

Abstract
The peptide antibiotic ramoplanin inhibits bacterial peptidoglycan (PG) biosynthesis by interrupting late-stage membrane-associated glycosyltransferase reactions catalyzed by the transglycosylase and MurG enzymes. The mechanism of ramoplanin involves sequestration of lipid-anchored PG biosynthesis intermediates, physically occluding these substrates from proper utilization by these enzymes. In this report, we describe the first molecular-level details of the interaction of ramoplanin with PG biosynthesis intermediates. NMR analysis in conjunction with chemical dissection of the PG monomer revealed that the ramoplanin octapeptide d-Hpg-d-Orn-d-alloThr-Hpg-d-Hpg-alloThr-Phe-d-Orn recognizes MurNAc-Ala-γ-d-Glu pyrophosphate, the minimum component of PG capable of high-affinity complexation and fibril formation. Ramoplanin therefore recognizes a PG binding locus different from the N-acyl-d-Ala-d-Ala moiety targeted by vancomycin. Because ramoplanin is structurally less complex than glycopeptide antibiotics such as vancomycin, peptidomimetic chemotherapeutics derived from this recognition sequence may find future use as antibiotics against vancomycin-resistant Enterococcus faecium, methicillin-resistant Staphylococcus aureus, and related pathogens.
Bacterial resistance to antibiotics has seriously impacted our ability to overcome infectious disease. Resistance to front line β-lactam, aminoglycoside, and glycopeptide antibiotics has become widespread, signaling the end of the golden age of antimicrobial chemotherapy (1). For almost thirty years, the glycopeptide antibiotic vancomycin has been the drug of last resort against infections from Gram-positive pathogens. Unfortunately, the utility of this drug class has been limited because of the recent onset of clinical cases of glycopeptide resistance, most notably in enterococcal and multidrug-resistant staphylococcal nosocomial pathogens. Given the dwindling arsenal of effective antibiotics against these infections, a replacement for vancomycin is urgently needed.
The lipoglycodepsipeptide antibiotic ramoplanin factor A2 (1, Fig. 1) is a promising candidate to replace vancomycin. Produced by fermentation of Actinoplanes ATCC 33076, ramoplanin A2 is highly active against numerous Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) (2–6), vancomycin-resistant Enterococcus faecium (VRE), and those resistant to ampicillin and erythromycin (6–11). Ramoplanin factor A2 is currently under Phase III clinical development for eradication of VRE and MRSA. No cases of clinical or laboratory-generated resistance to this antibiotic have been reported (12, 13).
Figure 1.

Chemical structures of ramoplanin factor A2 (25) and enduracidins A and B (27, 38).
A global view of the mechanism of action of ramoplanin is beginning to emerge. Reynolds and Somner determined that ramoplanin arrests cell wall biosynthesis at a late stage peptidoglycan (PG) biosynthesis step whereby the enzyme MurG catalyzes the conversion of undecaprenyl-pyrophosphoryl-N-acetylmuramyl-pentapeptide (Lipid I) and uridyl-pyrophosphoryl-N-acetylglucosamine (UDP-GlcNAc) to uridyl-diphosphate (UDP) and undecaprenyl-pyrophosphoryl-N-acetylmuramyl(N-acetylglucoseamine)-pentapeptide (Lipid II) (13). Reynolds further postulated that this mechanism of inhibition might involve complexation of Lipid I by ramoplanin, a mechanism that echoes the mode of action of both glycopeptide antibiotics and select lantibiotics. Work by van Heijenoort and colleagues verified inhibition of MurG by ramoplanin in vitro using recombinant, detergent solublized His-6-tagged enzyme and a synthetic Lipid I analogue (14). Brotz and coworkers demonstrated that ramoplanin altered the chromatographic migration profiles of both Lipid I and Lipid II, suggesting that the two form a complex with the antibiotic, thereby raising the possibility that ramoplanin might also interfere with transglycosylation via capture of Lipid II (1). Using citronellyl–Lipid II, a soluble synthetic analogue of Lipid II, Walker and coworkers (15) subsequently confirmed that ramoplanin indeed possessed this second PG biosynthesis inhibitory activity by directly showing its inhibition of extracellular transglycosylase activity. Importantly, it was also shown that on complexation with citronellyl–Lipid II, ramoplanin underwent a ligand-induced aggregation to produce insoluble fibrils, precluding molecular characterization of the inhibitory complex.
We report here the characterization of the complex of ramoplanin with PG precursors and related fragments. We find that the fibrils formed on solution complexation of ramoplanin with PG biosynthetic intermediates can be solublized with 20% dimethyl sulfoxide while preserving modest binding affinity, and thus allowing for the characterization of the binding interface by NMR-based methods. We have determined that ramoplanin utilizes an 8-aa sequence to recognize the muramyl carbohydrate and adjacent pyrophosphosphate moieties of PG, a different locus than the N-acyl-d-Ala-d-Ala dipeptide site targeted by vancomycin. Chemotherapeutics derived from this recognition sequence may find future use as antibiotics against VRE, MRSA, and related infections.
Experimental Methods
Methods and Materials.
Transmission electron micrographs were recorded on a Philips 400 electron microscope. HPLC was performed on a Thermo-Separations TSP 3000 system. Ramoplanin factor A2 (1) was kindly provided by Aventis Pharmaceuticals (Bridgewater, NJ). UDP-MurNAc-l-Ala-γ-d-Glu-meso-Dap-d-Ala-d-Ala [6, Park's nucleotide (16)], and UDP-MurNAc-l-Ala-γ-d-Glu-meso-Dap (7, UDP-MurNAc-tripeptide) were isolated from Bacillus subtilis according to the method of Holtje (17). 1-Phospho-MurNAc-l-Ala-γ-d-Glu-meso-Dap (8) was prepared according to the Van Heijenoort method (14). l-Ala-γ-d-Glu-d-Lys-d-Ala-d-Ala (4) and MurNAc-l-Ala-γ-d-Gln (10) were purchased from Bachem. P1-Citronellyl-P2-MurNAc-l-Ala-γ-d-Glu-l-Lys-d-Ala-d-Ala pyrophosphate (5) and the related MurNAc pyrophosphate 9 were prepared from benzyl N-acetyl-4,6-benzylidenemuramic acid as reported (18). Minimal inhibitory concentrations were performed according to standard methods, using B. subtilis strain ATCC 8037 (19).
Transmission Electron Micrographs (TEM).
To prepare fibrils for TEM measurements, a solution of ramoplanin (1, 1 mM) was mixed 1:1 with a solution of PG intermediate (5, 6, or 7, 1 mM) in 50 mM phosphate buffer (pH 6.0). Fibrils formed immediately if complexation ensued. Following fibril polymerization, aliquots for negative staining were diluted 1,000-fold into 0.05 M ammonium formate buffer (pH 6.0) containing 0.2 M NaCl. One drop (≈20 μl) of the mixture was immediately placed on the carbon film and negatively stained with a freshly prepared solution of 2% (wt/vol) uranyl acetate. Transmission electron micrographs were obtained at 80 kV with a magnification of ×20,000–60,000. Fibril dimensions were measured on a digitizing pad.
NMR Spectroscopy.
One- and two-dimensional NMR spectra were recorded on Varian Inova spectrometers equipped with a Varian 5-mm triple-resonance probe with 3-axis pulsed field gradients (PFGs) operating at 500 MHz and 600 MHz (1H) at 298 K. Proton chemical shifts were referenced to an external standard of DSS [3-(trimethylsilyl)-1-propanesulfonic acid sodium salt] in D2O. Standard pulse sequences were used for acquisition of homonuclear total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) experiments. The TOCSY data were recorded with a 150-ms mixing time, and an 8-kHz proton MLEV-16 isotropic mixing sequence. The NOESY data were recorded with a range of mixing times from 50 to 500 ms. Quadrature detection in the indirect time domain was accomplished using the method of hypercomplex reconstruction (20). For samples dissolved in H2O, selective solvent suppression was achieved using the double PFG excitation sculpting technique of Shaka (21, 22). All spectra were processed using the software package felix (Molecular Simulations, Waltham, MA).
Determination of Dissociation Constants.
The dissociation constants of the complexes between ramoplanin and PG precursors were determined by titration using 1H NMR in 20% DMSO-d6/D2O at 25°C. In these experiments, the concentration of PG precursors 5-7 was kept constant (0.25 mM), whereas the concentration of 1 was varied from 0.05–2.5 mM. The data were fit by nonlinear least-squares analysis to the following expression:
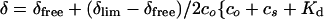 |
1 |
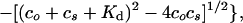 |
where δfree and δlim denote the 1H NMR chemical shifts of free and fully complexed 5-7, respectively; co is the concentration of compound 1; cs is the concentration of PG precursor; and Kd is the dissociation constant.
Results and Discussion
Ramoplanin Binds Lipid Intermediate I and Related Peptidoglycan Biosynthesis Intermediates.
Walker's verification of transglycosylase (TGase) inhibition by ramoplanin in vivo suggests that the primary mechanism of bacterial killing involves Lipid II sequestration (15). However, ramoplanin differs from transglycosylase inhibitors such as moenomycin and mersacidin in that it not only inhibits TGase activity, but also generates a metabolic block at the level of the MurG reaction, forcing a buildup of cellular pools of Lipid I (12, 13). Inhibition of MurG activity might be explained simply if the ramoplanin–Lipid II complex was an inhibitor of MurG, because the enzyme is peripherally associated with the inner face of the cytoplasmic membrane (23). Alternatively, should either ramoplanin or Lipid I encounter the other because of membrane translocation, an inhibitory complex could form. Thus, to ascertain whether Lipid I could serve as a ligand for ramoplanin, we synthesized citronellyl–Lipid I (5, Fig. 2), a soluble derivative of Lipid I, and examined its ability to bind to ramoplanin by using 1H NMR spectroscopy (Fig. 3; refs. 18 and 24).
Figure 2.

Chemical structures of synthetic and naturally occurring analogues of bacterial peptidoglycan monomers used in this study.
Figure 3.

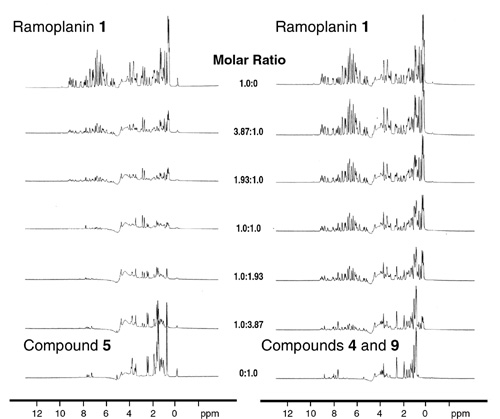
(A Left) 1H NMR titration spectra of ramoplanin (1) with citronellyl–Lipid intermediate I (5). As a 1:1 stoichiometry is reached (center trace), the intensity of the NMR signals is diminished and subjected to severe line broadening because of the formation of insoluble peptide fibrils. (A Right) 1H NMR titration spectra of ramoplanin (1) with equimolar P1-citronellyl-P2-MurNAc pyrophosphate (9) and l-Ala-γ-d-Glu-l-Lys-d-Ala-d-Ala (4) added in trans. At the 1:1:1 stoichiometric titration point (center trace), the NMR spectra of the mixture is the superposition of each individual component. No chemical shift changes or line broadening was observed during the titration, indicating that no interaction between the antibiotic and in trans peptidoglycan fragments occurred. (B) Transmission electron micrograph of the fibrils formed from the complexation of ramoplanin (1) with citronellyl–Lipid I (5). The fibrils formed from the interaction of 1 with 6 and 7, respectively, exhibited similar morphology by transmission electron micrograph (TEM). (C) Part of the aliphatic region of the 1H NMR spectrum of free ramoplanin (1), free Park's nucleotide (6), and a 1:1 mixture of the two depicting the chemical shift changes that occur on binding. (D) Representative Kd determination plot of the binding of ramoplanin (1) to Park's nucleotide (6) as obtained by 1H NMR titration. The curve denotes the experimental (o) and calculated (solid line) chemical shifts (δ) of the lactyl ether methyl protons (3a) of 6 as a function of ramoplanin concentration.
Indeed, titration of a solution of ramoplanin (1, 0.5 mM in D2O, pH 6.0) with 5 (0.5 mM in D2O, pH 6.0) resulted in the progressive loss of signal intensity because of alterations in the NMR relaxation properties of the fibrillar aggregate that formed during the titration (Fig. 3A). Similar aggregation was observed when ramoplanin was titrated against the related citronellyl–Lipid II (15). We have determined by NMR that ramoplanin anchors to membrane-mimicking unilamellar vesicles or micelles composed of physiologically relevant phospholipids (D.G.M., unpublished work), so it is very likely that the encounter occurs in the membrane bilayer. Although we note that fibril formation of ramoplanin with undecaprenyl lipid intermediates has not been observed in vivo, intercomplex association in vivo could prove beneficial for membrane anchoring or binding with PG monomers and/or associated PG synthase machinery.
Because ramoplanin formed fibrils with both Lipid I and II, we believed a minimal PG recognition motif exists for the antibiotic. We tested whether additional related PG biosynthetic intermediates were capable of binding to ramoplanin and forming fibrils. Ramoplanin was titrated against solutions of UDP-MurNAc-l-Ala-γ-d-Glu-l-Dap-d-Ala-d-Ala pentapeptide (6, Park's nucleotide) and UDP-MurNAc-l-Ala-γ-d-Glu-l-Dap tripeptide (7), two cytoplasmic biosynthetic precursors on the pathway for Lipid II biosynthesis. Both compounds bound the antibiotic and formed fibrils. As with the Lipid I analogue 5, fibril formation with 6 and 7 occurred instantaneously on mixing. Taken together with the titration studies with compound 5, these results indicate that ramoplanin can form a high-affinity complex with simpler PG intermediates, as well as Lipid I/II, and furthermore show that the presence of the undecaprenyl lipid is not an essential requirement for PG molecular recognition and fibril formation. Although Lipid II is likely the physiologically relevant target, these results suggest that simpler, more readily isolatable and soluble PG intermediates can serve as excellent models for studying the interaction of ramoplanin analogues with lipid intermediates.
The fibrils that formed from the interaction of ramoplanin with 5, 6, and 7 in water were examined using transmission electron microscopy (Fig. 3B). Fibrils formed from ramoplanin complexation with compounds 5-7 were found to be nearly identical in length and overall morphology. They formed flat or twisted ribbons ≈5 nm in width and 100 nm in length. The kinetics of fibril formation were extremely rapid, occurring within seconds after mixing homogeneous solutions of the two soluble precursors (0.1–0.5 mM each).
To further localize the site of antibiotic binding, we next examined by NMR whether the northern and southern halves of Lipid I, l-Ala-γ-d-Glu-l-Lys-d-Ala-d-Ala (4) and P1-citronellyl-P2-α-d-N-acetylmuramyl pyrophosphate (9) (18), were capable of binding to ramoplanin. When mixed with equimolar ramoplanin either separately or in combination, compounds 4 and 9 showed neither antibiotic binding nor fibril formation (Fig. 3A). Thus, the simultaneous presentation of both halves of 5 to ramoplanin resulted in complete disruption of the binding interaction and subsequent fibril formation. Because pentapeptide 4 contains the N-acyl-d-Ala-d-Ala moiety targeted by glycopeptide antibiotics, the mechanism of PG recognition must be completely different from that of vancomycin and related antibiotics. Furthermore, because ramoplanin binds to full length citronellyl–Lipid I (5) but not the component pieces (4, 9), binding and fibril formation absolutely requires an intact amide bond between the lactyl ether side chain of the carbohydrate and the pendant l-alanine residue of the pentapeptide.
Fibril Solublization and Molecular Dissection of the Ramoplanin–Lipid Intermediate Interaction.
Fibril formation in aqueous buffers precluded structural assessment of the recognition interface of ramoplanin with PG intermediates and assessment of relative affinities of PG precursors to the antibiotic. However, we discovered that addition of 20% DMSO drastically reduces or altogether eliminates fibril formation, yet preserves modest binding affinity. Fortuitously, Kurz and Guba's solution structure of ramoplanin was also performed in 20% DMSO-d6, so the starting conformation of the antibiotic was firmly established (25) and structural changes that ensued on ligand complexation could easily be observed by NMR. Furthermore, the 1H NMR spectrum of ramoplanin in 20% DMSO is nearly identical to that of the spectrum acquired in water (data not shown), so information gained regarding the initial encounter complex of ramoplanin with PG precursors in 20% DMSO may be extrapolated with confidence to encounters in aqueous systems.
Titrations of ramoplanin with 5, 6, or 7 in DMSO-d6/D2O (1:5 vol/vol) or DMSO-d6/H2O/D2O (20:72:8 vol/vol/vol) at 25°C resulted in widespread chemical shift changes in both antibiotic and PG ligand on binding (Fig. 3). For ramoplanin, the 1H chemical shift changes were primarily localized to the octapeptide stretch from hydroxyphenylglycine (Hpg)3 to ornithine (Orn)10. Similar chemical shift changes occurred in the PG intermediates on binding ramoplanin, such as the shifts of the protons of the C4-lactyl ether, the NHAc, the muramyl carbohydrate, and the Ala residues (Figs. 3C and 4A). Because each ramoplanin-PG intermediate complex was in fast chemical exchange on the NMR chemical shift time scale, equilibrium binding constants (Kd) could be directly obtained.
Figure 4.

(A) NMR localization of the binding interface between ramoplanin (1) and Park's nucleotide (6) (1:1 molar ratio). Protons that exhibit downfield chemical shifts on binding are colored green; those that shift upfield are depicted in orange. Protons that do not shift on binding are colorless. Intermolecular NOEs are depicted by red arrows. Possible anchoring electrostatic interactions between ramoplanin Orn4 and Orn10 residues and 6 are indicated by blue arrows. NMR experiments further localize the minimum PG structure recognized by ramoplanin to the intact pyrophosphate, the muramyl carbohydrate, and the first two amino acids of the pentapeptide. (B) Surface localization of the peptidoglycan monomer/lipid intermediate binding region of ramoplanin. Residues comprising the binding interface identified by NMR that lie on the same face of the antibiotic are colored yellow and mapped onto (C) the three-dimensional NMR structure of ramoplanin obtained in 20% DMSO (25).
Dissociation constants were measured for ramoplanin binding to compounds 6 and 7 by monitoring the chemical shift of the lactyl ether methyl proton (≈1.4 ppm) (3a position, see Fig. 4) and fitting this data to Eq. 1. As illustrated in Fig. 3D, the best nonlinear least squares statistical fit was obtained for the formation of a complex with a 1:1 stoichiometry. For Park's nucleotide (6), the dissociation constant of 180 ± 20 μM obtained was nearly an order of magnitude lower than the 2,380 ± 440 μM Kd obtained for UDP-MurNAc-tripeptide (7, ΔΔG = −1.53 kcal/mol). This ΔΔG value, consistent with loss of a partially buried or multiple solvent-exposed hydrogen bonds or a salt bridge, suggests that the terminal d-Ala carboxylate is an important binding component. For Lipid I analogue 5, an estimated Kd of ≤100 μM was obtained because of spectral overlap of the citronellyl methyl resonances with the diagnostic protons of 5 and because in titration experiments of >2 h duration a minute amount of fibrils precipitated in the NMR tube. Because each PG precursor interacted with ramoplanin with differing affinities in 20% DMSO, it is attractive to speculate that ramoplanin might be capable of discriminating between Lipid I and II in vivo. However, in aqueous solution, compounds 5, 6, and 7 were each equally capable of high-affinity association and rapid fibril formation with the antibiotic.
To further define a minimal binding determinant for the antibiotic–ligand interaction, we prepared truncated PG precursors and assessed their capacity for ramoplanin binding and fibril formation. Ramoplanin (1) is positively charged at physiological pH because of the presence of Orn4 and Orn10. We envisioned that these residues might interact with the anionic pyrophosphate and terminal carboxylate of Lipid I/II. We dissected the pyrophosphate bond of UDP-MurNAc-tripeptide (7) with snake venom nucleotide pyrophosphatase to form 1-phospho-MurNAc-l-Ala-γ-d-Glu-meso-Dap (8) and assessed its ability to interact with ramoplanin. Monophosphate 8 neither associated with ramoplanin nor formed fibrils. In addition, no binding or fibril formation was observed for ramoplanin with MurNAc-l-Ala-γ-d-Gln (10), UMP, or citronellyl-1-phosphate. These data suggest that the pyrophosphate is an important recognition element that is necessary but not sufficient to promote association. In sum, these data reflect the importance of key components for ramoplanin recognition: a MurNAc sugar containing a 1′-pyrophosphate, an intact amide bond between the 3′-lactyl side chain, and the pendant peptide moiety. Each substructure contributes to form a minimal recognition sequence capable of ramoplanin binding and ligand-induced fibril formation.
NMR Analysis of the Ramoplanin–Park's Nucleotide Complex.
To gain insight into the binding interface of ramoplanin with PG intermediates, we examined the binding of the antibiotic with Park's nucleotide (6) in 20% DMSO/8% D2O/72% H2O by two-dimensional NMR at 500 MHz. Park's nucleotide was chosen as a model for Lipid I and II for several reasons. Compound 6 was freely soluble during the NMR experiment, whereas concentrated solutions of ramoplanin with 5 in 20% DMSO would precipitate a small amount of fibrils during extended NOESY runs. Although 5 and 6 exhibited similar Kd values with ramoplanin, compound 6 could be readily isolated in multimilligram quantities from B. subtilis (17), whereas analogue 5 was prepared by a lengthy total synthesis (18).
Proton resonances of the complex of 1 with 6 were assigned using a combination of two-dimensional correlation spectroscopy (COSY) and total correlation spectroscopy (TOCSY) experiments and comparison to the parent spectra (25, 26). Two-dimensional NOESY spectra were obtained with a 500-ms mixing time and by using a 4-fold excess of 6 because of its modest binding constant. As compared with the spectrum of 1 and 6, the NOESY spectrum of the complex revealed numerous inter- and intramolecular NOEs that delineate the binding interface and conformational rearrangements in the antibiotic that ensue on complexation (Fig. 4). Most notable were the presence of five strong intermolecular NOEs that were distributed between ramoplanin residues Thr8 and Hpg6. The NH and aryl protons of Thr8–Hpg6 show short-range interactions with the uridyl protons, protons of the muramyl carbohydrate, and the lactyl ether side chain of 6. These five NOEs are sufficient to provide the orientation of the two molecules to each other (Fig. 4A). These residues are part of the Hpg3–Orn10 stretch that exhibits marked chemical shift changes on complexation (Fig. 4 A and B). Within this sequence stretch are several exchangeable ramoplanin side chain hydroxyls that likely assist in capture of the muramyl carbohydrate. Although significant chemical shift changes are seen in the terminal d-Ala-d-Ala α-CH and β-methyl protons, no intermolecular NOEs involving these residues were detected under these conditions.
In addition to new intermolecular NOEs that localize the antibiotic–PG ligand complex binding interface, several conformationally diagnostic NOEs in ramoplanin shift significantly or disappear altogether, signaling a flattening of the cup-shaped central two-stranded β-sheet of the antibiotic on ligand association (see the supporting information, which is published on the PNAS web site, www.pnas.org). The two-stranded sheet is held in an elongated closed loop defined by the depsipeptide lactone between Asn2 and chlorohydroxyphenylglycine (Chp)17 and the Thr8-Phe9 type I β-turn (Figs. 1 and 4). In 20% DMSO, a series of strong intramolecular NOEs are observed between the Thr8–Phe9 turn and Asn2–Chp17 depsipeptide lactone and surrounding residues, confirming their transannular relationship (Fig. 4C). However, on complexation to 6, many of these conformationally diagnostic transannular NOEs disappear, presumably because of a flattening of the cyclic depsipeptide architecture. Because the PG ligand binds to the solvent-exposed outer β-strand between Asn2 and Thr8 (Fig. 4B), flattening the cup-shaped two-stranded peptide backbone would result in exposure of the Phe9–Chp17–Hpg3 hydrophobic core to bulk solvent, creating a newly exposed hydrophobic face capable of dimerization and oligomerization with other complexes. This ligand-induced exposure of the ramoplanin hydrophobic core likely leads to the fibril formation that is observed in aqueous solution in the absence of 20% DMSO.
The Cell-Wall-Active Antibiotics Ramoplanin, Enduracidin, and Janiemycin Contain a Common Lipid Intermediate Recognition Motif.
For ramoplanin, the majority of induced chemical shifts and intermolecular NOEs were localized to an Hpg3–Orn10 octapeptide recognition motif. Thr8, Hpg7, and Hpg6 (and likely Thr5) play a key role in sequestration of the muramyl carbohydrate of PG precursors with Orn4 and Orn10 orienting and stabilizing the ligand in this complex (Fig. 4A). Our belief in a common recognition motif for lipid intermediates is further reinforced by our observation that a nearly identical motif is conserved in the enduracidins, a related family of cell-wall-active lipodepsipeptide antibiotics from Streptomyces fungicidicus (2-3, Fig. 1; ref. 27). In addition, although the structure of the lipodepsipeptide antibiotic janiemycin from Streptomyces macrosporeus has not yet been elucidated, its antibiotic function and amino acid composition are also nearly identical to ramoplanin and enduracidin (28). We predict that janiemycin also contains this PG recognition motif.
Ramoplanin and the Two Type-C Lantibiotics, Mersacidin and Actagardine, Share a Common Fold and Function.
In addition to the glycopeptides, a growing number of antibiotics have been found to sequester Lipid II as a fundamental part of their mechanism of action (29). These include two members of the pore-forming type-A lantibiotics, nisin and epidermin, and the two type-C lantibiotic transglycosylase inhibitors, mersacidin and actagardine (30, 31). Direct comparison of the primary and secondary structures of ramoplanin (25) with vancomycin (32, 33) reveals no obvious structural similarities between the two, suggesting that they bind Lipid II at different loci. The lack of apparent binding between ramoplanin and the d-Ala-d-Ala-containing pentapeptide 4 confirms this hypothesis. Comparison of ramoplanin with nisin and epidermin (30) indicates no conservation of primary or secondary structure, although these conformationally flexible pore-forming antibiotics are known to bind to Lipid II prior to membrane disruption (1, 34). However, comparison of the solution NMR structures of ramoplanin (25), mersacidin (35), and actagardine (36) reveals a conserved backbone fold (Fig. 5). The latter two antibiotics possess a unique mechanism of action involving binding to the MurNAc-GlcNAc disaccharide and pyrophosphate functionalities of Lipid II (37). Preservation of this overall fold between three Lipid II-binding antibiotics is suggestive of overlapping Lipid II binding sites and a common mechanism of action.
Figure 5.

Primary and backbone tertiary backbone structures of ramoplanin A2 (green) and two functionally related type-C lantibiotics, mersacidin (purple) and actagardine (blue). Nonstandard residues are denoted as follows: Abu, aminoisobutyric acid; 3-OH Asn, threo-3-hydroxy-l-asparagine; Chp, chlorohydroxyphenylglycine; Dha, dehydroalanine; FA, (2Z,4E)-7-methyl-octadienoic acid; Man, mannose. d-Amino acids are depicted by black circles. Ribbon representations of the tertiary structures were drawn using the program insight ii. Coordinates for ramoplanin (1DSR) and actagardine (1AJ1) NMR structures have been deposited in the Protein Data Bank. NMR coordinates for mersacidin were provided by Dr. Thomas Prasch.
Implications for Antibiotic Design.
Our structure/function studies of ramoplanin with PG intermediates and related synthetic analogues indicate that the Lipid I/II MurNAc-Ala-γ-d-Glu pyrophosphate core is the minimum structural element of the PG monomer required by ramoplanin for high-affinity binding and fibril formation in solution. Intriguingly, mersacidin exhibits a strikingly similar preference for binding to the GlcNAc-MurNAc-pyrophosphate moiety of Lipid II (37). Both ramoplanin and mersacidin (and likely actagardine) recognize a region of the PG monomer that is distinctly different than that of vancomycin. This discovery explains the effectiveness of ramoplanin for treatment of vancomycin-resistant pathogens and highlights its promising clinical potential. Furthermore, ramoplanin is more amenable to total synthesis and structure–activity analysis than is vancomycin or mersacidin because its chemical structure is less complex. Future structure–activity studies with ramoplanin will help dissect out the key structural features that promote high-affinity PG complexation and potent antimicrobial activity.
Supplementary Material
Acknowledgments
We thank Hoescht Marion Roussel for providing Ramoplanin; and Dr. Thomas Prasch (Johann Wolfgang Goethe-Universitate, Frankfurt) for providing the NMR coordinates for mersacidin. This work was supported by a grant from the McCabe Foundation (to D.G.M.), American Cancer Society Grant RPG-99-31-02-CCE (to D.G.M.), and National Institutes of Health Grants GM46611 (to D.G.M.), HL30954 (to J.W.W.), and DK39806 (to A.J.W.). J.K.K. is the recipient of National Institutes of Health National Research Service Award Fellowship GM20206.
Abbreviations
- PG
peptidoglycan
- NOESY
nuclear Overhauser effect spectroscopy
- Hpg
hydroxyphenylglycine
- Orn
ornithine
References
- 1.Brotz H, Josten M, Wiedemann I, Schneider U, Gotz F, Bierbaum G, Sahl H G. Mol Microbiol. 1998;30:317–327. doi: 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- 2.O'Hare M D, Ghosh G, Felmingham D, Grueneberg R N. J Antimicrob Chemother. 1990;25:217–220. doi: 10.1093/jac/25.2.217. [DOI] [PubMed] [Google Scholar]
- 3.O'Hare M D, Felmingham D, Gruneberg R N. Drugs Exp Clin Res. 1988;14:617–619. [PubMed] [Google Scholar]
- 4.Maple P A C, Hamilton-Miller J M T, Brumfitt W. J Antimicrob Chemother. 1989;23:517–525. doi: 10.1093/jac/23.4.517. [DOI] [PubMed] [Google Scholar]
- 5.Brumfitt W, Maple P A C, Hamilton-Miller J M T. Drugs Exp Clin Res. 1990;16:377–383. [PubMed] [Google Scholar]
- 6.Collins L A, Eliopoulos G M, Wennersten C B, Ferraro M J, Moellering R C., Jr Antimicrob Agents Chemother. 1993;37:1364–1366. doi: 10.1128/aac.37.6.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson C C, Taylor S, Pitsakis P, May P, Levison M E. Antimicrob Agents Chemother. 1992;36:2342–2345. doi: 10.1128/aac.36.10.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciabatti R, Kettenring J K, Winters G, Tuan G, Zerilli L, Cavalleri B. J Antibiot. 1989;42:254–267. doi: 10.7164/antibiotics.42.254. [DOI] [PubMed] [Google Scholar]
- 9.Cavalleri B, Pagani H, Volpe G, Selva E, Parenti F. J Antibiot. 1989;37:309–317. doi: 10.7164/antibiotics.37.309. [DOI] [PubMed] [Google Scholar]
- 10.Pallanza R, Berti M, Scotti R, Randishi E, Arioli V. J Antibiot. 1984;37:318–324. doi: 10.7164/antibiotics.37.318. [DOI] [PubMed] [Google Scholar]
- 11.Parenti F, Ciabatti R, Cavalleri B, Kettenring J. Drugs Exp Clin Res. 1990;16:451–455. [PubMed] [Google Scholar]
- 12.Reynolds P E, Somner E A. Drugs Exp Clin Res. 1990;16:385–389. [PubMed] [Google Scholar]
- 13.Somner E A, Reynolds P E. Antimicrob Agents Chemother. 1990;34:413–419. doi: 10.1128/aac.34.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Auger G, Crouvoisier M, Caroff M, van Heijenoort J, Blanot D. Lett Peptide Sci. 1997;4:371–376. [Google Scholar]
- 15.Lo M C, Men H, Branstrom A, Helm J, Yao N, Goldman R, Walker S. J Am Chem Soc. 2000;122:3540–3541. [Google Scholar]
- 16.Park J T. J Biol Chem. 1952;194:877–904. [PubMed] [Google Scholar]
- 17.Kohlrausch U, Holtje J V. FEMS Microbiol Lett. 1991;78:253–258. doi: 10.1016/0378-1097(91)90166-8. [DOI] [PubMed] [Google Scholar]
- 18.Cudic P, Behenna D C, Yu M K, Kruger R, Szewczuk L, McCafferty D G. Bioorg Med Chem Lett. 2001;11:3107–3110. doi: 10.1016/s0960-894x(01)00653-9. [DOI] [PubMed] [Google Scholar]
- 19.National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. 5th Ed. Wayne, PA: NCCLS; 2000. [Google Scholar]
- 20.Marion D, Kay L E, Sparks S W, Torchia D A, Bax A. J Am Chem Soc. 1989;111:1515–1517. [Google Scholar]
- 21.Hwang T L, Shaka A J. J Magn Reson A. 1995;112:275–279. [Google Scholar]
- 22.Stott K, Stonehouse J, Keeler J, Hwang T L, Shaka A J. J Am Chem Soc. 1995;117:4199–4200. [Google Scholar]
- 23.Bupp K, van Heijenhoort J. J Bacteriol. 1993;175:1841–1843. doi: 10.1128/jb.175.6.1841-1843.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Men H, Park P, Ge M, Walker S. J Am Chem Soc. 1998;120:2484–2485. [Google Scholar]
- 25.Kurz M, Guba W. Biochemistry. 1996;35:12570–12575. doi: 10.1021/bi961017q. [DOI] [PubMed] [Google Scholar]
- 26.Reddy S G, Waddell S T, Kuo D W, Wong K K, Pompliano D L. J Am Chem Soc. 1999;121:1175–1178. [Google Scholar]
- 27.Iwasaki H, Horii S, Asai M, Mizuno K, Ueyanagi J, Miyake A. Chem Pharm Bull. 1973;21:1184–1191. [Google Scholar]
- 28.Meyers E, Weisenborn F L, Pansy F E, Slusarchyk D S, Von Saltza M H, Rathnum M L, Parker W L. J Antibiot. 1970;23:502–507. doi: 10.7164/antibiotics.23.502. [DOI] [PubMed] [Google Scholar]
- 29.McCafferty D G, Cudic P, Yu M K, Behenna D C, Kruger R. Curr Op Chem Biol. 1999;3:672–680. doi: 10.1016/s1367-5931(99)00025-3. [DOI] [PubMed] [Google Scholar]
- 30.Bierbaum G. Chemother J. 1999;8:204–209. [Google Scholar]
- 31.Sahl H G, Bierbaum G. Annu Rev Microbiol. 1998;52:41–79. doi: 10.1146/annurev.micro.52.1.41. [DOI] [PubMed] [Google Scholar]
- 32.Loll P J, Bevivino A E, Korty B D, Axelsen P A. J Am Chem Soc. 1997;119:1516–1522. [Google Scholar]
- 33.Schafer M, Schneider T R, Sheldrick G M. Structure (London) 1996;4:1509–1515. doi: 10.1016/s0969-2126(96)00156-6. [DOI] [PubMed] [Google Scholar]
- 34.Wiedemann I, Breukink E, Van Kraaij C, Kuipers O P, Bierbaum G, De Kruijff B, Sahl H-G. J Biol Chem. 2001;276:1772–1779. doi: 10.1074/jbc.M006770200. [DOI] [PubMed] [Google Scholar]
- 35.Prasch T, Nauman T, Markert R L, Sattler M, Schubert M, Schaal S, Bauch M, Kogler H, Greisinger C. Eur J Biochem. 1997;244:501–512. doi: 10.1111/j.1432-1033.1997.00501.x. [DOI] [PubMed] [Google Scholar]
- 36.Zimmermann N, Jung G. Eur J Biochem. 1997;246:809–819. doi: 10.1111/j.1432-1033.1997.00809.x. [DOI] [PubMed] [Google Scholar]
- 37.Brotz H, Bierbaum G, Leopold K, Reynolds P E, Sahl H G. Antimicrob Agents Chemother. 1998;42:154–160. doi: 10.1128/aac.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hori M, Iwasaki H, Horii S, Yoshida I, Hongo T. Chem Pharm Bull. 1973;21:1175–1183. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}