

Abstract
Neutrophils, the most abundant leukocytes in human blood, have long been recognized as critical first responders in the innate immune system’s defense against pathogens. Some of the more notable innate anti-microbial properties of neutrophils include generation of superoxide free radicals like myeloperoxidase (MPO), production of proteases that reshape the extracellular matrix allowing for easier access to infected tissues, and release of neutrophil extracellular traps (NETs), extruded pieces of DNA that ensnare bacterial and fungi. These mechanisms developed to provide neutrophils with a vast array of specialized functions to provide the host defense against infection in an acute setting. However, emerging evidence over the past few decades has revealed a far more complex and nuanced role for these neutrophil-driven processes in various chronic conditions, particularly in cardiovascular diseases.
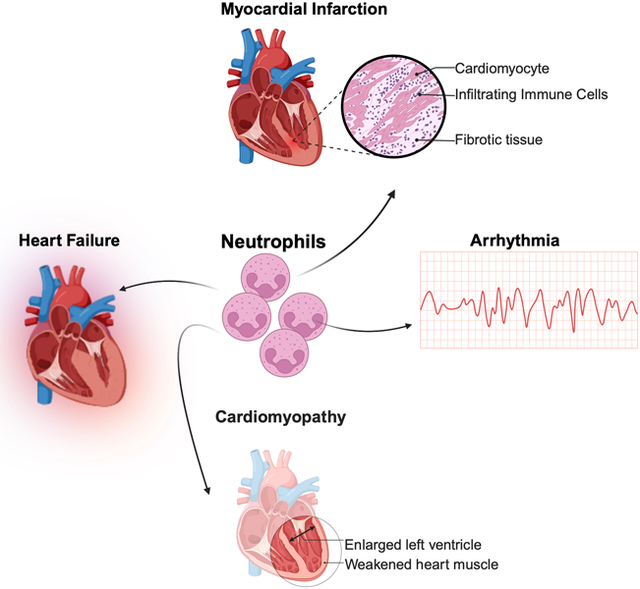
The pathophysiology of cardiac diseases involves a complex interplay of hemodynamic, neurohumoral, and inflammatory factors. Neutrophils, as key mediators of inflammation, contribute significantly to this intricate network. Their involvement extends far beyond their classical role in pathogen clearance, encompassing diverse functions that can both exacerbate tissue damage and contribute to repair processes. Here we consider the contributions of neutrophils to myocardial infarction, heart failure, cardiac arrhythmias and non-ischemic cardiomyopathies. Understanding these complex interactions is crucial for developing novel therapeutic strategies aimed at modulating neutrophil functions in these highly morbid cardiac diseases.
Keywords: Neutrophils, myocardial infarction, heart failure, arrhythmias, cardiac, cardiomyopathies
Graphical Abstract
SUMMARY SENTENCE:
In this review, we examine how neutrophils influence the pathobiology of myocardial infarction, heart failure, cardiac arrhythmias, and cardiomyopathies with attention to underlying mechanisms and potential novel therapeutic approaches targeting these processes.
MYOCARDIAL INFARCTION
Myocardial infarction (MI) remains a leading cause of morbidity and mortality worldwide[1]. In the United States alone, over 1 million people suffer from MI each year with an annual MI-associated mortality rate of over 35%[2]. MI results when atherosclerotic coronary artery plaque rupture provokes thrombus formation that limits or eliminates coronary blood flow. Myocardial infarction is a dynamic process that evolves over the course of hours, days to even weeks[3, 4]. The phases of MI evolution are defined in large part by underlying cellular events, including critical contributions from immune cell activation: acute phase (first 72 hours after MI), late repair and proliferative phase (4–14 days), and remodeling phase (>14 days)[5]. The role of neutrophils in MI has been extensively studied, revealing a complex and dynamic involvement that spans from the acute phase of injury through to long-term cardiac remodeling[3, 6, 7].
In the acute phase of MI, neutrophils are among the first immune cells to infiltrate the infarcted myocardium[8]. This rapid response is triggered by the release of damage-associated molecular patterns (DAMPs) from necrotic cardiomyocytes and the activation of the complement system[9, 10]. Within hours of the onset of ischemia, neutrophils begin to accumulate in the border zone between infarcted and viable myocardium. This initial infiltration is followed by a massive influx of neutrophils into the infarct core, peaking at around 24–48 hours post-MI[11].
The acute neutrophil response serves several important functions in the initial stages of MI. Neutrophils play a crucial role in clearing necrotic debris through phagocytosis and the release of proteolytic enzymes[12, 13]. This clearance is essential for initiating the wound healing process. Moreover, neutrophils secrete pro-inflammatory cytokines and chemokines that recruit additional immune cells, including monocytes and lymphocytes, to the site of injury[14–16]. This orchestration of the inflammatory response is critical for proper infarct healing and scar formation[17, 18].
While neutrophils play essential roles in the early response to MI, their massive influx contributes significantly to acute myocardial injury through several mechanisms. Neutrophil-derived reactive oxygen species (ROS) cause direct oxidative damage to cardiomyocytes[19, 20]. A review by Vinten-Johansen summarizes the evidence that neutrophil-mediated oxidative stress is a major contributor to reperfusion injury following MI[21]. The burst of ROS production upon reperfusion overwhelms the antioxidant defenses of cardiomyocytes, leading to lipid peroxidation, protein oxidation, and DNA damage. This oxidative damage can trigger cardiomyocyte apoptosis and expand the infarct size [22, 23].
The release of proteolytic enzymes by neutrophils, particularly neutrophil elastase and matrix metalloproteinases (MMPs), can degrade the extracellular matrix and weaken the structural integrity of the myocardium. Romson et al. showed that neutrophil depletion in dogs significantly reduced infarct size following coronary artery occlusion and reperfusion, highlighting the detrimental effects of neutrophil-derived proteases in acute MI[24]. It is worth noting that this and other early studies[25, 26] were carried out in large animal models, rather than mice, with techniques that are no longer in use. These technical differences may contribute to some apparent discrepancies between the contemporary and historical literature on the role of neutrophils in cardiac injury.
The formation of neutrophil extracellular traps (NETs) in the coronary microvasculature contributes to microvascular obstruction and the no-reflow phenomenon [27, 28]. NETs are web-like structures composed of decondensed chromatin scaffolds decorated with numerous bioactive proteins including myeloperoxidase (MPO), neutrophil elastase, cathepsin G, matrix metalloproteinases, and antimicrobial peptides such as defensins[29]. The DNA backbone is enriched with citrullinated and hyperacetylated histones that contribute to the chromatin decondensation necessary for NET formation. These structures initially were described as an antimicrobial mechanism but have since been implicated in numerous pathological processes[30, 31]. In the infarcted myocardium, NETs have primarily been identified through the presence of citrullinated histone H3, though definitive evidence of altered nuclear morphology and extracellular citrullinated DNA remains limited. The hypoxic environment characteristic of myocardial ischemia may influence the timing and extent of NETosis, as demonstrated in recent studies showing delayed NET formation under hypoxic conditions[32]. Savchenko et al. demonstrated that mice lacking peptidylarginine deiminase 4 (PAD4), an enzyme essential for NET formation, had significantly smaller infarcts and better preserved left ventricular function following ischemia-reperfusion injury[33]. While these findings underscore the importance of NETs in exacerbating acute myocardial damage[34–36]. It is worth noting that studies using PAD4 knockout models have limitations since PAD4 has multiple cellular functions beyond NET formation, including roles in transcriptional regulation and protein citrullination that could influence cardiac remodeling independently of NET formation. This complexity highlights the need for more selective approaches to studying NET biology in the context of myocardial injury.
The interaction between neutrophils and platelets in the setting of MI further amplifies tissue injury. Neutrophil-platelet aggregates form rapidly following coronary occlusion and contribute to microvascular obstruction[37]. Moreover, activated platelets enhance neutrophil activation and NET formation, creating a vicious cycle of thromboinflammation[38]. Siegel et al. showed that targeting the interaction between neutrophil Mac-1 and platelet GPIbα using a small molecule inhibitor reduced infarct size in a mouse model of MI, highlighting the therapeutic potential of disrupting neutrophil-platelet interactions[39].
As the acute phase of MI transitions into the healing and remodeling phases, the role of neutrophils becomes more nuanced. While the initial wave of neutrophil infiltration subsides within the first week post-MI, a low-grade neutrophil presence persists in the infarct and border zones for several weeks[40]. A study by Ma et al found that neutrophils isolated from the infarcted left ventricle (LV) of mice showed high expression of proinflammatory markers at Day 1 but rising expression of anti-inflammatory markers at Days 5 and 7 after MI. Flow cytometry demonstrated that although proinflammatory N1 neutrophils were always predominant the relative abundance of N2 neutrophils increased post-MI from 2.4 +/− 0.6% on Day 1 to 18.1 +/− 3.0% on Day 7[11]. These persistent neutrophils play important roles in regulating the reparative response and influencing long-term cardiac remodeling[41–44] through multiple mechanisms, including the production of specialized pro-resolving mediators and the orchestration of macrophage-mediated repair processes. For example, neutrophil-derived reactive oxygen species (ROS) trigger phenotypic changes in macrophages that favor tissue repair rather than inflammation [45]. These “repair-programming” neutrophils can modulate the local immune environment through the release of factors such as lactoferrin, lipoxins, and resolvins that actively promote the resolution of inflammation[46]. Additionally, neutrophils contribute to tissue repair through the clearance of dead cells and debris, the regulation of the extracellular matrix composition, and the secretion of growth factors that support tissue regeneration[47]. This dual role of neutrophils - as both mediators of acute injury and facilitators of repair - highlights the complex and context-dependent nature of neutrophil functions in myocardial infarction. These findings provide further evidence that neutrophils can act beyond their traditional destructive roles and suggest that certain neutrophil populations may contribute to cardiac repair. Understanding the factors that govern the transition between these opposing functions may provide new therapeutic opportunities for optimizing post-MI cardiac repair.
The resolution of neutrophil-mediated inflammation is crucial for proper infarct healing and prevention of adverse cardiac remodeling. Impaired neutrophil clearance or persistent neutrophil activation can lead to chronic inflammation, excessive fibrosis, and maladaptive ventricular remodeling[48, 49]. Horckmans et al. demonstrated that defective neutrophil clearance in a mouse model of MI led to impaired resolution of inflammation, exaggerated left ventricular dilation, and worse cardiac function[41]. This study highlighted the importance of timely neutrophil clearance in promoting optimal cardiac repair.
The prognostic value of neutrophil-related markers in MI has been shown in numerous clinical studies. Elevated neutrophil counts and neutrophil-to-lymphocyte ratios (NLR) at the time of hospital admission consistently have been associated with larger infarct sizes, increased risk of heart failure, and higher mortality rates in patients with acute MI[48, 50, 51]. A meta-analysis by Angkananard et al. found that high NLR was an independent predictor of short-term and long-term mortality in patients with ST-elevation myocardial infarction (STEMI)[52]. Moreover, circulating levels of NET components have emerged as potential biomarkers in MI. Mangold et al. found that plasma levels of cell-free DNA and myeloperoxidase-DNA complexes, markers of NET formation, were significantly elevated in STEMI patients and correlated with infarct size and left ventricular function[53]. These findings suggest that NET-related biomarkers may provide additional prognostic information beyond traditional risk factors in MI patients.
Therapeutic strategies aimed at limiting the inflammatory response post-MI historically yielded limited efficacy in clinical trials despite robust success in pre-clinical models[54], as is the unfortunate case for many novel therapeutics. Specifically, studies targeting CD/11/CD18[55] and C5[56] did not show any significant difference in infarct size between treatment and placebo. However, the monoclonal antibody inclacumab targeting P-selectin, an adhesion molecule on the surface of endothelial cells that modulates recruitment of inflammatory cells and platelets, showed decreased myocardial damage within 24 hours of MI in the SELECT-ACS trial[57]. Taken together, these mixed results suggest that inhibiting specific neutrophil functions with biologically plausible therapies may hold promise, but the clinical efficacy of these approaches is determined both by the choice of target and by a multitude of variables in clinical trial design and conduct.
Other novel approaches in the field aim to modulate specific aspects of neutrophil function. These include targeting neutrophil recruitment through CXCR2 antagonists, modulation of NET formation using PAD4 inhibitors, such as with Jubilant oral PAD4 inhibitor JBI-589, and pro-resolving mediators such as resolvins and lipoxins and targeting neutrophil-platelet interactions[58–63].
Future research focusing on the temporal and spatial dynamics of neutrophil functions in different stages of MI may lead to more effective therapeutic strategies. Moreover, the identification of distinct neutrophil subsets with potentially beneficial functions opens new avenues for developing targeted approaches that selectively modulate specific neutrophil populations while preserving their essential defensive functions.
HEART FAILURE
Heart failure (HF) is a highly morbid chronic condition characterized by the heart’s inability to to meet the body’s metabolic demands. HF is a final common pathway that can result from various cardiovascular disorders, including previous myocardial infarction, hypertension, and cardiomyopathies[64], as discussed elsewhere in this review. HF is divided broadly into HF with reduced ejection fraction (HFrEF, wherein the contractile function of the heart is impaired) and HF with preserved ejection fraction (HFpEF, wherein HF arises despite normal contractile function). In the United States alone, almost 7 million people have HF and the annual incidence of HF is over 1 million[2]. The role of inflammation in HF has gained significant attention in recent years, with neutrophils emerging as key players in the pathogenesis and progression of this complex syndrome [65].
In HFrEF, particularly in its acute decompensated form, there is often a marked increase in circulating neutrophils and elevated levels of neutrophil-recruiting chemokines in the myocardium[66, 67], leading to increased neutrophil infiltration into cardiac tissue. In chronic HFrEF a persistent low-grade inflammation characterized by ongoing neutrophil recruitment and activation is observed[68]. The distribution of neutrophils within the myocardium in HFrEF is often more diffuse compared to the localized infiltration seen in acute MI, reflecting the global nature of cardiac dysfunction in HFrEF [40, 69].
Several mechanisms contribute to the increased neutrophil presence and activation in HF. Heightened sympathetic nervous system activity and renin-angiotensin-aldosterone system activation characteristic of HFrEF can promote neutrophil mobilization from the bone marrow and enhance their recruitment to the heart[70, 71]. Increased oxidative stress in failing hearts can activate neutrophils and promote their adhesion to the endothelium[72, 73]. Impaired endothelial function in HFrEF facilitates neutrophil adhesion and transmigration into cardiac tissue[74]. Additionally, elevated levels of pro-inflammatory cytokines in HFrEF, such as TNF-α and IL-6, can prime neutrophils and enhance their effector functions[66].
Neutrophils contribute to the pathophysiology of HFrEF through several interrelated mechanisms. Activated neutrophils produce and secrete a range of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β. These cytokines play crucial roles in orchestrating the inflammatory response and can have direct effects on cardiomyocytes and other cardiac cell types[75, 76]. TNF-α has been shown to induce cardiomyocyte apoptosis, contribute to cardiac hypertrophy, and impair contractile function[77]. IL-6 is involved in the acute phase response and can promote cardiac fibrosis[78]. IL-1β is a potent pro-inflammatory cytokine that can induce cardiomyocyte death and contribute to adverse cardiac remodeling [79]. The sustained release of these cytokines by infiltrating neutrophils can lead to a chronic inflammatory state in the heart, contributing to the progression of heart failure [80].
In the context of HFrEF, neutrophil-derived ROS, primarily generated through the NADPH oxidase complex, can cause direct damage to cardiomyocytes, endothelial cells, and extracellular matrix proteins[81]. Oxidative stress can lead to lipid peroxidation, protein oxidation, and DNA damage, all of which can impair cellular function and promote cell death[82]. Moreover, ROS can activate various signaling pathways that contribute to adverse cardiac remodeling, including the activation of matrix MMPs and the induction of pro-fibrotic signals[83]. Carbone et al. demonstrated that neutrophil-derived ROS contribute to mitochondrial dysfunction in cardiomyocytes, leading to impaired energy production and contractile dysfunction in HFrEF[19].
The formation of NETs has emerged as an important mechanism by which neutrophils contribute to HFrEF pathogenesis. In the context of HFrEF, NETs have been implicated in the propagation of chronic inflammation and fibrosis[84]. The cytotoxic components of NETs, such as histones and MPO, can directly damage cardiomyocytes and endothelial cells[85–87]. Moreover, NETs can serve as a scaffold for the deposition of pro-inflammatory mediators, perpetuating the inflammatory response in the heart[88–91]. Recent studies have shown that inhibition of NET formation using PAD4 inhibitors or DNase treatment reduced cardiac fibrosis and improved cardiac function in animal models of pressure overload-induced heart failure[92].
Neutrophils are equipped with a potent arsenal of proteolytic enzymes, including neutrophil elastase, cathepsin G, and MMPs. While these enzymes play crucial roles in the antimicrobial functions of neutrophils, their release in the cardiac microenvironment can have significant pathological consequences in heart failure. Neutrophil elastase can degrade elastin and other structural proteins in the myocardium, contributing to adverse ventricular remodeling[40, 93, 94]. MMPs, particularly MMP-9, play crucial roles in extracellular matrix remodeling and can contribute to both adaptive and maladaptive processes in the heart. Excessive or prolonged MMP activation can lead to adverse remodeling, ventricular dilation, and progression of HFrEF[95].
Recent studies have highlighted the role of neutrophils in modulating cardiac metabolism and energetics in HFrEF. Neutrophil-derived factors can impair mitochondrial function in cardiomyocytes, leading to decreased ATP production and contractile dysfunction[20]. Moreover, the metabolic reprogramming of neutrophils themselves in the context of chronic inflammation may contribute to the overall metabolic derangements observed in HFrEF[96–98].
The prognostic value of neutrophil-related markers in HFrEF has been demonstrated in numerous clinical studies. Elevated neutrophil counts and high NLRs consistently have been linked to increased mortality, higher rates of hospitalization, and worse functional capacity in both acute and chronic HF[52, 99–101]. A study by Uthamalingam et al. found that patients with acute decompensated HFrEF and an NLR ≥ 5.1 had a significantly higher 30-day mortality rate compared to those with lower NLR values[102]. Clinical studies have have provided evidence for the involvement of NETs in HFrEF pathogenesis. Increased levels of circulating NET components, such as cell-free DNA and MPO-DNA complexes, have been observed in HFrEF patients and correlate with disease severity[103, 104]. A study by Ichimura et al. found that plasma levels of citrullinated histone H3, a marker of NET formation, were elevated in patients with acute decompensated HFrEF and associated with adverse outcomes[105].
The contributions of neutrophils to HFpEF are less well defined though recent studies suggest that there may be some shared pathobiology with HFrEF. NET formation was detected in a mouse model of HFpEF and NET inhibition improved the HFpEF phenotype[106]. Elevated NLRs[107–109] and circulating MPO[110, 111] have been repeatedly shown to correlate with adverse outcomes in HFpEF.
The emerging understanding of neutrophil involvement in HF has led to the exploration of novel therapeutic strategies. While global neutrophil depletion or inhibition may not be feasible due to the risk of compromising host defense, more targeted approaches are being investigated. Specific inhibitors of NADPH oxidase 2 (NOX2), the primary source of ROS in neutrophils, have shown cardioprotective effects in animal models of HF[112, 113]. Sirker et al. showed that genetic deletion of NOX2 in mice reduces oxidative stress and preserves cardiac function in a model of pressure overload-induced HFrEF [114]. MPO inhibitors have shown potential to reduce oxidative stress and improve outcomes in animal models of HFrEF. For example, Mollenhauer et al. found that MPO inhibition attenuates left ventricular remodeling and dysfunction in a mouse model of pressure overload[115]. Strategies aimed at inhibiting NET formation or promoting NET degradation also have shown promise in preclinical studies[116, 117]. These approaches may help mitigate the pro-inflammatory and pro-fibrotic effects of NETs in HFrEF.
Other clinical trials are focusing on targeting chemokine and cytokines that are downstream of neutrophil activation. The D-HART2 clinical trial, which investigated the effect the IL-1 inhibitor anakinra on HFrEF, showed decreased levels of the HF biomarker NT-proBNP after 12 weeks but no change in cardiorespiratory fitness[118]. The ongoing HERMES trial is looking at the efficacy and safety of IL-6 inhibition in HFpEF patients and the ENDEAVOR Phase 2–3 clinical trial will examine the efficacy of MPO inhibition on HFpEF outcomes[119]. Future studies examining the effect of modulating inflammatory transcription regulators, specifically kruppel-like factors, (Klf)[120] have been proposed. Klfs are zinc-finger transcription factors that negatively regulate proinflammatory activation. Pre-clinical studies have associated decreased levels of Klf2 in neutrophils with worsening cardiac dysfunction in an angiotensin II model of HF[121]. Therefore, overactivation of Klf2 could provide a protective mechanism against chronic neutrophil-driven inflammation in HF.
CARDIAC ARRHYTHMIAS
The role of inflammation in the pathogenesis of cardiac arrhythmias has gained attention in recent years, with neutrophils emerging as key players in this process (Figure 1). While much of the research has focused on atrial fibrillation (AF), the most common sustained arrhythmia in clinical practice, neutrophils have also been implicated in other types of arrhythmias, including life-threatening ventricular arrhythmias[122].
Figure 1. Neutrophil-associated mechanisms of arrhythmia.
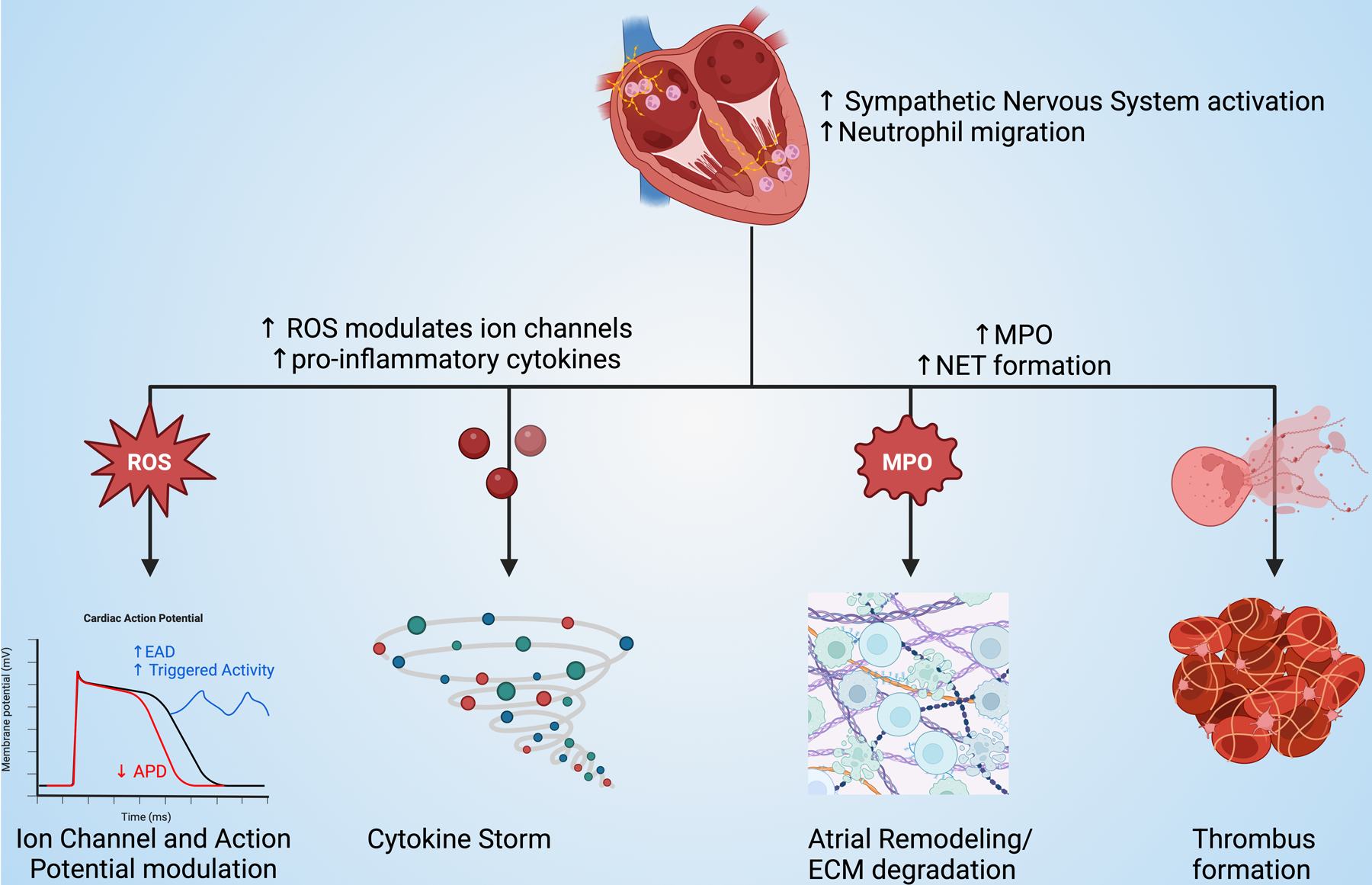
ROS: reactive oxygen species, EAD: early afterdepolarizations, APD: action potential duration, MPO: myeloperoxidase, NET: neutrophil extracellular trap, ECM: extracellular matrix
AF, characterized by rapid and irregular atrial activation, affects millions of people worldwide and is associated with significant morbidity and mortality[123]. In the United States alone nearly 3 million people have AF and the prevalence of AF is expected to increase to 9–12 million by 2030 [124]. The pathophysiology of AF is complex, involving electrical, structural, and autonomic remodeling of the atria[125]. Emerging evidence suggests that inflammation, and particularly neutrophil-mediated inflammation, plays a crucial role in these remodeling processes.
Neutrophil infiltration into the atrial tissue has been observed in both animal models of AF and human patients with the condition[126, 127]. Frustaci et al. were among the first to demonstrate the presence of inflammatory infiltrates, including neutrophils, in atrial biopsies from patients with AF[128]. Subsequent studies have corroborated these findings and further elucidated the mechanisms by which neutrophils contribute to the initiation and perpetuation of AF.
One of the primary mechanisms through which neutrophils promote arrhythmogenesis is through the generation of oxidative stress. Neutrophil-derived ROS can directly alter the electrophysiological properties of atrial cardiomyocytes by modulating ion channel function[129]. ROS have been shown to affect the function of several key ion channels involved in atrial repolarization, including potassium channels (IKur, IKACh) and calcium handling proteins (RyR2, SERCA2a)[130]. These alterations can lead to changes in action potential duration, early afterdepolarizations, and increased triggered activity, all of which contribute to the arrhythmogenic substrate in AF[131, 132].
Additionally, neutrophil-derived MPO has been implicated in atrial remodeling in AF. Rudolph et al. demonstrated that MPO-deficient mice were protected against angiotensin II-induced atrial fibrosis and vulnerability to AF[133]. They further showed that MPO was capable of oxidizing and activating MMPs, leading to enhanced degradation of extracellular matrix proteins and promoting structural remodeling of the atria. These findings highlight the complex interplay between neutrophil-derived factors and the atrial extracellular matrix in the pathogenesis of AF.
The formation of NETs has emerged as another important mechanism by which neutrophils contribute to the prothrombotic state in AF. He et al. found increased levels of circulating NET components in patients with AF compared to those in sinus rhythm[127]. Furthermore, NETs were observed in human atrial thrombi, suggesting a direct link between NET formation and thrombogenesis in AF[134, 135]. The procoagulant effects of NETs, combined with the endothelial dysfunction and blood stasis characteristic of AF, create an ideal environment for thrombus formation, potentially explaining the increased risk of cardioembolic events in AF patients.
Neutrophil-platelet interactions also play a significant role in the prothrombotic state associated with AF. Neutrophil-platelet aggregates have been found to be elevated in the peripheral blood of AF patients[136]. These aggregates not only contribute to thrombosis but also amplify the inflammatory response through bidirectional activation of neutrophils and platelets. Maugeri et al. showed that neutrophil-platelet aggregates were predictive of left atrial thrombus formation in patients with AF, suggesting their potential as a biomarker for assessing thromboembolic stroke risk[137].
The prognostic value of neutrophil-related markers in AF has been demonstrated in numerous clinical studies. Elevated neutrophil counts and high NLRs have been associated with an increased risk of AF development, AF recurrence after cardioversion or catheter ablation, and adverse outcomes in AF patients[138–141]. A meta-analysis by Lekkala et al. found that high NLR was significantly associated with an increased risk of recurrence following catheter ablation for AF, independent of traditional risk factors[142].
Recent research has also shed light on the role of neutrophils in ventricular arrhythmias, particularly in the context of myocardial ischemia and HF. In the setting of acute MI, neutrophil infiltration and the subsequent release of pro-inflammatory mediators and ROS can create a highly arrhythmogenic milieu[115, 143]. Grune et al. demonstrated that neutrophil depletion reduced the incidence of ventricular arrhythmias in a mouse model of myocardial ischemia-reperfusion injury, highlighting the potential of neutrophil-targeted therapies in preventing life-threatening arrhythmias in the acute phase of MI[144].
In chronic HF, neutrophil activation may contribute to the increased risk of ventricular arrhythmias. Neutrophil-derived factors can promote adverse electrical remodeling of the ventricles through various mechanisms, including direct effects on ion channel function and expression, promotion of cardiac fibrosis leading to conduction heterogeneity, impairment of gap junction function affecting intercellular electrical coupling, and modulation of autonomic innervation of the heart[122, 145]. These effects collectively contribute to the creation of an arrhythmogenic substrate in the failing heart, increasing the risk of both reentrant and triggered arrhythmias.
The emerging understanding of the role of neutrophils in cardiac arrhythmias has led to the exploration of novel therapeutic strategies. While global neutrophil depletion or inhibition may not be feasible due to the risk of compromising host defense, more targeted therapeutic approaches are being investigated. Selective inhibition of neutrophil recruitment targeting chemokine receptors or adhesion molecules involved in neutrophil trafficking to the heart may reduce excessive neutrophil infiltration while preserving systemic neutrophil function. For instance, inhibitors of CXCR2, a key chemokine receptor in neutrophil recruitment, have shown promise in preclinical models of AF[146, 147].
Modulation of NET formation through inhibitors of PAD4 or strategies to degrade extracellular DNA have shown potential in reducing the prothrombotic state associated with AF[58, 148]. However, the optimal timing and duration of such interventions need to be carefully considered. Targeting neutrophil-derived proteases using selective inhibitors of neutrophil elastase or MMPs may help mitigate adverse atrial remodeling in AF[149, 150]. Preliminary studies have shown that these approaches can reduce atrial fibrosis and vulnerability to AF in animal models.
Given the importance of oxidative stress in neutrophil-mediated arrhythmogenesis, targeted antioxidant therapies may prove beneficial. However, previous clinical trials with broad-spectrum antioxidants have yielded disappointing results, potentially highlighting the need for more targeted and potent antioxidant strategies[151, 152]. Novel approaches to modulate the inflammatory response, such as the use of resolvins and other pro-resolving mediators, may help promote the resolution of neutrophil-mediated inflammation and reduce the risk of arrhythmias[59, 153].
The translation of neutrophil-targeted therapies from preclinical models to clinical practice remains a challenge. The complex nature of neutrophil functions in cardiac electrophysiology necessitates careful consideration of the timing, duration, and specificity of interventions. Additionally, the heterogeneity of arrhythmia patients in terms of underlying pathology, comorbidities, and genetic factors may require personalized approaches to neutrophil modulation.
CARDIOMYOPATHIES
The term ‘cardiomyopathy’ refers to a group of diseases that are characterized by abnormal cardiac structure or function[154, 155]. Cardiomyopathies can lead to HF or arrhythmias (as discussed above). There are several types of cardiomyopathies, each with distinct causes, clinical presentations, and management strategies. Broadly, cardiomyopathies can be classified into two categories: ischemic (ICM) and non-ischemic cardiomyopathy (NICM). ICM arises due to coronary artery disease and MI; the contributions of neutrophils to these disease entities are discussed elsewhere in this review and have been characterized extensively previously[5, 40]. Here we will focus on an emerging literature that demonstrates multiple important roles for neutrophils in NICM, a diverse group of cardiomyopathies whose pathogenesis does not require ischemic injury[156–158]. This literature is based to a greater extent on small human studies and/or autopsy data as there are not well-developed animal models for many of these conditions. We will consider neutrophil contributions to NICM subtypes separately (Figure 2) and review the role of neutrophils in myocarditis and cardiomyopathy secondary to chemotherapy.
Figure 2. The roles of neutrophils in cardiomyopathy.
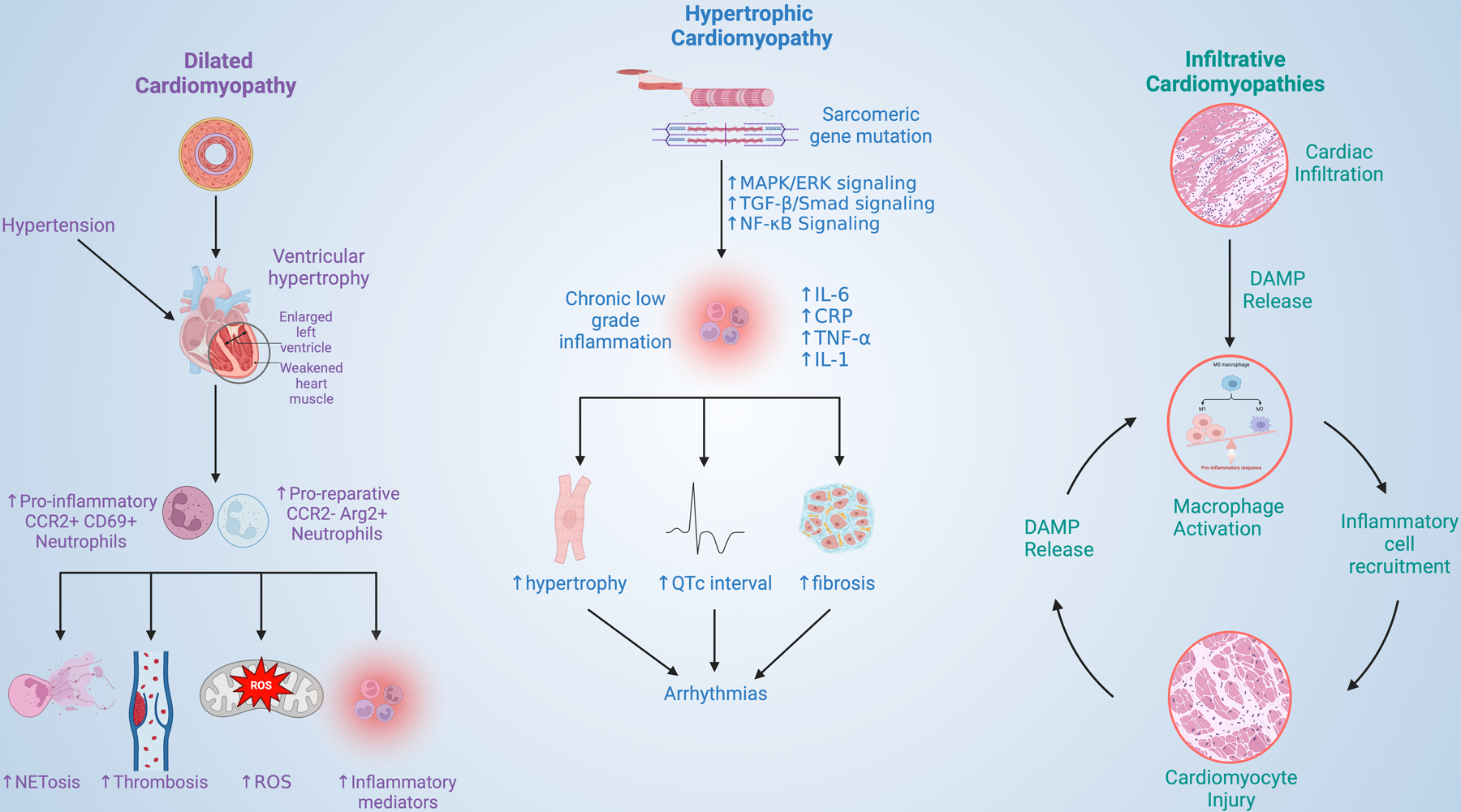
NET: neutrophil extracellular trap, ROS: reactive oxygen species, MAPK: mitogen-activated protein kinases, ERK: extracellular-signal related kinases, TGF-B: transforming growth factor beta, IL-6: Interleukin 6, CRP: C-reactive protein, TNF-a: tumor necrosis factor alpha, IL-1: interleukin 1, DAMP: damage associated molecular patterns
Hypertensive cardiomyopathy
Hypertension is the second most common cause of HF worldwide behind ischemic heart disease[156]. Patients with hypertension are twice as likely to develop HF compared to their normotensive counterparts[159]. Persistently pumping against elevated blood pressure causes the left ventricle to hypertrophy then progressively dilate, eventually leading to a DCM phenotype. Inflammation recently has been identified as one of the drivers behind progression of hypertensive cardiomyopathy. Using various murine models for hypertensive heart disease including transverse aortic constriction (TAC), studies have shown that a variety of both innate and adaptive immune cell subsets including neutrophils respond to ventricular pressure overload [160–162]. Martini et al. identified two distinct neutrophil populations within 4 weeks after TAC: one expressing high levels of CCR2 and CD69 associated with a proinflammatory phenotype and a smaller CCR2- subset expressing Arg2, a phenotype more consistent with repair[160]. Similarly, Wang et al. demonstrated Wnt5a-dependent expansion of the pathologic neutrophil compartment in a TAC model [161]. Other studies have shown that neutrophil recruitment to the heart is dependent on CCL2 signaling and negatively regulated by CX3CR1[163].
NET formation also been shown to contribute to adverse LV remodeling in hypertensive cardiomyopathy[105, 164]. NETs are thought to contribute to disease pathogenesis through thrombosis, altered mitochondrial respiration, and increased recruitment of inflammatory cells[105, 165]. NETosis in pressure overload models of hypertensive heart disease is related to developmental endothelial locus-1 (Del-1) which mediates neutrophil extravasation through ICAM-1 expression[164]. Endomyocardial biopsy samples of patients with DCM have shown increased NET production compared to healthy controls[105]. Additionally, patients with DCM and NET formation had overall worse contractile function and higher BNP (B-type natriuretic peptide) levels compared to DCM patients without NET formation, consistent with more severe disease.
Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM), the most common form of cardiomyopathy[156], is defined by the dilation and impaired contraction of one or both ventricles. At a cellular level, DCM is characterized by eccentrically hypertrophied cardiomyocytes with progressive energetic deficits and abnormal calcium handling. Over time prolonged cellular stress leads to cardiomyocyte necrosis and apoptosis with progressive interstitial fibrosis, further weakening the intrinsic function of the myocardial tissue[157]. DCM can be idiopathic, familial, or secondary to factors such as hypertension, alcohol abuse, myocarditis, pregnancy, and exposure to certain toxins and medications like chemotherapy. DCM can be asymptomatic but frequently leads to HF, characterized by fluid retention and shortness of breath, as it progresses. The pathophysiology of DCM involves a complex interplay of genetic, molecular, and environmental factors leading to structural changes and myocardial dysfunction. Some of these abnormal molecular and cellular mechanisms include neurohormonal overactivation, chronic inflammation, and oxidative stress. This review focuses on the role of immune activation, particularly within the neutrophil compartment, plays on disease pathogenesis[166].
Peripartum DCM
Peripartum cardiomyopathy (PPCM) is a distinct type of DCM that occurs in the late stages of pregnancy or shortly after delivery without other etiology. Some cases of PPCM resolve spontaneously whereas others progress to symptomatic HF[167, 168]. Many mechanisms have been proposed for the development of PPCM including hemodynamic changes, vasculo-hormonal factors and genetics. Inflammation has been implicated broadly; studies have shown elevated levels of CRP, IL-6, IFN-γ, TNF-α in patients with PPCM compared to controls[169–171].
There is evidence that neutrophils contribute to the pathogenesis of PPCM through enhanced activation and proliferation of fibroblasts[170]. Specifically, neutrophils in PPCM produce elevated levels of MPO which in turn activates MMPs, driving fibroblast activation and fibrosis. Proteomic analysis of serum samples from patients with PPCM has revealed dysregulation of inflammatory pathways in PPCM patients compared to controls and downregulation of pro-inflammatory pathways in patients with cardiac recovery[172]. Similarly, patients with PPCM who did not recover contractile function had higher NLRs compared to patients who recovered contractile function[173].
PPCM often is associated with hypertensive disorders of pregnancy. Specifically, one study showed 22% of patients with PPCM had pre-eclampsia, a complex multi-system disease diagnosed by onset of hypertension at > 20-weeks’ gestation[174, 175]. Emerging studies have implicated neutrophils as contributors to pre-eclampsia pathogenesis via similar pathways identified in PPCM. Increased neutrophil percentages have been associated with worse clinical outcomes in pre-eclamptic patients[176–178]. Markers of neutrophil activation, namely NET formation in the placenta and increased production of neutrophil gelatinase-associated lipocalin, were associated with severity of pre-eclamptic patients[179, 180] suggesting shared neutrophil pathobiology between these two important pregnancy-associated conditions.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant inherited condition that causes hypertrophy of the left ventricle. HCM frequently is asymptomatic but can induce angina, exertional dyspnea and potentially life-threatening arrhythmias. HCM is responsible for 33% of all sudden cardiac deaths in young athletes, particularly those who play basketball and football[181]. The pathophysiology of the condition is linked to mutations in genes encoding sarcomeric proteins, such as beta-myosin heavy chain (MYH7), myosin-binding protein C (MYBPC3), troponin T (TNNT2), and others[182]. These mutations lead to abnormal protein function and myofibrillar disarray[181]. Recent studies in the field suggest that low-grade inflammation contributes to cardiac remodeling in HCM. Specifically, studies have shown increased IL-6 and CRP levels in HCM patients compared to age matched controls[183]. Levels of proinflammatory cytokines like TNF-α and IL-1 were associated with increased myocardial fibrosis as detected on cardiac MRI[183].
Emerging evidence suggests that neutrophils contribute to the pathophysiology of HCM. Neutrophils and B-cells were highly enriched in a high-throughput sequencing dataset of HCM samples (GSE130036)[184]. HCM patients with an increased NLR had an increased risk of sudden cardiac death, prolonged QTc interval and worse contractile function, collectively consistent with a more severe disease phenotype[185, 186]. In the early stages of HCM, increased endocardial and vascular wall stress causes damage to endothelial cells. Damaged endothelial cells then in turn release factors like Von Willebrand Factor which can anchor NETs to the vessel wall and inflamed tissues in the heart. In a positive feedback loop, NET associated molecules including histones, DNA, and tissue factor further active both inflammatory and pro-thrombotic pathways causing further tissue and vessel injury[165, 183].
Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a autosomal dominant heritable disorder characterized by replacement of myocardium with fibrofatty tissue predominately impacting the right ventricle[187]. This fibrofatty replacement creates an arrhythmogenic substrate by disrupting the normal electrical conduction pathways causing potentially life-threatening ventricular arrhythmias. ARVC is caused by mutations in proteins that localize to the desmosome, the structure responsible for linking adjoining cardiac myocytes[187]. Patients with ARVC may be asymptomatic but also may experience arrhythmias that manifest as palpitations, syncope, and sudden cardiac death. ARVC, like HCM, is a common cause of malignant ventricular arrythmias and sudden death in young adults and athletes (11–22%)[188].
In a sudden cardiac death registry from Northern Italy, 22% of deaths were attributed to ARVC[189]. Within the ARVC group, 67% of patients had focal lymphocytic myocarditis prompting the authors of the report to propose the “inflammatory theory” of ARVC wherein the fibrofatty replacement is a response to chronic myocarditis. Postmortem heart samples from other patients with ARVC have confirmed the presence of an inflammatory milieu including T lymphocytes, macrophages, neutrophils and mast cells[190, 191]. Specifically, neutrophils were one of the most highly enriched cell types in the ARVC tissue samples. Further analysis of the data revealed a positive linear correlation between immune cell infiltration and myocardial fibrosis in ARVC myocardium. Analysis of RNA-seq data from the Gene Expression Omnibus (GEO) database demonstrated that a variety of immune cell transcriptional signatures were positively correlated with the ARVC phenotype[192]. Similarly, single cell RNAseq from postmortem ARVC cardiac samples demonstrated increased gene expression of neutrophil associated extracellular matrix degradation, activation of fibroblasts, and signaling through the pro-inflammatory NLRP3 pathway[190]. Myocardial fibrosis is the dominant pathogenic process in end-stage ARVC patients undergoing heart transplantation and it can be hypothesized that neutrophils contribute to myocardial fibrosis through degradation of the ECM and myocardial remodeling.
Takotsubo Cardiomyopathy
Takotsubo cardiomyopathy, also known as stress cardiomyopathy, often occurs in the setting of severe physical or emotional distress. The name is derived from the Japanese word, “takotsubo”, an octopus trap, which has a similar shape to the typical “ballooning” apex of the left ventricle in this condition. This characteristic morphology is thought to be due to the regional distribution of sympathetic nerve fibers that disproportionally affect the cardiac apex[193]. Takotsubo cardiomyopathy often presents similarly to MI with acute chest pain and wall motion abnormalities, but function often recovers after the acute stressor is removed[193].
One of the key diagnostic imaging findings associated with Takotsubo cardiomyopathy is an intense left ventricular/apical inflammatory signal on cardiac MRI[194]. Case reports describing Takotsubo cardiomyopathy in the setting of severe acute illness or emotional distress have provided insight into the immunological pathogenic mechanisms of this disease[195, 196]. Neutrophil abundance is increased in the peripheral blood of patients with Takotsubo cardiomyopathy[197] and, as with other types of cardiomyopathy, an elevated NLR ratio has been associated with increased risk of adverse events for patients with Takotsubo cardiomyopathy[198, 199]. One case report of Takotsubo associated with pheochromocytoma and adrenal crisis demonstrated neutrophil infiltrate and contraction band necrosis in an endomyocardial biopsy during acute presentation[196]. Other post-mortem biopsies of Takotsubo cardiomyopathy patients have shown a similar pattern of neutrophil granulocyte staining within the myocardium[195]. Mouse models of Takotsubo cardiomyopathy have shown peak neutrophil infiltrate at 6 hours which subsides by day 3 and is replaced predominantly by macrophages[200]. The characteristic neutrophil oxidative burst with nitric oxide is thought to contribute to the development of myocardial contractile dysfunction seen in this disorder[201].
Overview of Infiltrative Cardiomyopathies
The infiltrative cardiomyopathies are characterized by ectopic deposition of abnormal proteins, granulomas, iron, or other materials within the myocardium in the context of a systemic disorder, contributing initially to ventricular stiffness and ultimately to impaired contractile function and HF[202]. Many of these disorders often are referred to as restrictive cardiomyopathies given advanced abnormalities in diastolic ventricular filling. The underlying etiology of each infiltrative cardiomyopathy is distinct but evidence suggests shared pathobiology insofar as the chronic myocardial injury in these intractable infiltrative diseases causes tissue injury which promotes release of DAMPs that trigger a positive feedback loop activating macrophages which then recruit additional inflammatory cells such as neutrophils, causing further myocyte injury [203].
Cardiac amyloidosis
Amyloidosis is a systemic disorder characterized by extracellular deposition of insoluble proteins. The most common types of amyloidosis are amyloid light chain (AL), wherein the amyloid fibrils consist of misfolded monoclonal immunoglobulin light chains, and amyloid transthyretin (ATTR), wherein amyloid fibrils consist of misfolded and aggregated transthyretin[204, 205]. Many organs can be affected by amyloid deposition, but prognosis and outcomes are tied most closely to cardiac involvement. Cardiac amyloidosis, due to either AL or ATTR, is characterized by progressive HF and a susceptibility to multiple types of potentially life-threatening arrhythmias[205]. Definitive diagnosis of cardiac amyloidosis may require endomyocardial biopsy and the use of a specialized stain called Congo Red[206, 207]. Endomyocardial biopsies from patients with cardiac amyloidosis have shown significant inflammatory infiltrates[208]. One study from Hayashi et al. showed increased expression of inflammatory macrophage cell markers, especially CD68 and CD163, in diagnostic endomyocardial biopsies of amyloid patients, while another from Siegismund et al. highlighted increased CD45RO and CD3 infiltration [203, 208]. Older studies showed the neutrophil proteases, namely elastase and cathepsin G, have been associated with abnormally folded cardiac amyloid fibrils[209]. An elevated NLR also has been found in a AA amyloidosis, a rarer type of amyloidosis associated with chronic inflammatory conditions like familial mediterranean fever amyloidosis (FMF)[210]. Not all patients with FMF develop amyloidosis, but those who do have overproduction of the acute phase reactant, serum amyloid A, which can deposit into cardiac tissue in a similar manner to AL and ATTR[211]. These studies suggest that inflammation particularly within the macrophage and granulocyte compartments contribute to amyloid cardiac pathogenesis.
The mechanisms through which neutrophils contribute to cardiac amyloidosis pathobiology are not fully known. Studies suggest that chronic inflammation causes destabilization of the TTR tetramers causing disaggregation into more cardiotoxic TTR monomers and oligomers[212]. Additionally, neutrophil protease interaction with amyloid fibrils could cause abnormal folding and degradation exacerbating cardiac deposition of amyloid fibrils [209]. Collectively these findings suggest that neutrophils may have a secondary pathologic effect on protein folding rather than directly mediating disease pathogenesis.
Cardiac sarcoidosis
Sarcoidosis is multisystem disorder characterized by the formation of non-caseating granulation tissue most often affecting the lungs and mediastinal lymph nodes. However, sarcoidosis can affect the heart, often a harbinger of poor prognosis and severe disease[213]. Cardiac manifestations of sarcoid tend to be irregular and patchy, rendering endomyocardial biopsy less sensitive. Additionally, infiltrates tend to be on the basal and septal walls of the left ventricle which are difficult to access via biopsy. Previous analysis of endomyocardial biopsies of sarcoid patients revealed upregulation of numerous inflammatory cytokines including IL-1α, IL-2, IL-12 and IFN-γ and increased infiltration of immune cells[214]. One meta-analysis highlighted that the peripheral NLR ratio is consistently elevated in sarcoidosis patients compared to healthy controls and often correlates to severity of disease[215]. Similar results were found in bronchial alveolar lavage samples from sarcoid patients[216]. Interestingly, patients that developed pulmonary hypertension as a secondary complication from their sarcoidosis were also found to have an increased NLR[217], perhaps resulting from inflammation in cardiac sarcoid leading to fibrosis and compression of the microvasculature[218]. In one study, patients with more severe disease including development of cardiac sarcoid had higher levels of neutrophil gelatinase-associated lipocalin compared to patients with stable disease[219]. Sequencing of human cardiac sarcoidosis samples revealed upregulation of neutrophil degranulation and neutrophil activation in immune response genes[220]. Furthermore, sequencing revealed increased expression of canonical neutrophil-associated cytokines such as CCL5 and MMP9. Collectively these studies highlight that an increased abundance and activity of neutrophils in sarcoidosis is associated with disease severity and important secondary complications like pulmonary hypertension.
Cardiac Hemochromatosis
Hemochromatosis is a heritable disorder characterized by excessive iron retention. In addition to the liver and pancreas, one of the organs most affected by this disease is the heart. Excessive iron deposition in the heart can cause multiple manifestations of the disease including cardiomyopathy, arrhythmias, and sudden cardiac death[221]. Iron overload cardiomyopathy is initially characterized by impaired cardiac filling but as the disease progresses, the phenotype often transitions to DCM with impaired contractile function [222].
Iron plays an important role in neutrophil biology and is required for two core neutrophil functions including NADPH oxidase and MPO production. Studies have shown that an iron overload state such as hemochromatosis increases the respiratory/oxidative burst activity of neutrophils[223]. Similarly, studies have shown that iron overload increases circulating levels of TNF-α, increasing the levels of ICAM-1 and facilitating enhanced neutrophil adhesion and extravasation[223]. Neutrophils in an iron overload model have decreased expression of CD62L and increased phagocytotic activity. Conversely, inhibition of Timp3, which negatively regulates MMP expression, resulted in worsening cardiac systolic and diastolic dysfunction and over production of MMPs from inflammatory cells[224]. Taken together this evidence suggests that the excess iron in hemochromatosis likely results in hyperactive neutrophils potentiating damage to cardiac myocytes through generation of ROX and proteolytic enzymes. The hyperinflammatory state causes cardiac remodeling and fibrosis leading to restrictive cardiomyopathy.
Myocarditis
Myocarditis is an inflammatory heart disease that can cause DCM and HF. Myocarditis can be caused by various infections (including coxsackievirus B, adenovirus, streptococcal species, and Trypanosoma cruzi[225]), toxic agents, medications, and systemic conditions. Many systemic autoimmune conditions also can be associated with the development of myocarditis[226]. A recent JLB review very nicely highlighted the role neutrophils play in various type of myocarditis[227]. Specifically, neutrophil phagocytosis via the TLR8 pathway has been implicated in streptococcal and coxsackie B3 myocarditis. TNFα stimulates neutrophils to release MPO and trigger NET formation in coxsackie B3 infection [228]. Expansion of the neutrophil compartment via CXCL2/CXCL3 signaling in the coxsackie virus B3 myocarditis model on day 4 of infection correlates with decline of cardiac function[229]. Lipocalin-2, the neutrophil gelatinase-B-associated lipid transport protein is elevated in patients with myocarditis and in rat models but its exact role in potentiating the disease has yet to be identified[230].
Chemotherapy-induced cardiomyopathy
Multiple classes of chemotherapeutic agents are associated with cardiotoxicity[231]. Multiple in-depth reviews have detailed how the different classes of chemotherapeutic agents mediate cardiac injury[232, 233]. Here we will focus on the relatively understudied roles of neutrophils in chemotherapeutic cardiotoxicity (Figure 3).
Figure 3. Mechanisms of chemotherapy-Induced cardiomyopathy.

Dox: doxorubicin, ROS: reactive oxygen species, NE: neutrophil elastase, DAMPs: damage-associated molecular patterns, MMPs: matrix metalloproteinases, MPO: myeloperoxidase, IL-8: interleukin 8, NLR: neutrophil-lymphocyte ratio, TKI: tyrosine kinase inhibitors, NETs: neutrophil extracellular traps, VWF: von Willebrand factor, TLR: toll-like receptor, ER: endoplasmic reticulum
Anthracyclines
Anthracyclines, such as doxorubicin, have been used to treat various types of cancers including breast cancer, leukemia, lymphoma, and sarcoma for decades[234]. The primary dose-limiting adverse effect of anthracyclines is cardiotoxicity [235]. The mechanism of anthracycline cardiotoxicity is complex and likely involves direct toxicity to mitochondria, induction of free radical species and free ferric ions causing cardiomyocyte ferroptosis, and inhibition of the DNA repair enzyme topoisomerase IIb[234, 236]. Studies of mice treated with doxorubicin found that neutrophil influx and release of neutrophil elastase contributed to both early and late cardiac dysfunction[237]. Another study found that doxorubicin treatment increased production of CXCL-1 and IL-8, pro-inflammatory mediators of neutrophil recruitment[238]. Interestingly, the generation of NETs also was associated with the release of neutrophil elastase and DAMPs, further suggesting a role for a positive pro-inflammatory feedback loop in doxorubicin-induced cardiotoxicity[238]. Recruited neutrophils release MMPs which damage the endothelium and recruit activated platelets increasing the risk of thrombosis[236]. Other studies have shown that MPO contributes to cardiotoxicity by enhancing oxidation of sarcomeric proteins and cardiac apoptosis through p38 mitogen-activated protein kinase signaling[239]. Doxorubicin may also alter hematopoietic stem and progenitor cells such that they withstand genotoxic stress better. These precursor cells may then give rise to a mutant type of neutrophil that further augments cardiac toxicity[235].
As noted with other mechanisms of cardiac toxicity, human studies have shown that an increased NLR ratio predisposes patients to higher risks of developing cardiac toxicity. One study in breast cancer patients treated with anthracyclines found a NLR ≥ 2.6 during anthracycline treatment was associated with a fourfold increased risk of developing left ventricular dysfunction; incremental increases of 1-point NLR added an additional 15% risk[240]. Taken together these studies suggest that many facets of neutrophil biology including recruitment, neutrophil elastase, NETs, MMPs, and overall ratio may contribute to doxorubicin-induced cardiotoxicity.
HER2-targeted therapies
Breast cancer is the most common malignancy in women and up to 25% of breast cancers overexpress the human epidermal growth factor 2 (HER2) cell surface receptor[241]. Trastuzumab (Herceptin) is a monoclonal antibody against HER2 that is used to treat this aggressive type of cancer. However, cardiotoxicity is a well-recognized side effect of HER2-targeted therapies. The mechanism of injury of HER2 therapies is distinct in that HER2 (ErbB2) expression in cardiomyocytes conveys a protective role and inhibition via the monoclonal antibody disrupts these protective mechanisms. HER2 antagonist-mediated cardiomyopathy can often be reversible with cessation of treatment[242].
The MANTICORE trial investigated the pathophysiology of trastuzumab induced cardiotoxicity and found increased myocardial inflammation on cardiac MRI[243]. Further investigation showed increased expression of MMP-2 after cycle 4 of treatment. Longitudinal studies also found increased expression of neutrophil associated enzymes, namely MPO, following HER2-directed therapy[244]. Increased expression of MPO at baseline has been shown to convey elevated risk of cardiac toxicity in patients treated with the combination of trastuzumab and anthracycline[245]. As with anthracyclines, an elevated NLR during trastuzumab treatment was associated with increased risk of developing cardiac toxicity [246]. Less is known about the actual recruitment mechanism of neutrophils as it is thought that HER2-directed monoclonal antibodies do not cause direct toxicity and cell death to cardiac myocytes[247]. Further investigation into this field is important, though challenged by the fact that trastuzumab, as a humanized monoclonal antibody, cannot be studied readily in traditional animal models.
Tyrosine kinase inhibitors
Tyrosine kinase inhibitors (TKI) can be divided into two classes: small molecule inhibitors and monoclonal antibodies. These therapies have been widely successful in the treatment of melanoma, non-small cell lung cancers, chronic myeloid leukemia, and gastrointestinal tumors[242]. However, their use has been associated with a wide range of cardiovascular toxicities including acute coronary syndrome, fatal cardiac arrythmias, cardiomyopathy, and hypertension[248]. A diverse array of mechanisms have been implicated in TKI cardiotoxicity including oxidative stress, accumulation of drug metabolites disrupting sarcomere function, and inhibition of mitochondrial function leading to a variety of programmed cell death pathways[242]. As with anthracyclines, cardiac cell death is thought to activate TLR pathways and downstream pro-inflammatory mechanisms. Other proposed mechanisms include induced endoplasmic reticulum stress which causes downstream activation of NF-κB induced inflammation[249]. Rats treated with the TKI VEGF inhibitor, erlotinib, demonstrated increased neutrophil superoxide activity as compared to the controls[250]. Similar to other human studies, an NLR ratio ≥ 3 was associated with worse progression-free survival in NSCLC patients treated with the EGFR-inhibitor osimertinib[251, 252]. Neutrophils from CML patients demonstrate increased NET production and MPO generation, a phenotype that is augmented by TKI treatment[253, 254]. Ponatinib, one of the TKI inhibitors shown to increase NET formation, has also been associated with increased risk of thrombotic microangiopathy[255]. Mechanistically, NETs have been shown to cause increased activation of platelets and release of von-Willebrand factor, two key components in platelet adhesion events and coronary thrombosis ultimately leading to ischemic changes in cardiac function[255].
Immune checkpoint inhibitor myocarditis
Immune checkpoint inhibitors (ICI) are monoclonal antibodies that disrupt inhibitory signals placed on cytotoxic T-cells allowing for immune cell activation against tumor cells. ICIs have revolutionized the treatment of multiple types of cancer. One rare but life-threatening potential adverse effect of ICI treatment is myocarditis which is fatal in up to 50% of patients [256]. ICI myocarditis generally is thought to arise from activation of T lymphocyte subsets [256, 257] though other studies have highlighted a role for tissue resident macrophages in ICI myocarditis[258, 259]. The role of neutrophils in ICI myocarditis is less clear but an increased NLR prior to initiation of ICI increased the risk of cardiovascular toxicities including myocarditis, vascular toxicity, new onset hypertension, arrythmia and QTc prolongation[260]. Furthermore, patients who developed ICI myocarditis had a similar NLR to controls at baseline and an increasing NLR ratio prior to the onset of cardiac disease[261]. Therefore, while leading evidence in the field suggests that macrophages and T cells play a critical role in ICI myocarditis, emerging data suggest further investigation into the neutrophil compartment is warranted.
CONCLUSIONS
Neutrophils long have been known as critical mediators of innate immunity, protecting hosts from acute infection through a variety of targeted anti-microbial mechanisms. However, in the setting of chronic inflammation, neutrophils’ innate immune mechanisms can become maladaptive and contribute to disease pathogenesis rather than its resolution. With respect to cardiac disease, neutrophils were studied initially and extensively in the setting of acute myocardial infarction. More recently, however, the focus in the field has turned to the neutrophil functions in chronic cardiac disease how these roles may differ from the acute response to injury. This review highlights recent discoveries detailing how neutrophils contribute to the pathogenesis of ischemic heart disease, cardiac arrhythmias and non-ischemic causes of cardiomyopathy. In general, higher NLR ratios are associated with a more severe phenotype across most types of cardiac disease highlighted in this review. Generation of NETs has been shown to be associated with cardiac myocyte damage, fibrosis, and chronic inflammation in multiple models of cardiomyopathy. Increasing levels of fibrosis in both infiltrative and arrhythmogenic disease is associated with destabilizing and potentially life threatening arrythmias. Toxic exposures including excessive iron, chemotherapies, and immunotherapies have been shown to cause pathogenic release of neutrophil reactive oxygen species which damages cardiac myocytes.
Despite significant advances, the burden of morbidity and mortality remains very high for each of the selected disease entities. The pathobiology of each of these disease states is quite complex, affording many potential therapeutic targets including deleterious neutrophil functions. Novel treatment strategies clearly are required and no extant therapy for any cardiovascular disease directly targets neutrophils. However, new and ongoing efforts now focus on how selective inhibition of various aspects of neutrophil function could limit tissue damage and dampen pathogenic remodeling in these chronic cardiovascular disease states. Current clinical trials in the field are investigating therapies in transcriptional regulation, cytokine inhibition, and cellular recruitment to address how targeting neutrophils can decrease pathological remodeling to improve clinical outcomes.
KEY CONCEPTS.
Neutrophils long have been recognized as critical components of the acute response to myocardial infarction. The contributions of neutrophils to the pathobiology of chronic cardiac conditions such as heart failure, arrhythmias, and cardiomyopathies increasingly is recognized.
Multiple aspects of neutrophil biology and behavior contribute to cardiac disease development and progression including NET formation, thrombosis, generation of reactive oxygen species, extracellular matrix degradation, and release of inflammatory mediators.
Therapies targeting these neutrophil-mediated processes are under active investigation for the treatment of ischemic heart disease, heart failure and cardiac arrhythmias.
OPEN QUESTIONS.
The fidelity of large and small animal models of heart injury to the context of human clinical cardiovascular disease is uncertain.
The relative contributions of deleterious and reparative neutrophil functions to the acute and chronic response to cardiac injury remain incompletely defined.
Given the complex pathobiology of each of these cardiovascular disease processes, the promise of novel therapies directed at chronic neutrophil-mediated processes in the heart remains to be determined.
Footnotes
Conflicts of interest: None
REFERENCES
- 1.Salari N, Morddarvanjoghi F, Abdolmaleki A, Rasoulpoor S, Khaleghi AA, Hezarkhani LA, Shohaimi S & Mohammadi M (2023). The global prevalence of myocardial infarction: a systematic review and meta-analysis. BMC Cardiovasc Disord 23, 206. 10.1186/s12872-023-03231-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin SS, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Barone Gibbs B, Beaton AZ, Boehme AK, Commodore-Mensah Y, Currie ME, Elkind MSV, Evenson KR, Generoso G, Heard DG, Hiremath S, Johansen MC, Kalani R, Kazi DS, Ko D, Liu J, Magnani JW, Michos ED, Mussolino ME, Navaneethan SD, Parikh NI, Perman SM, Poudel R, Rezk-Hanna M, Roth GA, Shah NS, St-Onge MP, Thacker EL, Tsao CW, Urbut SM, Van Spall HGC, Voeks JH, Wang NY, Wong ND, Wong SS, Yaffe K, Palaniappan LP, American Heart Association Council on, E., Prevention Statistics, C. & Stroke Statistics, S. (2024). 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation. 149, e347–e913. 10.1161/CIR.0000000000001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prabhu SD & Frangogiannis NG (2016). The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119, 91–112. 10.1161/circresaha.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frangogiannis NG (2014). The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11, 255–265. 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kologrivova I, Shtatolkina M, Suslova T & Ryabov V (2021). Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front Immunol 12, 664457. 10.3389/fimmu.2021.664457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vafadarnejad E, Rizzo G, Krampert L, Arampatzi P, Arias-Loza AP, Nazzal Y, Rizakou A, Knochenhauer T, Bandi SR, Nugroho VA, Schulz DJJ, Roesch M, Alayrac P, Vilar J, Silvestre JS, Zernecke A, Saliba AE & Cochain C (2020). Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ Res 127, e232–e249. 10.1161/CIRCRESAHA.120.317200. [DOI] [PubMed] [Google Scholar]
- 7.Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K & Sano M (2013). Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 62, 24–35. 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Puhl SL & Steffens S (2019). Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front Cardiovasc Med 6, 25. 10.3389/fcvm.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y, Yabluchanskiy A & Lindsey ML (2013). Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 6, 11. 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soehnlein O, Steffens S, Hidalgo A & Weber C (2017). Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol 17, 248–261. 10.1038/nri.2017.10. [DOI] [PubMed] [Google Scholar]
- 11.Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY & Lindsey ML (2016). Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res 110, 51–61. 10.1093/cvr/cvw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daseke MJ 2nd, Chalise U, Becirovic-Agic M, Salomon JD, Cook LM, Case AJ & Lindsey ML (2021). Neutrophil signaling during myocardial infarction wound repair. Cell Signal. 77, 109816. 10.1016/j.cellsig.2020.109816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y (2021). Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells. 10. 10.3390/cells10071676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolaczkowska E & Kubes P (2013). Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13, 159–175. 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 15.Prame Kumar K, Nicholls AJ & Wong CHY (2018). Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res 371, 551–565. 10.1007/s00441-017-2753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soehnlein O, Lindbom L & Weber C (2009). Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood. 114, 4613–4623. 10.1182/blood-2009-06-221630. [DOI] [PubMed] [Google Scholar]
- 17.Chalise U, Becirovic-Agic M & Lindsey ML (2021). Neutrophil crosstalk during cardiac wound healing after myocardial infarction. Curr Opin Physiol 24. 10.1016/j.cophys.2022.100485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daseke MJ 2nd, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY & Lindsey ML (2019). Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res Cardiol 114, 37. 10.1007/s00395-019-0746-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carbone F, Bonaventura A & Montecucco F (2019). Neutrophil-Related Oxidants Drive Heart and Brain Remodeling After Ischemia/Reperfusion Injury. Front Physiol 10, 1587. 10.3389/fphys.2019.01587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Kazzi M, Rayner BS, Chami B, Dennis JM, Thomas SR & Witting PK (2020). Neutrophil-Mediated Cardiac Damage After Acute Myocardial Infarction: Significance of Defining a New Target Cell Type for Developing Cardioprotective Drugs. Antioxid Redox Signal. 33, 689–712. 10.1089/ars.2019.7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinten-Johansen J (2004). Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res 61, 481–497. 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Carbone F, Crowe LA, Roth A, Burger F, Lenglet S, Braunersreuther V, Brandt KJ, Quercioli A, Mach F, Vallee JP & Montecucco F (2016). Treatment with anti-RANKL antibody reduces infarct size and attenuates dysfunction impacting on neutrophil-mediated injury. J Mol Cell Cardiol 94, 82–94. 10.1016/j.yjmcc.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 23.Jolly SR, Kane WJ, Hook BG, Abrams GD, Kunkel SL & Lucchesi BR (1986). Reduction of myocardial infarct size by neutrophil depletion: effect of duration of occlusion. Am Heart J 112, 682–690. 10.1016/0002-8703(86)90461-8. [DOI] [PubMed] [Google Scholar]
- 24.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA & Lucchesi BR (1983). Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 67, 1016–1023. 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 25.Engler RL, Dahlgren MD, Morris DD, Peterson MA & Schmid-Schonbein GW (1986). Role of leukocytes in response to acute myocardial ischemia and reflow in dogs. Am J Physiol 251, H314–323. 10.1152/ajpheart.1986.251.2.H314. [DOI] [PubMed] [Google Scholar]
- 26.Litt MR, Jeremy RW, Weisman HF, Winkelstein JA & Becker LC (1989). Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation. 80, 1816–1827. 10.1161/01.cir.80.6.1816. [DOI] [PubMed] [Google Scholar]
- 27.Doring Y, Libby P & Soehnlein O (2020). Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ Res 126, 1228–1241. 10.1161/CIRCRESAHA.120.315931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, Zhao JH & Li YM (2015). Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol 308, H500–509. 10.1152/ajpheart.00381.2014. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Du C, Zhang Y & Zhu L (2024). Composition and Function of Neutrophil Extracellular Traps. Biomolecules. 14. 10.3390/biom14040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonaventura A, Liberale L, Carbone F, Vecchie A, Diaz-Canestro C, Camici GG, Montecucco F & Dallegri F (2018). The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Thromb Haemost 118, 6–27. 10.1160/TH17-09-0630. [DOI] [PubMed] [Google Scholar]
- 31.Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A & Fafutis-Morris M (2017). Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol 8, 81. 10.3389/fimmu.2017.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolling M, Eckstein M, Singh J, Schauer C, Schoen J, Shan X, Bozec A, Knopf J, Schett G, Munoz LE & Herrmann M (2022). Hypoxia Promotes Neutrophil Survival After Acute Myocardial Infarction. Front Immunol 13, 726153. 10.3389/fimmu.2022.726153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y & Wagner DD (2014). VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 123, 141–148. 10.1182/blood-2013-07-514992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helseth R, Knudsen EC, Eritsland J, Opstad TB, Arnesen H, Andersen GO & Seljeflot I (2019). Glucose associated NETosis in patients with ST-elevation myocardial infarction: an observational study. BMC Cardiovasc Disord 19, 221. 10.1186/s12872-019-1205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mangold A, Hofbauer TM, Ondracek AS, Artner T, Scherz T, Speidl WS, Krychtiuk KA, Sadushi-Kolici R, Jakowitsch J & Lang IM (2019). Neutrophil extracellular traps and monocyte subsets at the culprit lesion site of myocardial infarction patients. Sci Rep 9, 16304. 10.1038/s41598-019-52671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S & Ritis K (2015). Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J 36, 1405–1414. 10.1093/eurheartj/ehv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gawaz M (2004). Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res 61, 498–511. 10.1016/j.cardiores.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 38.Wienkamp AK, Erpenbeck L & Rossaint J (2022). Platelets in the NETworks interweaving inflammation and thrombosis. Front Immunol 13, 953129. 10.3389/fimmu.2022.953129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegel PM, Bojti I, Bassler N, Holien J, Flierl U, Wang X, Waggershauser P, Tonnar X, Vedecnik C, Lamprecht C, Stankova I, Li T, Helbing T, Wolf D, Anto-Michel N, Mitre LS, Ehrlich J, Orlean L, Bender I, Przewosnik A, Mauler M, Hollederer L, Moser M, Bode C, Parker MW, Peter K & Diehl P (2021). A DARPin targeting activated Mac-1 is a novel diagnostic tool and potential anti-inflammatory agent in myocarditis, sepsis and myocardial infarction. Basic Res Cardiol 116, 17. 10.1007/s00395-021-00849-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antipenko S, Mayfield N, Jinno M, Gunzer M, Ismahil MA, Hamid T, Prabhu SD & Rokosh G (2024). Neutrophils are indispensable for adverse cardiac remodeling in heart failure. J Mol Cell Cardiol 189, 1–11. 10.1016/j.yjmcc.2024.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O & Steffens S (2017). Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38, 187–197. 10.1093/eurheartj/ehw002. [DOI] [PubMed] [Google Scholar]
- 42.Lorchner H, Poling J, Gajawada P, Hou Y, Polyakova V, Kostin S, Adrian-Segarra JM, Boettger T, Wietelmann A, Warnecke H, Richter M, Kubin T & Braun T (2015). Myocardial healing requires Reg3beta-dependent accumulation of macrophages in the ischemic heart. Nat Med 21, 353–362. 10.1038/nm.3816. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Conrad C, Mills TW, Berg NK, Kim B, Ruan W, Lee JW, Zhang X, Yuan X & Eltzschig HK (2021). PMN-derived netrin-1 attenuates cardiac ischemia-reperfusion injury via myeloid ADORA2B signaling. J Exp Med 218. 10.1084/jem.20210008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eghbalzadeh K, Georgi L, Louis T, Zhao H, Keser U, Weber C, Mollenhauer M, Conforti A, Wahlers T & Paunel-Gorgulu A (2019). Compromised Anti-inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation After Myocardial Infarction. Front Immunol 10, 2313. 10.3389/fimmu.2019.02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, Fu L, Tian C, Yang J, He F & Tang L (2019). Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun 10, 1076. 10.1038/s41467-019-09046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizo-Tellez SA & Filep JG (2024). Beyond host defense and tissue injury: the emerging role of neutrophils in tissue repair. Am J Physiol Cell Physiol 326, C661–C683. 10.1152/ajpcell.00652.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J (2018). Neutrophils in tissue injury and repair. Cell Tissue Res 371, 531–539. 10.1007/s00441-017-2785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guasti L, Dentali F, Castiglioni L, Maroni L, Marino F, Squizzato A, Ageno W, Gianni M, Gaudio G, Grandi AM, Cosentino M & Venco A (2011). Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb Haemost 106, 591–599. 10.1160/TH11-02-0096. [DOI] [PubMed] [Google Scholar]
- 49.Okyere AD & Tilley DG (2020). Leukocyte-Dependent Regulation of Cardiac Fibrosis. Front Physiol 11, 301. 10.3389/fphys.2020.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C, Cong BL, Wang M, Abdullah M, Wang XL, Zhang YH, Xu SJ & Cui L (2018). Neutrophil to lymphocyte ratio as a predictor of myocardial damage and cardiac dysfunction in acute coronary syndrome patients. Integr Med Res 7, 192–199. 10.1016/j.imr.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedman GD, Klatsky AL & Siegelaub AB (1974). The leukocyte count as a predictor of myocardial infarction. N Engl J Med 290, 1275–1278. 10.1056/NEJM197406062902302. [DOI] [PubMed] [Google Scholar]
- 52.Angkananard T, Anothaisintawee T, McEvoy M, Attia J & Thakkinstian A (2018). Neutrophil Lymphocyte Ratio and Cardiovascular Disease Risk: A Systematic Review and Meta-Analysis. Biomed Res Int 2018, 2703518. 10.1155/2018/2703518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mangold A, Alias S, Scherz T, Hofbauer M, Jakowitsch J, Panzenbock A, Simon D, Laimer D, Bangert C, Kammerlander A, Mascherbauer J, Winter MP, Distelmaier K, Adlbrecht C, Preissner KT & Lang IM (2015). Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res 116, 1182–1192. 10.1161/CIRCRESAHA.116.304944. [DOI] [PubMed] [Google Scholar]
- 54.Rymer JA & Newby LK (2017). Failure to Launch: Targeting Inflammation in Acute Coronary Syndromes. JACC Basic Transl Sci 2, 484–497. 10.1016/j.jacbts.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F & Investigators H-M (2002). The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol 40, 1199–1204. 10.1016/s0735-1097(02)02136-8. [DOI] [PubMed] [Google Scholar]
- 56.Granger CB, Mahaffey KW, Weaver WD, Theroux P, Hochman JS, Filloon TG, Rollins S, Todaro TG, Nicolau JC, Ruzyllo W, Armstrong PW & Investigators C (2003). Pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation. 108, 1184–1190. 10.1161/01.CIR.0000087447.12918.85. [DOI] [PubMed] [Google Scholar]
- 57.Tardif JC, Tanguay JF, Wright SR, Duchatelle V, Petroni T, Gregoire JC, Ibrahim R, Heinonen TM, Robb S, Bertrand OF, Cournoyer D, Johnson D, Mann J, Guertin MC & L’Allier PL (2013). Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J Am Coll Cardiol 61, 2048–2055. 10.1016/j.jacc.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Du M, Yang W, Schmull S, Gu J & Xue S (2020). Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int Immunopharmacol 78, 106055. 10.1016/j.intimp.2019.106055. [DOI] [PubMed] [Google Scholar]
- 59.Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M & Halade GV (2015). Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol 84, 24–35. 10.1016/j.yjmcc.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kain V, Liu F, Kozlovskaya V, Ingle KA, Bolisetty S, Agarwal A, Khedkar S, Prabhu SD, Kharlampieva E & Halade GV (2017). Resolution Agonist 15-epi-Lipoxin A(4) Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci Rep 7, 9999. 10.1038/s41598-017-10441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaiser R, Escaig R, Erber J & Nicolai L (2021). Neutrophil-Platelet Interactions as Novel Treatment Targets in Cardiovascular Disease. Front Cardiovasc Med 8, 824112. 10.3389/fcvm.2021.824112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW & Kitsis RN (2003). Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia-reperfusion injury: recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 108, 2387–2392. 10.1161/01.Cir.0000093192.72099.9a. [DOI] [PubMed] [Google Scholar]
- 63.Deng H, Lin C, Garcia-Gerique L, Fu S, Cruz Z, Bonner EE, Rosenwasser M, Rajagopal S, Sadhu MN, Gajendran C, Zainuddin M, Gosu R, Sivanandhan D, Shelef MA, Nam B, Vogl DT, Gabrilovich DI & Nefedova Y (2022). A Novel Selective Inhibitor JBI-589 Targets PAD4-Mediated Neutrophil Migration to Suppress Tumor Progression. Cancer Res 82, 3561–3572. 10.1158/0008-5472.CAN-21-4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwinger RHG (2021). Pathophysiology of heart failure. Cardiovasc Diagn Ther 11, 263–276. 10.21037/cdt-20-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dumont BL, Neagoe PE, Charles E, Villeneuve L, Ninni S, Tardif JC, Rakel A, White M & Sirois MG (2024). Low-Density Neutrophils and Neutrophil Extracellular Traps (NETs) Are New Inflammatory Players in Heart Failure. Can J Cardiol 40, 1524–1535. 10.1016/j.cjca.2024.03.018. [DOI] [PubMed] [Google Scholar]
- 66.Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A & Aukrust P (2012). Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 122, 23–35. 10.1159/000338166. [DOI] [PubMed] [Google Scholar]
- 67.Tracchi I, Ghigliotti G, Mura M, Garibaldi S, Spallarossa P, Barisione C, Boasi V, Brunelli M, Corsiglia L, Barsotti A & Brunelli C (2009). Increased neutrophil lifespan in patients with congestive heart failure. Eur J Heart Fail. 11, 378–385. 10.1093/eurjhf/hfp031. [DOI] [PubMed] [Google Scholar]
- 68.Mesquita T, Lin YN & Ibrahim A (2021). Chronic low-grade inflammation in heart failure with preserved ejection fraction. Aging Cell. 20, e13453. 10.1111/acel.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD & Redfield MM (2010). Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol 56, 845–854. 10.1016/j.jacc.2010.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mendez-Ferrer S, Battista M & Frenette PS (2010). Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci 1192, 139–144. 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nicholls AJ, Wen SW, Hall P, Hickey MJ & Wong CHY (2018). Activation of the sympathetic nervous system modulates neutrophil function. J Leukoc Biol 103, 295–309. 10.1002/JLB.3MA0517-194RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niu XF, Ibbotson G & Kubes P (1996). A balance between nitric oxide and oxidants regulates mast cell-dependent neutrophil-endothelial cell interactions. Circ Res 79, 992–999. 10.1161/01.res.79.5.992. [DOI] [PubMed] [Google Scholar]
- 73.Niu XF, Smith CW & Kubes P (1994). Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ Res 74, 1133–1140. 10.1161/01.res.74.6.1133. [DOI] [PubMed] [Google Scholar]
- 74.Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM & Alcaide P (2016). Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J Am Heart Assoc 5, e003126. 10.1161/JAHA.115.003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hanna A & Frangogiannis NG (2020). Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc Drugs Ther 34, 849–863. 10.1007/s10557-020-07071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Chen C & Wang DW (2021). Inflammatory Cytokines, Immune Cells, and Organ Interactions in Heart Failure. Front Physiol 12, 695047. 10.3389/fphys.2021.695047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ & Feldman AM (1997). Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res 81, 627–635. 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 78.Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS & Brower GL (2010). Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 56, 225–231. 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gupta S, Markham DW, Drazner MH & Mammen PP (2008). Fulminant myocarditis. Nat Clin Pract Cardiovasc Med 5, 693–706. 10.1038/ncpcardio1331. [DOI] [PubMed] [Google Scholar]
- 80.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG & Mann DL (2001). Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 103, 2055–2059. 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 81.Mongirdiene A, Skrodenis L, Varoneckaite L, Mierkyte G & Gerulis J (2022). Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies. Biomedicines. 10. 10.3390/biomedicines10030602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, Jiang F & Peng ZY (2019). Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid Med Cell Longev 2019, 5080843. 10.1155/2019/5080843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishihara T, Tokitsu T, Sueta D, Oike F, Takae M, Fujisue K, Usuku H, Ito M, Kanazawa H, Araki S, Arima Y, Takashio S, Nakamura T, Sakamoto K, Suzuki S, Kaikita K, Yamamoto E & Tsujita K (2021). Clinical significance of reactive oxidative metabolites in patients with heart failure with reduced left ventricular ejection fraction. J Card Fail. 27, 57–66. 10.1016/j.cardfail.2020.07.020. [DOI] [PubMed] [Google Scholar]
- 84.He L, Liu R, Yue H, Zhu G, Fu L, Chen H, Guo Y & Qin C (2021). NETs promote pathogenic cardiac fibrosis and participate in ventricular aneurysm formation after ischemia injury through the facilitation of perivascular fibrosis. Biochem Biophys Res Commun 583, 154–161. 10.1016/j.bbrc.2021.10.068. [DOI] [PubMed] [Google Scholar]
- 85.Carmona-Rivera C, Zhao W, Yalavarthi S & Kaplan MJ (2015). Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis 74, 1417–1424. 10.1136/annrheumdis-2013-204837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ndrepepa G (2019). Myeloperoxidase - A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta 493, 36–51. 10.1016/j.cca.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 87.Nicholls SJ & Hazen SL (2005). Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 25, 1102–1111. 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 88.Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT & Engelmann B (2010). Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16, 887–896. 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 89.Michel JB & Ho-Tin-Noe B (2015). Thrombi and neutrophils. Circ Res 116, 1107–1108. 10.1161/CIRCRESAHA.115.306050. [DOI] [PubMed] [Google Scholar]
- 90.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B & Massberg S (2012). Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209, 819–835. 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yonekawa K, Neidhart M, Altwegg LA, Wyss CA, Corti R, Vogl T, Grigorian M, Gay S, Luscher TF & Maier W (2011). Myeloid related proteins activate Toll-like receptor 4 in human acute coronary syndromes. Atherosclerosis. 218, 486–492. 10.1016/j.atherosclerosis.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 92.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM & Wagner DD (2017). Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med 214, 439–458. 10.1084/jem.20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ogura Y, Tajiri K, Murakoshi N, Xu D, Yonebayashi S, Li S, Okabe Y, Feng D, Shimoda Y, Song Z, Mori H, Yuan Z, Aonuma K & Ieda M (2021). Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice. Int J Mol Sci 22. 10.3390/ijms22020722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rudolph V, Rudolph TK, Hennings JC, Blankenberg S, Schnabel R, Steven D, Haddad M, Knittel K, Wende S, Wenzel J, Munzel T, Heitzer T, Meinertz T, Hubner C & Baldus S (2007). Activation of polymorphonuclear neutrophils in patients with impaired left ventricular function. Free Radic Biol Med 43, 1189–1196. 10.1016/j.freeradbiomed.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 95.Rodrigues KE, Pontes MHB, Cantao MBS & Prado AF (2024). The role of matrix metalloproteinase-9 in cardiac remodeling and dysfunction and as a possible blood biomarker in heart failure. Pharmacol Res 206, 107285. 10.1016/j.phrs.2024.107285. [DOI] [PubMed] [Google Scholar]
- 96.Banerjee D, Tian R & Cai S (2023). The Role of Innate Immune Cells in Cardiac Injury and Repair: A Metabolic Perspective. Curr Cardiol Rep 25, 631–640. 10.1007/s11886-023-01897-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.DeBerge M, Chaudhary R, Schroth S & Thorp EB (2023). Immunometabolism at the Heart of Cardiovascular Disease. JACC Basic Transl Sci 8, 884–904. 10.1016/j.jacbts.2022.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun L, Yang X, Yuan Z & Wang H (2020). Metabolic Reprogramming in Immune Response and Tissue Inflammation. Arterioscler Thromb Vasc Biol 40, 1990–2001. 10.1161/ATVBAHA.120.314037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho JH, Cho HJ, Lee HY, Ki YJ, Jeon ES, Hwang KK, Chae SC, Baek SH, Kang SM, Choi DJ, Yoo BS, Kim KH, Kim JJ & Oh BH (2020). Neutrophil-Lymphocyte Ratio in Patients with Acute Heart Failure Predicts In-Hospital and Long-Term Mortality. J Clin Med 9. 10.3390/jcm9020557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Curran FM, Bhalraam U, Mohan M, Singh JS, Anker SD, Dickstein K, Doney AS, Filippatos G, George J, Metra M, Ng LL, Palmer CN, Samani NJ, van Veldhuisen DJ, Voors AA, Lang CC & Mordi IR (2021). Neutrophil-to-lymphocyte ratio and outcomes in patients with new-onset or worsening heart failure with reduced and preserved ejection fraction. ESC Heart Fail. 8, 3168–3179. 10.1002/ehf2.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Delcea C, Buzea CA & Dan GA (2019). The neutrophil to lymphocyte ratio in heart failure: a comprehensive review. Rom J Intern Med 57, 296–314. 10.2478/rjim-2019-0018. [DOI] [PubMed] [Google Scholar]
- 102.Uthamalingam S, Patvardhan EA, Subramanian S, Ahmed W, Martin W, Daley M & Capodilupo R (2011). Utility of the neutrophil to lymphocyte ratio in predicting long-term outcomes in acute decompensated heart failure. Am J Cardiol 107, 433–438. 10.1016/j.amjcard.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 103.Donkel SJ, Wolters FJ, Ikram MA & de Maat MPM (2021). Circulating Myeloperoxidase (MPO)-DNA complexes as marker for Neutrophil Extracellular Traps (NETs) levels and the association with cardiovascular risk factors in the general population. PLoS One. 16, e0253698. 10.1371/journal.pone.0253698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Polina IA, Ilatovskaya DV & DeLeon-Pennell KY (2020). Cell free DNA as a diagnostic and prognostic marker for cardiovascular diseases. Clin Chim Acta 503, 145–150. 10.1016/j.cca.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ichimura S, Misaka T, Ogawara R, Tomita Y, Anzai F, Sato Y, Miura S, Yokokawa T, Sato T, Oikawa M, Kobayashi A, Yoshihisa A & Takeishi Y (2024). Neutrophil Extracellular Traps in Myocardial Tissue Drive Cardiac Dysfunction and Adverse Outcomes in Patients With Heart Failure With Dilated Cardiomyopathy. Circ Heart Fail. 17, e011057. 10.1161/circheartfailure.123.011057. [DOI] [PubMed] [Google Scholar]
- 106.Zhang XL, Wang TY, Chen Z, Wang HW, Yin Y, Wang L, Wang Y, Xu B & Xu W (2022). HMGB1-Promoted Neutrophil Extracellular Traps Contribute to Cardiac Diastolic Dysfunction in Mice. J Am Heart Assoc 11, e023800. 10.1161/JAHA.121.023800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bai B, Cheng M, Jiang L, Xu J, Chen H & Xu Y (2021). High Neutrophil to Lymphocyte Ratio and Its Gene Signatures Correlate With Diastolic Dysfunction in Heart Failure With Preserved Ejection Fraction. Front Cardiovasc Med 8, 614757. 10.3389/fcvm.2021.614757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tamaki S, Nagai Y, Shutta R, Masuda D, Yamashita S, Seo M, Yamada T, Nakagawa A, Yasumura Y, Nakagawa Y, Yano M, Hayashi T, Hikoso S, Nakatani D, Sotomi Y, Sakata Y & Investigators OC-HF (2023). Combination of Neutrophil-to-Lymphocyte and Platelet-to-Lymphocyte Ratios as a Novel Predictor of Cardiac Death in Patients With Acute Decompensated Heart Failure With Preserved Left Ventricular Ejection Fraction: A Multicenter Study. J Am Heart Assoc 12, e026326. 10.1161/JAHA.122.026326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou R, Xia YY, Li Z, Wu LD, Shi Y, Ling ZY & Zhang JX (2024). HFpEF as systemic disease, insight from a diagnostic prediction model reminiscent of systemic inflammation and organ interaction in HFpEF patients. Sci Rep 14, 5386. 10.1038/s41598-024-55996-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hage C, Michaelsson E, Kull B, Miliotis T, Svedlund S, Linde C, Donal E, Daubert JC, Gan LM & Lund LH (2020). Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. 7, 1534–1546. 10.1002/ehf2.12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Michaelsson E, Lund LH, Hage C, Shah SJ, Voors AA, Saraste A, Redfors B, Grove EL, Barasa A, Richards AM, Svedlund S, Lagerstrom-Fermer M, Gabrielsen A, Garkaviy P, Gan LM & Lam CSP (2023). Myeloperoxidase Inhibition Reverses Biomarker Profiles Associated With Clinical Outcomes in HFpEF. JACC Heart Fail. 11, 775–787. 10.1016/j.jchf.2023.03.002. [DOI] [PubMed] [Google Scholar]
- 112.Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, Ratner AJ, Madesh M, Drosatos K & Morrow JP (2017). Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2. 10.1172/jci.insight.94248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szekeres FLM, Walum E, Wikstrom P & Arner A (2021). A small molecule inhibitor of Nox2 and Nox4 improves contractile function after ischemia-reperfusion in the mouse heart. Sci Rep 11, 11970. 10.1038/s41598-021-91575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sirker A, Murdoch CE, Protti A, Sawyer GJ, Santos CX, Martin D, Zhang X, Brewer AC, Zhang M & Shah AM (2016). Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J Mol Cell Cardiol 98, 11–17. 10.1016/j.yjmcc.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mollenhauer M, Friedrichs K, Lange M, Gesenberg J, Remane L, Kerkenpass C, Krause J, Schneider J, Ravekes T, Maass M, Halbach M, Peinkofer G, Saric T, Mehrkens D, Adam M, Deuschl FG, Lau D, Geertz B, Manchanda K, Eschenhagen T, Kubala L, Rudolph TK, Wu Y, Tang WHW, Hazen SL, Baldus S, Klinke A & Rudolph V (2017). Myeloperoxidase Mediates Postischemic Arrhythmogenic Ventricular Remodeling. Circ Res 121, 56–70. 10.1161/CIRCRESAHA.117.310870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ling S & Xu JW (2021). NETosis as a Pathogenic Factor for Heart Failure. Oxid Med Cell Longev 2021, 6687096. 10.1155/2021/6687096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu Y, Wei S, Wu X, Li Y & Han X (2023). Neutrophil extracellular traps in acute coronary syndrome. J Inflamm (Lond). 20, 17. 10.1186/s12950-023-00344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, Del Buono MG, Billingsley H, Wohlford G, Viscusi M, Oddi-Erdle C, Abouzaki NA, Dixon D, Biondi-Zoccai G, Arena R & Abbate A (2018). IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction. Circulation: Heart Failure. 11, e005036. 10.1161/CIRCHEARTFAILURE.118.005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lund LH, Lam CSP, Pizzato PE, Gabrielsen A, Michaelsson E, Nelander K, Ericsson H, Holden J, Folkvaljon F, Mattsson A, Collen A, Aurell M, Whatling C, Baldus S, Drelich G, Goudev A, Merkely B, Bergh N & Shah SJ (2023). Rationale and design of ENDEAVOR: A sequential phase 2b-3 randomized clinical trial to evaluate the effect of myeloperoxidase inhibition on symptoms and exercise capacity in heart failure with preserved or mildly reduced ejection fraction. Eur J Heart Fail. 25, 1696–1707. 10.1002/ejhf.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu Y, Song Y, Xie N, Zhao W, Lv J, Zhang T, Zhang Y, Chen H, Sun W, Luo Z, Cheng X, Jiang T, Wang Z, Chen X, Hu Y, Fang Y, Bai R, Liu X, He X, Ren Z, Huang J, Xiong H & Wang L (2024). KLF2-dependent transcriptional regulation safeguards the heart against pathological hypertrophy. Journal of Molecular and Cellular Cardiology. 10.1016/j.yjmcc.2024.12.010. [DOI] [PubMed] [Google Scholar]
- 121.Tang X, Wang P, Zhang R, Watanabe I, Chang E, Vinayachandran V, Nayak L, Lapping S, Liao S, Madera A, Sweet DR, Luo J, Fei J, Jeong HW, Adams RH, Zhang T, Liao X & Jain MK (2022). KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J Clin Invest 132. 10.1172/jci147191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hegemann N, Barth L, Doring Y, Voigt N & Grune J (2024). Implications for neutrophils in cardiac arrhythmias. Am J Physiol Heart Circ Physiol 326, H441–H458. 10.1152/ajpheart.00590.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li H, Song X, Liang Y, Bai X, Liu-Huo WS, Tang C, Chen W & Zhao L (2022). Global, regional, and national burden of disease study of atrial fibrillation/flutter, 1990–2019: results from a global burden of disease study, 2019. BMC Public Health. 22, 2015. 10.1186/s12889-022-14403-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kornej J, Borschel CS, Benjamin EJ & Schnabel RB (2020). Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ Res 127, 4–20. 10.1161/CIRCRESAHA.120.316340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Iwasaki YK, Nishida K, Kato T & Nattel S (2011). Atrial fibrillation pathophysiology: implications for management. Circulation. 124, 2264–2274. 10.1161/CIRCULATIONAHA.111.019893. [DOI] [PubMed] [Google Scholar]
- 126.Friedrichs K, Adam M, Remane L, Mollenhauer M, Rudolph V, Rudolph TK, Andrie RP, Stockigt F, Schrickel JW, Ravekes T, Deuschl F, Nickenig G, Willems S, Baldus S & Klinke A (2014). Induction of atrial fibrillation by neutrophils critically depends on CD11b/CD18 integrins. PLoS One. 9, e89307. 10.1371/journal.pone.0089307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.He L, Liu R, Yue H, Zhang X, Pan X, Sun Y, Shi J, Zhu G, Qin C & Guo Y (2023). Interaction between neutrophil extracellular traps and cardiomyocytes contributes to atrial fibrillation progression. Signal Transduct Target Ther 8, 279. 10.1038/s41392-023-01497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I, Sanna T, Bellocci F & Russo MA (2005). Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation. 112, 3680–3687. 10.1161/CIRCULATIONAHA.105.520999. [DOI] [PubMed] [Google Scholar]
- 129.Sovari AA & Dudley SC Jr. (2012). Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front Physiol 3, 311. 10.3389/fphys.2012.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pfenniger A, Yoo S & Arora R (2024). Oxidative stress and atrial fibrillation. J Mol Cell Cardiol 196, 141–151. 10.1016/j.yjmcc.2024.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nattel S & Harada M (2014). Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol 63, 2335–2345. 10.1016/j.jacc.2014.02.555. [DOI] [PubMed] [Google Scholar]
- 132.Wit AL & Boyden PA (2007). Triggered activity and atrial fibrillation. Heart Rhythm 4, S17–23. 10.1016/j.hrthm.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rudolph V, Andrie RP, Rudolph TK, Friedrichs K, Klinke A, Hirsch-Hoffmann B, Schwoerer AP, Lau D, Fu X, Klingel K, Sydow K, Didie M, Seniuk A, von Leitner EC, Szoecs K, Schrickel JW, Treede H, Wenzel U, Lewalter T, Nickenig G, Zimmermann WH, Meinertz T, Boger RH, Reichenspurner H, Freeman BA, Eschenhagen T, Ehmke H, Hazen SL, Willems S & Baldus S (2010). Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med 16, 470–474. 10.1038/nm.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Martinod K, Claessen A, Martens C, Krauel K, Velasquez Pereira LC, Witsch J & Witsch T (2024). NET burden in left atrial blood is associated with biomarkers of thrombosis and cardiac injury in patients with enlarged left atria. Clin Res Cardiol 10.1007/s00392-024-02464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martinod K & Wagner DD (2014). Thrombosis: tangled up in NETs. Blood. 123, 2768–2776. 10.1182/blood-2013-10-463646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Willoughby SR, Roberts-Thomson RL, Lim HS, Schultz C, Prabhu A, De Sciscio P, Wong CX, Worthley MI & Sanders P (2010). Atrial platelet reactivity in patients with atrial fibrillation. Heart Rhythm 7, 1178–1183. 10.1016/j.hrthm.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 137.Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, Rovere-Querini P & Manfredi AA (2014). Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 12, 2074–2088. 10.1111/jth.12710. [DOI] [PubMed] [Google Scholar]
- 138.Fagundes A Jr., Ruff CT, Morrow DA, Murphy SA, Palazzolo MG, Chen CZ, Jarolim P, Antman EM, Braunwald E & Giugliano RP (2023). Neutrophil-lymphocyte ratio and clinical outcomes in 19,697 patients with atrial fibrillation: Analyses from ENGAGE AF- TIMI 48 trial. Int J Cardiol 386, 118–124. 10.1016/j.ijcard.2023.05.031. [DOI] [PubMed] [Google Scholar]
- 139.Guo X, Zhang S, Yan X, Chen Y, Yu R, Long D, Sang C, Du X, Dong J & Ma C (2014). Postablation neutrophil/lymphocyte ratio correlates with arrhythmia recurrence after catheter ablation of lone atrial fibrillation. Chin Med J (Engl). 127, 1033–1038. [PubMed] [Google Scholar]
- 140.Paquissi FC (2016). The Predictive Role of Inflammatory Biomarkers in Atrial Fibrillation as Seen through Neutrophil-Lymphocyte Ratio Mirror. J Biomark 2016, 8160393. 10.1155/2016/8160393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang JL, Yang R, Zhu Y, Shao Y, Ji Y & Wang FF (2023). Association between the neutrophil-to-lymphocyte ratio and risk of in-hospital heart failure and arrhythmia in patients with acute myocardial infarction. Front Cardiovasc Med 10, 1275713. 10.3389/fcvm.2023.1275713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lekkala SP, Mellacheruvu SP, Gill KS, Khela PS, Singh G, Jitta SR, Patel M, Hingora MJ & Desai R (2024). Association between preablation and postablation neutrophil-lymphocyte ratio and atrial fibrillation recurrence: A meta-analysis. J Arrhythm 40, 214–221. 10.1002/joa3.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lim GB (2022). Macrophages and neutrophils modulate arrhythmia risk after myocardial infarction. Nat Rev Cardiol 19, 573. 10.1038/s41569-022-00758-x. [DOI] [PubMed] [Google Scholar]
- 144.Grune J, Lewis AJM, Yamazoe M, Hulsmans M, Rohde D, Xiao L, Zhang S, Ott C, Calcagno DM, Zhou Y, Timm K, Shanmuganathan M, Pulous FE, Schloss MJ, Foy BH, Capen D, Vinegoni C, Wojtkiewicz GR, Iwamoto Y, Grune T, Brown D, Higgins J, Ferreira VM, Herring N, Channon KM, Neubauer S, Oxford Acute Myocardial Infarction, S., Sosnovik DE, Milan DJ, Swirski FK, King KR, Aguirre AD, Ellinor PT & Nahrendorf M (2022). Neutrophils incite and macrophages avert electrical storm after myocardial infarction. Nat Cardiovasc Res 1, 649–664. 10.1038/s44161-022-00094-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grune J, Yamazoe M & Nahrendorf M (2021). Electroimmunology and cardiac arrhythmia. Nat Rev Cardiol 18, 547–564. 10.1038/s41569-021-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang YL, Cao HJ, Han X, Teng F, Chen C, Yang J, Yan X, Li PB, Liu Y, Xia YL, Guo SB & Li HH (2020). Chemokine Receptor CXCR-2 Initiates Atrial Fibrillation by Triggering Monocyte Mobilization in Mice. Hypertension. 76, 381–392. 10.1161/HYPERTENSIONAHA.120.14698. [DOI] [PubMed] [Google Scholar]
- 147.Zhang YL, Teng F, Han X, Li PB, Yan X, Guo SB & Li HH (2020). Selective blocking of CXCR2 prevents and reverses atrial fibrillation in spontaneously hypertensive rats. J Cell Mol Med 24, 11272–11282. 10.1111/jcmm.15694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT & Kaplan MJ (2014). Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 114, 947–956. 10.1161/CIRCRESAHA.114.303312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carai P, Gonzalez LF, Van Bruggen S, Spalart V, De Giorgio D, Geuens N, Martinod K, Jones EAV & Heymans S (2023). Neutrophil inhibition improves acute inflammation in a murine model of viral myocarditis. Cardiovasc Res 118, 3331–3345. 10.1093/cvr/cvac052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang X, Sun Y, Zhang Y, Fang F, Liu J, Xia Y & Liu Y (2022). Cardiac Biomarkers for the Detection and Management of Cancer Therapy-Related Cardiovascular Toxicity. Journal of Cardiovascular Development and Disease. 9, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Guo XY, Yan XL, Chen YW, Tang RB, Du X, Dong JZ & Ma CS (2014). Omega-3 fatty acids for postoperative atrial fibrillation: alone or in combination with antioxidant vitamins? Heart Lung Circ 23, 743–750. 10.1016/j.hlc.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 152.Violi F, Pastori D, Pignatelli P & Loffredo L (2014). Antioxidants for prevention of atrial fibrillation: a potentially useful future therapeutic approach? A review of the literature and meta-analysis. Europace. 16, 1107–1116. 10.1093/europace/euu040. [DOI] [PubMed] [Google Scholar]
- 153.Ajoolabady A, Nattel S, Lip GYH & Ren J (2022). Inflammasome Signaling in Atrial Fibrillation: JACC State-of-the-Art Review. J Am Coll Cardiol 79, 2349–2366. 10.1016/j.jacc.2022.03.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.McKenna WJ, Maron BJ & Thiene G (2017). Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circulation Research. 121, 722–730. 10.1161/CIRCRESAHA.117.309711. [DOI] [PubMed] [Google Scholar]
- 155.Ajmone Marsan N, Graziani F, Meucci MC, Wu HW, Lillo R, Bax JJ, Burzotta F, Massetti M, Jukema JW & Crea F (2024). Valvular heart disease and cardiomyopathy: reappraisal of their interplay. Nat Rev Cardiol 21, 37–50. 10.1038/s41569-023-00911-0. [DOI] [PubMed] [Google Scholar]
- 156.Bragazzi NL, Zhong W, Shu J, Abu Much A, Lotan D, Grupper A, Younis A & Dai H (2021). Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. European Journal of Preventive Cardiology. 28, 1682–1690. 10.1093/eurjpc/zwaa147. [DOI] [PubMed] [Google Scholar]
- 157.Heymans S, Lakdawala NK, Tschöpe C & Klingel K (2023). Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet. 402, 998–1011. 10.1016/s0140-6736(23)01241-2. [DOI] [PubMed] [Google Scholar]
- 158.Wang Y, Jia H & Song J (2023). Accurate Classification of Non-ischemic Cardiomyopathy. Curr Cardiol Rep 25, 1299–1317. 10.1007/s11886-023-01944-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Díez J & Butler J (2023). Growing Heart Failure Burden of Hypertensive Heart Disease: A Call to Action. Hypertension. 80, 13–21. 10.1161/HYPERTENSIONAHA.122.19373. [DOI] [PubMed] [Google Scholar]
- 160.Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, Carriero R, Termanini A, Colombo FS, Jachetti E, Panico C, Faggian G, Fumero A, Torracca L, Molgora M, Cibella J, Pagiatakis C, Brummelman J, Alvisi G, Mazza EMC, Colombo MP, Lugli E, Condorelli G & Kallikourdis M (2019). Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation. 140, 2089–2107. 10.1161/circulationaha.119.041694. [DOI] [PubMed] [Google Scholar]
- 161.Wang Y, Sano S, Oshima K, Sano M, Watanabe Y, Katanasaka Y, Yura Y, Jung C, Anzai A, Swirski FK, Gokce N & Walsh K (2019). Wnt5a-Mediated Neutrophil Recruitment Has an Obligatory Role in Pressure Overload-Induced Cardiac Dysfunction. Circulation. 140, 487–499. 10.1161/circulationaha.118.038820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kleiner JL, Köpke O, Faron A, Zhang Y, Cornelssen J, Coburn M, Frede S, Eichhorn L & Weisheit CK (2021). Characterization of Transverse Aortic Constriction in Mice Based on the Specific Recruitment of Leukocytes to the Hypertrophic Myocardium and the Aorta Ascendens. Mediators Inflamm 2021, 1376859. 10.1155/2021/1376859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weisheit CK, Kleiner JL, Rodrigo MB, Niepmann ST, Zimmer S, Duerr GD, Coburn M, Kurts C, Frede S & Eichhorn L (2021). CX3CR1 is a prerequisite for the development of cardiac hypertrophy and left ventricular dysfunction in mice upon transverse aortic constriction. PLoS One. 16, e0243788. 10.1371/journal.pone.0243788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao M, Zheng Z, Yin Z, Zhang J, Peng S, Liu J, Pan W, Wei C, Xu Y, Qin JJ, Wan J & Wang M (2023). DEL-1 deficiency aggravates pressure overload-induced heart failure by promoting neutrophil infiltration and neutrophil extracellular traps formation. Biochem Pharmacol 218, 115912. 10.1016/j.bcp.2023.115912. [DOI] [PubMed] [Google Scholar]
- 165.Becker RC, Phillip Owens A 3rd & Sadayappan S (2020). The potential roles of Von Willebrand factor and neutrophil extracellular traps in the natural history of hypertrophic and hypertensive cardiomyopathy. Thromb Res 192, 78–87. 10.1016/j.thromres.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Harding D, Chong MHA, Lahoti N, Bigogno CM, Prema R, Mohiddin SA & Marelli-Berg F (2023). Dilated cardiomyopathy and chronic cardiac inflammation: Pathogenesis, diagnosis and therapy. J Intern Med 293, 23–47. 10.1111/joim.13556. [DOI] [PubMed] [Google Scholar]
- 167.Schaufelberger M (2019). Cardiomyopathy and pregnancy. Heart. 105, 1543–1551. 10.1136/heartjnl-2018-313476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bala R, Mehta S, Roy VC, Kaur G & de Marvao A (2023). Peripartum cardiomyopathy: A review. Revista Portuguesa de Cardiologia 42, 917–924. 10.1016/j.repc.2023.01.029. [DOI] [PubMed] [Google Scholar]
- 169.Sliwa K, Förster O, Libhaber E, Fett JD, Sundstrom JB, Hilfiker-Kleiner D & Ansari AA (2006). Peripartum cardiomyopathy: inflammatory markers as predictors of outcome in 100 prospectively studied patients. Eur Heart J 27, 441–446. 10.1093/eurheartj/ehi481. [DOI] [PubMed] [Google Scholar]
- 170.Kryczka KE, Demkow M & Dzielińska Z (2024). Biomarkers in Peripartum Cardiomyopathy-What We Know and What Is Still to Be Found. Biomolecules. 14. 10.3390/biom14010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bauersachs J, König T, van der Meer P, Petrie MC, Hilfiker-Kleiner D, Mbakwem A, Hamdan R, Jackson AM, Forsyth P, de Boer RA, Mueller C, Lyon AR, Lund LH, Piepoli MF, Heymans S, Chioncel O, Anker SD, Ponikowski P, Seferovic PM, Johnson MR, Mebazaa A & Sliwa K (2019). Pathophysiology, diagnosis and management of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur J Heart Fail. 21, 827–843. 10.1002/ejhf.1493. [DOI] [PubMed] [Google Scholar]
- 172.Lovell JP, Bermea K, Yu J, Rousseau S, Cohen CD, Bhalodia A, Zita MD, Head RD, Blumenthal RS, Alharethi R, Damp J, Boehmer J, Alexis J, McNamara DM, Sharma G & Adamo L (2023). Serum Proteomic Analysis of Peripartum Cardiomyopathy Reveals Distinctive Dysregulation of Inflammatory and Cholesterol Metabolism Pathways. JACC Heart Fail. 11, 1231–1242. 10.1016/j.jchf.2023.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gürkan U, Akgöz H, Aksoy Ş, Can Gürkan Ö, Osken A, Unal Dayi S, Oz D & Haci R (2017). Value of the neutrophil-to-lymphocyte ratio in predicting left ventricular recovery in patients with peripartum cardiomyopathy. Wien Klin Wochenschr 129, 893–899. 10.1007/s00508-017-1227-6. [DOI] [PubMed] [Google Scholar]
- 174.Dimitriadis E, Rolnik DL, Zhou W, Estrada-Gutierrez G, Koga K, Francisco RPV, Whitehead C, Hyett J, da Silva Costa F, Nicolaides K & Menkhorst E (2023). Pre-eclampsia. Nature Reviews Disease Primers. 9, 8. 10.1038/s41572-023-00417-6. [DOI] [PubMed] [Google Scholar]
- 175.Davis MB, Arany Z, McNamara DM, Goland S & Elkayam U (2020). Peripartum Cardiomyopathy: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 75, 207–221. 10.1016/j.jacc.2019.11.014. [DOI] [PubMed] [Google Scholar]
- 176.Wang J, Zhu QW, Cheng XY, Liu JY, Zhang LL, Tao YM, Cui YB & Wei Y (2019). Assessment efficacy of neutrophil-lymphocyte ratio and monocyte-lymphocyte ratio in preeclampsia. J Reprod Immunol 132, 29–34. 10.1016/j.jri.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 177.Elmaradny E, Alneel G, Alkhattaf N, AlGadri T & Albriakan N (2022). Predictive values of combined platelet count, neutrophil-lymphocyte ratio, and platelet-lymphocyte ratio in preeclampsia. J Obstet Gynaecol 42, 1011–1017. 10.1080/01443615.2021.1986476. [DOI] [PubMed] [Google Scholar]
- 178.Kang Q, Li W, Yu N, Fan L, Zhang Y, Sha M, Xiao J, Wu J, Kang Q & Chen S (2020). Predictive role of neutrophil-to-lymphocyte ratio in preeclampsia: A meta-analysis including 3982 patients. Pregnancy Hypertens. 20, 111–118. 10.1016/j.preghy.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 179.Kim SM, Park JS, Norwitz ER, Jung HJ, Kim BJ, Park CW & Jun JK (2013). Circulating levels of neutrophil gelatinase-associated lipocalin (NGAL) correlate with the presence and severity of preeclampsia. Reprod Sci 20, 1083–1089. 10.1177/1933719113477480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Moodley M, Moodley J & Naicker T (2020). The Role of Neutrophils and Their Extracellular Traps in the Synergy of Pre-eclampsia and HIV Infection. Curr Hypertens Rep 22, 41. 10.1007/s11906-020-01047-z. [DOI] [PubMed] [Google Scholar]
- 181.Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS & Sherrid MV (2022). Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 79, 372–389. 10.1016/j.jacc.2021.12.002. [DOI] [PubMed] [Google Scholar]
- 182.Konno T, Chang S, Seidman JG & Seidman CE (2010). Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol 25, 205–209. 10.1097/HCO.0b013e3283375698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Becker RC, Owens AP 3rd & Sadayappan S (2020). Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J Thromb Thrombolysis. 49, 177–183. 10.1007/s11239-019-02026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zheng X, Yang Y, Huang Fu C & Huang R (2021). Identification and verification of promising diagnostic biomarkers in patients with hypertrophic cardiomyopathy associate with immune cell infiltration characteristics. Life Sci 285, 119956. 10.1016/j.lfs.2021.119956. [DOI] [PubMed] [Google Scholar]
- 185.Ozyilmaz S, Akgul O, Uyarel H, Pusuroglu H, Gul M, Satilmisoglu MH, Bolat I, Ozyilmaz I, Uçar H, Yildirim A & Bakir I (2017). The importance of the neutrophil-to-lymphocyte ratio in patients with hypertrophic cardiomyopathy. Rev Port Cardiol 36, 239–246. 10.1016/j.repc.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 186.Wang Z, Zhao L & He S (2020). Relation between neutrophil-to-lymphocyte ratio and mortality in patients with hypertrophic cardiomyopathy. Biomark Med 14, 1693–1701. 10.2217/bmm-2020-0463. [DOI] [PubMed] [Google Scholar]
- 187.Alblaihed L, Kositz C, Brady WJ, Al-Salamah T & Mattu A (2023). Diagnosis and management of arrhythmogenic right ventricular cardiomyopathy. The American Journal of Emergency Medicine. 65, 146–153. 10.1016/j.ajem.2022.12.010. [DOI] [PubMed] [Google Scholar]
- 188.Romero J, Mejia-Lopez E, Manrique C & Lucariello R (2013). Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC/D): A Systematic Literature Review. Clinical Medicine Insights: Cardiology. 7, CMC.S10940. 10.4137/cmc.S10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McKenna WJ & Caforio ALP (2022). Myocardial Inflammation and Sudden Death in the Inherited Cardiomyopathies. Can J Cardiol 38, 427–438. 10.1016/j.cjca.2022.01.004. [DOI] [PubMed] [Google Scholar]
- 190.Fu M, Hua X, Shu S, Xu X, Zhang H, Peng Z, Mo H, Liu Y, Chen X, Yang Y, Zhang N, Wang X, Liu Z, Yue G, Hu S & Song J (2024). Single-cell RNA sequencing in donor and end-stage heart failure patients identifies NLRP3 as a therapeutic target for arrhythmogenic right ventricular cardiomyopathy. BMC Medicine. 22, 11. 10.1186/s12916-023-03232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Campuzano O, Alcalde M, Iglesias A, Barahona-Dussault C, Sarquella-Brugada G, Benito B, Arzamendi D, Flores J, Leung TK, Talajic M, Oliva A & Brugada R (2012). Arrhythmogenic right ventricular cardiomyopathy: severe structural alterations are associated with inflammation. J Clin Pathol 65, 1077–1083. 10.1136/jclinpath-2012-201022. [DOI] [PubMed] [Google Scholar]
- 192.Lu W, Li Y, Dai Y & Chen K (2022). Dominant Myocardial Fibrosis and Complex Immune Microenvironment Jointly Shape the Pathogenesis of Arrhythmogenic Right Ventricular Cardiomyopathy. Front Cardiovasc Med 9, 900810. 10.3389/fcvm.2022.900810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pelliccia F, Kaski JC, Crea F & Camici PG (2017). Pathophysiology of Takotsubo Syndrome. Circulation. 135, 2426–2441. 10.1161/CIRCULATIONAHA.116.027121. [DOI] [PubMed] [Google Scholar]
- 194.Neil C, Nguyen TH, Kucia A, Crouch B, Sverdlov A, Chirkov Y, Mahadavan G, Selvanayagam J, Dawson D, Beltrame J, Zeitz C, Unger S, Redpath T, Frenneaux M & Horowitz J (2012). Slowly resolving global myocardial inflammation/oedema in Tako-Tsubo cardiomyopathy: evidence from T2-weighted cardiac MRI. Heart. 98, 1278. 10.1136/heartjnl-2011-301481. [DOI] [PubMed] [Google Scholar]
- 195.Bottari G, Trotta S, Marzullo A, Meliota G, Ciccone MM & Solarino B (2020). Sudden cardiac death after robbery: Homicide or natural death? J Forensic Leg Med 75, 102057. 10.1016/j.jflm.2020.102057. [DOI] [PubMed] [Google Scholar]
- 196.Iio K, Sakurai S, Kato T, Nishiyama S, Hata T, Mawatari E, Suzuki C, Takekoshi K, Higuchi K, Aizawa T & Ikeda U (2013). Endomyocardial biopsy in a patient with hemorrhagic pheochromocytoma presenting as inverted Takotsubo cardiomyopathy. Heart Vessels. 28, 255–263. 10.1007/s00380-012-0247-4. [DOI] [PubMed] [Google Scholar]
- 197.Yan J, Madina M, Deng C, Yuan Q, Cao S, Xie X & Ma Y (2021). Analysis of 9 Cases of Takotsubo Syndrome and an Analysis of the Clinical Characteristics of Takotsubo Syndrome From a Chinese Population. Front Cardiovasc Med 8, 732193. 10.3389/fcvm.2021.732193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zweiker D, Pogran E, Gargiulo L, Abd El-Razek A, Lechner I, Vosko I, Rechberger S, Bugger H, Christ G, Bonderman D, Kunschitz E, Czedik-Eysenberg C, Roithinger A, Weihs V, Kaufmann CC, Zirlik A, Bauer A, Metzler B, Lambert T, Steinwender C & Huber K (2022). Neutrophile-Lymphocyte Ratio and Outcome in Takotsubo Syndrome. Biology (Basel). 11. 10.3390/biology11081154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ahn HJ, Kang J, Lee SR, Park JJ, Lee HY, Choi DJ & Cho HJ (2023). Neutrophil-to-lymphocyte ratio as a predictor of in-hospital complications and overall mortality in Takotsubo syndrome preceded by physical triggers. BMC Cardiovasc Disord 23, 51. 10.1186/s12872-023-03078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Li X, Yang JJ & Xu D (2024). The role of inflammation in takotsubo syndrome: A new therapeutic target? J Cell Mol Med 28, e18503. 10.1111/jcmm.18503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu S, Chirkov YY & Horowitz JD (2018). Neutrophil-Initiated Myocardial Inflammation and Its Modulation by B-Type Natriuretic Peptide: A Potential Therapeutic Target. Int J Mol Sci 20. 10.3390/ijms20010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Muchtar E, Blauwet LA & Gertz MA (2017). Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res 121, 819–837. 10.1161/circresaha.117.310982. [DOI] [PubMed] [Google Scholar]
- 203.Hayashi Y, Hanawa H, Jiao S, Hasegawa G, Ohno Y, Yoshida K, Suzuki T, Kashimura T, Obata H, Tanaka K, Watanabe T & Minamino T (2015). Elevated Endomyocardial Biopsy Macrophage-Related Markers in Intractable Myocardial Diseases. Inflammation. 38, 2288–2299. 10.1007/s10753-015-0214-1. [DOI] [PubMed] [Google Scholar]
- 204.Falk RH, Alexander KM, Liao R & Dorbala S (2016). AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol 68, 1323–1341. 10.1016/j.jacc.2016.06.053. [DOI] [PubMed] [Google Scholar]
- 205.Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AW, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C & Hawkins PN (2016). Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 133, 2404–2412. 10.1161/circulationaha.116.021612. [DOI] [PubMed] [Google Scholar]
- 206.Morfino P, Aimo A, Panichella G, Rapezzi C & Emdin M (2022). Amyloid seeding as a disease mechanism and treatment target in transthyretin cardiac amyloidosis. Heart Fail Rev 27, 2187–2200. 10.1007/s10741-022-10237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gallegos C & Miller EJ (2020). Advances in PET-Based Cardiac Amyloid Radiotracers. Curr Cardiol Rep 22, 40. 10.1007/s11886-020-01284-3. [DOI] [PubMed] [Google Scholar]
- 208.Siegismund CS, Escher F, Lassner D, Kühl U, Gross U, Fruhwald F, Wenzel P, Münzel T, Frey N, Linke RP & Schultheiss H-P (2018). Intramyocardial inflammation predicts adverse outcome in patients with cardiac AL amyloidosis. European Journal of Heart Failure. 20, 751–757. 10.1002/ejhf.1039. [DOI] [PubMed] [Google Scholar]
- 209.Stone PJ, Campistol JM, Abraham CR, Rodgers O, Shirahama T & Skinner M (1993). Neutrophil Proteases Associated with Amyloid Fibrils. Biochemical and Biophysical Research Communications. 197, 130–136. 10.1006/bbrc.1993.2451. [DOI] [PubMed] [Google Scholar]
- 210.Uslu AU, Deveci K, Korkmaz S, Aydin B, Senel S, Sancakdar E & Sencan M (2013). Is neutrophil/lymphocyte ratio associated with subclinical inflammation and amyloidosis in patients with familial Mediterranean fever? Biomed Res Int 2013, 185317. 10.1155/2013/185317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Alsarah A, Alsara O & Laird-Fick HS (2017). Cardiac manifestations of Familial Mediterranean fever. Avicenna J Med 7, 158–163. 10.4103/ajm.AJM_78_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Griffin JM, Rosenblum H & Maurer MS (2021). Pathophysiology and Therapeutic Approaches to Cardiac Amyloidosis. Circulation Research. 128, 1554–1575. 10.1161/CIRCRESAHA.121.318187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tana C, Mantini C, Donatiello I, Mucci L, Tana M, Ricci F, Cipollone F & Giamberardino MA (2021). Clinical Features and Diagnosis of Cardiac Sarcoidosis. J Clin Med 10. 10.3390/jcm10091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Terasaki F, Ukimura A, Tsukada B, Fujita S, Katashima T, Otsuka K, Otsuka K, Kanzaki Y, Shimomura H, Fujita M, Tanaka T & Kitaura Y (2008). Enhanced expression of type 1 helper T-cell cytokines in the myocardium of active cardiac sarcoidosis. Circ J 72, 1303–1307. 10.1253/circj.72.1303. [DOI] [PubMed] [Google Scholar]
- 215.Ghasempour Alamdari M, Kalami N, Shojaan H, Aminizadeh S, Ghaedi A, Bazrgar A & Khanzadeh S (2023). Systematic review of the diagnostic role of neutrophil to lymphocyte ratio in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 40, e2023008. 10.36141/svdld.v40i1.13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kraaijvanger R, Janssen Bonás M, Vorselaars ADM & Veltkamp M (2020). Biomarkers in the Diagnosis and Prognosis of Sarcoidosis: Current Use and Future Prospects. Front Immunol 11, 1443. 10.3389/fimmu.2020.01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mirsaeidi M, Mortaz E, Omar HR, Camporesi EM & Sweiss N (2016). Association of Neutrophil to Lymphocyte Ratio and Pulmonary Hypertension in Sarcoidosis Patients. Tanaffos. 15, 44–47. [PMC free article] [PubMed] [Google Scholar]
- 218.Blankstein R & Waller AH (2016). Evaluation of Known or Suspected Cardiac Sarcoidosis. Circulation: Cardiovascular Imaging. 9, e000867. 10.1161/CIRCIMAGING.113.000867. [DOI] [PubMed] [Google Scholar]
- 219.Kato S, Inui N, Hozumi H, Inoue Y, Yasui H, Karayama M, Kono M, Suzuki Y, Furuhashi K, Enomoto N, Fujisawa T, Nakamura Y, Watanabe H & Suda T (2018). Neutrophil gelatinase-associated lipocalin in patients with sarcoidosis. Respiratory Medicine. 138, S20–S23. 10.1016/j.rmed.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 220.Liu J, Ma P, Lai L, Villanueva A, Koenig A, Bean GR, Bowles DE, Glass C, Watson M, Lavine KJ & Lin CY (2022). Transcriptional and Immune Landscape of Cardiac Sarcoidosis. Circ Res 131, 654–669. 10.1161/circresaha.121.320449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lubitz SA, Goldbarg SH & Mehta D (2008). Sudden cardiac death in infiltrative cardiomyopathies: sarcoidosis, scleroderma, amyloidosis, hemachromatosis. Prog Cardiovasc Dis 51, 58–73. 10.1016/j.pcad.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 222.Gulati V, Harikrishnan P, Palaniswamy C, Aronow WS, Jain D & Frishman WH (2014). Cardiac involvement in hemochromatosis. Cardiol Rev 22, 56–68. 10.1097/CRD.0b013e3182a67805. [DOI] [PubMed] [Google Scholar]
- 223.Renassia C, Louis S, Cuvellier S, Boussetta N, Deschemin JC, Borderie D, Bailly K, Poupon J, Dang PM, El-Benna J, Manceau S, Lefrère F, Vaulont S & Peyssonnaux C (2020). Neutrophils from hereditary hemochromatosis patients are protected from iron excess and are primed. Blood Adv 4, 3853–3863. 10.1182/bloodadvances.2020002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhabyeyev P, Das SK, Basu R, Shen M, Patel VB, Kassiri Z & Oudit GY (2018). TIMP3 deficiency exacerbates iron overload-mediated cardiomyopathy and liver disease. Am J Physiol Heart Circ Physiol 314, H978–h990. 10.1152/ajpheart.00597.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sagar S, Liu PP & Cooper LT Jr. (2012). Myocarditis. Lancet. 379, 738–747. 10.1016/s0140-6736(11)60648-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lampejo T, Durkin SM, Bhatt N & Guttmann O (2021). Acute myocarditis: aetiology, diagnosis and management. Clin Med (Lond). 21, e505–e510. 10.7861/clinmed.2021-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Liu K & Han B (2023). Role of immune cells in the pathogenesis of myocarditis. Journal of Leukocyte Biology. 115, 253–275. 10.1093/jleuko/qiad143. [DOI] [PubMed] [Google Scholar]
- 228.Rivadeneyra L, Charó N, Kviatcovsky D, de la Barrera S, Gómez RM & Schattner M (2018). Role of neutrophils in CVB3 infection and viral myocarditis. J Mol Cell Cardiol 125, 149–161. 10.1016/j.yjmcc.2018.08.029. [DOI] [PubMed] [Google Scholar]
- 229.Li H, Zhang M, Zhao Q, Zhao W, Zhuang Y, Wang J, Hang W, Wen Z, Wang L, Chen C & Wang DW (2023). Self-recruited neutrophils trigger over-activated innate immune response and phenotypic change of cardiomyocytes in fulminant viral myocarditis. Cell Discovery. 9, 103. 10.1038/s41421-023-00593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ding L, Hanawa H, Ota Y, Hasegawa G, Hao K, Asami F, Watanabe R, Yoshida T, Toba K, Yoshida K, Ogura M, Kodama M & Aizawa Y (2010). Lipocalin-2/neutrophil gelatinase-B associated lipocalin is strongly induced in hearts of rats with autoimmune myocarditis and in human myocarditis. Circ J 74, 523–530. 10.1253/circj.cj-09-0485. [DOI] [PubMed] [Google Scholar]
- 231.Zhang X, Gao Y, Yang B, Ma S, Zuo W & Wei J (2023). The mechanism and treatment of targeted anti-tumour drugs induced cardiotoxicity. Int Immunopharmacol 117, 109895. 10.1016/j.intimp.2023.109895. [DOI] [PubMed] [Google Scholar]
- 232.Morelli MB, Bongiovanni C, Da Pra S, Miano C, Sacchi F, Lauriola M & D’Uva G (2022). Cardiotoxicity of Anticancer Drugs: Molecular Mechanisms and Strategies for Cardioprotection. Front Cardiovasc Med 9, 847012. 10.3389/fcvm.2022.847012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Nagy A, Börzsei D, Hoffmann A, Török S, Veszelka M, Almási N, Varga C & Szabó R (2024). A Comprehensive Overview on Chemotherapy-Induced Cardiotoxicity: Insights into the Underlying Inflammatory and Oxidative Mechanisms. Cardiovascular Drugs and Therapy. 10.1007/s10557-024-07574-0. [DOI] [PubMed] [Google Scholar]
- 234.Veeder JA, Hothem LN, Cipriani AE, Jensen BC & Rodgers JE (2021). Chemotherapy-associated cardiomyopathy: Mechanisms of toxicity and cardioprotective strategies. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy. 41, 1066–1080. 10.1002/phar.2638. [DOI] [PubMed] [Google Scholar]
- 235.Sano S, Wang Y, Ogawa H, Horitani K, Sano M, Polizio AH, Kour A, Yura Y, Doviak H & Walsh K (2021). TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight. 6. 10.1172/jci.insight.146076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Todorova VK, Wei JY & Makhoul I (2021). Subclinical doxorubicin-induced cardiotoxicity update: role of neutrophils and endothelium. Am J Cancer Res 11, 4070–4091. [PMC free article] [PubMed] [Google Scholar]
- 237.Bhagat A, Shrestha P, Jeyabal P, Peng Z, Watowich SS & Kleinerman ES (2022). Doxorubicin-induced cardiotoxicity is mediated by neutrophils through release of neutrophil elastase. Front Oncol 12, 947604. 10.3389/fonc.2022.947604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhao P, Li Y, Xu X, Yang H, Li X, Fu S, Guo Z, Zhang J, Li H & Tian J (2024). Neutrophil extracellular traps mediate cardiomyocyte ferroptosis via the Hippo-Yap pathway to exacerbate doxorubicin-induced cardiotoxicity. Cell Mol Life Sci 81, 122. 10.1007/s00018-024-05169-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Nettersheim FS, Schlüter JD, Kreuzberg W, Mehrkens D, Grimm S, Nemade H, Braumann S, Hof A, Guthoff H, Peters V, Hoyer FF, Kargapolova Y, Lackmann J-W, Müller S, Pallasch CP, Hallek M, Sachinidis A, Adam M, Winkels H, Baldus S, Geißen S & Mollenhauer M (2023). Myeloperoxidase is a critical mediator of anthracycline-induced cardiomyopathy. Basic Research in Cardiology. 118, 36. 10.1007/s00395-023-01006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Baruch R, Zahler D, Zornitzki L, Arbel Y, Rozenbaum Z, Arnold JH, Raphael A, Khoury S, Banai S, Topilsky Y, Kapusta L & Laufer-Perl M (2023). High neutrophil-to-lymphocyte ratio as an early sign of cardiotoxicity in breast cancer patients treated with anthracycline. Clinical Cardiology. 46, 328–335. 10.1002/clc.23966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Copeland-Halperin RS, Liu JE & Yu AF (2019). Cardiotoxicity of HER2-targeted therapies. Curr Opin Cardiol 34, 451–458. 10.1097/hco.0000000000000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Shyam Sunder S, Sharma UC & Pokharel S (2023). Adverse effects of tyrosine kinase inhibitors in cancer therapy: pathophysiology, mechanisms and clinical management. Signal Transduction and Targeted Therapy. 8, 262. 10.1038/s41392-023-01469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kirkham AA, Pituskin E, Thompson RB, Mackey JR, Koshman SL, Jassal D, Pitz M, Haykowsky MJ, Pagano JJ, Chow K, Tsui AK, Ezekowitz JA, Oudit GY & Paterson DI (2022). Cardiac and cardiometabolic phenotyping of trastuzumab-mediated cardiotoxicity: a secondary analysis of the MANTICORE trial. Eur Heart J Cardiovasc Pharmacother. 8, 130–139. 10.1093/ehjcvp/pvab016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Putt M, Hahn VS, Januzzi JL, Sawaya H, Sebag IA, Plana JC, Picard MH, Carver JR, Halpern EF, Kuter I, Passeri J, Cohen V, Banchs J, Martin RP, Gerszten RE, Scherrer-Crosbie M & Ky B (2015). Longitudinal Changes in Multiple Biomarkers Are Associated with Cardiotoxicity in Breast Cancer Patients Treated with Doxorubicin, Taxanes, and Trastuzumab. Clin Chem 61, 1164–1172. 10.1373/clinchem.2015.241232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Demissei BG, Hubbard RA, Zhang L, Smith AM, Sheline K, McDonald C, Narayan V, Domchek SM, DeMichele A, Shah P, Clark AS, Fox K, Matro J, Bradbury AR, Knollman H, Getz KD, Armenian SH, Januzzi JL, Tang WHW, Liu P & Ky B (2020). Changes in Cardiovascular Biomarkers With Breast Cancer Therapy and Associations With Cardiac Dysfunction. Journal of the American Heart Association. 9, e014708. 10.1161/JAHA.119.014708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Pivatto Júnior F, Santos ÂBS, Englert EF, Mazzutti G, Costa GOM, Saffi MAL, Liedke PER, Fritsch VH & Biolo A (2023). Monocyte-to-lymphocyte ratio as predictor of cancer therapy-related cardiotoxicity in patients with breast cancer: a pilot cohort study. Breast Cancer Research and Treatment. 200, 355–362. 10.1007/s10549-023-06979-z. [DOI] [PubMed] [Google Scholar]
- 247.Bouwer NI, Jager A, Liesting C, Kofflard MJM, Brugts JJ, Kitzen J, Boersma E & Levin MD (2020). Cardiac monitoring in HER2-positive patients on trastuzumab treatment: A review and implications for clinical practice. Breast. 52, 33–44. 10.1016/j.breast.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kaddoura R, Dabdoob WA, Ahmed K & Yassin MA (2023). A practical guide to managing cardiopulmonary toxicities of tyrosine kinase inhibitors in chronic myeloid leukemia. Front Med (Lausanne). 10, 1163137. 10.3389/fmed.2023.1163137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wang H, Wang Y, Li J, He Z, Boswell SA, Chung M, You F & Han S (2023). Three tyrosine kinase inhibitors cause cardiotoxicity by inducing endoplasmic reticulum stress and inflammation in cardiomyocytes. BMC Medicine. 21, 147. 10.1186/s12916-023-02838-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mak IT, Kramer JH, Chmielinska JJ, Spurney CF & Weglicki WB (2015). EGFR-TKI, erlotinib, causes hypomagnesemia, oxidative stress, and cardiac dysfunction: attenuation by NK-1 receptor blockade. J Cardiovasc Pharmacol 65, 54–61. 10.1097/fjc.0000000000000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Park JY, Jang SH, Lee CY, Kim T, Chung SJ, Lee YJ, Kim HI, Kim JH, Park S, Hwang YI & Jung KS (2022). Pretreatment Neutrophil-to-Lymphocyte Ratio and Smoking History as Prognostic Factors in Advanced Non-Small Cell Lung Cancer Patients Treated with Osimertinib. Tuberc Respir Dis (Seoul). 85, 155–164. 10.4046/trd.2021.0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chen K-C, Huang Y-H, Hsu K-H, Tseng J-S, Chang G-C & Yang T-Y (2023). The Role of Neutrophil-to-Lymphocyte Ratio in Advanced EGFR-Mutant NSCLC Patients Treated with First-Line Osimertinib. OncoTargets and Therapy. 16, 317–326. 10.2147/OTT.S407301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Telerman A, Granot G, Leibovitch C, Yarchovsky-Dolberg O, Shacham-Abulafia A, Partouche S, Yeshurun M, Ellis MH, Raanani P & Wolach O (2022). Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors. Cancers. 14, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wolach O, Telerman A, Leibovitch C, Yarchovsky-Dolberg O, Shacham-Abulafia A, Partouche S, Yeshurun M, Ellis M, Raanani P & Granot G (2021). CML-271: Neutrophil Extracellular Traps Are Increased in Chronic Myeloid Leukemia and Are Differentially Affected by Tyrosine Kinase Inhibitors. Clinical Lymphoma Myeloma and Leukemia. 21, S332. 10.1016/S2152-2650(21)01777-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, Ozawa K, Shentu W, Brown E, Shirai T, McCarty OJ, Ruggeri Z, Moslehi J, Chen J, Druker BJ, López JA & Lindner JR (2019). Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood. 133, 1597–1606. 10.1182/blood-2018-10-881557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhu H, Galdos FX, Lee D, Waliany S, Huang YV, Ryan J, Dang K, Neal JW, Wakelee HA, Reddy SA, Srinivas S, Lin LL, Witteles RM, Maecker HT, Davis MM, Nguyen PK & Wu SM (2022). Identification of Pathogenic Immune Cell Subsets Associated With Checkpoint Inhibitor-Induced Myocarditis. Circulation. 146, 316–335. 10.1161/circulationaha.121.056730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Baik AH, Oluwole OO, Johnson DB, Shah N, Salem J-E, Tsai KK & Moslehi JJ (2021). Mechanisms of Cardiovascular Toxicities Associated With Immunotherapies. Circulation Research. 128, 1780–1801. 10.1161/CIRCRESAHA.120.315894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jimenez J, Kostelecky N, Mitchell JD, Zhang KW, Lin C-Y, Lenihan DJ & Lavine KJ (2023). Clinicopathological classification of immune checkpoint inhibitor-associated myocarditis: possible refinement by measuring macrophage abundance. Cardio-Oncology. 9, 14. 10.1186/s40959-023-00166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ma P, Liu J, Qin J, Lai L, Heo GS, Luehmann H, Sultan D, Bredemeyer A, Bajapa G, Feng G, Jimenez J, He R, Parks A, Amrute J, Villanueva A, Liu Y, Lin C-Y, Mack M, Amancherla K, Moslehi J & Lavine KJ (2024). Expansion of Pathogenic Cardiac Macrophages in Immune Checkpoint Inhibitor Myocarditis. Circulation. 149, 48–66. 10.1161/CIRCULATIONAHA.122.062551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Haj-Yehia E, Mincu RI, Korste S, Lampe L, Margraf SM, Michel L, Mahabadi AA, Ferdinandy P, Rassaf T & Totzeck M (2024). High neutrophil-to-lymphocyte ratio is associated with cancer therapy-related cardiovascular toxicity in high-risk cancer patients under immune checkpoint inhibitor therapy. Clin Res Cardiol 113, 301–312. 10.1007/s00392-023-02327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Drobni ZD, Zafar A, Zubiri L, Zlotoff DA, Alvi RM, Lee C, Hartmann S, Gilman HK, Villani AC, Nohria A, Groarke JD, Sullivan RJ, Reynolds KL, Zhang L & Neilan TG (2020). Decreased Absolute Lymphocyte Count and Increased Neutrophil/Lymphocyte Ratio With Immune Checkpoint Inhibitor-Associated Myocarditis. J Am Heart Assoc 9, e018306. 10.1161/jaha.120.018306. [DOI] [PMC free article] [PubMed] [Google Scholar]