

Abstract
Purpose
To detect alterations in amacrine cells associated with retinal ganglion cell (RGC) depletion caused by experimental optic nerve transection and glaucoma.
Methods
Intraocular pressure (IOP) was elevated unilaterally in 18 rats by translimbal trabecular laser treatment, and eyes were studied at 1 (n = 6), 2 (n = 5), and 3 (n = 7) months. Complete optic nerve transection was performed unilaterally in nine rats with survival for 1 (n = 4) and 3 (n = 5) months. Serial cryosections (five per eye) were immunohistochemically labeled with rabbit anti-γ-aminobutyric acid (GABA) and anti-glycine antibodies. Cells in the ganglion cell and inner nuclear layers that labeled for GABA or glycine were counted in a masked fashion under bright-field microscopy. Additional labeling with other RGC and amacrine antigens was also performed. RGC loss was quantified by axon counts.
Results
Amacrine cells identified by GABA and glycine labeling were not significantly affected by experimental glaucoma, with a mean decrease of 15% compared with bilaterally untreated control cells (557 ± 186 neurons/mm [glaucoma] versus 653.9 ± 114.4 neurons/mm [control] of retina; P = 0.15, t-test). There was no significant trend for amacrine cell counts to be lower in eyes with fewer RGCs (r = −0.39, P = 0.11). By contrast, there was highly significant loss of GABA and glycine staining 3 months after nerve transection, both in the treated and the fellow eyes (P < 0.0001, t-test). However, there was a substantial number of remaining amacrine cells in transected retinas, as indicated by labeling for calretinin and calbindin.
Conclusions
Experimental glaucoma causes minimal change in amacrine cells and their expression of neurotransmitters. After nerve transection, neurotransmitter presence declines, but many amacrine cell bodies remain. Differences among optic nerve injury models, as well as effects on “untreated” fellow eyes, should be recognized.
A macrine cells of the vertebrate retina are interneurons that modify the visual signal as it moves through the photoreceptor-bipolar-retinal ganglion cell (RGC) chain. They are synaptically active in the inner plexiform layer (IPL), with cell bodies in both the inner portion of the inner nuclear layer (INL) and in the ganglion cell layer (GCL). Amacrine cells integrate, modulate, and interpose a temporal domain in the visual message presented to the RGC.1 Amacrine cells are the most diverse retinal neuronal type, with more than 20 classes, based on size, shape, stratification pattern, and neurotransmitter.2 Most amacrine cells are inhibitory neurons, using the inhibitory neurotransmitters γ-aminobutyric acid (GABA) or glycine. Amacrine cells expressing either GABA or glycine comprise 95% of all amacrine cells in the retina.3 Among human retinal amacrine cells, the GABAergic comprise 55% and the glycinergic 40% of the total.
Several studies have evaluated the effect of RGC depletion on amacrine cells, but most research has focused on the neonatal eye. GABAergic and glycinergic amacrine cell distribution was altered by unilateral removal at birth of the superior colliculus, pretectum, and optic tract in rats.4 This experiment found a decrease in the number of INL cells immunoreactive for the GABA isoforms GAD-65 and GAD-67. At the same time, cells expressing the GAD-65 isoform increased in the GCL. In two other investigations, 1-day-old rats underwent unilateral optic nerve transection with survival for 1 to 2 months. There was no change in labeling for tyrosine hydroxylase, choline acetyltransferase, and substance P among amacrine cells.5,6 In the neonatal ferret, early RGC elimination caused a 25% reduction in presumed amacrine cells that were immunoreactive for glycine transporter and a 34% reduction in GABAergic amacrine cells.7
Few studies have specifically quantified a potential change in amacrine cells after optic nerve injury in adult animals or humans. Dijk et al.8 studied the effects of ischemia on amacrine cell subtype-specific transcript levels in the adult rat retina by real-time quantitative polymerase chain reaction. By 4 weeks after ischemic injury, glycinergic amacrine cell markers recovered to 65% to 75% of the normal control level, whereas GABAergic markers completely recovered. There was a substantial reduction 4 weeks after ischemia in cells labeling for parvalbumin, glycine transporter 1, and choline acetyltransferase.9
To our knowledge, there are no studies on the effect of amacrine cells by transection or crush of the nerve. In studies of experimental and human glaucoma, there are qualitative and some quantitative studies of amacrine cells, but the data are modest. Vickers et al.10 and Harwerth et al.11 examined a small number of retinal specimens from monkeys with experimental glaucoma and detected no loss of amacrine cells. A report of another study of four glaucomatous monkey eyes stated that there was a qualitative loss of presumed amacrine cells labeling for NADPH-diaphorase.12 May and Mittag13 studied amacrine cells in DBA/2NNia (DBA) mice with a condition exhibiting some features of glaucoma. Amacrine cell subpopulations had no significant staining differences compared with nonglaucomatous mice when antibodies against tyrosine hydroxylase, GABA, and vesicular acetylcholine transporter (VAChT) were used. Moon et al.14 labeled amacrine cells in normal mice and in DBA/2J mice GABA, glycine and choline acetyltransferase, and nitric oxide synthase. They observed no changes in the population of glycine-immunoreactive cells, but reported increases in some GABA-labeled cells in one layer and decreases in another. No statistical comparisons were presented. In the past, our laboratory has shown preservation of photoreceptor and INL cells in rat,15 monkey,16 and human17 glaucoma eyes, but we did not attempt quantification of amacrine cells. Whether amacrine cells are affected by glaucoma is still a controversial issue.
Thus, it is important to study amacrine cells in a mature animal model and to determine quantitatively whether RGC loss results in amacrine cell death. It may be that concomitant loss of amacrine cells would potentiate secondary degeneration of RGC in diseases such as glaucoma and other optic neuropathies.18–20 Amacrine cells frequently synapse with RGCs and may provide some trophic or functional support to them. In addition, there are initial suggestions that progenitor cells may be used to replace lost RGCs in eye diseases, including glaucoma.21 If neuronal replacement is to be contemplated, it is important to establish how many cell types are needed.
Our hypothesis is that RGC elimination alters the number or functional status of amacrine cells in the adult rat retina. We sought to identify a large proportion of amacrine cells by immunohistochemically labeling them for GABA and glycine. Investigators in earlier studies, who used antibodies against other molecules, were able to study only particular subpopulations of amacrine cells. We used two rat models known to affect RGCs: surgical optic nerve transection, in which severe ganglion cell loss occurs, and experimental glaucoma, which was produced by translimbal laser photocoagulation of the trabecular meshwork, which produces variable ganglion cell loss.
Methods
Animals
Thirty-three male Wistar rats (375–425 g) were treated in accordance with the ARVO Statement for Use of Animals in Ophthalmic and Vision Research, in protocols approved and monitored by the Animal Care Committee of the Johns Hopkins University School of Medicine. Animals were housed with a 14-hour light/10-hour dark cycle, with standard chow and water available ad libitum.
Experimental Glaucoma
Elevated intraocular pressure (IOP) was induced in one eye of 18 rats by treating the aqueous outflow area in an external approach with a 532-nm diode laser, in a protocol developed by Levkovitch-Verbin et al.15 Briefly, animals were anesthetized with intraperitoneal ketamine (75 mg/kg) and xylazine (5 mg/kg) and topical proparacaine 1% eye drops. Laser energy was delivered to the trabecular meshwork at the slit lamp perpendicular to the trabeculae and parallel to the iris. Initial treatment was 40 to 50 spots of 50-μm size, 0.4 W, and 0.6-second duration. Treatment was repeated at 1 week if the difference in the IOP between the two eyes was less than 6 mm Hg. IOP was measured in rats under anesthesia as the average of 10 readings with a handheld tonometer (Tonopen XL; Mentor Ophthalmics, Norwell, MA). IOP measurements were taken immediately before laser treatment, 1 day after, and then weekly for the duration of the experiment. Rats were killed for immunohistochemical analysis at 1, 2, and 3 months after the first laser treatment (five to seven rats in each group).
Rat Optic Nerve Transection
Optic nerve transection was performed unilaterally in 10 male Wistar rats under anesthesia with intraperitoneal ketamine (75 mg/kg) and xylazine (5 mg/kg) and topical proparacaine 1% eye drops. With an operating microscope, the superior conjunctiva was incised, the muscles and connective tissue were separated, and the intraorbital optic nerve was exposed. The optic nerve was transected with a diamond knife approximately 2 mm behind the globe, with care taken to avoid occlusion of the central retinal artery. The retina was examined ophthalmoscopically to assure blood vessel integrity. Erythromycin ointment was applied to the eye with the transected nerve. Five rats each were killed at 1 month and 3 months after transection (data were not obtained from one rat in the 1-month group because of poor tissue preservation). Five untreated rats served as bilateral control subjects.
Preparation of Tissues
Rats were killed by exsanguination while under deep ketamine/xylazine anesthesia. Intracardiac perfusion was performed with 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) at a rate of 20 mL/min for 20 minutes. Both eyes were enucleated, and optic nerves were postfixed in 4% paraformaldehyde before processing for grading and quantification of axonal loss. After removal of the anterior segments, eyes were processed by serial exposure to 2% paraformaldehyde in 5% sucrose in 0.2 M phosphate buffer (pH 7.2), for 60 minutes. They were taken through graded sucrose solutions, 5%, 10%, 12.5%, and 15%, for 30 minutes each, and then placed in 20% sucrose overnight. Eye cups were embedded in optimal cutting temperature (OCT) compound (Sakura Finetek USA, Torrance, CA). Cryosections 8 μm thick were collected onto slides (SuperFrost Plus; Fisher Scientific, Pittsburgh, PA) and stored at −80°C before immunolabeling.
Immunohistochemistry
Primary antibodies to detect amacrine cells were rabbit anti-GABA (Calbiochem/Oncogene Research Products, San Diego, CA), rabbit anti-glycine (Chemicon International, Temecula, CA), rabbit anti-calbindin (Calbiochem/Oncogene Research Products), and rabbit anti-calretinin (Calbiochem/Oncogene Research Products). Retinal ganglion cells were identified by using a mouse anti-Thy1.1 primary antibody (Chemicon International). Primary antibodies were used at a dilution of 1:50 for both anti-GABA and anti-glycine, 1:100 for anti-calbindin, 1:3000 for anti-calretinin, and 1:500 for anti-Thy1.1. Cryosections were fixed in methanol at −20°C for 5 minutes and washed with Tris-buffered saline (TBS) containing 0.3% Triton X-100, with the exception of anti-Thy1.1 for which the methanol step was omitted. Endogenous peroxidase activity was quenched by immersion in 3% hydrogen peroxide for 15 minutes. Nonspecific binding was blocked with 10% normal goat serum in TBS and 0.3% Triton X-100 for 1 hour before incubation with primary antibody overnight at 4°C in a humid chamber. Sections were incubated at 1:500 with biotinylated goat anti-rabbit secondary antibody (Kirkegaard and Perry Laboratories, Inc., Gaithersburg, MD) for anti-GABA and anti-glycine or biotinylated goat anti-mouse secondary (Kirkegaard and Perry Laboratories, Inc.) for anti-Thy1.1. Slides were washed and incubated with the tertiary reagent from the avidin-biotin complex (ABC) kit, using 3-amino-9-ethylcarbazole as the chromogen. Only sections stained for rabbit anti-GABA and rabbit anti-glycine were counterstained with Harris’ hematoxylin stain (Polysciences Inc., Warrington, PA). All sections were mounted with Kaiser’s glycerol jelly. Negative control experiments included nonimmune serum of the same species as the primary antibody at the same protein concentration and incubation in buffer alone. Images of slides were captured digitally with standardized microscope and camera settings (Axioskop and Axiocam with AxioVision ver. 3.1 Software; Carl Zeiss Meditec, Inc., Dublin, CA).
RGC Axon-Counting Procedure
A 1-mm thick cross-section of the optic nerve from both eyes of each animal was removed beginning 1.5 mm posterior to the eye, postfixed in 1% osmium tetroxide in phosphate buffer, processed into epoxy resin, sectioned at 1-μm thickness, and stained with 1% toluidine blue. The total nerve fiber count in treated eyes was estimated for each animal from these tissue sections by measuring the overall nerve area, determining the density of axons at high power in 10 randomly selected areas15 (MetaMorph System; Universal Imaging Corp., West Chester, PA), and multiplying density times area to yield axon count. For this study, the number of axons in treated glaucomatous eyes was compared with the mean axon estimate (85,511 ± 11,040 [SD]) from 205 normal Wistar rat nerves collected as part of other studies and counted in an identical fashion.
Immunohistochemical Amacrine Cell Counting
Five cryosections from each rat eye, sectioned at 8 μm, underwent immunohistochemical staining with both the rabbit anti-GABA and anti-glycine primary antibodies. The first section was representative of the first 50 to 100 μm of the retina, and each successive section was 100 μm deeper, to ensure that the sections were representative of a broad sample of the rat retina. The immunolabeled sections were counted under 40× magnification with a bright-field microscope. Only cells that were clearly immunostained for GABA or glycine and had a visible, hematoxylin-stained nucleus were identified as GABA- or glycine-expressing amacrine cells. All sections were masked before counting. The distance along the retina that was evaluated in each section was measured on an image-analysis system. In this way, the number of positive cells was expressed as labeled neurons per linear millimeters of retina.
Statistical Analysis
We used t-tests, correlation analysis, and linear regression for evaluation of study results.
Results
Normal Distribution
We determined the number of neurons labeling for GABA and glycine in the GCL and INL in both eyes of five untreated rats. In the remainder of this analysis, we used the data from these 10 eyes as the data representative of bilateral control animals. The analyses were unchanged if only one eye from each of the bilateral control animals was included. Of all the amacrine cells labeled by the two antibodies, 83% were present in the INL and 17% in the GCL (Table 1). Among the amacrine cells identified, 54% were GABAergic and 46% were glycinergic. Proportionately more of the GABAergic amacrine cells resided in the GCL than in the INL (22% compared with 13% of glycinergic amacrines).
Table 1.
Normal Amacrine Cell Counts in 10 Normal Rat Eyes
| GCL | INL | Total | |
|---|---|---|---|
| GABA+ | 76.3 ± 14.2 | 276.5 ± 46.5 | 352.8 ± 57.0 |
| Glycine+ | 37.8 ± 17.2 | 263.0 ± 59.7 | 301.1 ± 74.8 |
| Total amacrine | 114.1 | 539.5 | 653.9 |
Units are labeled neurons per millimeter of retina.
Change in Amacrine Density after Glaucoma or Transection
The peak IOP levels in glaucoma-treated eyes observed for 1, 2, and 3 months after initial laser treatment were 33.4 ± 6.8, 36.8 ± 1.6, and 34.0 ± 6.1 mm Hg, respectively (mean ± SD), which represents a mean IOP higher than that of the fellow untreated eyes of 17.1 to 20.9 mm Hg. The exposure to IOP over time in treated eyes compared with fellow eyes was judged by calculating the positive IOP integral (area under the IOP curve over time). The means were 124.0 ± 95.4, 107.3 ± 28.3, and 118.0 ± 97.9 mm Hg in the 1-, 2-, and 3-month glaucoma groups, respectively (differences not statistically significant from each other, all P > 0.05).
Among the 18 animals that were subjected to experimentally induced glaucoma, the mean total number of amacrine cells labeled as either GABA or glycine positive was 557 ± 186 neurons/mm of retina (Fig. 1). This was 15% lower than the corresponding number in bilateral control rat retina (Table 2), but the difference was not statistically significant (P = 0.15, t-test). A power calculation determined that with the number of specimens studied and their variances, we had a 99.7% power to determine that the difference between glaucoma and the control was 50%, and a 71.2% power to detect a 25% difference. When the degree of glaucoma injury was estimated by the remaining number of axons in the optic nerve cross-section, the association of glaucoma damage with amacrine cells expressing either GABA or glycine showed a modest trend toward fewer positive cells in glaucomatous eyes with greater damage, but this association explained only a small amount of the variability in the number of labeled amacrine cells, and was not statistically significant (correlation coefficient, r = −0.39, P = 0.11, Fig. 2).
Figure 1.
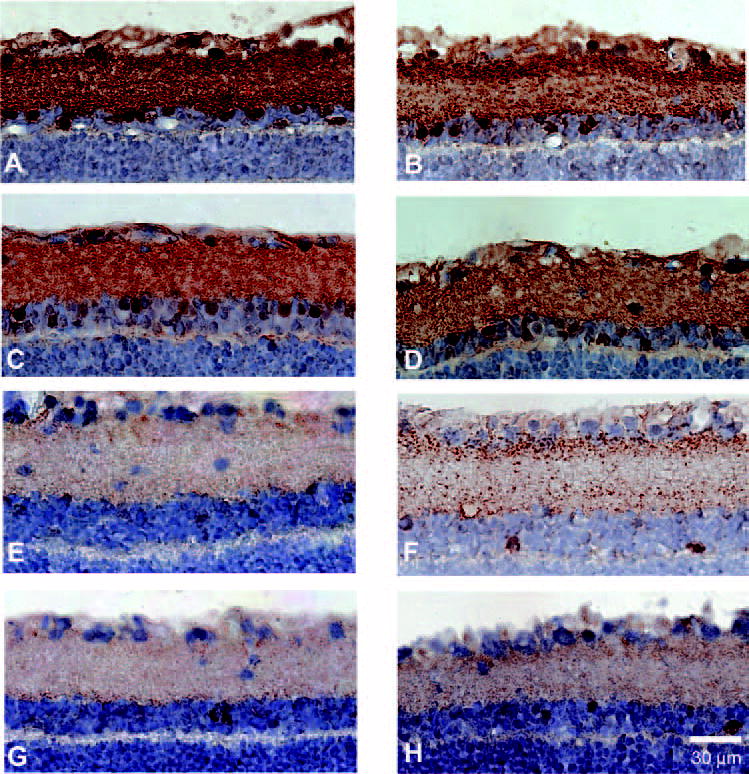
Immunohistochemical labeling for GABA and glycine in eyes of 3-month posttransection and 3-month glaucoma rats. GABA labeling was qualitatively similar in the GCL and IPL in a 3-month glaucoma eye (A) and in a matched fellow eye (B). Similarly, glycine labeling was similar in the GCL and IPL in a 3-month glaucoma eye (C) and in a matched fellow eye (D). By contrast, GABA staining was greatly decreased 3 months after transection in both treated (E) and fellow (F) eyes. Similarly, glycine labeling was significantly reduced 3 months after transection in both treated (G) and fellow (H) eyes.
Table 2.
Total Amacrine Counts in Treatment Groups
|
P |
||||||
|---|---|---|---|---|---|---|
| Treatment Group | Treated Eye | Fellow Eye | Treated vs. Control | Treated vs. Fellow | Fellow vs. Control | n |
| Glaucoma | ||||||
| 1 month | 548.1 ± 202.9 | 582.0 ± 184.5 | 0.29 | 0.77 | 0.35 | 6 |
| 2 months | 604.5 ± 232.9 | 642.5 ± 206.0 | 0.95 | 0.68 | 0.89 | 5 |
| 3 months | 521.1 ± 121.8 | 496.4 ± 101.1 | 0.04 | 0.69 | 0.01 | 7 |
| Transection | ||||||
| 1 month | 494.7 ± 62.9 | 543.2 ± 47.3 | 0.02 | 0.26 | 0.09 | 4 |
| 3 months | 186.3 ± 59.8 | 256.0 ± 49.6 | <0.0001* | 0.08 | <0.0001* | 5 |
| Bilateral Control | 653.9 ± 114.4 | 10 | ||||
Data are the mean neurons per linear millimeter of retina ± SD.
Statistically significant difference.
Figure 2.
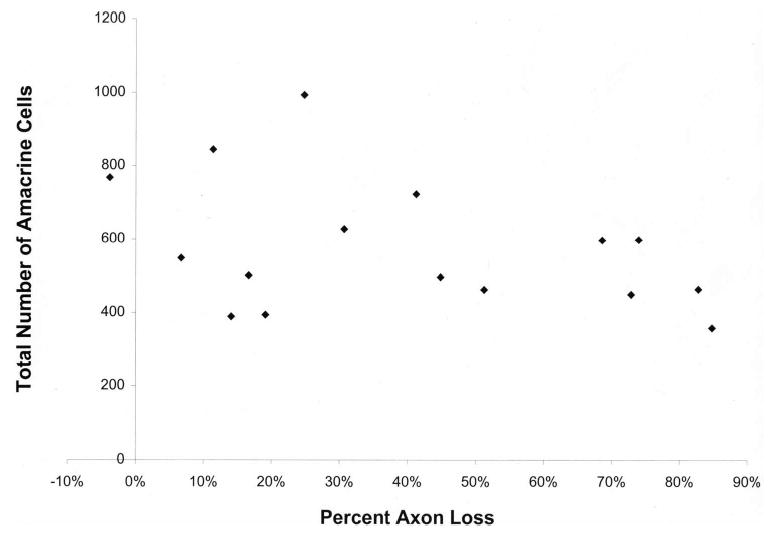
Total amacrine cells labeled by both GABA and glycine in the GCL and INL compared with axon loss in glaucomatous eyes. Amacrine cell count compared with the degree of RGC axon loss in rats with glaucoma shows a statistically non-significant trend toward fewer amacrine cells with greater damage (P = 0.11).
The mean total amacrine count for each subgroup of treated eyes and their fellow eyes were also compared to each other and to the bilateral controls (Table 2). There were 15 comparisons for the three glaucoma durations (1, 2, and 3 months) and two transection durations (1 and 3 months) against the bilateral control data and between the two eyes of each animal. We adjusted for these multiple comparisons, treating a probability of 0.0004 or less as significant, with this level indicated by both the least-significant-difference method and the Bonferroni approach.22 There was a slight trend toward fewer amacrine cells in glaucomatous eyes in the 3-month duration group (treated compared with bilateral control, and fellow eyes compared with bilateral control), but no difference between treated and fellow eyes. None of these differences achieved the P < 0.0004 level of significance.
By contrast, the 3 month transection group had dramatic loss of labeling among amacrine cells, with a 72% decrease in the transected eyes and a 61% loss in the fellow eyes (both P < 0.0001, t-test). The proportion of amacrine cells that were labeled for either GABA or glycine was lower by 71% in the GCL and 72% in the INL compared with bilateral control cells. The 1-month transection eyes had an 18% decrease that was not statistically significant (Table 2).
There are at least two explanations for the significant decreases in labeling in eyes 3 months after optic nerve transection. One possibility is that amacrine cells are dead, and another explanation is that they are still present, but have stopped producing GABA or glycine at levels that are detectable in our immunolabeling. We sought to distinguish the two possibilities by examining the 3-month posttransection eyes in more detail compared with control tissues. In the following studies, we were particularly interested in findings in the GCL. In the INL, amacrine cells are intermixed with displaced RGCs, as well as bipolar and Müller cells. The ability to study detailed effects of the treatments is, therefore, clearer in the GCL. As noted earlier, the loss of GABA and glycine labeling was similar in the GCL and the INL, and so GCL findings are relevant to the change in neurotransmitter labeling.
There are two neuronal cell types in the GCL—amacrine cells and RGCs—in approximately equal proportion in normal eyes. Because we observed cells with the morphology of neurons (by size and nuclear shape) in the GCL, one might question whether there were still RGCs alive 3 months after transection. This would be one explanation of neurons in the GCL that did not label for GABA or glycine. The cross-sections of the optic nerve in our 3-month posttransection eyes showed no intact axons, whereas the fellow eyes of this group had normal axon counts (Fig. 3). It might be argued that that RGC bodies would survive in the retina, even with completely atrophic axons in the nerve. We have strong evidence that this is not the case. Even 2 weeks after complete nerve transection, there were no remaining RGC bodies in the GCL, as judged by backfilling RGCs with fluorescent gold tracer (Fluorogold; Fluorochrome, Denver, CO) and later transecting the nerve (Fig. 4). The only cells containing fluorescent tracer in the GCL at 2 weeks after transection were glial macrophages containing the tracer from the then-dead RGCs.
Figure 3.
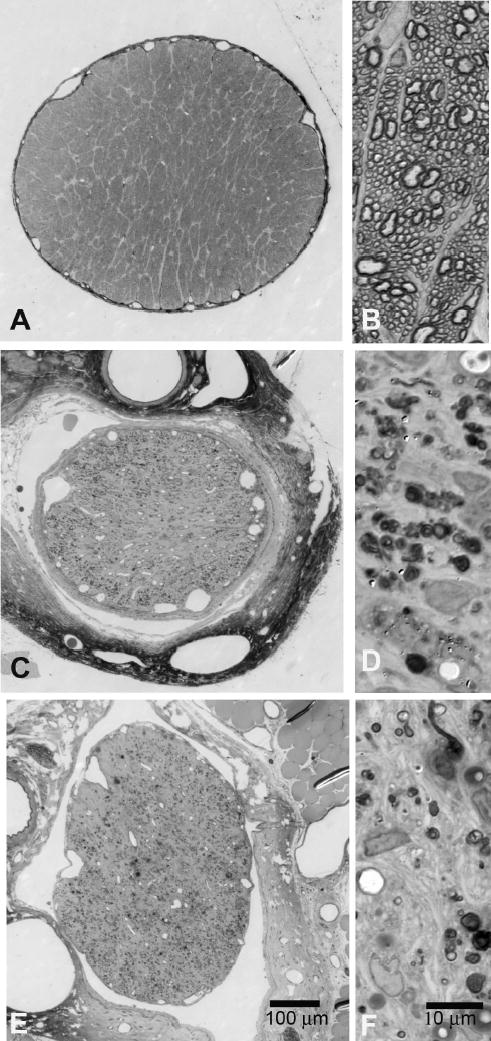
Optic nerve axon loss in transected rats at 1 month and 3 months. Rat optic nerve in cross-section. (A) Normal optic nerve at low magnification. (B) Normal optic nerve with many axons at higher magnification. (C, D) Optic nerve 1 month after transection showing no axons with normal morphology remaining. (E, F) Optic nerve 3 months after transection with no intact axons. Toluidine blue stain.
Figure 4.
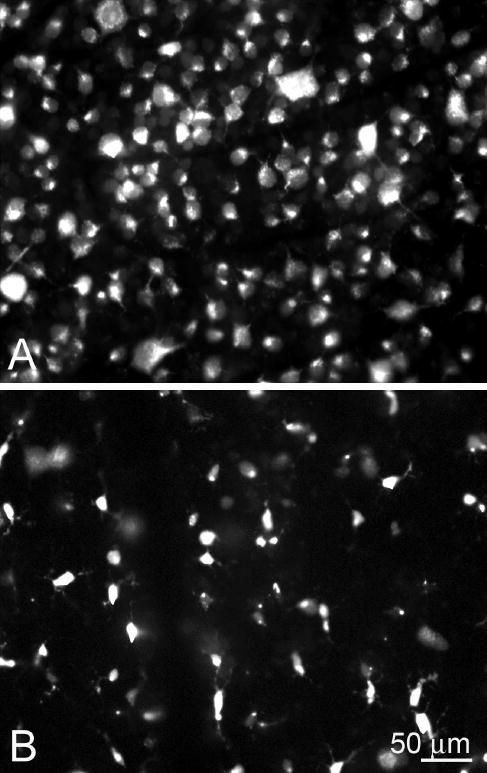
RGC loss after nerve transection. (A) Normal appearance of rat retina with many RGCs that are specifically identified by their morphology, backfilled by injection of fluorescent tracer into the superior colliculus. (B) Two weeks after optic nerve transection, there were no RGCs remaining, and the fluorescent tracer was present within phagocytes that had engulfed the dead RGCs and had a different morphology. Published, with permission, from Blair M, Pease ME, Hammond J, et al. Effect of glatiramer acetate on primary and secondary degeneration of retinal ganglion cells in the rat. Invest Ophthalmol Vis Sci. 2005;46:884–890. © Cadmus Professional Communications.
To confirm further that all RGCs were indeed gone from the GCL in the 3-month posttransection eyes, we labeled sections with antibodies against Thy-1.1 and neurofilament, both highly specific for RGCs. Control and fellow eyes had prominent label, but 3-month posttransection eyes showed no remaining Thy-1.1 label (Fig. 5). The findings for neurofilament labeling were similar (data not shown).
Figure 5.
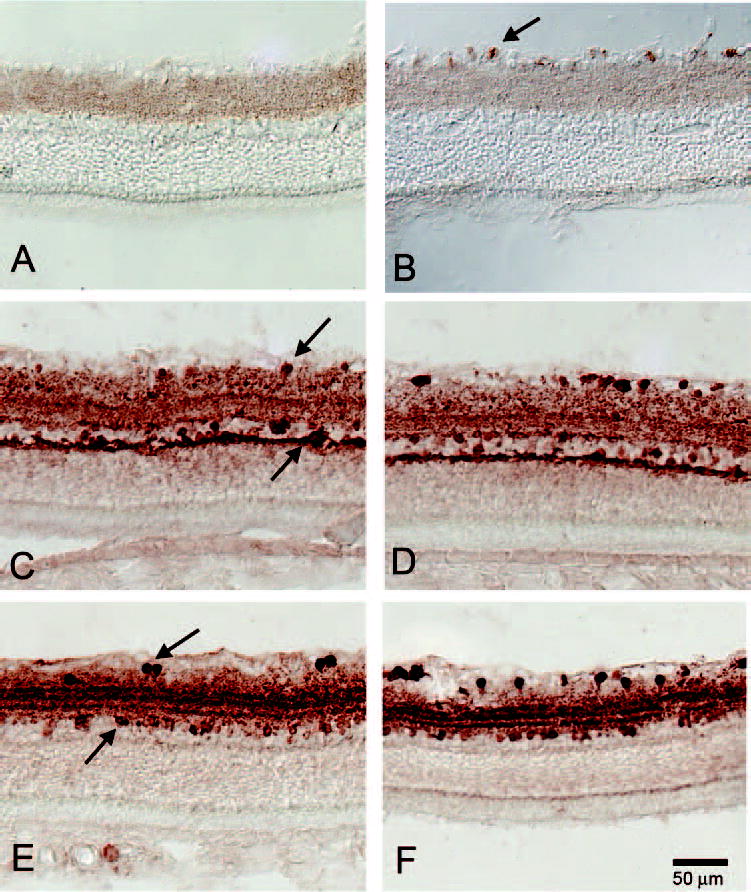
Three-month transection rat retinas labeled for Thy1, calbindin, and calretinin. (A) Three-month transection rat eye with no retinal labeling for Thy-1.1, indicating loss of RGCs. (B) The control eye of the 3-month posttransection had abundant labeling for Thy-1.1 in the GCL (arrows). (C) Three months after transection, many calbindin-positive amacrine cells were noted in the GCL and INL (arrows). (D) The control eye, 3 months after full transection, had many calbindin-positive amacrine cells in the GCL and INL. (E) Three-month full transection with substantial calretinin-labeling of amacrine cells in the GCL and INL (arrows). (F) The control eye of the 3-month fully transected optic nerve with labeling for calretinin for amacrine cells in the GCL and INL.
To demonstrate that the remaining cells in the GCL were amacrine cells, though they did not label as frequently for GABA or glycine, we immunolabeled these specimens and the control for other antigens known to be found in amacrine cells, including calbindin and calretinin (Fig. 5). Because each of these markers is found in only a minority of amacrine cells, it was not possible to use these labeling examples to quantify further the amacrine cell population.23 However, it is clear that there was residual labeling with multiple amacrine markers in the face of profound loss of GABA and glycine labeling.
Change in GABA-Labeled Amacrine Cells Alone
When the data for GABA-labeled amacrine cells were considered separately from the total of all amacrine cells, the result was nearly identical with that for the overall data (Table 3). There were no statistically significant changes in GABA labeling in most treatment groups, though the trend was for slightly fewer labeled cells in all groups compared with bilateral controls. The one group with very significant loss of labeling was the 3-month posttransection group, in which both transected and fellow eyes were lower in labeling than was the control (75% and 66% lower, respectively).
Table 3.
Total GABA-Labeled Cells
|
P |
||||||
|---|---|---|---|---|---|---|
| Treatment Group | Treated Eye | Fellow Eye | Treated vs. Control | Treated vs. Fellow | Fellow vs. Control | n |
| Glaucoma | ||||||
| 1 Month | 311.0 ± 34.5 | 313.4 ± 35.6 | 0.13 | 0.92 | 0.15 | 6 |
| 2 Months | 328.0 ± 35.5 | 292.6 ± 36.6 | 0.39 | 0.94 | 0.05 | 5 |
| 3 Months | 304.5 ± 18.2 | 325.0 ± 28.3 | 0.05 | 0.13 | 0.25 | 7 |
| Transection | ||||||
| 1 Month | 306.8 ± 18.4 | 317.8 ± 21.6 | 0.15 | 0.46 | 0.26 | 4 |
| 3 Months | 87.4 ± 19.2 | 119.2 ± 23.9 | <0.0001 | 0.05 | <0.0001 | 5 |
| Bilateral control | 352.8 ± 57.0 | 10 | ||||
Data are the mean GABA-labeled cells per retinal section ± SD.
When the GABA-labeled cells were compared to the control by retinal layer, there was a significant decrease in the GABA+ amacrine cells of the GCL in both glaucomatous and fellow eyes at 1 month, but not at other time points (Table 4). The glaucoma groups did not differ from the control in labeled cells in the INL at any time point. Among the transection groups, the counts in the 1- and 3-month posttransection eyes were both significantly different from the control, as was the 3-month fellow eye group (Table 4).
Table 4.
GABA-Labeled Amacrine Cells by Retinal Location
|
GCL |
INL |
|||
|---|---|---|---|---|
| Treatment Group | Treated Eye | Fellow Eye | Treated Eye | Fellow Eye |
| Glaucoma | ||||
| 1 Month | 37.0 ± 6.3* | 41.8 ± 5.9* | 274.0 ± 28.1 | 271.7 ± 30.2 |
| 2 Months | 56.9 ± 8.7 | 61.0 ± 11.6 | 267.2 ± 39.7 | 231.7 ± 26.1 |
| 3 Months | 59.9 ± 3.9 | 60.2 ± 8.6 | 244.6 ± 14.9 | 264.8 ± 21.5 |
| Transection | ||||
| 1 Month | 41.4 ± 3.2 | 62.7 ± 3.2 | 265.4 ± 18.3 | 255.1 ± 19.3 |
| 3 Months | 15.2 ± 3.5* | 20.9 ± 3.7* | 72.2 ± 15.7* | 96.7 ± 20.4* |
| Bilateral control | 76.3 ± 14.2 | 276.5 ± 46.5 | ||
Difference from bilateral controls significant at P < 0.0004.
Change in Glycine-Labeled Amacrine Cells Alone
The glaucoma groups had no statistically significant changes in total glycine-labeled cells in treated eyes, though there was a significant decrease in 3-month glaucoma fellow eyes (Table 5). The 3-month posttransection eyes were significantly decreased in glycine label in both treated and fellow eyes compared with the control.
Table 5.
Total Glycine-Labeled Cells
|
P |
||||||
|---|---|---|---|---|---|---|
| Treatment Group | Treated Eye | Fellow Eye | Treated vs. Control | Treated vs. Fellow | Fellow vs. Control | |
| Glaucoma | ||||||
| 1 Month | 203.2 ± 27.2 | 230.2 ± 43.2 | 0.009 | 0.22 | 0.05 | 6 |
| 2 Months | 278.5 ± 12.8 | 276.3 ± 39.8 | 0.48 | 0.9 | 0.47 | 6 |
| 3 Months | 216.5 ± 21.3 | 171.4 ± 29.6 | 0.01 | 0.007 | 0.0006 | 7 |
| Transection | ||||||
| 1 Month | 189.7 ± 25.7 | 216.4 ± 3.9 | 0.01 | 0.09 | 0.05 | 4 |
| 3 Months | 99.0 ± 12.9 | 136.5 ± 16.2 | <0.0001* | 0.004 | 0.0004* | 5 |
| Bilateral control | 301.1 ± 74.8 | 10 | ||||
Difference from bilateral controls significant at P < 0.0004.
Whereas GABA amacrine cells had significant decreases in labeling in the GCL, this was not true of glycinergic amacrine cells in any glaucoma or transection group (Table 6). The data show some decrease in labeling, which was greatest in the 1-month glaucoma group, with 33% less than the control (not significant). The inability to detect a change in glycinergic amacrine cells in the GCL could have resulted, at least in part, from the small number of glycinergic amacrine cells in this layer. In the INL, glycine labeling was significantly decreased in the 3-month posttransection group, in both treated (68% decrease) and fellow eyes (55% decrease). In addition, the fellow eyes of the 3-month glaucoma group also showed a significant decrease, though the value in treated eyes in this group did not achieve significance.
Table 6.
Glycine-Labeled Amacrine Cells by Retinal Location
|
GCL |
INL |
|||
|---|---|---|---|---|
| Treatment Group | Treated Eye | Fellow Eye | Treated Eye | Fellow Eye |
| Glaucoma | ||||
| 1 Month | 25.4 ± 8.9 | 30.1 ± 13.4 | 177.8 ± 19.4 | 200.1 ± 30.3 |
| 2 Months | 32.4 ± 5.6 | 45.6 ± 10.5 | 246.2 ± 10.7 | 228.8 ± 31.8 |
| 3 Months | 27.2 ± 5.2 | 26.1 ± 8.9 | 189.3 ± 21.0 | 145.4 ± 19.9* |
| Transection | ||||
| 1 Month | 25.4 ± 3.4 | 28.0 ± 3.4 | 162.4 ± 21.7 | 188.5 ± 5.9 |
| 3 Months | 18.5 ± 3.7 | 24.6 ± 6.2 | 80.4 ± 12.0* | 111.9 ± 12.9* |
| Bilateral control | 37.8 ± 17.2 | 263.0 ± 59.7 | ||
Difference from bilateral controls significant at P < 0.0004.
Discussion
Previous studies have not detected quantitative loss of any retinal cells other than RGCs in human, monkey, or rodent eyes with spontaneous or experimental glaucoma.10–17 There have been two reports that photoreceptors may be affected by glaucoma. One report found swelling of some cones, but no quantitative loss was documented.24 An investigation of the outer nuclear layer in blind eyes with glaucoma secondary to ocular trauma suggested that these eyes may have fewer photoreceptors.25 Such trauma is known to lead to outer retinal injury independent of glaucomatous damage.26 In a detailed, quantitative study of primary human glaucomatous eyes, we found no loss of photoreceptors, nor any thinning of the inner nuclear layer.17 In a rat model of experimental glaucoma, there was no loss of cells in the inner or outer nuclear layer.15
One important finding from this investigation is that there was no statistically significant change in amacrine cell label in the experimental glaucomatous eyes. Although there were tendencies in stratified analysis of subgroups toward fewer labeled amacrine cells, none of these achieved significance. Many of the glaucomatous eyes lost a substantial number of RGCs—an average loss of more than 40%—and they had been followed up for 3 months after initiation of glaucomatous injury. If the effect of glaucoma on amacrine cells were dependent on the degree of RGC loss, it would be expected that there would be a correlation between the two parameters, which we did not detect. We think it is appropriate to conclude that amacrine cells are not sensitive to chronic elevation of IOP. Nor are they significantly affected by the severity and course of RGC loss in our glaucoma model. It will be interesting to attempt to study monkey or human eyes with experimental or spontaneous glaucoma, respectively, for effects on amacrine cells. Such investigation could be performed more easily in monkey eyes, because the specific techniques used in the current study require ideal fixation with glutaraldehyde fixative.
It is not a simple matter to ensure that some amacrine cells have not died in experimental glaucoma. In the GCL and INL, RGCs and amacrine cells are intermixed. RGCs can be definitively labeled by backfilling them with dye, but no such conclusive method can be applied to amacrine cells. Because there are more than 20 different types of amacrine cells, no single label method provides sufficient coverage of the entire group for quantitative study. We chose to use labeling for GABA and glycine, which covers more than 90% of all amacrine cells. Of labeled amacrine cells in normal eyes, 83% were found in the INL, and 17% were in the GCL. Of the cells found in the GCL, we estimated that nearly half were amacrine cells (data not shown). This was a conclusion derived from nuclear staining (to count all neurons) and the number remaining after transection (when RGCs would be removed).
A second finding was a decrease in amacrine cell labeling in the animals that had undergone optic nerve transection 3 months before death. The effect was significant and was present in both transected and fellow eyes. The substantial loss of GABA and glycine labeling could have resulted either from the death of amacrine cells, from a fall in neurotransmitter levels in cells that were still present, or from a combination of the two events. As seen in our results, the more likely conclusion is that amacrine cells were still largely present, but had ceased to contain neurotransmitter at levels detectable in our immunoassay. We reach this conclusion after determining that RGCs had indeed been eliminated from the eyes of nerve-transection animals by both axon counts and specific immunolabeling in the GCL. These data show that there were no axonless RGC bodies in the retina, unless they had simultaneously ceased to produce detectable amounts of Thy 1.1 and neurofilament. Yet, there were clearly neuron-shaped cells in the GCL 3 months after transection that did not label for either GABA or glycine. Many of these cells labeled with calretinin and calbindin, molecules that are typical of some amacrine cell subtypes.27 Hence, the loss of GABA and glycine labeling is best interpreted, not as a loss of amacrine cells, but as a change in expression of their neurotransmitters.
It is logical that a decrease in amacrine neurotransmitters might result from the precipitous loss of RGC bodies within 2 weeks after nerve transection. Teleologically, if there are no RGC with which to synapse, the amacrine cells would undergo dendritic retraction and would downregulate neurotransmitter production, but there are contradictory features of the events that cannot be so easily explained. The fellow eyes of transection animals 3 months after injury also had a profound loss of amacrine labeling despite the presence of a normal number of RGC in these retinas. Perhaps, surgical damage to the optic nerve leads to biochemical alterations in the fellow eye. This phenomenon has been documented by Bodeutsch et al.28 After unilateral optic nerve crush, they reported bilateral activation of the early gene c-jun as well as microglial activation in both the retina of the eye with the crushed nerve and in the fellow retina. This result, taken with our own, points out that the fellow eye may be affected in experiments in which the optic nerve and its sheath are violated by crushing or cutting. Suitable bilaterally untreated control data should be included in any such investigation. We speculate that at least some of the contralateral effects result from the proximity of the fellow eye to the opened dural sheath at the transection and the attendant alterations in the cerebrospinal fluid.
We conclude that there is no definitive evidence that glaucomatous damage leads to quantitative loss of cells other than RGCs in the retina. This finding is remarkable when we consider that there are outer retinal diseases that not only lead to loss of the cells of primary pathology, but also loss of their subsequent synaptic partners. In retinitis pigmentosa and in Batten disease, human eyes showed histopathologically the loss of rods and cones and significant decrements in cells of the INL and GCL.29,30 Along with cell loss, abnormal new neurite formation was detected in remaining cells of patients with retinitis pigmentosa.31 It is interesting that outer retinal disease leads to such profound change in the remainder of the retina, whereas damage from glaucoma seems restricted to RGCs. Primary death of RGCs seems to lead to secondary degeneration of other RGCs,19,20 but not to such successive loss in other retinal cell types. The reason for this relatively one-way trans-synaptic degeneration is not immediately apparent, but may be of great significance in our understanding of retinal neuronal death.
Acknowledgments
The authors thank John Hammond for surgical assistance, Danielle Valenta for technical assistance, Aimee Broman for help with the statistical analyses, and Donald Zack for helpful suggestions.
Footnotes
Supported by National Eye Institute Grants EY02120 and EY01765.
Disclosure: J.L. Kielczewski, None; M.E. Pease, None; H.A. Quigley, None
References
- 1.Kolb H. Amacrine cells of the mammalian retina: neurocircuitry and functional roles. Eye. 1997;11:904–923. doi: 10.1038/eye.1997.230. [DOI] [PubMed] [Google Scholar]
- 2.Strettoi E, Masland RH. The number of unidentified amacrine cells in the mammalian retina. Proc Natl Acad Sci USA. 1996;93:14906–14911. doi: 10.1073/pnas.93.25.14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacNeil MA, Masland RH. Extreme diversity among amacrine cells: implications for function. Neuron. 1998;20:971–982. doi: 10.1016/s0896-6273(00)80478-x. [DOI] [PubMed] [Google Scholar]
- 4.Yamasaki E, Andrade de Costa B, Barbosa VD, Hokoc J. Retinal ganglion depletion alters the phenotypic expression of GABA and GAD in the rat retina. Eur J Neurosci. 1997;9:1885–1890. doi: 10.1111/j.1460-9568.1997.tb00755.x. [DOI] [PubMed] [Google Scholar]
- 5.Osborne NN, Perry VH. Effect of neonatal optic nerve transection on some classes of amacrine cells in the rat retina. Brain Res. 1985;343:230–235. doi: 10.1016/0006-8993(85)90739-5. [DOI] [PubMed] [Google Scholar]
- 6.Wu DK, Cepko CL. Development of dopaminergic neurons is insensitive to optic nerve section in the neonatal rat retina. Dev Brain Res. 1993;74:253–260. doi: 10.1016/0165-3806(93)90011-x. [DOI] [PubMed] [Google Scholar]
- 7.Williams RR, Cusato K, Raven MA, Reese BE. Organization of the inner retina following early ganglion cell elimination of the retinal ganglion cell population: effects on cell numbers and stratification patterns. Vis Neurosci. 2001;18:233–244. doi: 10.1017/s0952523801182088. [DOI] [PubMed] [Google Scholar]
- 8.Dijk F, Leeuwen SV, Kamphuis W. Differential effects of ischemia/reperfusion on amacrine cell subtype-specific transcript levels in the rat retina. Brain Res. 2004;1026:194–204. doi: 10.1016/j.brainres.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 9.Dijk F, Kamphuis W. An immunocytochemical study on specific amacrine cell subpopulations in the rat retina after ischemia. Brain Res. 2004;1026:205–217. doi: 10.1016/j.brainres.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Vickers JC, Schumer RA, Podos SM, Wang RF, Riederer BM, Morrison JH. Differential vulnerability of neurochemically identified subpopulations of retinal neurons in a monkey model of glaucoma. Brain Res. 1995;680:23–35. doi: 10.1016/0006-8993(95)00211-8. [DOI] [PubMed] [Google Scholar]
- 11.Harwerth RS, Carter-Dawson L, Shen F, Smith EL, Crawford MLJ. Ganglion cell losses underlying visual field defects from experimental glaucoma. Invest Ophthalmol Vis Sci. 1999;40:2242–2250. [PubMed] [Google Scholar]
- 12.May CA, Hayreh SS, Furuyoshi N, Ossoinig K, Kaufman PL, Lütjen-Drecoll E. Choroidal ganglion cell plexus and retinal vasculature in monkeys with laser-induced glaucoma. Ophthalmologica. 1997;211:161–171. doi: 10.1159/000310784. [DOI] [PubMed] [Google Scholar]
- 13.May CA, Mittag T. Neuronal nitric oxide synthase (nNOS) positive retinal amacrine cells are altered in the DBA/2NNia mouse, a murine model for angle-closure glaucoma. J Glaucoma. 2004;13:496–499. doi: 10.1097/01.ijg.0000137435.83307.fd. [DOI] [PubMed] [Google Scholar]
- 14.Moon J, Kim I, Gwon J, et al. Changes in retinal neuronal populations in the DBA/2J mouse. Cell Tissue Res. 2005;320:51–59. doi: 10.1007/s00441-004-1062-8. [DOI] [PubMed] [Google Scholar]
- 15.Levkovitch-Verbin H, Quigley HA, Martin KRG, Valenta D, Kerrigan-Baumrind LA, Pease ME. Translimbal laser photocoagulation to the trabecular meshwork as a model of glaucoma in rats. Invest Ophthalmol Vis Sci. 2002;43:402–410. [PubMed] [Google Scholar]
- 16.Wygnanski T, Desatnik H, Glovinsky Y, Quigley HA. Comparison of ganglion cell loss and cone loss in experimental glaucoma. Am J Ophthalmol. 1995;120:184–189. doi: 10.1016/s0002-9394(14)72606-6. [DOI] [PubMed] [Google Scholar]
- 17.Kendell KR, Quigley HA, Kerrigan LA, Pease ME, Quigley EN. Primary open-angle glaucoma is not associated with photoreceptor loss. Invest Ophthalmol Vis Sci. 1995;36:200–205. [PubMed] [Google Scholar]
- 18.Schwartz M. Neurodegeneration and neuroprotection in glaucoma: development of a therapeutic neuroprotective vaccine. The Friedenwald lecture. Invest Ophthalmol Vis Sci. 2003;44:1407–1411. doi: 10.1167/iovs.02-0594. [DOI] [PubMed] [Google Scholar]
- 19.Levkovitch-Verbin H, Quigley HA, Kerrigan-Baumrind LA, D’Anna S, Kerrigan DF, Pease ME. Optic nerve transection in monkeys may result in secondary degeneration of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2001;42:975–982. [PubMed] [Google Scholar]
- 20.Levkovitch-Verbin H, Quigley HA, Martin KRG, Zack DJ, Pease ME, Valenta DF. A model to study differences between primary and secondary degeneration of retinal ganglion cells in rats by partial optic nerve transection. Invest Ophthalmol Vis Sci. 2003;44:3388–3395. doi: 10.1167/iovs.02-0646. [DOI] [PubMed] [Google Scholar]
- 21.Klassen HJ, Ng TF, Kurimoto Y, et al. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest Ophthalmol Vis Sci. 2004;45:4167–4173. doi: 10.1167/iovs.04-0511. [DOI] [PubMed] [Google Scholar]
- 22.Hsu JC. Multiple Comparisons: Theory and Methods. London: Chapman & Hall; 1996.
- 23.Massey SC, Mills SL. Antibody to calretinin stains AII amacrine cells in the rabbit retina: double-label and confocal analyses. J Comp Neurol. 1999;411:3–18. [PubMed] [Google Scholar]
- 24.Nork TM, Ver Hoeve JN, Poulsen GL, et al. Swelling and loss of photoreceptors in chronic human and experimental glaucomas. Arch Ophthalmol. 2000;118:235–245. doi: 10.1001/archopht.118.2.235. [DOI] [PubMed] [Google Scholar]
- 25.Panda S, Jonas JB. Decreased photoreceptor count in human eyes with secondary angle-closure glaucoma. Invest Ophthalmol Vis Sci. 1992;33:2532–2536. [PubMed] [Google Scholar]
- 26.Sipperley J, Quigley HA, Gass JDM. Traumatic retinopathy in primates: the explanation of commotio retinae. Arch Ophthalmol. 1978;96:2267–2273. doi: 10.1001/archopht.1978.03910060563021. [DOI] [PubMed] [Google Scholar]
- 27.Kolb H, Zhang L, Dehorvver L, Cuenca N. A new look at calretinin-immunoreactive amacrine cell types in the monkey retina. J Comp Neurol. 2002;453:168–184. doi: 10.1002/cne.10405. [DOI] [PubMed] [Google Scholar]
- 28.Bodeutsch N, Siebert H, Dermon C, Thanos S. Unilateral injury to the adult rat optic nerve causes multiple cellular responses in the contralateral site. J Neurobiol. 1999;38:116–128. doi: 10.1002/(sici)1097-4695(199901)38:1<116::aid-neu9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 29.Humayun MS, Prince M, de Juan E, Jr, et al. Morphometric analysis of the extramacular retina from postmortem eyes with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1999;40:143–148. [PubMed] [Google Scholar]
- 30.Bensaoula T, Shibuya H, Katz ML, et al. Histopathologic and immunocytochemical analysis of the retina and ocular tissues in Batten disease. Ophthalmology. 2000;107:1746–1753. doi: 10.1016/s0161-6420(00)00264-5. [DOI] [PubMed] [Google Scholar]
- 31.Fariss RN, Li ZY, Milam AH. Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol. 2000;129:215–223. doi: 10.1016/s0002-9394(99)00401-8. [DOI] [PubMed] [Google Scholar]