

Abstract
The thermodynamic properties of aqueous nitroxyl (HNO) and its anion (NO−) have been revised to show that the ground state of NO− is triplet and that HNO in its singlet ground state has much lower acidity, pKa(1HNO/3NO−) ≈ 11.4, than previously believed. These conclusions are in accord with the observed large differences between 1HNO and 3NO− in their reactivities toward O2 and NO. Laser flash photolysis was used to generate 1HNO and 3NO− by photochemical cleavage of trioxodinitrate (Angeli's anion). The spin-allowed addition of 3O2 to 3NO− produced peroxynitrite with nearly diffusion-controlled rate (k = 2.7 × 109 M−1⋅s−1). In contrast, the spin-forbidden addition of 3O2 to 1HNO was not detected (k ≪ 3 × 105 M−1⋅s−1). Both 1HNO and 3NO− reacted sequentially with two NO to generate N3O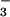 as a long-lived intermediate; the rate laws of N3O
as a long-lived intermediate; the rate laws of N3O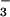 formation were linear in concentrations of NO and 1HNO (k = 5.8 × 106 M−1⋅s−1) or NO and 3NO− (k = 2.3 × 109 M−1⋅s−1). Catalysis by the hydroxide ion was observed for the reactions of 1HNO with both O2 and NO. This effect is explicable by a spin-forbidden deprotonation by OH− (k = 4.9 × 104 M−1⋅s−1) of the relatively unreactive 1HNO into the extremely reactive 3NO−. Dimerization of 1HNO to produce N2O occurred much more slowly (k = 8 × 106 M−1⋅s−1) than previously suggested. The implications of these results for evaluating the biological roles of nitroxyl are discussed.
formation were linear in concentrations of NO and 1HNO (k = 5.8 × 106 M−1⋅s−1) or NO and 3NO− (k = 2.3 × 109 M−1⋅s−1). Catalysis by the hydroxide ion was observed for the reactions of 1HNO with both O2 and NO. This effect is explicable by a spin-forbidden deprotonation by OH− (k = 4.9 × 104 M−1⋅s−1) of the relatively unreactive 1HNO into the extremely reactive 3NO−. Dimerization of 1HNO to produce N2O occurred much more slowly (k = 8 × 106 M−1⋅s−1) than previously suggested. The implications of these results for evaluating the biological roles of nitroxyl are discussed.
Nitroxyl (HNO, also known as nitrosyl hydride) and its anion, NO−, are the simplest molecules with nitrogen in the +1 oxidation state and yet their aqueous chemistry is not well understood. Recent suggestions that these redox neighbors of the biologically important NO radical may play a role in cellular metabolism (1–4) and in aerobic environments may be precursors to cytotoxic peroxynitrite, ONOO−, (5, 6) have engendered considerable interest in the chemistry of HNO/NO−. The characterization of these species is complicated by their instability with respect to formation of nitrous oxide (7, 8). In most cases where nitroxyl has been invoked as an intermediate, the rate-determining step was its generation, a situation that allows little insight into the properties and reactivities of HNO/NO− themselves. The NO− anion is isoelectronic with O2 and, like O2, should have a triplet ground state, whereas the ground state of HNO should be a singlet. Indeed, these ground state assignments have been well established for HNO/NO− in the gas phase (9, 10).
A frequently used source for aqueous HNO/NO− is trioxodinitrate (N2O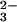 , also known as Angeli's anion), whose conjugate acid (H2N2O3) has consecutive pKa values of 2.5 and 9.7 (11). It is widely accepted (7, 8) that slow decomposition of the monoprotonated anion occurs through heterolytic N—N bond cleavage
, also known as Angeli's anion), whose conjugate acid (H2N2O3) has consecutive pKa values of 2.5 and 9.7 (11). It is widely accepted (7, 8) that slow decomposition of the monoprotonated anion occurs through heterolytic N—N bond cleavage
 |
1 |
Subsequent addition of O2 could yield peroxynitrite
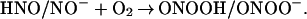 |
2 |
However, nitrate, which is the peroxynitrite decomposition product, was not detected among the end products of HN2O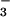 decay (12). This result was interpreted as evidence against the occurrence of reaction 2. On the other hand, the same researchers reported peroxynitrite formation during N2O
decay (12). This result was interpreted as evidence against the occurrence of reaction 2. On the other hand, the same researchers reported peroxynitrite formation during N2O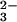 photolysis in alkaline solution (13). To reconcile the data, it was suggested that thermal reaction 1 followed by deprotonation of HNO produces singlet NO−, which is the ground state in water, and that 1NO− is unreactive toward O2. In contrast, photochemical cleavage of N2O
photolysis in alkaline solution (13). To reconcile the data, it was suggested that thermal reaction 1 followed by deprotonation of HNO produces singlet NO−, which is the ground state in water, and that 1NO− is unreactive toward O2. In contrast, photochemical cleavage of N2O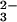 was thought to generate the long-lived triplet excited state of NO−, which reacted with O2. However, it seems unlikely that hydration can reverse a gas-phase energy gap of about 70 kJ/mol between the ground state 3NO− and the excited state 1NO− (10). Moreover, by analogy with 1O2, whose lifetime in water is only 4 μs (14), the existence of a long-lived excited state of NO− also appears highly unlikely. Thus, reaction 2 remains obscure, particularly because others have reported O2 consumption during spontaneous decomposition of HN2O
was thought to generate the long-lived triplet excited state of NO−, which reacted with O2. However, it seems unlikely that hydration can reverse a gas-phase energy gap of about 70 kJ/mol between the ground state 3NO− and the excited state 1NO− (10). Moreover, by analogy with 1O2, whose lifetime in water is only 4 μs (14), the existence of a long-lived excited state of NO− also appears highly unlikely. Thus, reaction 2 remains obscure, particularly because others have reported O2 consumption during spontaneous decomposition of HN2O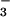 (2, 15) and detection of small amounts of NO
(2, 15) and detection of small amounts of NO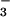 upon completion of the reaction (16).
upon completion of the reaction (16).
In the early 1970s two research groups used pulse radiolysis to produce HNO/NO− by reaction between the hydrated electrons and NO (17, 18). This approach precluded examination of HNO/NO− reactivities toward O2 because of a very rapid addition of two NO radicals to form N3O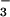 , which then slowly decomposed to nitrous oxide and nitrite
, which then slowly decomposed to nitrous oxide and nitrite
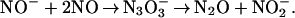 |
3 |
A pKa value of 4.7 for HNO was reported, although the spin states of NO− and HNO were not specified (17). The estimates for the reduction potential of the NO/NO− couple based on this value (19) are almost a volt higher than those obtained by electrochemical techniques (20, 21); this discrepancy has never been addressed. Recent ab initio calculations placed the pKa of HNO at 7.2 (22).
In the hope of clarifying the HNO/NO− chemistry, we have used UV laser flash photolysis to produce HNO/NO− species by photochemical cleavage of Angeli's anion and to investigate their reactivities. Here we present evidence that HNO has a much weaker acidity than previously believed. We show that the deprotonation of HNO is a slow spin-forbidden process that controls the observed chemistry in alkaline solutions. The reactivities of HNO and NO− toward O2 and NO have been investigated and a quantitative mechanistic description of these reactions is presented.
Materials and Methods
Sample Solutions.
All chemicals were of analytical grade and were used as received. Milli-Q purified water was used throughout. Stock solutions of Na2N2O3 (Cayman Chemical, Ann Arbor, MI) in 10 mM NaOH were prepared daily. Relatively stable N2O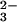 [ɛ248 = 8,300 M−1⋅cm−1 (23)] sample solutions at pH 11–14.3 were prepared by diluting the Na2N2O3 stock. Unstable HN2O
[ɛ248 = 8,300 M−1⋅cm−1 (23)] sample solutions at pH 11–14.3 were prepared by diluting the Na2N2O3 stock. Unstable HN2O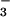 [ɛ237 = 6,100 M−1⋅cm−1 (24)] sample solutions at pH 4–10 were prepared by flow-mixing equal volumes of the Na2N2O3 stock and 0.2 M phosphate, acetate, or borate buffers as described below. Nitric oxide (Matheson) was purified by passing through a scrubbing column with 2 M KOH and then through water. The various NO/Ar and O2/Ar mixtures were produced by combining the gas streams of NO or O2 with Ar passed through calibrated flowmeters. All solutions were thoroughly purged with argon before introducing the NO-containing mixtures. The NO and O2 solubilities at 1 atm (1 atm = 101.3 kPa) were taken as 1.9 and 1.3 mM, respectively. A 100-W xenon arc beam reflected at 45o from a broadband dielectric mirror (220–300 nm) was used for UV steady-state photolysis. All experiments were performed at ambient temperature (22 ± 2°C).
[ɛ237 = 6,100 M−1⋅cm−1 (24)] sample solutions at pH 4–10 were prepared by flow-mixing equal volumes of the Na2N2O3 stock and 0.2 M phosphate, acetate, or borate buffers as described below. Nitric oxide (Matheson) was purified by passing through a scrubbing column with 2 M KOH and then through water. The various NO/Ar and O2/Ar mixtures were produced by combining the gas streams of NO or O2 with Ar passed through calibrated flowmeters. All solutions were thoroughly purged with argon before introducing the NO-containing mixtures. The NO and O2 solubilities at 1 atm (1 atm = 101.3 kPa) were taken as 1.9 and 1.3 mM, respectively. A 100-W xenon arc beam reflected at 45o from a broadband dielectric mirror (220–300 nm) was used for UV steady-state photolysis. All experiments were performed at ambient temperature (22 ± 2°C).
Laser Flash Photolysis with Flow Premixing.
Transient absorption spectra were recorded by using a computerized kinetic spectrometer system as described (25). Briefly, a 266-nm Nd:YAG laser operating at 20 Hz was used to photolyse the samples in a 0.4 × 1-cm quartz flow cell. To allow for solution replacement between the laser shots and monitoring slower kinetics, the sample excitation frequency was reduced to either 1 or 0.1 Hz by using a computer-controlled electromechanical shutter. Two solutions (e.g., alkaline N2O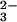 and buffer), each saturated with a desired gas mixture, were forced by a positive gas pressure into a 12-jet tangential mixer and then through the flow cell with a flow rate of ≈12 ml/min; the laser excitation occurred within less than 1 s after mixing. The transient absorption was probed along a 1-cm optical path by a light beam from a 75-W xenon arc lamp, which was pulsed for short time scales, and the kinetic traces obtained were averaged over 10–20 laser pulses. For the quantum yield estimates, the laser pulse energies (30–40 mJ/cm2) were measured by a calibrated thermoelectric bolometer.
and buffer), each saturated with a desired gas mixture, were forced by a positive gas pressure into a 12-jet tangential mixer and then through the flow cell with a flow rate of ≈12 ml/min; the laser excitation occurred within less than 1 s after mixing. The transient absorption was probed along a 1-cm optical path by a light beam from a 75-W xenon arc lamp, which was pulsed for short time scales, and the kinetic traces obtained were averaged over 10–20 laser pulses. For the quantum yield estimates, the laser pulse energies (30–40 mJ/cm2) were measured by a calibrated thermoelectric bolometer.
Results and Discussion
Energetics and Spin States.
To facilitate the analysis of the kinetic data, we begin with a thermodynamic description of the intermediates that can be generated by photolysis of the trioxodinitrate solutions. Fig. 1 presents the free energies of formation, ΔfG0, for the NO/HNO/NO− species in aqueous solution, estimated by using published data. The accurate value for ΔfG0(NOaq) = 102 kJ/mol has been derived (19). The enthalpy of 1HNO formation in the gas phase has been reviewed and the value of 107 kJ/mol has been recommended (26). From this value and the tabulated entropy S0(1HNOgas) = 220.7 J/(mol·K) (27), we calculate ΔfG0(1HNOgas) = 120 kJ/mol. The free energy of HNO hydration is unknown, but is expected to be small by analogy with neutral molecules of similar dimensions and compositions, e.g., HCN and H2CO, for which ΔhydrG0 are approximately −5 and −1.7 kJ/mol, respectively. The value ΔfG0(1HNOaq) ≈115 kJ/mol is, therefore, a reasonable estimate, which is also close to a 109 kJ/mol value derived previously (19).
Figure 1.

Energy diagram for NO/HNO/NO− species in aqueous solution at 298 K and 1 mol/kg standard states. Note the energy axis break at 120 kJ/mol. The spectroscopic designations for the electronic states are given in parenthesis. Only the lowest excited states are shown for HNO and NO−.
The reduction potential of NO measured by the photoelectrochemical technique has been reported as E0(NO/NO−) = −0.81 V vs. NHE without specifying the NO− spin state (21). This value is consistent with the upper limit E0(NO/NO−) < −0.7 V vs. NHE that can be inferred from the onset of the irreversible NO reduction wave observed in controlled-potential coulometry (20). In both experiments the reducing electrons were supplied by the metal electrodes, a process for which there is no spin prohibition regardless of the product spin state. Assuming, therefore, that these measurements pertain to the NO reduction in the ground 3NO− state, ΔfG0(3NO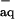 ) ≈180 kJ/mol can be estimated. Finally, the values for ΔfG0(3HNOaq) ≈190 kJ/mol and for ΔfG0(1NO
) ≈180 kJ/mol can be estimated. Finally, the values for ΔfG0(3HNOaq) ≈190 kJ/mol and for ΔfG0(1NO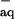 ) ≈248 kJ/mol can be assigned under a reasonable presumption that hydration does not appreciably alter the gas-phase energy gaps of 75 kJ/mol between 3HNO and 1HNO (9) and 68 kJ/mol between 1NO− and 3NO− (10).
) ≈248 kJ/mol can be assigned under a reasonable presumption that hydration does not appreciably alter the gas-phase energy gaps of 75 kJ/mol between 3HNO and 1HNO (9) and 68 kJ/mol between 1NO− and 3NO− (10).
From the energy diagram, we estimate pKa(1HNO/3NO−) ≈11.4 and pKa(1HNO/1NO−) ≈23, i.e., the acidity of HNO is very low. This result attests to the closer chemical similarity between HNO and an aldehyde than between HNO and an oxyacid. In its triplet state, HNO becomes a strong acid, pKa(3HNO/3NO−) ≈−1.8.
Peroxynitrite Formation upon Addition of O2.
Alkaline solutions.
In strongly alkaline solutions trioxodinitrate is completely deprotonated and stable for hours. Steady-state UV photolysis of N2O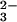 in air-equilibrated solutions resulted in the rapid disappearance of the N2O
in air-equilibrated solutions resulted in the rapid disappearance of the N2O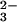 absorption band at 248 nm and in the simultaneous appearance of characteristic absorption of the peroxynitrite anion (ONOO−) around 300 nm and nitrite at λ < 230 nm (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). This result is in qualitative accord with the previous communication (13). The same spectral changes were observed in O2-saturated solutions. The presence of two well-defined isosbestic points at 225 and 283 nm indicated the constancy of reaction stoichiometry during the photolysis. From the spectral changes, a chemical yield of peroxynitrite (defined as −Δ[ONOO−]/Δ[N2O
absorption band at 248 nm and in the simultaneous appearance of characteristic absorption of the peroxynitrite anion (ONOO−) around 300 nm and nitrite at λ < 230 nm (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). This result is in qualitative accord with the previous communication (13). The same spectral changes were observed in O2-saturated solutions. The presence of two well-defined isosbestic points at 225 and 283 nm indicated the constancy of reaction stoichiometry during the photolysis. From the spectral changes, a chemical yield of peroxynitrite (defined as −Δ[ONOO−]/Δ[N2O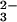 ]) of 0.95 was calculated, i.e., within the uncertainties in molar absorptivities used for this estimate, the stoichiometry of photochemical reaction was
]) of 0.95 was calculated, i.e., within the uncertainties in molar absorptivities used for this estimate, the stoichiometry of photochemical reaction was
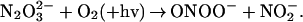 |
4 |
Purging O2 from the solution did not alter the rate of N2O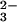 decay, but completely suppressed ONOO− formation. These observations suggest that ONOO− is produced by the reaction of O2 with the nascent products of the N2O
decay, but completely suppressed ONOO− formation. These observations suggest that ONOO− is produced by the reaction of O2 with the nascent products of the N2O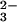 photochemical decomposition, rather than by direct scavenging of the N2O
photochemical decomposition, rather than by direct scavenging of the N2O excited state by O2.
excited state by O2.
This conclusion is in accord with the kinetics of peroxynitrite formation, which was monitored by flash photolysis of N2O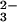 in O2-saturated solutions. The prompt bleaching of the N2O
in O2-saturated solutions. The prompt bleaching of the N2O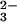 spectrum at λ < 300 nm induced by the laser flash was followed by the absorption growth around 300 nm, corresponding to ONOO− formation. At pH ≥12, the rise of absorbance conformed well to a single exponential growth, ΔAt = ΔA∞{1 − exp(−kformt)}. The values of kform, determined from kinetic traces recorded at different pH values, sharply increased with the solution alkalinity as shown in Fig. 2. At pH 12.5 and above, kform was essentially proportional to [OH−]. At the same time, the peroxynitrite signal amplitude at the end of the kinetic run (ΔA∞) showed a sigmoidal dependence on pH (Fig. 2). A salient kinetic feature of the ONOO− formation is that, at a fixed pH, its rate constant was independent of oxygen concentration throughout the experimentally accessible range (Fig. 7, which is published as supporting information on the PNAS web site). Thus, the rate-determining step of this process is the reaction between the N2O
spectrum at λ < 300 nm induced by the laser flash was followed by the absorption growth around 300 nm, corresponding to ONOO− formation. At pH ≥12, the rise of absorbance conformed well to a single exponential growth, ΔAt = ΔA∞{1 − exp(−kformt)}. The values of kform, determined from kinetic traces recorded at different pH values, sharply increased with the solution alkalinity as shown in Fig. 2. At pH 12.5 and above, kform was essentially proportional to [OH−]. At the same time, the peroxynitrite signal amplitude at the end of the kinetic run (ΔA∞) showed a sigmoidal dependence on pH (Fig. 2). A salient kinetic feature of the ONOO− formation is that, at a fixed pH, its rate constant was independent of oxygen concentration throughout the experimentally accessible range (Fig. 7, which is published as supporting information on the PNAS web site). Thus, the rate-determining step of this process is the reaction between the N2O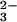 photolysis product and OH −; the reaction with O2 occurs in a step that is not rate limiting.
photolysis product and OH −; the reaction with O2 occurs in a step that is not rate limiting.
Figure 2.

Dependencies on the solution pH of the peroxynitrite formation rate constant (kform, ○) and the normalized absorbance amplitude recorded at 315 nm after completion of the reaction (□). Conditions were: 0.15 mM N2O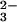 in O2-saturated solution; pH ≥ 11 were maintained with NaOH, pH < 11 with 0.1 M borate. The solid lines show theoretical dependencies with k7 = 4.9 × 104 M−1⋅s−1, k9 = 8 × 106 M−1⋅s−1, and [1HNO]0 = 0.05 mM, calculated as described in the text and Note 1.
in O2-saturated solution; pH ≥ 11 were maintained with NaOH, pH < 11 with 0.1 M borate. The solid lines show theoretical dependencies with k7 = 4.9 × 104 M−1⋅s−1, k9 = 8 × 106 M−1⋅s−1, and [1HNO]0 = 0.05 mM, calculated as described in the text and Note 1.
Collectively, the results described are consistent with the reaction sequence that begins with heterolytic photochemical cleavage of N2O , producing NO− in its singlet state
, producing NO− in its singlet state
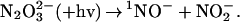 |
5 |
As described above, 1NO− is an extremely strong base (pKa ≈23) and, therefore, should be protonated by water nearly instantaneously
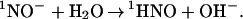 |
6 |
In fact, the lifetime of 1NO− is expected to be so short that not even its diffusion-controlled reactions with O2 should be able to compete with reaction 6. Here we suggest that the rate-determining step in the peroxynitrite formation is the deprotonation of 1HNO to 3NO−, which is a much weaker base (pKa ≈11.4) than 1NO−
 |
7 |
Completing the process is the rapid, spin-allowed addition reaction
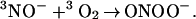 |
8 |
This mechanism accounts for the major reactivity features. Specifically, it predicts the independence of kform of [O2], as well as the linear dependence of kform on pH. The deviation from linearity of the latter and the signal amplitude decrease in solutions with pH <13 (Fig. 2) suggest the existence of an additional channel for 1HNO disappearance, which competes with reaction 7. We could not satisfactorily account for these effects by introducing a first-order channel for the 1HNO decay. Moreover, a falloff in the ONOO− yield with decreasing pH analogous to that shown in Fig. 2 was not observed under steady-state photolysis, where the transitory concentration of 1HNO is much smaller than in the flash photolysis experiments. We therefore conclude that the reaction competing with reaction 7 is a bimolecular recombination of 1HNO
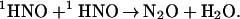 |
9 |
This reaction has been invariably invoked previously to explain the stoichiometry and the isotopic composition of end products of the spontaneous decay of Angeli's salt (7, 8, 28). The competition between reactions 7 and 9 is readily amenable to analytical solution, which gives dependencies of the relative amplitude and the ONOO− formation rate constant upon k7[OH−] and k9[1HNO]0 (see Note 1, which is published as supporting information on the PNAS web site). The initial concentration [1HNO]0 ≈0.05 mM was determined from the peroxynitrite signal amplitudes around pH 14, where it was assumed that [1HNO]0 = [ONOO−]∞. Fitting the theoretical dependencies to the data in Fig. 2 yielded k7 = (4.9 ± 0.5) × 104 M−1⋅s−1 and k9 = (8 ± 3) × 106 M−1⋅s−1. The value of k7 closely corresponds to the slope of the kform vs. pH curve at pH >13 in Fig. 2 and is rather insensitive to the value of k9. In contrast, the position of the inflection point in the calculated relative amplitude curve is sensitive to the magnitude of k9; this, together with the significant scatter of the data points, results in the relatively large uncertainty in the derived value for k9.
Our suggestion of 1HNO deprotonation (reaction 7) as the rate-determining step of ONOO− formation is further supported by the observation of the H/D isotope effect on the reaction rate. The ONOO− formation rate constants were measured in H2O and D2O under conditions of Fig. 2 at 0.25 M alkali adjusted with NaOH and NaOD, respectively. At this alkalinity, reaction 7 dominates reaction 9 and kform ≈ k7[OH−]. The ratio kform(H2O)/kform(D2O) = 3.0 ± 0.1 was obtained, which is consistent with proton involvement in the rate-determining step.
The quantum yield of ONOO− upon 266-nm laser pulse irradiation of N2O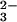 in O2-saturated 0.25 M NaOH was estimated to be ≈0.5. The stoichiometry of peroxynitrite formation given by reaction 4 shows that there are no channels for the N2O
in O2-saturated 0.25 M NaOH was estimated to be ≈0.5. The stoichiometry of peroxynitrite formation given by reaction 4 shows that there are no channels for the N2O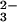 photochemical decomposition other than reaction 5. Thus, the quantum yield of the N2O
photochemical decomposition other than reaction 5. Thus, the quantum yield of the N2O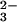 photochemical cleavage is also close to 0.5.
photochemical cleavage is also close to 0.5.
Neutral solutions.
In neutral solutions, the HN2O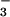 monoanion is the predominant trioxodinitrate species. The transient absorption spectra recorded after laser flash photolysis of O2-saturated HN2O
monoanion is the predominant trioxodinitrate species. The transient absorption spectra recorded after laser flash photolysis of O2-saturated HN2O solution showed a characteristic peroxynitrite band around 300 nm (Fig. 3). This absorption decayed in seconds, which is consistent with peroxynitrite lifetime at pH 7. To further verify that the 300-nm band in Fig. 3 belongs to peroxynitrite, we resorted to a highly specific test based on the rapid decay of peroxynitrite in bicarbonate solutions. At 20 mM HCO
solution showed a characteristic peroxynitrite band around 300 nm (Fig. 3). This absorption decayed in seconds, which is consistent with peroxynitrite lifetime at pH 7. To further verify that the 300-nm band in Fig. 3 belongs to peroxynitrite, we resorted to a highly specific test based on the rapid decay of peroxynitrite in bicarbonate solutions. At 20 mM HCO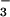 and pH 7, the half-life of the 300-nm band decreased to 20 ms, which corresponds to the half-life of authentic peroxynitrite under these conditions (29).
and pH 7, the half-life of the 300-nm band decreased to 20 ms, which corresponds to the half-life of authentic peroxynitrite under these conditions (29).
Figure 3.

Transient absorption spectra measured at the indicated delay times after laser pulse excitation of 0.6 mM HN2O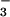 at pH 7 (0.1 M phosphate). Open symbols: O2-saturated solution; closed symbols: Ar-saturated solution. The solid line is calculated by using Eq. 13 with y = 0.25 and ΔC = 0.052 mM; the dashed lines are visual aids only. (Inset) Kinetic traces recorded at 315 nm for O2- and Ar-saturated solutions; the short negative-going signal at the time origin is caused by laser stray light.
at pH 7 (0.1 M phosphate). Open symbols: O2-saturated solution; closed symbols: Ar-saturated solution. The solid line is calculated by using Eq. 13 with y = 0.25 and ΔC = 0.052 mM; the dashed lines are visual aids only. (Inset) Kinetic traces recorded at 315 nm for O2- and Ar-saturated solutions; the short negative-going signal at the time origin is caused by laser stray light.
The kinetics of peroxynitrite formation in nearly neutral solutions differed from those in alkali in two major ways. First, the formation rate constant (kform) did not depend on pH in the 6–9 range and was orders of magnitude greater than at alkalinities above pH 11. Second, kform linearly depended on the O2 concentration (Fig. 7), suggesting that the rate-determining step of peroxynitrite formation in neutral solutions involves a direct reaction between a nascent product of the HN2O photolysis and O2. Apparently, a mechanism given by reactions 5–8 cannot account for the observations when the light-absorbing species is HN2O
photolysis and O2. Apparently, a mechanism given by reactions 5–8 cannot account for the observations when the light-absorbing species is HN2O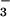 .
.
To explain the data, we suggest two channels of the heterolytic photochemical cleavage of HN2O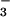 , one of which results in the formation of HNO in its triplet state, i.e., the reactions
, one of which results in the formation of HNO in its triplet state, i.e., the reactions
 |
10 |
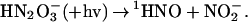 |
11 |
The protonation of N2O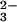 reduces the N—N bond order from 2 to 1, because an H atom in HN2O
reduces the N—N bond order from 2 to 1, because an H atom in HN2O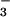 resides on a nitrogen atom (7). This difference in the N—N bonding may conceivably modulate the primary photochemical cleavage pathways. Branching into multiple pathways has been well documented for the photochemical decompositions of oxyanions; moreover, many of them very rapidly expel 3O as a result of the formally spin-forbidden heterolytic bond cleavage (30–32). It is also possible that reaction 10 is, in fact, spin-allowed. The triplet state of NO
resides on a nitrogen atom (7). This difference in the N—N bonding may conceivably modulate the primary photochemical cleavage pathways. Branching into multiple pathways has been well documented for the photochemical decompositions of oxyanions; moreover, many of them very rapidly expel 3O as a result of the formally spin-forbidden heterolytic bond cleavage (30–32). It is also possible that reaction 10 is, in fact, spin-allowed. The triplet state of NO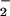 is only 2.3 eV above the ground state (33) and the 266-nm photon energy (4.7 eV) is sufficient to produce 3HNO and 3NO
is only 2.3 eV above the ground state (33) and the 266-nm photon energy (4.7 eV) is sufficient to produce 3HNO and 3NO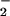 . As we argued previously, 3HNO is a strong acid and, therefore, should dissociate very rapidly in a buffered solution
. As we argued previously, 3HNO is a strong acid and, therefore, should dissociate very rapidly in a buffered solution
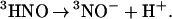 |
12 |
Peroxynitrite formation via reaction 8, which becomes the rate-limiting step, then follows. It should be noted that any alternative chain of events that leads to 3NO− within a few nanoseconds upon laser light absorption will explain the data.
The kinetics of absorbance increase at 315 nm in O2-saturated solution consisted of a fast rise, which could not be time resolved, and a slower growth with first-order rate constant kform (Fig. 3 Inset). The transient spectrum at the end of the fast rise (0.05 μs in Fig. 3) could be assigned to the superposition of absorption by 3NO− and bleaching of HN2O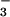 apparent below 280 nm. Indeed, very similar spectra appeared on this time scale upon flash photolysis of Ar-saturated HN2O
apparent below 280 nm. Indeed, very similar spectra appeared on this time scale upon flash photolysis of Ar-saturated HN2O solutions (Fig. 3), where no peroxynitrite was formed. The slower absorption growth at 315 nm corresponded to reaction 8; from the linear dependence of kform on [O2] the rate constant k8 = (2.7 ± 0.2) × 109 M−1⋅s−1 was obtained (see Fig. 7).
solutions (Fig. 3), where no peroxynitrite was formed. The slower absorption growth at 315 nm corresponded to reaction 8; from the linear dependence of kform on [O2] the rate constant k8 = (2.7 ± 0.2) × 109 M−1⋅s−1 was obtained (see Fig. 7).
The transient absorbance amplitude at 315 nm upon the completion of peroxynitrite formation was independent of oxygen concentration in the 0.3 to 1.3 mM range, which indicates that 3NO− was quantitatively scavenged by O2. The spectrum at the end of peroxynitrite accumulation can be described by the equation
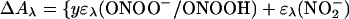 |
13 |
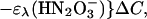 |
where ΔC is the concentration of HN2O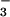 decomposed by the laser flash and y = ([ONOO−] + [ONOOH])/ΔC is the stoichiometric yield of peroxynitrite. The effective peroxynitrite spectrum, ɛλ(ONOO−/ONOOH), was constructed from the spectra of ONOO− and ONOOH (pKa = 6.6) according to their contribution at pH 7. The peroxynitrite yield y ≈0.25 was estimated by fitting this equation to the data in Fig. 3. Thus, the yield of 3NO− is also about 0.25 and the yield of 1HNO should be close to 0.75. The estimated quantum yield of peroxynitrite (≈0.2) is nearly the same as its stoichiometric yield.
decomposed by the laser flash and y = ([ONOO−] + [ONOOH])/ΔC is the stoichiometric yield of peroxynitrite. The effective peroxynitrite spectrum, ɛλ(ONOO−/ONOOH), was constructed from the spectra of ONOO− and ONOOH (pKa = 6.6) according to their contribution at pH 7. The peroxynitrite yield y ≈0.25 was estimated by fitting this equation to the data in Fig. 3. Thus, the yield of 3NO− is also about 0.25 and the yield of 1HNO should be close to 0.75. The estimated quantum yield of peroxynitrite (≈0.2) is nearly the same as its stoichiometric yield.
Formation of N3O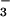 by Reactions of HNO/NO− with NO.
by Reactions of HNO/NO− with NO.
Neutral solutions.
In neutral solutions saturated with NO, the prompt bleaching of HN2O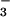 below 280 nm was followed by the growth of an absorption band at 380 nm (Fig. 4), which subsequently decayed with a 2.5-ms half-life (see Fig. 8, which is published as supporting information on the PNAS web site). Both the absorption spectrum and the lifetime were characteristic of the N3O
below 280 nm was followed by the growth of an absorption band at 380 nm (Fig. 4), which subsequently decayed with a 2.5-ms half-life (see Fig. 8, which is published as supporting information on the PNAS web site). Both the absorption spectrum and the lifetime were characteristic of the N3O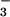 anion reported previously (17, 18, 34); reaction 3 was suggested for the N3O
anion reported previously (17, 18, 34); reaction 3 was suggested for the N3O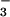 formation and decay in these reports.
formation and decay in these reports.
Figure 4.

Transient absorption spectra recorded at the indicated delay times after flash photolysis of 0.3 mM HN2O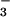 in NO-saturated solution at pH 7 (0.1 M phosphate). (Inset) Kinetic trace recorded at 380 nm and showing formation of N3O
in NO-saturated solution at pH 7 (0.1 M phosphate). (Inset) Kinetic trace recorded at 380 nm and showing formation of N3O ; note the time axis scale change.
; note the time axis scale change.
The kinetics of N3O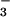 formation consisted of two exponential components well separated in time (Fig. 4 Inset). The pseudofirst-order rate constants for both components (kfast and kslow) increased linearly with NO concentration. We have attributed the fast process to the production of N3O
formation consisted of two exponential components well separated in time (Fig. 4 Inset). The pseudofirst-order rate constants for both components (kfast and kslow) increased linearly with NO concentration. We have attributed the fast process to the production of N3O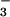 by the rapid sequential additions of two NO radicals to the 3NO− anion generated in reactions 10 and 12, i.e., the reactions
by the rapid sequential additions of two NO radicals to the 3NO− anion generated in reactions 10 and 12, i.e., the reactions
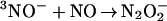 |
14 |
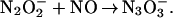 |
15 |
The effective bimolecular rate constant of (2.3 ± 0.2) × 109 M−1⋅s−1, which approximately corresponds to the slower of these reactions, was determined from the slope of the kfast vs. [NO] plot (see Fig. 9, which is published as supporting information on the PNAS web site). The relative contributions of the fast and slow pathways to the N3O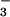 formation measured from the amplitudes of these components (Fig. 4 Inset) were about 25% and 75%, respectively. These values are in agreement with the yields of 3NO− and 1HNO estimated above for the reaction sequence 10–12 from the data on peroxynitrite formation. We have, therefore, concluded that the rate-determining reaction in the slow process of the N3O
formation measured from the amplitudes of these components (Fig. 4 Inset) were about 25% and 75%, respectively. These values are in agreement with the yields of 3NO− and 1HNO estimated above for the reaction sequence 10–12 from the data on peroxynitrite formation. We have, therefore, concluded that the rate-determining reaction in the slow process of the N3O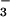 formation is the addition of NO to 1HNO
formation is the addition of NO to 1HNO
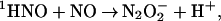 |
16 |
which is followed by rapid reaction 15. The rate constant k16 = (5.8 ± 0.4) × 106 M−1⋅s−1 was obtained from the kslow vs [NO] dependence (see Fig. 9). At a constant concentration of NO, kslow was independent of pH in the 4 to 10 range (Fig. 5 Inset).
Figure 5.

Dependence of the N3O formation rate constant (kapp) on the solution pH (adjusted with NaOH at pH ≥ 12 and 0.1 M borate at pH 10). (Inset) pH independence of the slow step rate constant (kslow) for N3O
formation rate constant (kapp) on the solution pH (adjusted with NaOH at pH ≥ 12 and 0.1 M borate at pH 10). (Inset) pH independence of the slow step rate constant (kslow) for N3O formation in buffered solutions. Both solid lines correspond to Eq. 17 with a = 1.1 × 104 s−1 and b = 5.3 × 104 M−1⋅s−1. All data are for NO-saturated solutions.
formation in buffered solutions. Both solid lines correspond to Eq. 17 with a = 1.1 × 104 s−1 and b = 5.3 × 104 M−1⋅s−1. All data are for NO-saturated solutions.
Alkaline solutions.
In alkaline solutions, the fast process of N3O formation was not observed and only the slow N3O
formation was not observed and only the slow N3O accumulation occurred as a single exponential step. Both the yield of N3O
accumulation occurred as a single exponential step. Both the yield of N3O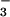 and the rate of its subsequent decay were pH-independent above pH 10 and remained the same as at pH 7. In contrast, the apparent pseudo-first-order rate constant for N3O
and the rate of its subsequent decay were pH-independent above pH 10 and remained the same as at pH 7. In contrast, the apparent pseudo-first-order rate constant for N3O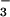 formation (kapp) steeply increased with alkalinity (Fig. 5) in a manner similar to that shown in Fig. 2 for ONOO− formation. The pH dependence of kapp conformed to the expression
formation (kapp) steeply increased with alkalinity (Fig. 5) in a manner similar to that shown in Fig. 2 for ONOO− formation. The pH dependence of kapp conformed to the expression
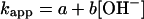 |
17 |
with a = (1.1 ± 0.1) × 104 s−1 and b = (5.3 ± 0.5) × 104 M−1⋅s−1. The second term describes the effect of alkalinity and can be attributed to the generation of 3NO− by deprotonation of 1HNO (reaction 7). Indeed, within the experimental accuracy, the b value is identical to the k7 value determined above from the peroxynitrite formation experiments. The formation of 3NO− is followed by very rapid reactions 14 and 15, leading to N3O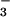 . Likewise, the term a corresponds to reaction 16, i.e., k16[NO] = a in the NO-saturated solutions. Under these conditions, side reaction 9 is too slow to play a role. Thus, the flash photolysis data for both O2- and NO-containing solutions could be accounted for on the basis of the common reactions 5–7 and 10–12.
. Likewise, the term a corresponds to reaction 16, i.e., k16[NO] = a in the NO-saturated solutions. Under these conditions, side reaction 9 is too slow to play a role. Thus, the flash photolysis data for both O2- and NO-containing solutions could be accounted for on the basis of the common reactions 5–7 and 10–12.
Mechanistic Considerations.
The mechanistic information obtained in this work is summarized in Scheme S1, where reactions are numbered as in the text. Their common feature is that at least one of the reaction partners is an uncharged species; consequently, the effects of medium ionic strength on the rate constants reported here are expected to be small. The interconversion between 3NO− and 1HNO (reaction 7) is of special interest because it gives a unique example of a spin-forbidden (nonadiabatic) proton transfer between a pair of small molecules. The equilibrium constant K7 = 400 M−1 can be calculated from pKa(1HNO/3NO−) = 11.4. Using the experimental value for k7 = 4.9 × 104 M−1⋅s−1, we estimate the rate constant for the 3NO− decay through the spin-forbidden protonation by water
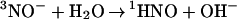 |
-7 |
to be k−7 = 1.2 × 102 s−1, i.e., the 3NO− lifetime in water is several milliseconds. This long lifetime allows the 3NO− anion to participate in reactions with O2 and NO. Without the spin prohibition, anions with similar basicity equilibrate with water much more rapidly. Recently, the spin-forbidden protonation of 3NO− has been directly observed in the gas phase, albeit for much stronger acids than water (35). The independent estimates for the upper limit of pKa(1HNO/3NO−) < 14.3 and for the lower limit of E0(NO/3NO−) > −1 V, which are in agreement with the values in Fig. 1, can be made by using our kinetic data (see Note 2, which is published as supporting information on the PNAS web site).
Scheme 1.

Another potentially important spin-forbidden reaction could lead to peroxynitrite
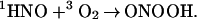 |
18 |
Although this reaction should be exoergonic by 100 kJ/mol, we found no evidence for its occurrence, which allowed an estimate of the upper limit for k18. From the relative amplitudes in Fig. 2 it follows that k18[O2] ≪ k9[1HNO]0 and, therefore, k18 ≪ 3 × 105 M−1 ⋅s−1. A more plausible mode of oxygen reactivity is hydrogen atom transfer
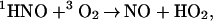 |
19 |
which is not only energetically favorable by 25 kJ/mol, but is also spin-allowed. Both reactions 9 and 19 have been studied in the gas phase and the ratio (k9/k19)gas ≈2 × 105 at 298 K can be calculated from the published data (36). If the ratio were the same in water, reaction 9 would always dominate under our experimental conditions, making reaction 19 undetectable. For more on water vs. gas rate comparison see Note 3, which is published as supporting information on the PNAS web site.
The mechanistic interpretation of the experimental data based on Fig. 1 and shown in Scheme S1 provides a novel view on the interplay between the spin states and reactivities in the HNO/NO− system. At the same time, it challenges the interpretation of the early pulse radiolysis work (17, 18). The disagreements pertain to pKa of HNO, to the existence of rapid equilibrium N2O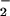 ⇋ NO + NO−, and to the rate of N3O
⇋ NO + NO−, and to the rate of N3O formation from NO− in NO-saturated solutions. It appears that reexamination of the related experiments in the literature is required to resolve these matters. Similarly, the pKa(1HNO/3NO−) value of 7.2 derived from the quantum mechanical calculations (22) is incompatible with our data, suggesting that further computational refinements may be necessary. Finally, a value that is almost 1,000 times greater than determined in present work was suggested for k9 (28). However, this value is based largely on the early pulse radiolysis results that we are now questioning.
formation from NO− in NO-saturated solutions. It appears that reexamination of the related experiments in the literature is required to resolve these matters. Similarly, the pKa(1HNO/3NO−) value of 7.2 derived from the quantum mechanical calculations (22) is incompatible with our data, suggesting that further computational refinements may be necessary. Finally, a value that is almost 1,000 times greater than determined in present work was suggested for k9 (28). However, this value is based largely on the early pulse radiolysis results that we are now questioning.
Biological Implications.
If nitrogen(+1) is indeed produced in biological systems, it must be present in the form of 1HNO at physiological pH. As a small neutral molecule, 1HNO should be able to freely traverse biological membranes, a property that can make a physiological role of 1HNO very significant. We estimate the reduction potentials E0(NO, H+/1HNO) ≈ −0.14 V and E0(1HNO, 2H+/NH2OH) ≈ 0.7 V; the corresponding values for pH 7 are −0.55 V and 0.3 V, all vs. NHE. With these potentials, 1HNO can engage in redox reactions with a number of diverse biological reductants and oxidants. However, in NO-rich environments, consecutive addition of NO to form N3O (reactions 16 and 15) can be envisioned as the major sink for 1HNO. In this respect the spontaneous decomposition of Angeli's salt provides a convenient method to generate 1HNO in the absence of NO. At the same time, our results underscore the importance of using freshly prepared alkaline stocks of Angeli's salt to avoid contamination with peroxynitrite through reactions 7 and 8 upon aging of the stock.
(reactions 16 and 15) can be envisioned as the major sink for 1HNO. In this respect the spontaneous decomposition of Angeli's salt provides a convenient method to generate 1HNO in the absence of NO. At the same time, our results underscore the importance of using freshly prepared alkaline stocks of Angeli's salt to avoid contamination with peroxynitrite through reactions 7 and 8 upon aging of the stock.
Can peroxynitrite be produced through intermediacy of 3NO− and 1HNO in oxygenated tissues? Although our results do not rule out this possibility, they impose limitations on the reaction conditions that may lead to peroxynitrite. We have shown that addition of O2 to 3NO− is essentially diffusion controlled and gives peroxynitrite. However, generation of 3NO− requires a very strong reductant, which is unlikely to survive other reactions in biological milieus, unless embedded in a hydrophobic environment with limited access to dissolved species. Such environments may include hydrophobic pockets in proteins and biological membranes. It is much more probable that biological systems can generate 1HNO rather than 3NO− because it requires only a moderate reductant. Direct addition of O2 to 1HNO that could yield peroxynitrite is spin-forbidden and, as we have shown, cannot be rapid, if it occurs at all. It appears that the possibility of peroxynitrite formation depends greatly on the magnitude of the rate constant for this reaction, which remains unknown. In this regard, coordination to paramagnetic metal ions (including those in proteins) could conceivably facilitate formally spin-forbidden peroxynitrite formation from 1HNO and O2.
Supplementary Material
Acknowledgments
We thank Drs. Merenyi and Poskrebyshev for insightful discussions. Work at New York University was supported by a grant from the Kresge Foundation. Research at Brookhaven National Laboratory was carried out under the auspices of the U.S. Department of Energy under Contract DE-AC02-98CH10886 from the Division of Chemical Sciences, Office of Basic Energy Sciences.
References
- 1.Murphy M E, Sies H. Proc Natl Acad Sci USA. 1991;88:10860–10864. doi: 10.1073/pnas.88.23.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukuto J M, Hobbs A J, Ignarro L J. Biochem Biophys Res Commun. 1993;196:707–713. doi: 10.1006/bbrc.1993.2307. [DOI] [PubMed] [Google Scholar]
- 3.Hobbs A J, Fukuto J M, Ignarro L J. Proc Natl Acad Sci USA. 1994;91:10992–10996. doi: 10.1073/pnas.91.23.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt H H, Hofmann H, Schindler U, Shutenko Z S, Cunningham D D, Feelisch M. Proc Natl Acad Sci USA. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharpe M A, Cooper C E. Biochem J. 1998;332:9–19. doi: 10.1042/bj3320009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liochev S I, Fridovich I. J Biol Chem. 2001;276:35253–35257. doi: 10.1074/jbc.M104237200. [DOI] [PubMed] [Google Scholar]
- 7.Bonner F T, Hughes M N. Comments Inorg Chem. 1988;7:215–234. [Google Scholar]
- 8.Bonner F T, Stedman G. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. Chichester: Wiley; 1996. pp. 3–18. [Google Scholar]
- 9.Ellis H B, Jr, Ellison G B. J Chem Phys. 1983;78:6541–6558. [Google Scholar]
- 10.Tronc M, Huetz A, Landau M, Pichou F, Reinhardt J. J Phys B. 1975;8:1160–1169. [Google Scholar]
- 11.Sturrock P E, Ray J D, McDowell J, Hunt H R. Inorg Chem. 1963;2:649–650. [Google Scholar]
- 12.Hughes M N. Biochim Biophys Acta. 1999;1411:263–272. doi: 10.1016/s0005-2728(99)00019-5. [DOI] [PubMed] [Google Scholar]
- 13.Donald C E, Hughes M N, Thompson J M, Bonner F T. Inorg Chem. 1986;25:2676–2677. [Google Scholar]
- 14.Rodgers M A J, Snowden P T. J Am Chem Soc. 1982;104:5541–5543. [Google Scholar]
- 15.Miranda K M, Espey M G, Yamada K, Krishna M, Ludwick N, Kim S, Jourd'heuil D, Grisham M B, Feelisch M, Fukuto J M, Wink D A. J Biol Chem. 2001;276:1720–1727. doi: 10.1074/jbc.M006174200. [DOI] [PubMed] [Google Scholar]
- 16.Reif A, Zecca L, Riederer P, Feelisch M, Schmidt H H. Free Radical Biol Med. 2001;30:803–808. doi: 10.1016/s0891-5849(01)00477-4. [DOI] [PubMed] [Google Scholar]
- 17.Grätzel M, Taniguchi S, Henglein A. Ber Bunsenges Phys Chem. 1970;74:1003–1010. [Google Scholar]
- 18.Seddon W A, Fletcher J W, Sopchyshyn F C. Can J Chem. 1973;51:1123–1130. [Google Scholar]
- 19.Stanbury D M. Adv Inorg Chem. 1989;33:69–138. [Google Scholar]
- 20.Ehman D L, Sawyer D T. J Electroanal Chem. 1968;16:541–549. [Google Scholar]
- 21.Benderskii V A, Krivenko A G, Ponomarev E A. Sov Electrochem. 1989;25:154–161. [Google Scholar]
- 22.Bartberger M D, Fukuto J M, Houk K N. Proc Natl Acad Sci USA. 2001;98:2194–2198. doi: 10.1073/pnas.041481598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Addison, C. C., Gamlen, G. A. & Thompson, R. (1952) J. Chem. Soc. 338–345.
- 24.Maragos C M, Morley D, Wink D A, Dunams T M, Saavedra J E, Hoffman A, Bove A A, Isaac L, Hrabie J A, Keefer L K. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 25.Shafirovich V, Dourandin A, Huang W, Luneva N P, Geacintov N E. J Phys Chem B. 1999;103:10924–10933. [Google Scholar]
- 26.Andersson W R. Combust Flame. 1999;117:394–403. [Google Scholar]
- 27.Chase M W, Jr, Davies C A, Downey J R, Jr, Frurip D J, McDonald R A, Syverud A N. J Phys Chem Ref Data. 1985;14, Suppl. 1., Part II:927–1856. [Google Scholar]
- 28.Bazylinski D A, Hollocher T C. Inorg Chem. 1985;24:4285–4288. [Google Scholar]
- 29.Lymar S V, Hurst J K. J Am Chem Soc. 1995;117:8867–8868. [Google Scholar]
- 30.Treinin A. Isr J Chem. 1970;8:103–113. [Google Scholar]
- 31.Buxton G V, Subhani M S. J Chem Soc Faraday Trans 1. 1972;68:958–969. [Google Scholar]
- 32.Klaening U K, Sehested K, Wolff T. J Chem Soc Faraday Trans 1. 1984;80:2969–2979. [Google Scholar]
- 33.Treinin A, Hayon E. J Am Chem Soc. 1976;98:3884–3891. [Google Scholar]
- 34.Czapski G, Holcman J, Bielski B H J. J Am Chem Soc. 1994;116:11465–11469. [Google Scholar]
- 35.Janaway G A, Brauman J I. J Phys Chem A. 2000;104:1795–1798. [Google Scholar]
- 36.Bryukov M G, Kachanov A A, Timonnen R, Seetula J, Vandoren J, Sarkisov O M. Chem Phys Lett. 1993;208:392–398. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}