

Abstract
SNAP receptor (SNARE)-mediated fusion is regarded as a core event in exocytosis. Exocytosis is supported by other proteins that set up SNARE interactions between secretory vesicle and plasma membranes or facilitate fusion pore formation. Secretory carrier membrane proteins (SCAMPs) are candidate proteins for functioning in these events. In neuroendocrine PC12 cells, SCAMP2 colocalizes on the cell surface with three other proteins required for dense-core vesicle exocytosis: phospholipase D1 (PLD1), the small GTPase Arf6, and Arf6 guanine nucleotide exchange protein ARNO. Arf6 and PLD1 coimmunoprecipitate (coIP) with SCAMP2. These associations have been implicated in exocytosis by observing enhanced coIP of Arf6 with SCAMP2 after cell depolarization and in the presence of guanosine 5′-O-(3-thio)triphosphate and by inhibition of coIP by a SCAMP-derived peptide that inhibits exocytosis. The peptide also suppresses PLD activity associated with exocytosis. Using amperometry to analyze exocytosis, we show that expression of a point mutant of SCAMP2 that exhibits decreased association with Arf6 and of mutant Arf6 deficient in activating PLD1 have the same inhibitory effects on early events in membrane fusion. However, mutant SCAMP2 also uniquely inhibits fusion pore dilation. Thus, SCAMP2 couples Arf6-stimulated PLD activity to exocytosis and links this process to formation of fusion pores.
INTRODUCTION
Neuroendocrine cells such as PC12 cells have been used widely to define the molecular machinery controlling exocytosis. Their secretory granules, dense core vesicles (DCVs), undergo rapid fusion with the plasma membrane in response to stimulation. This process involves several steps, including docking/tethering DCVs to the plasma membrane, priming the secretory machinery, assembling trans-SNARE complexes, and triggering fusion (Chapman, 2002; Jahn et al., 2003; Martin, 2003). Furthermore, experiments using PC12 cells have provided considerable insight concerning the molecular organization of DCV fusion sites (Lang et al., 2001; Holroyd et al., 2002; Liu et al., 2002; Han et al., 2004) and interactions between SNARE proteins and synaptotagmins, candidate calcium sensors and effectors of membrane fusion (Zhang et al., 2002; Tucker et al., 2003; Bai et al., 2004b).
Several studies have been instrumental in showing that phosphatidic acid (PA) and inositol phospholipids, especially phosphatidylinositol 4,5-bisphosphate (PIP2), make critical contributions in promoting DCV exocytosis (Hay et al., 1995; Holz et al., 2000; Vitale et al., 2001, 2002; Tucker et al., 2003; Bai et al., 2004a). PIP2 synthesis at the cell surface is supported by type I phosphatidylinositol 4-phosphate 5-kinase (PIP5K-1γ), which is activated by Arf6 and by PA (Honda et al., 1999; Aikawa and Martin, 2003). PA is produced by phospholipase D (PLD), which is also activated by Arf6 and by PIP2 (Sciorra et al., 1999; Dana et al., 2000), and like PIP5K-1γ is concentrated at the PC12 cell's plasma membrane (Vitale et al., 2001, 2002). Thus, Arf6, PIP5K, PLD, PA, and PIP2 form a network that supports exocytosis; yet, how they might interact and collaborate with the core machinery is only beginning to be understood. Furthermore, addressing how these lipids and their regulators are distributed and how they function seems essential for understanding events involved in reorganizing membrane bilayers during opening and expansion of fusion pores.
We have been interested in the function of SCAMPs in exocytosis. This family of proteins with four transmembrane spans resides in the cell surface recycling system (Brand and Castle, 1993). SCAMPs 1 and 2 have been implicated as participants in a late step of exocytosis although their exact roles are not known (Fernandez-Chacon et al., 1999; Guo et al., 2002; Liu et al., 2002). In PC12 cells, a portion of SCAMP2 is concentrated in the plasma membrane at exocytotic sites that are marked by docked DCVs and by syntaxin 1, an exocytotic SNARE and complexin, a SNARE complex binding protein (Pabst et al., 2000; Liu et al., 2002). The function of SCAMP2 involves a conserved segment of the protein known as E peptide, which is positioned at the cytoplasmic membrane surface linking the second and third transmembrane spans (Hubbard et al., 2000; Guo et al., 2002; Liu et al., 2002). E peptide is basic and aromatic and associates electrostatically with polyphosphoinositides, e.g., PIP2, causing their sequestration (Ellena et al., 2004; Gambhir et al., 2004). Thus, we have wondered whether SCAMP2 might serve to focus the actions of Arf6, PLD1, PA, and PIP2 at sites of SNARE-mediated DCV exocytosis. Our colocalizations, protein interaction studies, and assay of PLD activity all support this possibility. Furthermore, we show using amperometry that mutant SCAMP2 inhibits exocytosis not only by interfering with events leading to fusion pore opening in a very similar manner to a PLD activation-deficient mutant of Arf6 but also by interfering with dilation of the opened fusion pores.
MATERIALS AND METHODS
Reagents (Including Plasmids and Antibodies)
Noradrenalin, poly-d-lysine, GDP, doxycycline, and protein A-Sepharose were from Sigma-Aldrich (St. Louis, MO); guanosine 5′-O-(3-thio)triphosphate (GTPγS) was from Roche Diagnostics (Indianapolis, IN); lysophosphatidylcholine (LPC; α-palmitoyl) and phosphatidylbutanol (1,2-dioleoyl) were from Avanti Polar Lipids (Alabaster, AL); [3H]oleic acid was from PerkinElmer Life and Analytical Sciences (Boston, MA); and Lipofectamine Plus was from Invitrogen (Carlsbad, CA). Enhanced chemiluminescence (ECL) reagents (Super Signal) and NHS-biotin were from Pierce Endogen (Rockford, IL). Carbon fiber electrodes (CPE-1) for amperometry were from ALA Scientific (Westbury, NY). SCAMP peptides were synthesized, purified, and analyzed by high-performance liquid chromatography and mass spectroscopy at the UVA Biomolecular Research Facility. Antibodies were obtained as follows: rabbit anti-SCAMP antibodies (SCAMPs 1–3) characterized previously (Hubbard et al., 2000; Guo et al., 2002; Liu et al., 2002); monoclonal anti-SCAMP 7C12 (Brand et al., 1991); rabbit anti-PLD1, from Michael Frohman (SUNY, Stonybrook, NY); rabbit anti-ARF6, from Julie Donaldson (National Institutes of Health, Bethesda, MD); myc-epitope monoclonal 9E10 (Wu and Castle, 1997); and secondary antibodies (Alexa conjugates) and neutra-Avidin from Molecular Probes (Eugene, OR). Plasmid pTRE2 was from BD Biosciences Clontech (Palo Alto, CA); bicistronic MSCV-based pIRES-EGFP vector was from Derek Persons (St. Jude's Children's Research Hospital, Memphis, TN). pCGN-hPLD1 encoding PLD1 with N-terminal hemagglutinin (HA) tag was from Michael Frohman (SUNY, Stonybrook); it was subcloned into pTRE2 using Nhe1 and EcoRV. Constructs encoding Arf6 and variants T27N, Q67L, N48R, and Arf1 all with C-terminal HA epitope were in pTRE2. Arf6 and its N48R mutant also were subcloned into pIRES-EGFP (EcoRI and SmaI sites). N-terminally Myc-tagged SCAMP2, wild type and W202A (rat sequence) (Liu et al., 2002) were subcloned into pIRES-EGFP (NheI and XhoI) sites.
Cell Culture, Transfection, and Expression
Rat pheochromocytoma PC12 cells were from Edwin Chapman (University of Wisconsin, Madison, WI) and tetracycline-regulated PC12 cells (tet-off) were from BD Biosciences Clontech. PC12 cells were cultured in DMEM containing 5% horse serum, 5% iron-supplemented calf serum (Hyclone Laboratories, Logan, UT); tet-off PC12 cells were cultured in 10% horse serum, 5% fetal bovine serum (BD Biosciences Clontech). Both were cultured at 10% CO2. Cells were transfected by electroporation (single 8-ms pulse, 255 V, 4-mm cuvette; ECM830 electrosquare porator) using 50 μg of DNA per 2 × 107 cells in 750 μl of electroporation buffer (Wang et al., 2001). Tet-off PC12 cells were transfected using Lipofectamine Plus (Liu et al., 2002), and to achieve similar levels of overexpression, samples were incubated for different time periods before adding doxycycline (2 μg/ml). Levels of overexpression were determined as before (Liu et al., 2002) by Western blotting in combination with estimating the fraction of transfected cells by fluorescence microscopy.
Immunofluorescence Labeling of Cells and Plasma Membrane Sheets
For immunofluorescence, cells were plated on poly-d-lysine-coated coverslips. Intact cells were incubated at 37°C for 5 min in low-K+ buffer (5.6 mM KCl, 145 mM NaCl) or stimulated with high K+ buffer (56 mM KCl, 95 mM NaCl) each also containing 2.2 mM CaCl2, 0.5 mM MgCl2, 15 mM HEPES, pH 7.4, and 5.6 mM glucose. Cells were fixed in formaldehyde and immunolabeled as described previously (Liu et al., 2002). Alternatively, unstimulated or stimulated cells were chilled on ice, washed three times with ice-cold 25 mM HEPES, pH 7.0, 25 mM KCl, 2.5 mM magnesium acetate, and 0.5 mM dithiothreitol (DTT) and sonicated (single pulse) to prepare plasma membrane sheets (Liu et al., 2002). Sheets were fixed, washed, blocked, and immunolabeled as described previously (Wu and Castle, 1997; Liu et al., 2002). Cell and plasma membrane samples were imaged and digitally deconvolved using OpenLab software. For double labeling with two rabbit antibodies, staining was done in series: first primary antibody, first fluorescent secondary antibody, biotinylated second primary antibody, fluorescent neutravidin conjugate. 2τ was biotinylated while bound to a column of immobilized epitope peptide and was eluted with glycine, pH 2.2. Colocalization of SCAMP2 with other proteins was assessed quantitatively on deconvolved images of at least 12 plasma membrane sheets using a cross-correlation algorithm in which the range of values for the cross-correlation (Pearson) coefficient extends from –1.0 (not colocalized) to +1.0 (fully colocalized) (Manders et al., 1993). True colocalization was distinguished from random overlap by demonstrating loss of colocalization after relative displacement of red and green components in pixel increments (van Steensel et al., 1996).
Coimmunoprecipitation Studies
Coimmunoprecipitation experiments of SCAMPs and Arfs were performed on cells solubilized in 1% Triton X-100, 0.5% deoxycholate (DOC), 0.1% SDS, 50 mM Tris, 100 mM NaCl, 2 mM MgCl2, 10% glycerol, and proteinase inhibitor cocktail. For coimmunoprecipitations (coIPs) of PLD1, cells were solubilized in medium containing 1% NP-40 in place of Triton, DOC, and SDS. Lysates were clarified by centrifugation (50,000 rpm, 10 min, TLA120.2 rotor) and then incubated overnight at 4°C with SCAMP2 antibody bound to protein A-Sepharose. Immune complexes were washed three times in solubilization medium without DOC and SDS, once in phosphate-buffered saline, and eluted in sample buffer containing 0.2 M DTT for SDS-PAGE and Western blotting (ECL or 125I secondary antibody detection). In experiments in which monoclonal antibody (mAb) 7C12 was used to estimate levels of SCAMPs 1–3 present on blots, measurements were corrected for the differing avidities of the antibody for SCAMPs 1:2:3 (1.0:0.6:0.14; Singleton et al., 1997). In certain experiments, PC12 cells were permeabilized in KEGPMA buffer (137 mM potassium glutamate, 5 mM EGTA, 20 mM PIPES, pH 6.6, 1 mM MgCl2, and 2 mM ATP) containing 20 μM digitonin for 5 min at 25°C (Holz et al., 1992). Permeabilized cells were washed once with ice-cold KEGPMA buffer, incubated on ice with 5–100 μM SCAMP-derived peptide (as specified) in the same buffer, and then for 10 min at 37°C after adding 100 μM GTPγS (or 1 mM GDP). After incubation, samples were solubilized and immunoprecipitated as described above.
PLD Activity
PLD activity was assayed by transphosphatidylation using a variation on previous strategies (Caumont et al., 1998; Santy and Casanova, 2001). PC12 cells were labeled 16–20 h with 2 μCi/ml [3H]oleic acid in culture medium, washed in ice-cold Na-GB buffer (137 mM Na-glutamate, 2 mM MgCl2, 20 mM PIPES, and 1 mg/ml bovine serum albumin, pH 6.8) and then incubated 15 min 4°C in 3 IU/ml streptolysin-O (SLO) in Na-GB buffer. After replacing the buffer, samples were permeablized 3 min, at 37°C, and changed to K-GB buffer (137 mM K-glutamate replacing Na-glutamate) supplemented with 0.2 mM EGTA and 0.8 mM ATP. Where indicated, peptides (≥100× stocks in 0.3 M PIPES, pH 6.8, were added and incubated 10 min on ice. After addition of 0.3% (final) 1-butanol, either 3 mM EGTA or 3 mM Ca-EGTA (pCa = 5), and GTPγS (100 μM final, where specified), samples were incubated 5 min at 37°C. Incubations were terminated by chilling on ice and adding in succession equal volumes of ice-cold methanol, 0.1 N HCl-0.1% SDS, and CHCl3. The lower phase was dried, dissolved in CHCl3, spotted on silica gel plates, and chromatographed in ethyl acetate:isooctane:acetic acid (50:25:10) (Santy and Casanova, 2001). Phosphatidylbutanol spots were excised, quantitated by scintillation counting, and results were recorded as percentage of total radioactivity in the CHCl3 extract.
Amperometry
Procedures for amperometry were modified from Wang et al. (2001). PC12 cells 44 h posttransfection with pIRES-EGFP constructs were plated on poly-d-lysine/collagen (5 μg/cm2 of each)-coated dishes (1 × 105 cell/35-mm dish) and were incubated 15 h in medium containing 1.25 mM noradrenalin, 0.25 mM ascorbate. After loading and at least 1-h chase in culture medium, cells were transferred to 142 mM NaCl, 4.2 mM KCl, 1 mM Na2HPO4, 0.7 mM MgCl2, 2 mM CaCl2, and 10 mM HEPES, pH 7.3, selected by green fluorescent protein content, and stimulated by local perfusion (3 psi from an Eppendorf Transjector 5246) of K+ depolarizing medium (105 mM KCl, 45 mM NaCl, 1 mM NaH2PO4, 0.7 mM MgCl2, 2 mM CaCl2, and 10 mM HEPES, pH 7.3) from a 2-μm-diameter micropipet placed ∼10 μm away from the cell surface. Perfusion start times were standardized to 1 s after initiating recording. Release of noradrenalin was detected by a cut 5-μm carbon fiber lightly contacting the cell surface and at a potential of +650 mV. Current signals were fed to an Axopatch 200 amplifier, low-pass filtered at 1 kHz and digitized at 3.3 kHz by a computer running Lab Man (a custom data acquisition program developed in the Szabo laboratory). Each cell was stimulated one to three times for 8 s at 30-s intervals with K+ depolarizing medium. In experiments testing the effects of LPC on the SAF/spikes ratio, K+ depolarizing medium containing 15 μM LPC was used in the perfusion pipet.
Individual experiments were performed on nontransfected cells along with parallel sets of cells transfected with wild-type and mutant SCAMP2 or wild-type and mutant Arf6 such that all cells were handled consistently. The morphology of all types of cells recorded was indistinguishable. In particular, there was no change in the surface or shape of the cells expressing SCAMP2-W202A that were recorded. Thus contributions of small spikes to recordings reflecting increased distance from the electrode are anticipated to be comparable in all samples; therefore, the ability to distinguish SAF is equivalent in all types of cells. Recordings were analyzed two ways: 1) manually from printouts to detect stand-alone feet and to compare their number to the number of spikes and 2) using computer programs provided by Meyer Jackson and Payne Chang (University of Wisconsin) to identify and time SAF and spikes poststimulation and to determine durations of prespike feet (Wang et al., 2001; Bai et al., 2004b). Events ≥3–4 pA having sharp upward/downward deflections were counted as spikes. The recordings selected for presentation (Figure 5A) were filtered at 100 Hz to reduce noise (Wang et al., 2003). To control for cell-to-cell variability in secretory response, results were calculated as means for all cells in an experimental sample (15–40 cells per group from multiple transfections for each protein) (Colliver et al., 2000; Bai et al., 2004b). Distributions were fitted to exponentials using the Origin program (MicroCal Software, Northampton, MA). Prespike foot duration used to calculate mean open time of newly opened fusion pores was analyzed for feet ≥1 ms (3 times the data sampling rate), and the data were pooled as in Bai et al. (2004b). Ratios of SAFs:spikes were quantitiated for each time interval to calculate means for each group. All results are reported as means ± SE, and one-way analysis of variance (ANOVA) was used to evaluate statistical significance.
Figure 5.

Effects of expressed SCAMP2-W202A, Arf6-N48R and their wild-type counterparts on NA secretion from PC12 cells measured by amperometry. (A) Representative recordings (30 s) of nontransfected cells (control) and cells transfected with SCAMP2 wild type (SC2-WT), mutant W202A (SC2-W202A), Arf6 wild type (Arf6-WT), and Arf6-N48R; depolarization (105 mM K+) was for the first 8 s. In SC2-W202A, the inset shows a SAF (recording filtered at 100 Hz). (B) Plots of cumulative release events (spikes + SAF) over 30 s in each type of sample determined by calculating the means for all cells; depolarization at zero time. Plots were fitted to exponentials y = A(1 – e–k(x–L)), L >0; derived values of k (rate constant of fusion pore opening), L (lag), and A (maximum observable events) are shown in bar graphs (C–E). (F) SAF/spikes ratio shown as a bar graph; values are slopes of best-fit lines for scatter plots of number SAF versus number of spikes counted manually for each cell within each sample (Supplemental Figure S1). For C–F, data are from 25 to 30 cells for each type of sample. (G) Lifetimes of prespike feet τ0 determined from amperometry recordings. Feet were analyzed according to previous criteria (Chow and von Ruden, 1995) using a program provided by M. Jackson and P. Chang (University of Wisconsin). Analysis was on >120 spikes in each type of sample except 40 spikes for SC2-W202A (due to limited events). All results are mean + SEM and one-way ANOVA was used to evaluate statistical significance; * indicates p < 0.05; **p < 0.01.
RESULTS
SCAMP2 Colocalizes with PLD1, Arf6, and Arf6 Exchange Protein ARNO at the Plasma Membrane of PC12 Cells
PLD1 is localized to the plasma membrane in neuroendocrine cells (Vitale et al., 2001; Du et al., 2003). This distribution contrasts with its intracellular concentration and regulated relocation to the cell surface in mast cells (Brown et al., 1998; Choi et al., 2002; Powner et al., 2002), adipocytes (Emoto et al., 2000), and serum-starved/fed fibroblasts (Du et al., 2003). Because PLD1 and SCAMP2 function late in exocytosis and both have potential interactions with PIP2, we were interested whether they might act at a common level. Initially, we compared the localizations of PLD1 and SCAMP2. Although SCAMP2 is present in cytoplasmic foci, there is substantial concentration at the cell periphery where most of PLD1 is localized (Figure 1A). This suggested possible colocalization at the plasma membrane. Indeed, when we compared distributions on plasma membrane sheets, SCAMP2 and PLD1 seemed extensively colocalized (Figure 1B). Colocalization was documented quantitatively on multiple membrane sheets using a cross-correlation algorithm. Plots in Figure 1, E–G, show that offsetting the red and green images from each other decreases correlation, indicating that colocalization does not reflect random overlap (van Steensel et al., 1996). Because the bulk of SCAMP2 is within the plasma membrane and not on organelles, particularly DCVs, associated with the membrane sheets (Liu et al., 2002), we conclude that SCAMP2 and PLD1 are closely apposed. Furthermore, their proximity was not detectably altered when examined on membranes from depolarized cells. Colocalization also was observed between SCAMP2 and overexpressed HA-tagged PLD1, although overexpressed PLD1 also occurred in additional separate foci (our unpublished data).
Figure 1.

Colocalization of SCAMP2 (SC2) with endogenous PLD1, Arf6, and ARNO. (A) Immunofluorescence of PC12 cells labeled with anti-PLD1 and biotinylated anti-SCAMP2. A digitally deconvolved section through the centers of the cells is shown illustrating SCAMP2 within the cytoplasm, PLD1 concentrated more peripherally, and substantial overlap mainly at the periphery (merged image). (B–D) Examples of plasma membrane sheets from PC12 cells labeled with biotinylated anti-SCAMP2 and anti-PLD1 (B), anti-Arf6 (C), and anti-ARNO (D). Bars correspond to 5 μm. (E–G) Plots of cross-correlation coefficients as a function of image offset illustrating colocalization of SCAMP2 with PLD1 (E), Arf6 (F), and ARNO (G). Pearson cross-correlation coefficients (+ SEM) measured from 12 to 15 plasma membrane sheets for each of the samples were PLD1, 0.67 ± 0.02; Arf6, 0.34 ± 0.04; ARNO, 0.50 ± 0.04. For comparison, the cross-correlation for SCAMP2 versus secretogranin is 0.67 + 0.02. No significant difference in colocalization was observed on plasma membrane sheets from unstimulated and depolarized cells.
In stimulated PC12 cells, activation of PLD1 involves redistribution of Arf6 from peripheral cytoplasmic compartments to the plasma membrane where GDP/GTP exchange is catalyzed by Arf nucleotide binding site opener (ARNO) or a similar exchange protein (Caumont et al., 2000; Powner et al., 2002; Vitale et al., 2002). Because SCAMP2 is colocalized with PLD1, we compared its distribution to that of Arf6. Arf6 also colocalized with SCAMP2 on plasma membrane lawns (Figure 1C). Although the extent of overlap was less than for PLD1 as indicated by the decreased cross-correlation, it was authentic colocalization based on image offset analysis (Figure 1F). On cell depolarization, we did not detect a measurable increase in the extent to which Arf6 colocalized with SCAMP2 (our unpublished data), even though depolarization has been reported to redistribute Arf6 to the cell periphery (Vitale et al., 2002). Because Arf6 regulates multiple events at the cell surface, it is possible that any fractional change in colocalization with SCAMP2 is too small to detect. Overlap also was observed for SCAMP2 and exogenous HA-tagged Arf6, which generally mimics the distribution of endogenous Arf6 (Donaldson, 2003) (our unpublished data). Finally, we found that ARNO colocalized well with SCAMP2 on plasma membrane lawns (Figure 1, D and G), suggesting that SCAMP2 might be a site on the plasma membrane where Arf6 is activated by GDP/GTP exchange. As for PLD1 and Arf6, colocalization did not change in lawns from depolarized cells.
Previously, we have used plasma membrane lawns to determine that ∼85% of secretogranin, a marker of DCVs that are retained on the lawns, colocalized with plasma membrane-associated SCAMP2 (Liu et al., 2002). In our present immunolocalization studies, we have determined that 83% of plasma membrane SCAMP2 foci are PLD1-positive. Thus, it seems likely that DCVs undergo exocytosis at sites that are marked by both SCAMP2 and PLD1.
SCAMP2 Associates with Arf6 and PLD1 as Demonstrated by coIP
Because the plasma membrane fraction of SCAMP2 colocalized with PLD1, Arf6, and ARNO, we decided to probe for possible interactions of SCAMP2 with these proteins by coimmunoprecipitation. We used an anti-SCAMP2 antibody (2τ) for immunoprecipitation (IP) from cells expressing exogenous Arf6 or PLD1, both epitope-tagged with HA. As shown in Figure 2A, 2τ is SCAMP2 specific, recognizing a single band at ∼39,000 in cell lysates. Arf6 was coimmunoprecipitated (coIPd) with SCAMP2. After correcting for the efficiency of IP of SCAMP2 (20%), the amount of Arf6 coIPd was relatively small (<0.5% of total); however, the interaction seemed specific based on criteria presented below. The association was not affected by inclusion of SDS in the detergent medium used for coIP, suggesting a strong interaction. The amount coIPd increased after depolarization of the cells in high K+ medium (Figure 2B), suggesting that interaction might be related to exocytosis. HA-PLD1 also coIPd with SCAMP2, and the fraction of total PLD1 recovered in the IP was 0.5–2%. In contrast to Arf6, the interaction seemed sensitive to the presence of SDS in the IP buffer. For PLD1, the amount recovered was comparable from cells that were unstimulated or stimulated by depolarization (Figure 2C). This suggested a constitutive association with SCAMP2. When coIP of several mutants of Arf6 (all HA-tagged) with SCAMP2 was examined in depolarized cells, association was comparable for wild-type Arf6, Arf6-Q67L (a GTPase-deficient mutant), and Arf6-N48R (which binds GTP but fails to activate PLD1 (Honda et al., 1999; Dana et al., 2000; Vitale et al., 2001). However, binding of Arf6-T27N (not nucleotide-loaded) was modest, although still above background (Figure 2D), suggesting that interaction with SCAMP2 was stabilized by activated Arf6 but might not be strictly GTP dependent. We confirmed the preferential association of Arf6-GTP with SCAMP2 by coIP from digitonin-permeabilized cells. CoIP of HA-Arf6 was robust from samples incubated with GTPγS but was greatly decreased when GDP was included (Figure 2E). SCAMP associations with Arf6 and PLD1 were also interesting in that they were selective when tested among SCAMP isoforms 1–3. In PC12 cells, SCAMPs 1, 2, and 3 are present in relative amounts of ∼1:0.5:2 as determined from western blots using anti-SCAMP mAb 7C12. CoIP of HA-Arf6 was ∼25-fold selective for SCAMP2 based on the amount of Arf6 recovered in each coIP and normalized to the amount of each SCAMP isoform IPd (Figure 2F). CoIP of HA-PLD1 was approximately nine-fold selective for SCAMP2 compared with SCAMP1 and more than threefold compared with SCAMP3 (Figure 2F). Finally, association with SCAMP2 was clearly preferential for Arf6 as compared with Arf 1 (Figure 2F).
Figure 2.

Association of SCAMP2 (SC2) with Arf6 and PLD1 detected by immunoprecipitation. IP was carried out with anti-SCAMP2 (or as indicated below each lane) and nonspecific rabbit IgG (IG) as control. Lysate (L) samples are 0.2% of total (B–D) and 0.5% of total (E). Blots were probed with antibodies as indicated in each panel. (A) Western blot of PC12 cell lysate documenting the specificity of anti-SCAMP2. (B) Cells transfected with HA-Arf6 were treated (5 min) with low K+ buffer or with high K+ buffer (depolarization). The IPs were probed sequentially with anti-HA (top) and anti-SCAMP mAb 7C12 (below); the level of SCAMP2 in the lysate sample was too low to be detected by 7C12. (C) CoIP of HA-tagged PLD1 with SCAMP2 with or without K+ depolarization as in B. (D) Western blot of IPs from cells transfected with HA-tagged wild-type Arf6, Arf6-T27N, Arf6-Q67L, and Arf6-N48R. (E) Cells transfected with HA-Arf6 were permeabilized with 20 μM digitonin, incubated with 100 μM GTPγS or 1 mM GDP, lysed and IPd. (F) Selectivity of SCAMP–Arf interactions among SCAMP and Arf isoforms. IPs with isoform-specific anti-SCAMP 1–3 antibodies from cells transfected with HA-Arf6 (top left), HA-Arf1 (top right), and HA-PLD1 (beneath). Selectivity of coIP among SCAMPs was calculated as Arf6:SCAMP, Arf1:SCAMP and PLD1:SCAMP ratios present in each IP and is shown in the bar graph. Amounts of Arf6, Arf1, and PLD1 coIPd were evaluated from the blots shown, whereas the amount of SCAMP in each IP was evaluated by reblotting with mAb 7C12 and 125I-secondary antibody.
To address further the potential functional importance of the SCAMP2–Arf6 interaction, we compared coIP of endogenous Arf6 with expressed wild-type SCAMP2 and a SCAMP2 point mutant, W202A, in the E peptide segment (previously known as mutant B; Liu et al., 2002), that is a dominant inhibitor of DCV exocytosis (Liu et al., 2002). Association was decreased by ∼50% in the mutant, suggesting that it might be relevant to exocytosis (Figure 3A). Additional support for this possibility was provided by the observation that the E peptide segment of SCAMP2, a potent inhibitor of exocytosis (Guo et al., 2002; Liu et al., 2002), interfered with GTPγS-dependent association of SCAMP2 and HA-Arf6. Figure 3B shows that it effectively blocked coIP of Arf6, whereas an inactive structural variant of E peptide (WY replaced by AA as second and third residues; Liu et al., 2002) and the cytoplasmic C-terminal peptide both had only modest effects. Strikingly, the dose-response for E peptide blockade of coIP mimicked closely the dose-response for E peptide inhibition of DCV exocytosis from permeabilized cells (Liu et al., 2002) (Figure 3C). Association of HA-PLD1 with SCAMP2 was not significantly affected by 50 μM E peptide, the same as for the C-terminal peptide (Figure 3B). This finding clearly distinguished PLD1's association with SCAMP2 from Arf6's association with SCAMP2. We have not detected coIP of expressed ARNO with SCAMP2 to date, possibly because the level of ARNO expression was insufficient or the association was too weak to sustain during coIP.
Figure 3.

Perturbation of Arf6 and PLD1 associations with SCAMP2. (A) Expressed myc-tagged SCAMP2 wild type and W202A mutant (expressed in 2.4:1 ratio) were IPd with anti-myc antibody, and the blot was probed with anti-Arf antibody. The amounts of myc-tagged SCAMP2 wild-type and W202A mutant recovered in the IP (determined by Western blotting using 125I-secondary antibody) were the same. Quantitation of Arf6 indicated that mutant SCAMP2 coIPd 50% less Arf6 than wild-type SCAMP2. * identifies a nonspecific band. Lysate (L) samples are 1% of total. (B) Top and middle, CoIP of HA-Arf6 by anti-SCAMP2 from digitonin-permeabilized cells treated with 100 μM GTPγS is essentially completely competed by SCAMP2-E peptide (CWYRPIYKAFRSDNS) but only modestly by C-terminal peptide (HRAASSAAQGAFQGN) and by mutated E peptide (CAARPIYKAFRSDNS). Bottom, CoIP of HA-PLD1 by anti-SCAMP2 is only modestly decreased by 50 μM E or C-terminal peptide. Lysate (L) samples: 1% of total (top) and 0.5% of total (middle and bottom). (C) Dose response for E peptide inhibition of coIP of HA-Arf6 by anti-SCAMP2.
Effects of the E Peptide of SCAMP2 on PLD Activity
Further evidence that the association of SCAMP2 with Arf6 and PLD1 is relevant to DCV exocytosis has come from examining the effects of the E peptide on PLD activity. Previously, Ca2+ and GTPγS have been used to stimulate PLD activity in SLO-permeabilized chromaffin cells, and activation has been linked to Arf6-GTP function and to exocytosis (Caumont et al., 1998). Using SLO-permeabilized PC12 cells, we have identified analogous PLD activity stimulated by Ca2+ and GTPγS. Notably, E peptide but not mutated E peptide (as described above) or C-terminal peptide of SCAMP2 substantially reduced activity (Figure 4A), and the extent of inhibition (40–45%) was comparable with that observed previously (∼60%) using myristoylated N-terminal Arf6 peptide as blocking agent (Caumont et al., 1998). Furthermore, titration of E peptide concentration indicated a half-maximal inhibitory effect at 10–15 μM (Figure 4B). This is the same concentration range that caused half-maximal inhibition of coIP of Arf6 with SCAMP2 (Figure 3) and of DCV exocytosis (Liu et al., 2002). Although it would be advantageous to examine whether expression of SCAMP2-W202A also inhibits PLD activation, this experiment was not feasible due to insufficient transfection efficiency of the mutant in PC12 cells.
Figure 4.

E peptide of SCAMP2 inhibits PLD activity stimulated by Ca2+ and GTPγS in SLO-permeabilized PC12 cells. (A) Results from four separate experiments showing that 100 μM E peptide, but not mutated E peptide or C-terminal peptide, partially inhibits Ca2+ and GTPγS-stimulated PLD activity. Error bars indicate SEM; **p < 0.01 compared by t test to control. (B) Titration of inhibitory effect of E peptide on PLD activity; results are the mean ± SEM from four separate experiments. The half-maximal effect occurred at 10–15 μM E peptide.
Effects of SCAMP2 and Arf6 Mutants on Noradrenalin Secretion Analyzed by Amperometry
Carbon fiber amperometry has been advantageous for following exocytotic events in real time and has helped to clarify the roles of proteins comprising the exocytotic machinery in specific steps of fusion, including opening, stabilizing, or dilating fusion pores (Graham and Burgoyne, 2000; Wang et al., 2001, 2003; Bai et al., 2004b; Graham et al., 2004; Han et al., 2004). Overexpressed SCAMP2-W202A and PLD activation-deficient Arf6-N48R have each been characterized as inhibitors of late exocytotic events in PC12 cells by their effects on secretion of growth hormone and noradrenalin (NA), respectively (Liu et al., 2002; Vitale et al., 2002). As several of our observations pointed to functional associations of SCAMP2, Arf6, and PLD1 in DCV exocytosis, we decided to use amperometry to compare the effects of the SCAMP2 and Arf6 mutants and their wild-type counterparts on exocytosis to discern whether the two mutants might affect the same step(s). Using a bicistronic vector, PC12 cells were transfected with SCAMP2 or Arf6 constructs along with green fluorescent protein (GFP) to identify transfected cells for recording. Transfection resulted in approximately fivefold overexpression of each construct as determined previously (Liu et al., 2002) (see Materials and Methods). Examples of traces from NA-loaded cells that have been depolarized by locally perfusing with 105 mM K+ from a glass pipette (Wang et al., 2001) are shown in Figure 5A. As noted previously (Wang et al., 2003), two types of events were observed: spikes—sharp peaks of several picoamperes corresponding to opening and dilation of fusion pores and stand-alone feet (SAF)—broadened events with amplitudes smaller than 2 pA, corresponding to opening/closing flicker without pore dilation (Chow and von Ruden, 1995; Wang et al., 2003). Responses for nontransfected cells and cells expressing either wild-type construct typically contain numerous spikes, whereas responses for Arf6-N48R and especially SCAMP2-W202A contained fewer spikes. Interestingly, the majority of events observed for SCAMP2-W202A were SAF. Because electrical signals provide information about different states of the fusion pore, we used the following kinetic model (Wang et al., 2001; Bai et al., 2004b) to analyze our data:
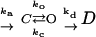 |
C and O represent closed and open fusion pores, respectively, and D represents dilated fusion pores leading to amperometric spikes. ko, kc, kd, respectively, are rate constants for opening, closing, and dilating fusion pores, and ka is an aggregate rate constant for upstream steps. The irreversible O → D (dilation of fusion pore) is reflected as spikes, whereas the reversible C ↔ O (flickering of fusion pore) is reflected as SAF and prespike feet. The responses were compared quantitatively using several parameters and were interpreted using the model. First, we plotted cumulative appearance of exocytotic events (spikes + SAF) averaged from all recordings of each type of sample (Colliver et al., 2000; Wang et al., 2001; Bai et al., 2004b) (Figure 5B). Because there is a delay in onset of release events after the start of depolarization, the data were fitted by single exponential curves: y = A(1 – e–k(x–L)), L ≥0. Here, k reflects ko, the frequency of fusion pore opening, whereas L (lag) is the delay of response. A is the maximal number of DCVs releasable in response to depolarization. The curves clearly show the inhibitory effects of SCAMP2-W202A and Arf6-N48R. Values of k, L, and A are plotted in bar graphs (Figure 5, C–E); in all cases, values for A are consistent with actual total number of events detected (our unpublished data). The data show that both mutants decrease k and A and increase L to very similar extents. Wild-type SCAMP2 decreased k and increased L but to a lesser extent than the mutant, whereas wild-type Arf6 did not alter k but nearly abolished L. Neither wild-type protein affected A as seen from the cumulative event curves.
The total number of exocytotic events as reflected in the values for A are the same for SCAMP2-W202A and Arf6-N48R (Figure 5E); however, the majority of events for SCAMP2 mutant B were SAF (Figure 5A). This observation suggested that expression of SCAMP2-W202A had changed the ratio of SAF/spikes. To check this, we plotted number of spikes versus number of SAF in each cell's response for all cells of each type, and from these scatter plots (Supplemental Figure S1), we determined the SAF/spikes ratio from the slopes of best-fit lines (Figure 5F). According to the kinetic model, the SAF/spikes ratio is a reflection of kc/kd. It is an index of fusion pore dilation (Wang et al., 2003), and its value reflects the tendency of a newly opened fusion pore to either close or dilate. SCAMP2-W202A was unique in substantially increasing the SAF/spikes ratio; the ratio for Arf6-N48R was like that of the other samples (Figure 5F).
We also compared the duration of prespike feet (τ0; Figure 5G). Most spikes are preceded by a prespike foot, which is interpreted as detection of NA leaking through an initially opened fusion pore before it has dilated (Chow and von Ruden, 1995; Travis and Wightman, 1998). τ0 = 1/(kc + kd), and it is an index of stability of the newly opened fusion pore. We found negligible differences in lifetimes for the different types of samples. The τ0 results in combination with the SAF/spikes ratio (reflecting kc/kd) enabled us to deduce that for SCAMP2-W202A, kc is significantly increased and kd is decreased, indicating that expression of the W202A mutant causes newly opened fusion pores to be much more likely to close than dilate. For Arf6-N48R, the unchanged SAF/spikes ratio (Figure 5F) meant that kc and kd are unchanged relative to nontransfected cells.
Effect of SCAMP2-W202A on NA Loading of DCVs
Although SCAMP2 is not detectable in DCVs but is distributed elsewhere intracellularly as well as on the plasma membrane (Liu et al., 2002), we felt it was necessary to consider whether the mutant SCAMP2 might affect either the formation of DCVs or their loading with NA. Thus, we compared spike areas (total charge from NA oxidation) for exocytotic events from all types of samples. Because the radii of DCVs from PC12 cells have a Gaussian distribution, the cube root of total charge should have a similar distribution (Travis and Wightman, 1998). We confirmed this and have plotted the means of Q1/3 as a bar graph in Supplemental Figure S2. As can be seen, the values for cells expressing SCAMP2-W202A were slightly but significantly lower than for all other types of samples. Substantially reduced NA storage potentially could result in an increased SAF/spikes ratio during exocytosis due to misidentifying spikes as SAF. We used lysophosphatidylcholine (LPC) addition to demonstrate that this was not the case. Previously, we showed that addition of LPC could substantially reverse inhibition of depolarization-induced secretion of human growth hormone from PC12 cells expressing SCAMP2-W202A without altering unstimulated secretion (or depolarization-induced secretion from cells expressing wild-type SCAMP2) (Liu et al., 2002). Thus, we included 15 μM LPC in the perfusion pipette containing 105 mM K+ and tested its effect on the SAF/spikes ratio. LPC restored the SAF/spikes ratio of SCAMP2-W202A to the values obtained for nontransfected cells and cells expressing wild-type SCAMP2 (Supplemental Figure S1). Furthermore, LPC addition did not alter the SAF/spikes ratio for Arf6-N48R-expressing cells (Supplemental Figure S1) or for nontransfected cells (our unpublished data). Because the Q1/3 values are the same in the presence and absence of LPC yet the SAF/spikes ratio changes so dramatically, it is clear that expression of the W202A mutant does not impair the ability to distinguish between spikes and SAF. We conclude that SCAMP2-W202A does decrease NA storage somewhat; however, the extent is sufficiently small that it is not a contributing factor to the change in SAF/spikes ratio elicited by the mutant SCAMP.
Effects of the SCAMP2 and Arf6 mutants on parameters of individual spikes such as rise time and half-width are a potential source of additional insight regarding common mechanisms of action. However, due to complications arising from decreased NA storage selectively in cells expressing SCAMP2-W202A, we have decided that these measures are unlikely to provide a reliable comparison and thus we have not pursued them.
DISCUSSION
Although SNAREs and associated proteins are widely regarded as central machinery of DCV exocytosis with synaptotagmins providing a key link between Ca2+ signaling and the final steps of formation of fusion pores (Wang et al., 2001; Rothman, 2002; Bai et al., 2004b; Han et al., 2004; Tucker et al., 2004), we have built a case that SCAMP2 also supports fusion pore formation. Not only is SCAMP2 concentrated at DCV docking sites in PC12 cells as defined by colocalization with syntaxin 1 and complexin (Liu et al., 2002) but also it colocalizes on the plasma membrane and associates with PLD1 and Arf6 (Figures 1 and 2), two other proteins that function late in exocytosis (Vitale et al., 2001; Vitale et al., 2002). Although it could be argued that interactions involving SCAMP2 might occur elsewhere than on the plasma membrane due to SCAMP2's presence in internal organelles (Liu et al., 2002), several of our findings argue that the associations with Arf6 and PLD1 relate to exocytosis. First, the bulk of PLD1 in PC12 cells is concentrated on the plasma membrane (Vitale et al., 2001) where colocalization with SCAMP2 is very thorough (Figure 1) and where DCVs are docked (Liu et al., 2002). Second, SCAMP2 also colocalizes at the plasma membrane with ARNO, which supports DCV exocytosis presumably by activating Arf6 (Caumont et al., 2000). Third, coIP of SCAMP2 and Arf6 is enhanced by GTPγS and by Arf6-GTP, which both support PC12 cell exocytosis (Banerjee et al., 1996; Aikawa and Martin, 2003), and strikingly, coIP is increased upon stimulating PC12 cells by depolarization (Figure 2). Fourth, coIP of Arf6 by SCAMP2-W202A is about half as efficient as for wild-type SCAMP2 (Figure 3), even though both forms of SCAMP2 have comparable localizations (Liu et al., 2002). Thus, decreased association correlates with inhibition of exocytosis. Finally, coIP is antagonized by E peptide with a dose response (Figure 3) that is essentially the same as the dose response for E peptide blockade of exocytosis (Guo et al., 2002; Liu et al., 2002).
Two other types of studies have enabled us to move beyond these interesting correlations to clarify a functional role for the association of SCAMP2 with Arf6 and PLD1 in DCV exocytosis. First, we have found that E peptide interferes with the activity of PLD stimulated by Ca2+ and GTPγS (Figure 4), an activity that seems necessary for DCV exocytosis (Caumont et al., 1998; Vitale et al., 2001). The dose response for the partial inhibitory effect closely resembles the dose response for inhibition of secretion, even though E peptide does not significantly alter PLD's association with SCAMP2 (Figure 3). Second, we have shown by amperometry that PLD activation-deficient Arf6-N48R and SCAMP2-W202A perturb the kinetics of exocytosis of individual DCVs in very similar manners. Both mutants cause the same decrease in frequency of fusion pore opening, same increase in the delay of response after stimulation, and same decrease in total NA release events (spikes + SAF) (Figure 5). Together, our results argue convincingly that the two mutants affect the same step of the fusion process and that appropriate activation or coupling of PLD for DCV exocytosis involves both Arf6 and SCAMP2.
How might these proteins be functioning together? As an integral membrane protein, SCAMP2 may serve as a platform for activating Arf6 (through ARNO) and focusing it in proximity to PLD, which is already associated with SCAMP2 at exocytotic sites. PLD action produces PA, which may promote exocytosis directly through effects on membrane curvature or indirectly through activation of PIP5K (Honda et al., 1999); PIP5K in turn produces PIP2, which activates PLD (Du et al., 2003) and supports exocytosis (Aikawa and Martin, 2003). Previous studies have shown that E peptide of SCAMP2 binds to phospholipid membranes and is positioned deep within the surface where it sequesters polyanionic phospholipids such as PIP2 by electrostatic attraction (Ellena et al., 2004). Because the E peptide segment in full-length SCAMP2 is flanked by transmembrane spans, it may have the same membrane positioning and function as the synthetic peptide. Thus, SCAMP2 may couple both Arf6-GTP and PIP2 components of PLD activation and ensure the linkage of activity to DCV exocytosis. In Arf6, association with SCAMP2 may reflect a direct interaction with E peptide segment or an indirect interaction linked to PIP2-dependent ARNO activity. Exogenous E peptide would disrupt these components, and mutation of W202 to A in SCAMP2 may decrease Arf6 binding (Figure 2) and alter PIP2 sequestration. According to this scenario, overexpression of Arf6-N48R and SCAMP2-W202A each would attenuate PLD activity, respectively, by decreasing activation via functional Arf6 and by decreasing coupling of both Arf6 and PIP2. Reduced PLD activity would then be the common action of the two mutants resulting in identical changes in k, L, and total NA release events (Figure 5). Overexpression of wild-type SCAMP2 leads to modest decreases in the rate constant leading to fusion pore opening and the lag. This may reflect a competitive effect, e.g., one in which active Arf6 is diluted into a larger SCAMP2 pool, slowing but not otherwise affecting SCAMP2 coupling to PLD and to the core machinery of exocytosis. Where wild-type Arf6 is overexpressed, DCV exocytosis is expedited possibly by increasing the amount of Arf6 that can colocalize with endogenous SCAMP2 and PLD1 and thereby serve in exocytotic coupling. We suspect that Arf6 overexpression facilitates priming of DCVs because it decreases the lag preceding NA release but does not alter other parameters in relation to nontransfected cells (Figure 5).
Interestingly, our analysis of DCV exocytosis by amperometry also uncovered a unique and more distal effect of SCAMP2-W202A involving impaired dilation of already opened fusion pores. Because overexpression of wild-type SCAMP2 at a similar level does not cause the same changes, it seems unlikely that the effect is an overexpression artifact, although we cannot rule out the possibility that mutant SCAMP2 might disproportionately alter membrane tension. The effect of the mutant places the perturbation in intimate association with the fusion pore and suggests that SCAMP2 may link upstream events such as PLD activation directly to proteins that contribute to the pore boundary or to pore regulation. Dilation of fusion pores involves radial dispersion of membrane proteins and lipids and requires substantial energy input (Cohen and Melikyan, 2004). It is striking that the single amino acid changed in mutant SCAMP2 profoundly affects this process, and it could signify that SCAMP2 might constitute part of the pore boundary. Although SNAREs are likely fusion pore components (Rothman, 2002; Han et al., 2004; Tucker et al., 2004), the structural similarity of SCAMP2 to the V0 sector of H+ ATPase, another candidate pore component (Peters et al., 2001), has been noted previously (Liu et al., 2002; Bayer et al., 2004). Alternatively, mutant SCAMP2 might perturb pores less directly. For example, if wild-type SCAMP2 normally functions to sequester PIP2 as suggested from studies of E peptide (Ellena et al., 2004), then the mutant may compromise this action locally at fusion DCV sites. Even though PIP2 supports DCV exocytosis (Aikawa and Martin, 2003), restricting its distribution ultimately may be essential during fusion pore formation to regulate membrane curvature. In addition, it is intriguing that abrogation of SCAMP1 expression in mice causes decreased stability of exocytotic fusion pores in mast cells (Fernandez-Chacon et al., 1999). SCAMPs 1 and 2 are near neighbors in the plasma membranes of PC12 cells and mast cells (Guo et al., 2002; Liu et al., 2002); although SCAMP1 associates with Arf6 and PLD much less effectively than SCAMP2, it is possible that the two isoforms collaborate with one another and have complementary roles in regulating late events in DCV exocytosis. Finally, potential associations of SCAMP2 with synaptotagmin may be of interest. Other amperometric studies (Wang et al., 2001; Bai et al., 2004b) analyzed using the same kinetic model that we have used indicate that overexpressed synaptotagmins alter both kc and kd. Thus, SCAMP2 and synaptotagmin may collaborate in controlling fusion pore dynamics and given that the two proteins reside in distinct membranes, their triggered association during stimulation may be especially interesting to explore.
In conclusion, SCAMP2 has been implicated as a protein that functions in DCV exocytosis in PC12 cells (Liu et al., 2002). Our present data show that SCAMP2 associates with Arf6 and PLD1 and is likely to couple their actions to exocytosis. Although it is not yet known whether Arf6 and PLD1 bind directly to SCAMP2, we have established that perturbation of association of Arf6 with SCAMP2, activation of PLD, and exocytosis may entail a common mechanism involving SCAMP2's E peptide domain. Finally, mutant SCAMP2 clearly interferes with dilation of exocytotic fusion pores, suggesting that distal to its coupling of Arf6 and PLD1, SCAMP2 may link or contribute to the fusion pore itself. Our model of SCAMP2 as a membrane platform for Arf6 and PLD1 and a prospective link to the fusion pore is depicted in Figure 6.
Figure 6.

Model depicting SCAMP2 as a plasma membrane platform on which Arf6 is activated by ARNO, PLD1 is activated by Arf6-GTP and PIP2, and PLD activity is coupled to DCV exocytosis. PIP2 (cylinder-shaped lipid) is shown as an activator of PLD1 and sequestered by the E peptide of SCAMP2; PA (cone-shaped lipid) produced by PLD1 in the fusion zone may destabilize the bilayer and facilitate fusion. SCAMP2 is also shown in proximity to syntaxin-1 (Syn1) and to complexin (Cpx). These features reflect its prospective function in fusion pore dilation and its excellent colocalization with complexin.
Supplementary Material
Acknowledgments
We thank the following people for contributions to this work: Zhijian Liu for advice and assistance with data analysis; Drs. Meyer Jackson and Payne Chang (University of Wisconsin) for advice on amperometry and for providing the computer program used for data analysis; Dr. Chih-Tien Wang (University of California, San Diego, San Diego, CA) for advice on amperometry; Charles Hubbard for molecular biology; Dr. Lorraine Santy for advice on PLD assay and use of Arf6 constructs; Dr. Angela Otero for assistance with statistical analyses; Dr. Attila Szabo for computer support, and Dr. Edwin Chapman and laboratory members for providing PC12 cells and advice on culture conditions. The following individuals kindly provided antibodies and constructs: Dr. Michael Frohman (SUNY, Stonybrook); Dr. Derek Persons (St. Jude's Childrens Research Hospital), Drs. Ian Macara and Lorraine Santy (University of Virginia, Charlottesville, VA) and Dr. Julie Donaldson (National Heart, Lung, and Blood Institute, National Institutes of Health). This work was supported by Grants DE09655 and AI47150 from the National Institutes of Health.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–03–0231) on July 19, 2005.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Aikawa, Y., and Martin, T. F. (2003). ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J. Cell Biol. 162, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, J., Tucker, W. C., and Chapman, E. R. (2004a). PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44. [DOI] [PubMed] [Google Scholar]
- Bai, J., Wang, C. T., Richards, D. A., Jackson, M. B., and Chapman, E. R. (2004b). Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron 41, 929–942. [DOI] [PubMed] [Google Scholar]
- Banerjee, A., Kowalchyk, J. A., DasGupta, B. R., and Martin, T.F.J. (1996). SNAP-25 is required for a late postdocking step in Ca2+-dependent exocytosis. J. Biol. Chem. 271, 20227–20230. [DOI] [PubMed] [Google Scholar]
- Bayer, M. J., Reese, C., Buhler, S., Peters, C., and Mayer, A. (2004). Vacuole membrane fusion: Vo functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J. Cell Biol. 162, 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, S. H., and Castle, J. D. (1993). SCAMP 37, a new marker within the general cell surface recycling system. EMBO J. 12, 3753–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, S. H., Laurie, S. M., Mixon, M. B., and Castle, J. D. (1991). Secretory carrier membrane proteins 31–35 define a common protein composition among secretory carrier membranes. J. Biol. Chem. 266, 18949–18957. [PubMed] [Google Scholar]
- Brown, F. D., Thompson, N., Saqib, K. M., Clark, J. M., Powner, D., Thompson, N. T., Solari, R., and Wakelam, M. J. (1998). Phospholipase D1 localises to secretory granules and lysosomes and is plasma-membrane translocated on cellular stimulation. Curr. Biol. 8, 835–838. [DOI] [PubMed] [Google Scholar]
- Caumont, A. S., Galas, M. C., Vitale, N., Aunis, D., and Bader, M. F. (1998). Regulated exocytosis in chromaffin cells. J. Biol. Chem. 273, 1373–1379. [DOI] [PubMed] [Google Scholar]
- Caumont, A. S., Vitale, N., Gensse, M., Galas, M. C., Casanova, J. E., and Bader, M. F. (2000). Identification of a plasma membrane-associated guanine nucleotide exchange factor for ARF6 in chromaffin cells. Possible role in the regulated exocytotic pathway. J. Biol. Chem. 275, 15637–15644. [DOI] [PubMed] [Google Scholar]
- Chapman, E. R. (2002). Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat. Rev. Mol. Cell. Biol. 3, 498–508. [DOI] [PubMed] [Google Scholar]
- Choi, W. S., Kim, Y. M., Combs, C., Frohman, M. A., and Beaven, M. A. (2002). Phospholipases D1 and D2 regulate different phases of exocytosis in mast cells. J. Immunol. 168, 5682–5689. [DOI] [PubMed] [Google Scholar]
- Chow, R. H., and von Ruden, L. (1995). Electrochemical detection of secretion from single cells. In: Single Channel Recording, ed. B. Sakmann and E. Neher, New York: Plenum, 245–275.
- Cohen, F. S., and Melikyan, G. B. (2004). The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 199, 1–14. [DOI] [PubMed] [Google Scholar]
- Colliver, T. L., Hess, E. J., Pothos, E. N., Sulzer, D., and Ewing, A. G. (2000). Quantitative and statistical analysis of the shape of amperometric spikes recorded from two populations of cells. J. Neurochem. 74, 1086–1097. [DOI] [PubMed] [Google Scholar]
- Dana, R. R., Eigsti, C., Holmes, K. L., and Leto, T. L. (2000). A regulatory role for ADP-ribosylation factor 6 (ARF6) in activation of the phagocyte NADPH oxidase. J. Biol. Chem. 275, 32566–32571. [DOI] [PubMed] [Google Scholar]
- Donaldson, J. G. (2003). Multiple roles for Arf 6, sorting, structuring, and signaling at the plasma membrane. J. Biol. Chem. 278, 41573–41576. [DOI] [PubMed] [Google Scholar]
- Du, G., Altshuller, Y. M., Vitale, N., Huang, P., Chasserot-Golaz, S., Morris, A. J., Bader, M. F., and Frohman, M. A. (2003). Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J. Cell Biol. 162, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellena, J. F., Malthroup, J., Wu, J., Rauch, M., Jayasinghe, S., Castle, J. D., and Cafiso, D. S. (2004). Membrane position of a basic aromatic peptide that sequesters phosphatidylinositol 4,5 bisphosphate determined by site-directed spin labeling and high resolution NMR. Biophys. J. 87, 3221–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto, M., Klarlund, J. K., Waters, S. B., Hu, V., Buxton, J. M., Chawla, A., and Czech, M. P. (2000). A role for phospholipase D in GLUT4 glucose transporter translocation. J. Biol. Chem. 275, 7144–7151. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon, R., Toledo, G. A., Hammer, R. E., and Sudhof, T. C. (1999). Analysis of SCAMP1 function in secretory vesicle exocytosis by means of gene targeting in mice. J. Biol. Chem. 274, 32551–32554. [DOI] [PubMed] [Google Scholar]
- Gambhir, A., Hangyas-Mihalyne, G., Zaitseva, I., Cafiso, D. S., Wang, J., Murray, D., Pentyala, S. N., Smith, S. O., and McLaughlin, S. (2004). Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86, 2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, M. E., Barclay, J. W., and Burgoyne, R. D. (2004). Syntaxin/Munc18 Interactions in the Late Events during Vesicle Fusion and Release in Exocytosis. J. Biol. Chem. 279, 32751–32760. [DOI] [PubMed] [Google Scholar]
- Graham, M. E., and Burgoyne, R. D. (2000). Comparison of cysteine string protein (Csp) and mutant alpha-SNAP overexpression reveals a role for csp in late steps of membrane fusion in dense-core granule exocytosis in adrenal chromaffin cells. J. Neurosci. 20, 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., Liu, L., Cafiso, D., and Castle, D. (2002). Perturbation of a very late step of regulated exocytosis by a secretory carrier membrane protein (SCAMP2)-derived peptide. J. Biol. Chem. 277, 35357–35363. [DOI] [PubMed] [Google Scholar]
- Han, X., Wang, C. T., Bai, J., Chapman, E. R., and Jackson, M. B. (2004). Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289–292. [DOI] [PubMed] [Google Scholar]
- Hay, J. C., Fisette, P. L., Jenkins, G. H., Fukami, K., Takenawa, T., Anderson, R. A., and Martin, T. F. (1995). ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature 374, 173–177. [DOI] [PubMed] [Google Scholar]
- Holroyd, P., Lang, T., Wenzel, D., De Camilli, P., and Jahn, R. (2002). Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc. Natl. Acad. Sci. USA 99, 16806–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz, R. W., Bittner, M. A., and Senter, R. A. (1992). Regulated exocytotic fusion I: chromaffin cells and PC12 cells. Methods Enzymol. 219, 165–177. [DOI] [PubMed] [Google Scholar]
- Holz, R. W., Hlubek, M. D., Sorensen, S. D., Fisher, S. K., Balla, T., Ozaki, S., Prestwich, G .D., Stuenkel, E. L., and Bittner, M. A. (2000). A pleckstrin homology domain specific for phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5-P2) and fused to green fluorescent protein identifies plasma membrane PtdIns-4,5-P2 as being important in exocytosis. J. Biol. Chem. 275, 17878–17885. [DOI] [PubMed] [Google Scholar]
- Honda, A., et al. (1999). Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99, 521–532. [DOI] [PubMed] [Google Scholar]
- Hubbard, C., Singleton, D., Rauch, M., Jayasinghe, S., Cafiso, D., and Castle, D. (2000). The secretory carrier membrane protein family: structure and membrane topology. Mol. Biol. Cell 11, 2933–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, R., Lang, T., and Sudhof, T. C. (2003). Membrane fusion. Cell 112, 519–533. [DOI] [PubMed] [Google Scholar]
- Lang, T., Bruns, D., Wenzel, D., Riedel, D., Holroyd, P., Thiele, C., and Jahn, R. (2001). SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 20, 2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L., Guo, Z., Tieu, Q., Castle, A., and Castle, D. (2002). Role of secretory carrier membrane protein SCAMP2 in granule exocytosis. Mol. Biol. Cell 13, 4266–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders, E. M. M., Verbeek, F. J., and Aten, J. A. (1993). Measurement of co-localization of objects in dual-colour confocal images. J. Microscopy 169, 375–382. [DOI] [PubMed] [Google Scholar]
- Martin, T. F. (2003). Tuning exocytosis for speed: fast and slow modes. Biochim. Biophys. Acta 1641, 157–165. [DOI] [PubMed] [Google Scholar]
- Pabst, S., Hazzard, J. W., Antonin, W., Sudhof, T. C., Jahn, R., Rizo, J., and Fasshauer, D. (2000). Selective interaction of complexin with the neuronal SNARE complex. J. Biol. Chem. 275, 19808–19818. [DOI] [PubMed] [Google Scholar]
- Peters, C., Bayer, M. J., Buhler, S., Andersen, J. S., Mann, M., and Mayer, A. (2001). Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 409, 581–588. [DOI] [PubMed] [Google Scholar]
- Powner, D. J., Hodgkin, M. N., and Wakelam, M. J. (2002). Antigen-stimulated activation of phospholipase D1b by Rac1, ARF6, and PKCα in RBL-2H3 cells. Mol. Biol. Cell 13, 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, J. E. (2002). Lasker Basic Medical Research Award. The machinery and principles of vesicle transport in the cell. Nat. Med. 8, 1059–1062. [DOI] [PubMed] [Google Scholar]
- Santy, L. C., and Casanova, J. E. (2001). Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J. Cell Biol. 154, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciorra, V. A., Rudge, S. A., Prestwich, G. D., Frohman, M. A., Engebrecht, J., and Morris, A. J. (1999). Identification of a phosphoinositide binding motif that mediates activation of mammalian and yeast phospholipase D isoenzymes. EMBO J. 20, 5911–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, D. R., Wu, T. T., and Castle, J. D. (1997). Three mammalian SCAMPs (secretory carrier membrane proteins) are highly related products of distinct genes having similar subcellular distributions. J. Cell Sci. 110, 2099–2107. [DOI] [PubMed] [Google Scholar]
- Travis, E. R., and Wightman, R. M. (1998). Spatio-temporal resolution of exocytosis from individual cells. Annu. Rev. Biophys. Biomol. Struct. 27, 77–103. [DOI] [PubMed] [Google Scholar]
- Tucker, W. C., Edwardson, J. M., Bai, J., Kim, H. J., Martin, T. F., and Chapman, E. R. (2003). Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J. Cell Biol. 162, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, W. C., Weber, T., and Chapman, E. R. (2004). Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438. [DOI] [PubMed] [Google Scholar]
- van Steensel, B., van Binnendijk, E. P., Hornsby, C. D., van der Voort, H. T. M., Krozowski, Z. S., de Kloet, E. R., and van Driel, R. (1996). Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 109, 787–792. [DOI] [PubMed] [Google Scholar]
- Vitale, N., Caumont, A. S., Chasserot-Golaz, S., Du, G., Wu, S., Sciorra, V. A., Morris, A. J., Frohman, M. A., and Bader, M. F. (2001). Phospholipase D 1, a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 20, 2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale, N., Chasserot-Golaz, S., Bailly, Y., Morinaga, N., Frohman, M. A., and Bader, M. F. (2002). Calcium-regulated exocytosis of dense-core vesicles requires the activation of ADP-ribosylation factor (ARF)6 by ARF nucleotide binding site opener at the plasma membrane. J. Cell Biol. 159, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. T., Grishanin, R., Earles, C. A., Chang, P. Y., Martin, T. F., Chapman, E. R., and Jackson, M. B. (2001). Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science 294, 1111–1115. [DOI] [PubMed] [Google Scholar]
- Wang, C. T., Lu, J. C., Bai, J., Chang, P. Y., Martin, T. F., Chapman, E. R., and Jackson, M. B. (2003). Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature 424, 943–947. [DOI] [PubMed] [Google Scholar]
- Wu, T. T., and Castle, J. D. (1997). Evidence for colocalization and interaction between 37 and 39 kDa isoforms of secretory carrier membrane proteins (SCAMPs). J. Cell Sci. 110, 1533–1541. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Kim-Miller, M. J., Fukuda, M., Kowalchyk, J. A., and Martin, T. F. (2002). Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron 34, 599–611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.