

Abstract
The hypomethylated fraction of plant genomes is usually enriched in genes and can be selectively cloned using methylation filtration (MF). Therefore, MF has been used as a gene enrichment technology in sorghum and maize, where gene enrichment was proportional to genome size. Here we apply MF to a broad variety of plant species spanning a wide range of genome sizes. Differential methylation of genic and non-genic sequences was observed in all species tested, from non-vascular to vascular plants, but in some cases, such as wheat and pine, a lower than expected level of enrichment was observed. Remarkably, hexaploid wheat and pine show a dramatically large number of gene-like sequences relative to other plants. In hexaploid wheat, this apparent excess of genes may reflect an abundance of methylated pseudogenes, which may thus be more prevalent in recent polyploids.
Methylation is a common DNA modification among eukaryotes, typically in the form of cytosine methylated on carbon-5 (5-methylcytosine, 5mC). Saccharomyces cerevisiae has no 5mC, and in other fungi methylation is targeted to the scarce repeated DNA (Selker 1990; Rossignol and Faugeron 1994). The levels and patterns of DNA methylation are highly variable in animals, ranging from no detectable 5mC in the nematode Caenorhabditis elegans and limited, developmentally restricted methylation in Drosophila melanogaster (Lyko et al. 2000) to widespread genomic methylation in vertebrates (Tweedie et al. 1997). On the other hand, DNA methylation seems to be ubiquitous among plants, where it is found at symmetric CpNpG and asymmetric CpNpN sites, as well as the CpG sites frequent in mammals (Gruenbaum et al. 1981; Meyer et al. 1994). The level of 5mC is variable in plants, from 6% of cytosines in Arabidopsis (Kakutani et al. 1999) to 25% in maize (Papa et al. 2001).
The distribution of 5mC in mammals varies during development (Monk et al. 1987), although it is conspicuous in repetitive sequences (Walsh et al. 1998). Genes are also methylated (Rabinowicz et al. 2003b), but CpG islands are essentially unmethylated (Cross and Bird 1995). In plants, inactive transposons are densely methylated (Chandler and Walbot 1986; Flavell 1994; Martienssen 1998), while genes rarely have any DNA methylation (Bennetzen et al. 1994; Rabinowicz et al. 2003b), and when they do it is restricted to the 3′ and 5′ ends (Walbot and Warren 1990; Patterson et al. 1993; Lippman et al. 2004).
The function of DNA methylation is still controversial, although it can clearly silence genes (Colot and Rossignol 1999; Martienssen and Colot 2001; Bird 2002). In plants, transposons are methylated upon silencing (Chandler and Walbot 1986; Chomet et al. 1987), which can result in regulation of nearby genes (Martienssen et al. 1990). Further, transposons can be activated in mutants defective in a chromatin remodeling ATPase (Miura et al. 2001; Singer et al. 2001) and in DNA methyltransferases (Kato et al. 2003; Lippman et al. 2003), in which DNA methylation levels are reduced by 70%-80%. Microarray profiling has revealed that most DNA methylation in plants is found in transposons, suggesting this may be its primary function (Lippman et al. 2004). In animals, it has also been suggested that DNA methylation silences transposons, which are expressed in methylation-defective mutant mice (Yoder et al. 1997; Walsh et al. 1998). However, methylation of most retrotransposable elements is also lost during normal mammalian embryogenesis, so the significance of these results is not clear (Monk et al. 1987; Reik et al. 2001). An alternative hypothesis proposes that methylation targets both genes and repetitive DNA in order to decrease transcriptional “noise.” Consistent with this idea, most mammalian genes are methylated, and only CpG islands are protected from methylation to permit access to promoters (Bird 1995; Tornaletti and Pfeifer 1995; Tada et al. 1997; Rabinowicz et al. 2003b).
Many plant genomes—including those of important crops— are very large and repetitive (Arumuganathan and Earle 1991; Kumar and Bennetzen 1999). For this reason, high-quality plant genome sequences have been obtained only from small plant genomes such as those of Arabidopsis and rice (The Arabidopsis Genome Initiative 2000; Sasaki and Burr 2000; Feng et al. 2002; Sasaki et al. 2002; The Rice Chromosome 10 Sequencing Consortium 2003). These projects represent an invaluable resource for plant biology in general. However, the extremely large size of other plant genomes such as those of pine and wheat effectively prohibits the determination of their complete sequence in this way (Rabinowicz et al. 2003a).
Several gene-enriched sequencing strategies have been used to try to overcome the problem posed by large genomes, the most popular of which is the sequencing of cDNA libraries to produce expressed sequence tags (ESTs) (Adams et al. 1991). Although useful for gene annotation, EST sequencing is limited to genes that are detectably expressed, and misses non-coding sequences such as introns and promoters (Palmer et al. 2003). Another approach has exploited the tendency of some DNA transposons to insert preferentially in genes (Hanley et al. 2000; Raizada et al. 2001). However, transposon insertion is not random (Greenblatt 1984; May et al. 2003) and yields a partial representation of genes when attempted on a large scale (Palmer et al. 2003; Fernandes et al. 2004).
A third approach takes advantage of the differential reassociation kinetics of low- and high-copy DNA. Isolating and cloning the slowly annealing low-copy or high-C0t (HC) fraction of plant genomic DNA results in enrichment for gene sequences (Yuan et al. 2003). This technology has been successfully used in maize (Whitelaw et al. 2003) although, theoretically, it may select against large gene families that could be normalized in the process, and the extensive manipulation of the DNA required to construct HC libraries resulted in a high proportion of mutated sequences when applied to maize (Fu et al. 2004). Furthermore, the HC maize sequences are on average 43% GC versus 50% GC for WGS (whole genome shotgun) (http://www.tigr.org/tdb/tgi/maize/release4.0/assembly_summary.shtml), which may reflect a selection for AT-rich sequences, which reanneal more slowly than GC-rich sequences do.
The difference in methylation between plant genes and repeats is the basis for another gene-enrichment technique, called methylation filtration (MF) (Rabinowicz 2003). MF takes advantage of the McrBC (modified cytosine restricton system), a bacterial methylation-dependent restriction endonuclease (Raleigh and Wilson 1986; Dila et al. 1990) that has minimal sequence requirements for restriction (Sutherland et al. 1992). Therefore, using a mcrBC+ Eschericia coli host strain to construct genomic shotgun libraries, repetitive DNA can be largely excluded, preserving the low-copy (i.e., genic) DNA (Supplemental Fig. 1). In a pilot study, MF applied to the maize genome yielded a sixfold enrichment for genes relative to a WGS library used as a control (Rabinowicz et al. 1999).
Gene-enrichment techniques constitute an effective approach to selectively clone and sequence genes from large plant genomes, in which the majority of the DNA is composed of repetitive elements that can total up to 80% of the genome (Hake and Walbot 1980). MF has been applied comprehensively to maize and sorghum, reaching nearly 1× coverage of the unmethylated fraction of these genomes. As a result, ∼95% of the genes in either genome were tagged, and it was estimated that most genes and regulatory elements are unmethylated in these two plants. Further analyses also provided insights into the biology of transposable element methylation and activity (Palmer et al. 2003; Whitelaw et al. 2003; Bedell et al. 2005).
We report the results of pilot MF sequencing projects in several plant species including moss, fern, pine, and various economically important angiosperms. Hypomethylated DNA is enriched in genes in all plants tested, and there is a correlation between genome size and the degree of gene enrichment (gene enrichment factor, or GEF), particularly among the grasses. The data also highlight differences in genome organization and suggest that some species, often recent polyploids, may have undergone a process of gene amplification that resulted in the generation of pseudogenes, while a process of gene loss may have occurred in older polyploids as part of a process of diploidization.
Results and Discussion
We selected a diverse group of plant species in order to test the efficiency of MF relative to WGS (Fig. 1). We included economically important crops and trees as well as moss and fern. For comparison, we included a random subset of maize, sorghum, and diploid wheat MF and WGS sequences submitted to GenBank by others and us, as well as a set of Arabidopsis and cabbage (Brassica oleracea) WGS sequences (Supplemental Table 1). Genomic sequencing has shown that the model dicotyledoneous (dicot) plant Arabidopsis has substantially fewer genes than rice, the monocotyledoneous (monocot) model (The Arabidopsis Genome Initiative 2000; Goff et al. 2002; Yu et al. 2002). Therefore, we separately grouped monocots (rice, sorghum, maize, wild diploid wheat, barley, and bread wheat) and dicots (Arabidopsis, oilseed rape, cabbage, soybean, potato, tomato, and cotton) in this study. Non-angiosperms (fern, moss, and pine) were placed in a third group.
Figure 1.

Phylogenetic tree showing approximate evolutionary distances among the plant species used in this study. The corresponding gene enrichment factor (GEF) is shown in parenthesis.
The number of plant genes
We first estimated the gene content in each genome by analyzing the WGS sequence set for each species. In order to reliably identify genes, we used a carefully curated database of a subset of known plant proteins. This subset is much smaller than the number of annotated “known” proteins, but is much more reliable (see Methods). We compared all WGS sequences to this database using BLASTX. We then plotted the observed and the expected frequencies of genic database matches for each species, based on their relative genome or sub-genome sizes (Fig. 2). The frequency of gene matches in monocot WGS sequences consistently decreases as genome size increases (Fig. 2A). Using rice as a reference, we estimated the gene number in each genome or sub-genome. This was achieved by first dividing the rice genome size by each of the other genome sizes, and then multiplying the relative gene density in each genome by the rice gene number, which is estimated at 41,000 (Feng et al. 2002; Sasaki et al. 2002; The Rice Chromosome 10 Sequencing Consortium 2003). The observed number of genes in each species is remarkably similar among the grasses (Table 1), presumably because the evolutionary divergence is relatively small (Kellogg 2001). Rice has a slight excess of genes, probably due to the presence of large gene families (Lai et al. 2004a).
Figure 2.

Percentage of genes in each WGS set of sequences. (A) Monocots. The expected number is calculated relative to rice (see Methods). (B) Dicots. The expected number is calculated relative to Arabidopsis (C) Non-angiosperms. The expected number is calculated relative to rice.
Table 1.
Genomic features of the studied plants
| Monocots | Genome size (Mbp)a | Sub-genome size (Mbp) | Ploidy level | Percent protein database matches | Relative gene densityb | Gene number per sub-genome | Genes per genome |
|---|---|---|---|---|---|---|---|
| Rice | 400 | 400 | 2× | 8.39 | 1 | NA | 41,000 |
| Sorghum | 750 | 750 | 2× | 3.64 | 0.43 | NA | 33,400 |
| Maize | 2670 | 2670 | 2× | 1.23 | 0.15 | NA | 40,300 |
| Wild wheat | 4000 | 4000 | 2× | 0.67 | 0.08 | NA | 32,800 |
| Barley | 5400 | 5400 | 2× | 0.51 | 0.06 | NA | 33,600 |
| Bread wheat | 16,800 | 5600 | 6× | 1.44 | 0.17 | 98,640 | 295,900 |
| Dicots | |||||||
| Arabidopsis | 135 | 135 | 2× | 22.22 | 1 | NA | 26,300 |
| Soybean | 1100 | 550 | 4× | 6.03 | 0.27 | 29,110 | 58,200 |
| Oilseed rape | 1100 | 550 | 4× | 6.78 | 0.30 | 32,720 | 65,400 |
| Cabbage | 760 | 760 | 2× | 8.49 | 0.38 | NA | 56,600 |
| Potato | 2000 | 1000 | 4× | 4.27 | 0.19 | 37,450 | 74,900 |
| Tomato | 1000 | 1000 | 2× | 2.84 | 0.13 | NA | 24,900 |
| Cotton | 3000 | 1500 | 4× | 4.07 | 0.18 | 53,550 | 107,100 |
| Non-Angiosperms | |||||||
| Moss | 380 | 380 | 2× | 4.44 | 0.53 | NA | 20,600 |
| Fern | 9000 | 9000 | 2× | 0.49 | 0.06 | NA | 42,300 |
| Pine | 21,600 | 21,600 | 2× | 0.85 | 0.10 | NA | 224,300 |
Genome sizes were taken from the Plant DNA C-values Database, Royal Botanic Gardens, Kew, UK (http://www.rbgkew.org.uk/cva1/database1.html), except for wild wheat (Li et al. 2004), fern (J. Banks, pers. comm.), Arabidopsis (http://www.arabidopsis.org; Round et al. 1997), and rice (http://www.rgp.dna.affrc.go.jp/cgi-bin/statusdb/irgsp-status.cgi). (NA) not applicable
The relative gene density per genome or sub-genome was calculated relative to rice in monocots and non-angiosperms, and relative to Arabidopsis in dicots
A major exception is hexaploid bread wheat, which has a much larger number of genes than expected, given that the sub-genome size is comparable to wild wheat and barley, which are close relatives (Kellogg 2001). The bread wheat sub-genomes contain nearly two and a half times more genes than rice, which brings the total gene number in the hexaploid genome to almost 300,000 (Table 1). This apparent excess of genes in each sub-genome in hexaploid wheat is surprising, as there is a common consensus that grasses contain similar numbers of genes (Bennetzen 2000).
Another interesting case is maize. If it is considered as an ancient tetraploid, then it has less than half the expected number of genes, so that extensive gene loss has occurred since genome duplication (Gaut and Doebley 1997), a process known as “diploidization” (Ilic et al. 2003; Lai et al. 2004b; Langham et al. 2004). Consistent with this hypothesis, the duplicated maize genome contains nearly as many genes as diploid rice (Table 1). The recent estimate of 59,000 genes in maize by analyzing BAC end sequences may have overestimated the number of hypothetical genes, many of which are transposons (Messing et al. 2004). For these reasons, we considered maize a diploid for the analysis.
In the dicot group we used shotgun sequences of the Landsberg erecta ecotype of Arabidopsis as a reference. Among these sequences, 22.22% had a match in our protein database. Tomato, the only diploid genome among those tested, has a similar number of genes as Arabidopsis (Table 1) and similar expected and observed gene frequencies (Fig. 2B). The remaining dicot genomes are polyploid, and some insight into their history can be inferred from the density of genes. The Brassica diploid genome underwent triplication after its divergence from Arabidopsis at least 14.5 million years ago (Mya) (Lagercrantz 1998; Yang et al. 1999). Further, oilseed rape is an allopolyploid formed by hybridization of Brassica rapa (A genome) and Brassica oleracea (C genome), which occurred during domestication in the last few thousand years (Prakash and Hinata 1980; Parkin et al. 1995). If the number of genes in all six genomes included in this polyploid had remained unchanged, one would expect six times as many genes as Arabidopsis. However, oilseed rape has only three times as many genes (Table 1). Substantial gene loss must have occurred between the ancient triplication and formation of the allopolyploid hybrid, because cabbage (Brassica oleracea) has only twice as many genes as Arabidopsis, rather than three times. Thus, we considered oilseed rape a tetraploid with a sub-genome size of 550 Mbp, and cabbage a diploid with a 760-Mbp genome.
The tetraploids cotton and potato have a larger than expected number of genes per sub-genome. While cotton is an ancient polyploid that originated 1.5 Mya (Senchina et al. 2003), potato is believed to have become tetraploid during domestication, only a few thousand years ago (Cribb and Hawkes 1986). Soybean is an ancient tetraploid that has become further duplicated and undergone extensive diploidization (Zhu et al. 1994; Shoemaker et al. 1996; Krishnan et al. 2001). If considered an octaploid, each of its 275-Mbp sub-genomes would have much lower than the expected frequency of genes. Thus, we considered it a tetraploid genome with a sub-genome size of 550 Mbp (Table 1).
The numbers of genes in ferns are comparable to those of most angiosperms, consistent with the presence of many known genes in these plants (Banks 1999; Floyd and Bowman 2004). However, their degree of polyploidy is uncertain; it has been proposed that most ferns are ancient polyploids, and abundant pseudogenes have been reported (Pichersky et al. 1990; Gastony 1991). If the fern genome is in fact polyploid, the gene number per sub-genome will be less than in angiosperms. Moss, on the other hand, has fewer genes than angiosperms, and even fewer considering that it is believed to be a tetraploid (R. Quatrano, pers. comm.). Nevertheless, most known gene functions are present in its genome (Banks 1999; Floyd and Bowman 2004). Like bread wheat, the gymnosperm pine has an extremely high gene density (Fig. 2C) and almost as many genes (Table 1). The explanation for the high gene content in pine remains elusive, as evidence of polyploidy in pine has not been reported. However, tandem gene duplications frequently found in pine (Krutovsky et al. 2004) could be part of the explanation. Nevertheless, it is striking that a diploid organism can have >200,000 genes.
Gene enrichment by MF
We applied MF to the three groups of plants in order to evaluate the GEF (calculated as the percentage of genes in the MF library divided by the percentage of genes in the WGS library) in each genome. MF enriched for genes in all plant libraries tested (Fig. 3). We then used the observed GEF to calculate additional parameters, based on the assumption that almost all genes are unmethylated. If this assumption is true, then 1/GEF is a reasonable approximation for the proportion of the genome that is unmethylated, or UM/N, where UM is the size of the unmethylated genome and N is the total number of sequences (Palmer et al. 2003). First, we calculated UM by dividing the genome size by the GEF (Table 2). Next, we calculated the proportion of unmethylated repeats (r/R). This can be calculated by dividing the fraction of repeat matches in MF reads (r/UM) by the GEF, as well as by the fraction of repeat matches in WGS reads [r/R = (r/UM) × (UM/N) × (N/R)]. We then estimated the size of the “gene space” by subtracting the fraction of unmethylated repeats from UM (UM - r).
Figure 3.

Gene enrichment for each species. (A) Monocots. The expected GEF is calculated by extrapolation of the gene frequency found in rice to a genome of the corresponding size. (B) Dicots. The expected GEF is calculated by extrapolation of the gene frequency found in soybean to a genome of the corresponding size. (C) Same as B using tomato as a reference. (D) Non-angiosperms. The expected GEF is calculated by extrapolation of the gene frequency found in rice to a genome of the corresponding size. The GEF values are listed in Supplemental Table 1, and a list of all gene matches is shown in Supplemental Table 2.
Table 2.
Gene space size and level of gene-enrichment of the studied plants
| Monocots | Gene enrichment factor (GEF) | Unmethylated repeats (%r/R) | Unmethylated space | Gene space | Percent repeats in MF sequences |
|---|---|---|---|---|---|
| Rice | 1.89 | 11.3 | 212 | 195 | 8.03 |
| Sorghum | 3.54 | 7.4 | 212 | 184 | 13.27 |
| Maize | 13.15 | 2 | 203 | 161 | 20.53 |
| Barley | 18.68 | 3 | 289 | 181 | 37.37 |
| Bread wheat | 4.71 | 7.2 | 1189 | 942 | 20.8 |
| Wild wheat | 3.8 | 21.4 | 1052 | 510 | 51.54 |
| Dicots | |||||
| Soybean | 2.47 | 7 | 223 | 216 | 3.46 |
| Oilseed rape | 2.83 | 6.1 | 194 | 186 | 3.97 |
| Potato | 2.51 | 12.9 | 398 | 363 | 8.66 |
| Tomato | 3.02 | 9.7 | 331 | 291 | 11.96 |
| Cotton | 3.15 | 9.1 | 476 | 450 | 5.42 |
| Non-Angiosperms | |||||
| Moss | 2.51 | 45.5 | 151 | 138 | 8.7 |
| Fern | 5.19 | 12.1 | 1349 | 1122 | 16.82 |
| Pine | 3.26 | 18.2 | 6626 | 5784 | 12.7 |
In general, GEF increases proportionately with genome size, as expected (Fig. 3A). As a result, monocot genomes have a comparable gene space between 160 and 200 Mbp. Maize has a GEF of 13.15, which is higher than previously reported (Palmer et al. 2003), possibly due to improvements in transposon annotation and sequence quality, although sampling error may also contribute. One exception is bread wheat, which has a low GEF and a huge gene space at least five times larger than that of other grasses (Table 2). This is due to a very high gene density in unfiltered reads, implying a very high gene number. We considered several explanations. First, the effective genome size may be smaller than 16.8 Gbp if a large fraction of the genome was composed of homopolymeric repeats and palindromes, which cannot be cloned in plasmid vectors. This seems unlikely given that bacteriophage libraries yield single-copy genes at expected frequencies (Martienssen and Baulcombe 1989). Second, it is possible that wheat is far more polyploid than initially suspected, so that diploids are actually ancient hexaploids. However, WGS sampling of wild wheat progenitors of hexaploid wheat suggests a more reasonable number of genes, comparable to that of rice (Table 1). A third possibility is that repeats are frequently unmethylated, so that methylation filtering has little impact. This contributes to the low GEF in wild wheat, in which at least 21% of repeats are unmethylated (Table 2), but the proportion of unmethylated repeats in bread wheat (7%) was comparable to that of other grasses (Table 2).
Thus, hexaploid wheat has an unusually large number of genes. If they were functional, this would lead to genetic redundancy beyond even that expected for a hexaploid, which seems unlikely. Rather, our results indicate the vast majority of these “genes” are methylated, and likely to be pseudogenes that have been recently amplified and silenced (Bedell et al. 2005). This is not the explanation for low filtering in diploid wild wheat, which has normal numbers of genes but a high proportion of unmethylated transposons, which may therefore be active. It should be noted that different DNA preparations were used for MF and WGS libraries in this case, introducing potential experimental error (Li et al. 2004). Also, sequences corresponding to 300 WGS clones from bread wheat have been recently submitted to GenBank (CW511369-CW512048) and have many fewer gene matches than our larger sample, but this may be due to sampling error.
The observed GEF in dicots was somewhat lower than in monocots based on ploidy and genome size (Fig. 3B), resulting in a larger but comparable gene space. Soybean has a GEF of 2.5, which is reasonable if we consider soybean a tetraploid with a sub-genome size of 550 Mbp. For this reason and because it is the smallest genome or sub-genome, we used soybean as the reference genome size. With the exception of tomato, the only diploid analyzed, all dicot plants also have a higher than expected number of genes (Fig. 2B). This is probably because of cryptic polyploidization that would increase the number of genes in each genome. If tomato is used as the reference genome instead, the GEF of the partial polyploids oilseed rape and soybean are higher than expected, while cotton and potato have the opposite trend, and the observed levels of GEF are closer to the expected ones (Fig. 3C). In either case, potato and cotton show the lowest observed to expected GEF ratio. One explanation is that these genomes have a higher number of methylated pseudogenes, similar to that of bread wheat but not as exaggerated. Cotton is a much older polyploid than bread wheat and potato.
Remarkably, genes and repeats are differentially methylated also in primitive plants. Moss has a higher GEF than rice, but fern has a GEF of 5.19, which is lower than expected for a 9000-Mbp genome (Fig. 3D). Polyploidy is unlikely to be the entire explanation, as overall gene number is similar to those of diploid angiosperms (Table 1). The GEF of 3.26 found in pine is also very low for a 21,600-Mbp genome. We cannot explain this low level of enrichment. If a fraction of the large number of genes found in pine WGS sequences were methylated pseudogenes, it would result in a low GEF, but we do not know if this is the case.
We also analyzed the frequency of McrBC restriction sites that overlap potentially methylated sequences both in genes and repeats for each dataset (see Methods). Sequences depleted of McrBC sites are expected to be enriched in MF libraries regardless of methylation and often correspond to repeats that accumulated C to T transitions (Palmer et al. 2003). Consistent with the large-scale analysis in maize (Palmer et al. 2003), all monocot repeats have a lower frequency of McrBC sites in MF libraries than WGS, suggesting that many repeats recovered in filtered libraries are probably ancient, mutated copies of repetitive elements. In contrast, as genes are mostly unmethylated in plants, genic MF reads are not expected to have a lower McrBC site frequency. In fact, genes have generally higher frequencies of McrBC sites in MF libraries than genic sequences found in WGS (Fig. 4).
Figure 4.

Frequency of McrBC recognition sites in genes and repeats in each WGS or MF set of sequences. (A) Monocots, (B) dicots, (C) non-angiosperms. A classification of repeat content in each genome is described in Supplemental Table 3.
Polyploidy, gene duplication, and gene loss
We have estimated the gene content of a wide variety of plant genomes, as well as the size of the unmethylated gene space. Gene content varies widely reflecting extensive genomic duplication through polyploidy. Even those plants that are considered diploids may have descended from a polyploid ancestor (Leitch and Bennett 1997; Blanc et al. 2003) following extensive genome rearrangements (Song et al. 1995; Liu et al. 1998; Osborn et al. 2003). In contrast, the size of the unmethylated sub-genome is remarkably similar from species to species, despite huge differences in genome size, suggesting that methylation of repeats has played a major role in the expansion of plant genomes (Martienssen 1998), accounting in part for the C-value paradox.
Among the monocots, maize and bread wheat have the largest differences in gene content per sub-genome relative to rice; while maize has lost half of its genes, wheat has gained more than twice as many. This difference may be related to the timing of polyploidization in each species. The hexaploid bread wheat genome has three components A, B, and D, and the wild wheat Ae. tauschii is thought to have contributed the D genome. As wild wheat has a normal gene number, the pseudogene expansion in bread wheat occurred either in the AB tetraploid progenitor generated ∼0.5 Mya (Huang et al. 2002) or after the hexaploidization event that occurred only 8000 years ago (Feldman et al. 1995). Our analysis suggests that it was accompanied by a dramatic amplification of genes that were subsequently silenced by methylation. Pseudogene duplication may have involved transposable elements, as recently documented in rice (Jiang et al. 2004), consistent with gene reactivation by transposons in synthetic allopolyploids (Kashkush et al. 2003). In contrast, maize is a much more ancient polyploid in which genes that were duplicated (and potentially silenced) have been lost over evolutionary time, returning it to a diploid gene content (Ilic et al. 2003; Lai et al. 2004b; Langham et al. 2004).
Among the dicots examined, the two Brassica genomes are closely related and have also undergone gene loss following polyploidy. Oilseed rape is a recent allopolyploid accounting for the slightly larger number of genes per sub-genome. Cabbage is derived from the same ancestral hexaploid as the parents of oilseed rape, but has lost fewer genes per sub-genome, possibly because it has not undergone recent hybridization. Potato is also a recent tetraploid and has an excess of genes in each sub-genome (Table 1). Cotton is a relatively old (1.5 Myr) tetraploid (Senchina et al. 2003), but it still has a large number of genes per sub-genome, perhaps due to a slow process of gene loss consistent with a proposal that duplicated cotton genes evolve independently (Cronn et al. 1999).
Moss, fern, and pine, included in the third group, are evolutionarily very distant from the angiosperms. It is possible that sequence divergence, together with a database bias, may result in an underestimation of gene content of these species. This may be the case in fern, which shows only a slightly higher than expected number of genes in spite of most ferns being paleopolyploids (Gastony 1991). The extremely high number of pine genes is puzzling. Significant sequence data and a detailed evolutionary analysis would be required to have a more complete picture of the evolution of this and other complex plant genomes. The higher frequency of McrBC sites in MF genes versus WGS genes observed in some plant species is also intriguing (Fig. 4). Genes poor in McrBC sites found in WGS libraries could be pseudogenes, which are thought to be abundant in rice (Bennetzen et al. 2004), but more extensive sequence data will be necessary to resolve this issue.
Conclusions
Our results demonstrate that, as in maize and sorghum, the unmethylated portion of most plant genomes is very gene-rich, while a high proportion of transposons is heavily methylated. Most dicot species have an excess of genes compared with Arabidopsis and appear to have undergone one or more rounds of polyploidization, accompanied by dramatic changes in gene content. Furthermore, there is some evidence of changes in DNA methylation during polyploidization, in agreement with other studies (Lee and Chen 2001; Shaked et al. 2001; Madlung et al. 2002; Adams et al. 2003, 2004). In the monocot plants studied, there is a good correlation between GEF and genome size, with the exception of the two wheat species. The relatively low GEF observed in each case appears to be explained by different phenomena. In wild wheat, unmethylated repetitive elements are recovered in MF libraries. Due to its large genome, even a small fraction of the total repetitive content may represent a large portion of the sequences in an MF library, reducing the frequency of genes. In cases like this, other gene enrichment approaches such as HC (Yuan et al. 2003) could be attempted to achieve extensive gene representation, because a large number of unmethylated repeats could be eliminated in an HC library. However, no such wild wheat library has been sequenced to our knowledge. In bread wheat, however, a large number of methylated pseudogenes appears to be the explanation, rather than seven- to ninefold more functional genes than other grasses. In agreement with this conclusion, sequences from an HC library of bread wheat, deposited in Genbank (CL900626-CL902992 and CW991694-CW991860), have a comparable (actually slightly lower) gene density than our MF library (data not shown). The lower frequency might result from hybridization of adjacent duplicates of pseudogene sequences in the snap-back fraction, which is eliminated from these libraries. Thus, if extensive gene coverage is achieved in hexaploid wheat by a combination of MF and HC, it will be possible to determine the methylation status of the recovered genes, providing additional epigenetic information.
From a practical point of view, although incorrect genome size estimations, differences in sequence read lengths, and database biases may be sources of errors in our analysis, MF is a tool for methylation analysis of plant genomes, and a gene-enriched sequencing strategy when applied to large plant genomes.
Methods
Plant material
Bread wheat, rice, barley, soybean, potato, tomato, oilseed rape, and cotton were grown in a greenhouse and mature leaves were collected for nuclear DNA preparation. Inflorescence tissue was used for oilseed rape nuclear DNA preparation. Mature greenhouse-grown fern sporophytes (Carolina Biological Supply Company), whole moss plants, and mature pine needles were used for nuclear DNA preparations. Inbred lines and cultivars are listed in Supplemental Table 1.
Library construction and sequencing
Nuclear DNA was purified, mechanically sheared, and cloned into pUC19 using 3-nucleotide overhang adaptors as described (Rabinowicz 2003) or into pBCSK as described (Bedell et al. 2005). DNA ligation reactions were transformed into mcrBC+ E. coli strains (DH5α or JM107). WGS libraries were constructed in the same way except that they were transformed into the mcrBC–strains DH10b or JM107MA2. Clones were sequenced using Big Dye Terminator chemistry (Applied Biosystems, Inc.) and ABI 3700 sequencers. The number and average read length of the sequences are reported in Supplemental Table 1.
BLAST analysis
Sequences were first trimmed for vector and low-quality sequences. Then, all sequences <100 bp or that had a match on our organelle database (Palmer et al. 2003) at E < 10–30 were excluded from any further analysis. The remaining sequences were first compared using BLASTN to our (Palmer et al. 2003) and TIGR's (http://www.tigr.org/tdb/e2k1/plant.repeats) repeat databases, which exclude MITES and microsatellites due to their association with genes that may result in misclassification of genic sequences as repetitive. The percentage of sequencing with a repetitive match at E < 10–10 was recorded (Supplemental Table 1). Then sequences were compared using BLASTX to a protein database of known genes (Palmer et al. 2003). Any sequence matching a protein at E < 10–7 that does not match any repeat was considered genic.
McrBC sites content
McrBC cuts methylated DNA in the sequence [A/G]C, where the C is methylated, and most methylation in plants occurs in the sequences CG or CNG. So we calculated the frequency of the McrBC sites overlapping potentially methylated sequences: [A/G]CNG, [A/G]CG, CG[C/T], and CNG[C/T].
Gene content calculations
It is difficult to estimate the number of genes in an organism by sampling the genome with a few hundred or thousand sequencing reads. However, it is possible to estimate the number of genes relative to a known genome that has been completely sequenced and annotated, and which has a similar set of gene products and similar gene architecture. For example, we know that the rice genome is 400 Mbp and contains approximately 41,000 genes. By taking about a thousand random reads from rice and performing a BLAST search against a given database, we can estimate the relative “gene density” found in rice as the percent matches at a given cutoff. Similarly, we can estimate the relative “gene density” of another species by sequencing a comparable number of random reads and calculating the percent matches to the same database. The ratio of these two numbers provides a fraction (e.g., twice as dense or half as dense as rice), which can be multiplied by the total number of genes in rice (41,000), and by the relative genome size, to estimate the gene number. If the BLAST database is very carefully curated to remove repeats, this estimate will be much more accurate than if GenBank, for example, is used. Therefore, we calculated the monocots' gene numbers as follows: We considered the number of genes (G) in rice to be 41,000, as estimated from completely sequenced chromosomes and extensive annotation and analysis (Feng et al. 2002; Sasaki et al. 2002; The Rice Chromosome 10 Sequencing Consortium 2003). Let the genome size of species A be S(A), and the “gene density” (g) be the proportion of WGS reads that match genes from our curated, known plant gene set (smaller than the actual set of plant genes). This is related to the real gene density by some constant k, which is the same for all plant species. Then g(A) = k G(A)/S(A). In Species A, the number of genes can be calculated in the following way:
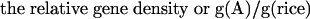 |
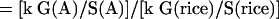 |
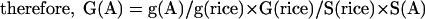 |
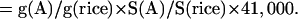 |
For example, in Table 1, the Arabidopsis genome is less than 1/3 the size of the rice genome, but is more than twice as gene dense (22% rather than 8.5%). It would therefore be expected to have 2/3 as many genes, or 27,000. This is close to the observed value (26,300). Of course, gene family architecture (number of copies per family) and other factors will not be constant between species and will impact these estimates to varying degrees, but they are useful estimates nonetheless.
In order to ensure the most uniform gene architecture and gene content in our reference genomes compared to our sample genomes, monocot sample sequences were compared with rice, while dicot sample sequences were compared with Arabidopsis. For non-angiosperms, we also used rice as the reference genome. We used published reference genome sizes and published reference gene numbers for rice and Arabidopsis, respectively, in our calculations (Table 1). Clearly, as these reference genomes are updated, the estimated gene numbers in our sample genomes will change.
The expected percent of gene-matching reads in Figure 2, A and C were calculated using the 400-Mbp genome size and the 8.4% gene matches observed in rice as the reference genome (assuming that each diploid sub-genome has the same gene number as rice, 41,000, we can calculate the “expected” level of gene enrichment for any given genome). First, the relative genome size was calculated by dividing each actual genome size by the rice genome size (400 Mbp). Then, the 8.4% gene matches (found in rice) were divided by the relative genome sizes to obtain the corresponding expected percent of gene-matching reads. Numbers in Figure 2B were calculated in the same way, but using Arabidopsis as a reference.
Supplementary Material
Acknowledgments
We thank the following researchers and institutions, who kindly provided seeds or plant tissue: Most seed stocks were kindly provided by the USDA National Plant Germplasm System. Andris Kleinhofs (Washington State University), Feridoon Mehdizadegan (Maine Seed Potato Board), Bikram Gill (Kansas State University), John Mullet (Texas A&M University), and Ben Burr (Brookhaven National Laboratory) kindly provided barley, potato, bread wheat, sorghum, and cotton seed, respectively. Allyson Schwartz (North Carolina Botanical Gardens, Chapel Hill) and Ralph Quatrano (Washington University) kindly provided pine and moss tissue, respectively. This work was supported in part by grants from the NSF Plant Genome Research Program (DBI-0110143) and USDA IFAFS (2001-52100-11331) to R.A.M. and W.R.M, and a grant from the NSF PGRP on Functional Genomics of Polyploids (DBI-0077774).
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.4100405.
Footnotes
[Supplemental material is available on line at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. CZ897387-CZ899108 and CZ904997-CZ905001 for rice; CZ885111-CZ886935 and CZ904956-CZ904957 for barley; CZ888291-CZ891417 and CZ904958-CZ904975 for bread wheat; CZ899109-CZ902001 and CZ905002-CZ905005 for soybean; CZ886936-CZ888290 for oilseed rape; CZ896505-CZ897386 for potato; CZ902002-CZ904955 and CZ905006-CZ905009 for tomato; CZ891418-CZ892414 and CZ904976-CZ904980 for cotton; CZ893553-CZ894712 for moss; CZ892415-CZ893552 and CZ904981-CZ904996 for fern; and CZ894713-CZ896504 for pine. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: USDA National Plant Germplasm System, A. Kleinhofs, F. Mehdizadegan, B. Gill, J. Mullet, B. Burr, A. Schwartz, and R. Quatrano.]
References
- Adams, M.D., Kelley, J.M., Gocayne, J.D., Dubnick, M., Polymeropoulos, M.H., Xiao, H., Merril, C.R., Wu, A., Olde, B., Moreno, R.F., et al. 1991. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 252: 1651-1656. [DOI] [PubMed] [Google Scholar]
- Adams, K.L., Cronn, R., Percifield, R., and Wendel, J.F. 2003. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. 100: 4649-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, K.L., Percifield, R., and Wendel, J. 2004. Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 168: 2217-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumuganathan, K. and Earle, E.D. 1991. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9: 208-218. [Google Scholar]
- The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815. [DOI] [PubMed] [Google Scholar]
- Banks, J.A. 1999. Gametophyte development in ferns. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50: 163-186. [DOI] [PubMed] [Google Scholar]
- Bedell, J.A., Budiman, M.A., Nunberg, A., Citek, R.W., Robbins, D., Jones, J., Flick, E., Rholfing, T., Fries, J., Bradford, K., et al. 2005. Sorghum genome sequencing by methylation filtration. PLoS Biol. 3: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen, J.L. 2000. Comparative sequence analysis of plant nuclear genomes: Microcolinearity and its many exceptions. Plant Cell 12: 1021-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen, J.L., Schrick, K., Springer, P.S., Brown, W.E., and SanMiguel, P. 1994. Active maize genes are unmodified and flanked by diverse classes of modified, highly repetitive DNA. Genome 37: 565-576. [DOI] [PubMed] [Google Scholar]
- Bennetzen, J.L., Coleman, C., Liu, R., Ma, J., and Ramakrishna, W. 2004. Consistent over-estimation of gene number in complex plant genomes. Curr. Opin. Plant Biol. 7: 732-736. [DOI] [PubMed] [Google Scholar]
- Bird, A.P. 1995. Gene number, noise reduction and biological complexity. Trends Genet. 11: 94-100. [DOI] [PubMed] [Google Scholar]
- ———. 2002. DNA methylation patterns and epigenetic memory. Genes & Dev. 16: 6-21. [DOI] [PubMed] [Google Scholar]
- Blanc, G., Hokamp, K., and Wolfe, K.H. 2003. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 13: 137-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler, V.L. and Walbot, V. 1986. DNA modification of a maize transposable element correlates with loss of activity. Proc. Natl. Acad. Sci. 83: 1767-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomet, P.S., Wessler, S., and Dellaporta, S.L. 1987. Inactivation of the maize transposable element Activator (Ac) is associated with its DNA modification. EMBO J. 6: 295-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colot, V. and Rossignol, J.L. 1999. Eukaryotic DNA methylation as an evolutionary device. Bioessays 21: 402-411. [DOI] [PubMed] [Google Scholar]
- Cribb, P.J. and Hawkes, J.G. 1986. Experimental evidence for the origin of Solanum tuberosum subspecies andigena. In Solanaceae: Biology and systematics (ed. W.G. D'Arcy), pp. 383-404. Columbia University Press, New York.
- Cronn, R.C., Small, R.L., and Wendel, J.F. 1999. Duplicated genes evolve independently after polyploid formation in cotton. Proc. Natl. Acad. Sci. 96: 14406-14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, S.H. and Bird, A.P. 1995. CpG islands and genes. Curr. Opin. Genet. Dev. 5: 309-314. [DOI] [PubMed] [Google Scholar]
- Dila, D., Sutherland, E., Moran, L., Slatko, B., and Raleigh, E.A. 1990. Genetic and sequence organization of the mcrBC locus of Escherichia coli K-12. J. Bacteriol. 172: 4888-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman, M., Lupton, F., and Miller, T. 1995. Wheats. In Evolution of crop plants (eds. J. Smartt and N. Simmonds), pp. 184-192. Longman Scientific and Technical Press, London.
- Feng, Q., Zhang, Y., Hao, P., Wang, S., Fu, G., Huang, Y., Li, Y., Zhu, J., Liu, Y., Hu, X., et al. 2002. Sequence and analysis of rice chromosome 4. Nature 420: 316-320. [DOI] [PubMed] [Google Scholar]
- Fernandes, J., Dong, Q., Schneider, B., Morrow, D.J., Nan, G.L., Brendel, V., and Walbot, V. 2004. Genome-wide mutagenesis of Zea mays L. using RescueMu transposons. Genome Biol. 5: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell, R.B. 1994. Inactivation of gene expression in plants as a consequence of specific sequence duplication. Proc. Natl. Acad. Sci. 91: 3490-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd, S.K. and Bowman, J.L. 2004. Gene regulation: Ancient microRNA target sequences in plants. Nature 428: 485-486. [DOI] [PubMed] [Google Scholar]
- Fu, Y., Hsia, A.P., Guo, L., and Schnable, P.S. 2004. Types and frequencies of sequencing errors in methyl-filtered and high c0t maize genome survey sequences. Plant Physiol. 135: 2040-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastony, G.J. 1991. Gene silencing in a polyploid homosporous fern: Paleopolyploidy revisited. Proc. Natl. Acad. Sci. 88: 1602-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut, B.S. and Doebley, J.F. 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. 94: 6809-6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff, S.A., Ricke, D., Lan, T.H., Presting, G., Wang, R., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92-100. [DOI] [PubMed] [Google Scholar]
- Greenblatt, I.M. 1984. A chromosome replication pattern deduced from pericarp phenotypes resulting from movements of the transposable element, modulator, in maize. Genetics 108: 471-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum, Y., Naveh-Many, T., Cedar, H., and Razin, A. 1981. Sequence specificity of methylation in higher plant DNA. Nature 292: 860-862. [DOI] [PubMed] [Google Scholar]
- Hake, S. and Walbot, V. 1980. The genome of Zea mays, its organization and homology to related grasses. Chromosoma 79: 251-270. [Google Scholar]
- Hanley, S., Edwards, D., Stevenson, D., Haines, S., Hegarty, M., Schuch, W., and Edwards, K.J. 2000. Identification of transposon-tagged genes by the random sequencing of Mutator-tagged DNA fragments from Zea mays. Plant J. 23: 557-566. [DOI] [PubMed] [Google Scholar]
- Huang, S., Sirikhachornkit, A., Su, X., Faris, J., Gill, B., Haselkorn, R., and Gornicki, P. 2002. Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc. Natl. Acad. Sci. 99: 8133-8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic, K., SanMiguel, P.J., and Bennetzen, J.L. 2003. A complex history of rearrangement in an orthologous region of the maize, sorghum, and rice genomes. Proc. Natl. Acad. Sci. 100: 12265-12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, N., Bao, Z., Zhang, X., Eddy, S.R., and Wessler, S.R. 2004. Pack-MULE transposable elements mediate gene evolution in plants. Nature 431: 569-573. [DOI] [PubMed] [Google Scholar]
- Kakutani, T., Munakata, K., Richards, E.J., and Hirochika, H. 1999. Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics 151: 831-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush, K., Feldman, M., and Levy, A.A. 2003. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 33: 102-106. [DOI] [PubMed] [Google Scholar]
- Kato, M., Miura, A., Bender, J., Jacobsen, S.E., and Kakutani, T. 2003. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr. Biol. 13: 421-426. [DOI] [PubMed] [Google Scholar]
- Kellogg, E.A. 2001. Evolutionary history of the grasses. Plant Physiol. 125: 1198-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan, P., Sapra, V.T., Soliman, K.M., and Zipf, A. 2001. FISH mapping of the 5S and 18S-28S rDNA loci in different species of Glycine. J. Hered. 92: 295-300. [DOI] [PubMed] [Google Scholar]
- Krutovsky, K.V., Troggio, M., Brown, G.R., Jermstad, K.D., and Neale, D.B. 2004. Comparative mapping in the Pinaceae. Genetics 168: 447-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A. and Bennetzen, J.L. 1999. Plant retrotransposons. Annu. Rev. Genet. 33: 479-532. [DOI] [PubMed] [Google Scholar]
- Lagercrantz, U. 1998. Comparative mapping between Arabidopsis thaliana and Brassica nigra indicates that Brassica genomes have evolved through extensive genome replication accompanied by chromosome fusions and frequent rearrangements. Genetics 150: 1217-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, J., Dey, N., Kim, C.S., Bharti, A.K., Rudd, S., Mayer, K.F., Larkins, B.A., Becraft, P., and Messing, J. 2004a. Characterization of the maize endosperm transcriptome and its comparison to the rice genome. Genome Res. 14: 1932-1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, J., Ma, J., Swigonova, Z., Ramakrishna, W., Linton, E., Llaca, V., Tanyolac, B., Park, Y.J., Jeong, O.Y., Bennetzen, J.L., et al. 2004b. Gene loss and movement in the maize genome. Genome Res. 14: 1924-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langham, R.J., Walsh, J., Dunn, M., Ko, C., Goff, S.A., and Freeling, M. 2004. Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 166: 935-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H.S. and Chen, Z.J. 2001. Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc. Natl. Acad. Sci. 98: 6753-6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch, I.J. and Bennett, M.D. 1997. Polyploidy in angiosperms. Trends Plant Sci. 2: 470-476. [Google Scholar]
- Li, W., Zhang, P., Fellers, J.P., Friebe, B., and Gill, B.S. 2004. Sequence composition, organization, and evolution of the core Triticeae genome. Plant J. 40: 500-511. [DOI] [PubMed] [Google Scholar]
- Lippman, Z., May, B., Yordan, C., Singer, T., and Martienssen, R. 2003. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 1: E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman, Z., Gendrel, A.V., Black, M., Vaughn, M.W., Dedhia, N., McCombie, W.R., Lavine, K., Mittal, V., May, B., Kasschau, K.D., et al. 2004. Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471-476. [DOI] [PubMed] [Google Scholar]
- Liu, B., Vega, J.M., and Feldman, M. 1998. Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. II. Changes in low-copy coding DNA sequences. Genome 41: 535-542. [DOI] [PubMed] [Google Scholar]
- Lyko, F., Ramsahoye, B.H., and Jaenisch, R. 2000. DNA methylation in Drosophila melanogaster. Nature 408: 538-540. [DOI] [PubMed] [Google Scholar]
- Madlung, A., Masuelli, R.W., Watson, B., Reynolds, S.H., Davison, J., and Comai, L. 2002. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 129: 733-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen, R. 1998. Transposons, DNA methylation and gene control. Trends Genet. 14: 263-264. [DOI] [PubMed] [Google Scholar]
- Martienssen, R.A. and Baulcombe, D.C. 1989. An unusual wheat insertion sequence (WIS1) lies upstream of an α-amylase gene in hexaploid wheat, and carries a “minisatellite” array. Mol. Gen. Genet. 217: 401-410. [DOI] [PubMed] [Google Scholar]
- Martienssen, R.A. and Colot, V. 2001. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293: 1070-1074. [DOI] [PubMed] [Google Scholar]
- Martienssen, R., Barkan, A., Taylor, W.C., and Freeling, M. 1990. Somatically heritable switches in the DNA modification of Mu transposable elements monitored with a suppressible mutant in maize. Genes & Dev. 4: 331-343. [DOI] [PubMed] [Google Scholar]
- May, B.P., Liu, H., Vollbrecht, E., Senior, L., Rabinowicz, P.D., Roh, D., Pan, X., Stein, L., Freeling, M., Alexander, D., et al. 2003. Maize-targeted mutagenesis: A knockout resource for maize. Proc. Natl. Acad. Sci. 100: 11541-11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing, J., Bharti, A.K., Karlowski, W.M., Gundlach, H., Kim, H.R., Yu, Y., Wei, F., Fuks, G., Soderlund, C.A., Mayer, K.F., et al. 2004. Sequence composition and genome organization of maize. Proc. Natl. Acad. Sci. 101: 14349-14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, P., Niedenhof, I., and ten Lohuis, M. 1994. Evidence for cytosine methylation of non-symmetrical sequences in transgenic Petunia hybrida. EMBO J. 13: 2084-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, A., Yonebayashi, S., Watanabe, K., Toyama, T., Shimada, H., and Kakutani, T. 2001. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411: 212-214. [DOI] [PubMed] [Google Scholar]
- Monk, M., Boubelik, M., and Lehnert, S. 1987. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99: 371-382. [DOI] [PubMed] [Google Scholar]
- Osborn, T.C., Butrulle, D.V., Sharpe, A.G., Pickering, K.J., Parkin, I.A., Parker, J.S., and Lydiate, D.J. 2003. Detection and effects of a homeologous reciprocal transposition in Brassica napus. Genetics 165: 1569-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, L.E., Rabinowicz, P.D., O'Shaughnessy, A.L., Balija, V.S., Nascimento, L.U., Dike, S., de la Bastide, M., Martienssen, R.A., and McCombie, W.R. 2003. Maize genome sequencing by methylation filtration. Science 302: 2115-2117. [DOI] [PubMed] [Google Scholar]
- Papa, C.M., Springer, N.M., Muszynski, M.G., Meeley, R., and Kaeppler, S.M. 2001. Maize chromomethylase Zea methyltransferase2 is required for CpNpG methylation. Plant Cell 13: 1919-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin, I.A., Sharpe, A.G., Keith, D., and Lydiate, D.J. 1995. Identification of the A and C genomes of amphidiploid Brassica nupus (oilseed rape). Genome 38: 1122-1131. [DOI] [PubMed] [Google Scholar]
- Patterson, G.I., Thorpe, C.J., and Chandler, V.L. 1993. Paramutation, an allelic interaction, is associated with a stable and heritable reduction of transcription of the maize b regulatory gene. Genetics 135: 881-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichersky, E., Soltis, D., and Soltis, P. 1990. Defective chlorophyll a/b-binding protein genes in the genome of a homosporous fern. Proc. Natl. Acad. Sci. 87: 195-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash, S. and Hinata, K. 1980. Taxonomy, cytogenetics and origin of crop Brassica, a review. Opera Bot. 55: 1-57. [Google Scholar]
- Rabinowicz, P.D. 2003. Constructing gene-enriched plant genomic libraries using methylation filtration technology. Meth. Mol. Biol. 236: 21-36. [DOI] [PubMed] [Google Scholar]
- Rabinowicz, P.D., Schutz, K., Dedhia, N., Yordan, C., Parnell, L.D., Stein, L., McCombie, W.R., and Martienssen, R.A. 1999. Differential methylation of genes and retrotransposons facilitates shotgun sequencing of the maize genome. Nat. Genet. 23: 305-308. [DOI] [PubMed] [Google Scholar]
- Rabinowicz, P.D., McCombie, W.R., and Martienssen, R.A. 2003a. Gene enrichment in plant genomic shotgun libraries. Curr. Opin. Plant Biol. 6: 150-156. [DOI] [PubMed] [Google Scholar]
- Rabinowicz, P.D., Palmer, L.E., May, B.P., Hemann, M.T., Lowe, S.W., McCombie, W.R., and Martienssen, R.A. 2003b. Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res. 13: 2658-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raizada, M.N., Nan, G.L., and Walbot, V. 2001. Somatic and germinal mobility of the RescueMu transposon in transgenic maize. Plant Cell 13: 1587-1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh, E.A. and Wilson, G. 1986. Escherichia coli K-12 restricts DNA containing 5-methylcytosine. Proc. Natl. Acad. Sci. 83: 9070-9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik, W., Dean, W., and Walter, J. 2001. Epigenetic reprogramming in mammalian development. Science 293: 1089-1093. [DOI] [PubMed] [Google Scholar]
- The Rice Chromosome 10 Sequencing Consortium. 2003. In-depth view of structure, activity, and evolution of rice chromosome 10. Science 300: 1566-1569. [DOI] [PubMed] [Google Scholar]
- Rossignol, J.L. and Faugeron, G. 1994. Gene inactivation triggered by recognition between DNA repeats. Experientia 50: 307-317. [DOI] [PubMed] [Google Scholar]
- Round, E.K., Flowers, S.K., and Richards, E.J. 1997. Arabidopsis thaliana centromere regions: Genetic map positions and repetitive DNA structure. Genome Res. 7: 1045-1053. [DOI] [PubMed] [Google Scholar]
- Sasaki, T. and Burr, B. 2000. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 3: 138-141. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., Matsumoto, T., Yamamoto, K., Sakata, K., Baba, T., Katayose, Y., Wu, J., Niimura, Y., Cheng, Z., Nagamura, Y., et al. 2002. The genome sequence and structure of rice chromosome 1. Nature 420: 312-316. [DOI] [PubMed] [Google Scholar]
- Selker, E.U. 1990. Premeiotic instability of repeated sequences in Neurospora crassa. Annu. Rev. Genet. 24: 579-613. [DOI] [PubMed] [Google Scholar]
- Senchina, D.S., Alvarez, I., Cronn, R.C., Liu, B., Rong, J., Noyes, R.D., Paterson, A.H., Wing, R.A., Wilkins, T.A., and Wendel, J.F. 2003. Rate variation among nuclear genes and the age of polyploidy in Gossypium. Mol. Biol. Evol. 20: 633-643. [DOI] [PubMed] [Google Scholar]
- Shaked, H., Kashkush, K., Ozkan, H., Feldman, M., and Levy, A.A. 2001. Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13: 1749-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker, R.C., Polzin, K., Labate, J., Specht, J., Brummer, E.C., Olson, T., Young, N., Concibido, V., Wilcox, J., Tamulonis, J.P., et al. 1996. Genome duplication in soybean (Glycine subgenus soja). Genetics 144: 329-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, T., Yordan, C., and Martienssen, R.A. 2001. Robertson's Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1). Genes & Dev. 15: 591-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, K., Lu, P., Tang, K., and Osborn, T.C. 1995. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. 92: 7719-7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, E., Coe, L., and Raleigh, E.A. 1992. McrBC: A multisubunit GTP-dependent restriction endonuclease. J. Mol. Biol. 225: 327-348. [DOI] [PubMed] [Google Scholar]
- Tada, M., Tada, T., Lefebvre, L., Barton, S.C., and Surani, M.A. 1997. Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. EMBO J. 16: 6510-6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti, S. and Pfeifer, G.P. 1995. Complete and tissue-independent methylation of CpG sites in the p53 gene: Implications for mutations in human cancers. Oncogene 10: 1493-1499. [PubMed] [Google Scholar]
- Tweedie, S., Charlton, J., Clark, V., and Bird, A. 1997. Methylation of genomes and genes at the invertebrate-vertebrate boundary. Mol. Cell. Biol. 17: 1469-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walbot, V. and Warren, C. 1990. DNA methylation in the alcohol dehydrogenase-1 gene of maize. Plant Mol. Biol. 15: 121-125. [DOI] [PubMed] [Google Scholar]
- Walsh, C.P., Chaillet, J.R., and Bestor, T.H. 1998. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20: 116-117. [DOI] [PubMed] [Google Scholar]
- Whitelaw, C.A., Barbazuk, W.B., Pertea, G., Chan, A.P., Cheung, F., Lee, Y., Zheng, L., van Heeringen, S., Karamycheva, S., Bennetzen, J.L., et al. 2003. Enrichment of gene-coding sequences in maize by genome filtration. Science 302: 2118-2120. [DOI] [PubMed] [Google Scholar]
- Yang, Y.W., Lai, K.N., Tai, P.Y., and Li, W.H. 1999. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J. Mol. Evol. 48: 597-604. [DOI] [PubMed] [Google Scholar]
- Yoder, J.A., Walsh, C.P., and Bestor, T.H. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13: 335-340. [DOI] [PubMed] [Google Scholar]
- Yu, J., Hu, S., Wang, J., Wong, G.K., Li, S., Liu, B., Deng, Y., Dai, L., Zhou, Y., Zhang, X., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79-92. [DOI] [PubMed] [Google Scholar]
- Yuan, Y., SanMiguel, P.J., and Bennetzen, J.L. 2003. High-Cot sequence analysis of the maize genome. Plant J. 34: 249-255. [DOI] [PubMed] [Google Scholar]
- Zhu, T., Schupp, J.M., Oliphant, A., and Keim, P. 1994. Hypomethylated sequences: Characterization of the duplicate soybean genome. Mol. Gen. Genet. 244: 638-645. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.tigr.org/tdb/tgi/maize/release4.0/assembly_summary.shtml; TIGR maize AZM assembly summary.
- http://www.tigr.org/tdb/e2k1/plant.repeats; TIGR plant repeat database.
- http://rgp.dna.affrc.go.jp/cgi-bin/statusdb/irgsp-status.cgi International Rice Genome Sequencing Project status.
- http://www.rbgkew.org.uk/cval/database1.html; Plant DNA C-values Database, Royal Botanic Gardens, Kew, UK.
- http://www.arabidopsis.org; The Arabidopsis Information Resource homepage.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.