kirchhoff JE, Speerschneider T, Abildgaard L, Edvardsson N, Sørensen US et al. The KCa2 channel inhibitor AP30663 selectively increases atrial refractoriness, converts vernakalant-resistant atrial fibrillation and prevents its reinduction in conscious pigs. Front Pharmacol 2020;11:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603. Gal P, Klaassen ES, Bergmann KR, Saghari M, Burggraaf J, Kemme MJB et al. First clinical study with AP30663—a KCa 2 channel inhibitor in development for conversion of atrial fibrillation. Clin Transl Sci 2020;13:1336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. Acesion Pharma . A Double-Blind, Randomised, Placebo-Controlled, Parallel-Group Study of AP30663 Given Intravenously for Cardioversion in Patients With Atrial Fibrillation. clinicaltrials.gov; 2023 Apr. Report No.: NCT04571385.
- 605. Mason JW, Elliott GT, Romano SJ, Mendzelevski B, Allgren R, Gillings M et al. HBI-3000: a novel drug for conversion of atrial fibrillation—phase 1 study results. Circulation 2019;140:A11495–A11495. [Google Scholar]
- 606. Guo D, Liu Q, Liu T, Elliott G, Gingras M, Kowey PR et al. Electrophysiological properties of HBI-3000: a new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes. J Cardiovasc Pharmacol 2011;57:79–85. [DOI] [PubMed] [Google Scholar]
- 607. Schmidt C, Wiedmann F, Zhou X-B, Heijman J, Voigt N, Ratte A et al. Inverse remodelling of K2P3.1 K+ channel expression and action potential duration in left ventricular dysfunction and atrial fibrillation: implications for patient-specific antiarrhythmic drug therapy. Eur Heart J 2017;38:1764–74. [DOI] [PubMed] [Google Scholar]
- 608. Wiedmann F, Beyersdorf C, Zhou XB, Kraft M, Paasche A, Jávorszky N et al. Treatment of atrial fibrillation with doxapram: TASK-1 potassium channel inhibition as a novel pharmacological strategy. Cardiovasc Res 2022;118:1728–41. [DOI] [PubMed] [Google Scholar]
- 609. Kraft M, Büscher A, Wiedmann F, L’hoste Y, Haefeli WE, Frey N et al. Current drug treatment strategies for atrial fibrillation and TASK-1 inhibition as an emerging novel therapy option. Front Pharmacol 2021;12:638445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610. Aleong RG, Sauer WH, Davis G, Murphy GA, Port JD, Anand IS et al. Prevention of atrial fibrillation by bucindolol is dependent on the beta₁389 Arg/Gly adrenergic receptor polymorphism. JACC Heart Fail 2013;1:338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611. Piccini JP, Abraham WT, Dufton C, Carroll IA, Healey JS, van Veldhuisen DJ et al. Bucindolol for the maintenance of sinus rhythm in a genotype-defined HF population: the GENETIC-AF trial. JACC Heart Fail 2019;7:586–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612. Piccini JP, Dufton C, Carroll IA, Healey JS, Abraham WT, Khaykin Y et al. Bucindolol decreases atrial fibrillation burden in patients with heart failure and the ADRB1 Arg389Arg genotype. Circ Arrhythm Electrophysiol 2021;14:e009591. [DOI] [PubMed] [Google Scholar]
- 613. Abraham WT, Piccini JP, Dufton C, Carroll IA, Healey JS, O’Connor CM et al. Dose-limiting, adverse event-associated bradycardia with β-blocker treatment of atrial fibrillation in the GENETIC-AF trial. Heart Rhythm O2 2022;3:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Ezekowitz MD, Nagarakanti R, Lubinski A, Bandman O, Canafax D, Ellis DJ et al. A randomized trial of budiodarone in paroxysmal atrial fibrillation. J Interv Card Electrophysiol 2012;34:1–9. [DOI] [PubMed] [Google Scholar]
- 615. Zhang D, Wu C-T, Qi X, Meijering RAM, Hoogstra-Berends F, Tadevosyan A et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014;129:346–58. [DOI] [PubMed] [Google Scholar]
- 616. Roy D, Pratt CM, Torp-Pedersen C, Wyse DG, Toft E, Juul-Moller S et al. Vernakalant hydrochloride for rapid conversion of atrial fibrillation: a phase 3, randomized, placebo-controlled trial. Circulation 2008;117:1518–25. [DOI] [PubMed] [Google Scholar]
- 617. Kowey PR, Dorian P, Mitchell LB, Pratt CM, Roy D, Schwartz PJ et al. Vernakalant hydrochloride for the rapid conversion of atrial fibrillation after cardiac surgery: a randomized, double-blind, placebo-controlled trial. Circ Arrhythm Electrophysiol 2009;2:652–9. [DOI] [PubMed] [Google Scholar]
- 618. Pratt CM, Roy D, Torp-Pedersen C, Wyse DG, Toft E, Juul-Moller S et al. Usefulness of vernakalant hydrochloride injection for rapid conversion of atrial fibrillation. Am J Cardiol 2010;106:1277–83. [DOI] [PubMed] [Google Scholar]
- 619. Stiell IG, Roos JS, Kavanagh KM, Dickinson G. A multicenter, open-label study of vernakalant for the conversion of atrial fibrillation to sinus rhythm. Am Heart J 2010;159:1095–101. [DOI] [PubMed] [Google Scholar]
- 620. Tsiachris D, Doundoulakis I, Pagkalidou E, Kordalis A, Deftereos S, Gatzoulis KA et al. Pharmacologic cardioversion in patients with paroxysmal atrial fibrillation: a network meta-analysis. Cardiovasc Drugs Ther 2021;35:293–308. [DOI] [PubMed] [Google Scholar]
- 621. Alboni P, Botto GL, Baldi N, Luzi M, Russo V, Gianfranchi L et al. Outpatient treatment of recent-onset atrial fibrillation with the “pill-in-the-pocket” approach. N Engl J Med 2004;351:2384–91. [DOI] [PubMed] [Google Scholar]
- 622. Hauser TH, Pinto DS, Josephson ME, Zimetbaum P. Safety and feasibility of a clinical pathway for the outpatient initiation of antiarrhythmic medications in patients with atrial fibrillation or atrial flutter. Am J Cardiol 2003;91:1437–41. [DOI] [PubMed] [Google Scholar]
- 623. Steeds RP, Birchall AS, Smith M, Channer KS. An open label, randomised, crossover study comparing sotalol and atenolol in the treatment of symptomatic paroxysmal atrial fibrillation. Heart 1999;82:170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624. Turner N, Thwaites BC. Exercise induced widening of the QRS complex in a patient on flecainide. Heart 2001;85:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625. Lehmann MH, Hardy S, Archibald D, quart B, MacNeil DJ. Sex difference in risk of torsade de pointes with d,l-sotalol. Circulation 1996;94:2535–41. [DOI] [PubMed] [Google Scholar]
- 626. Gössinger HD, Siostrzonek P, Mösslacher H. Combined sotalol and flecainide given at low dosage in patients with the Wolff-Parkinson-White syndrome. Int J Cardiol 1990;26:380–2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.