

Abstract
A fundamental question in RNA folding is the nature of the rate-limiting step. Folding of large RNAs often is trapped by the need to undo misfolded structures, which precludes the study of the other, potentially more interesting aspects in the rate-limiting step, such as conformational search, metal ion binding, and the role of productive intermediates. The catalytic domain of the Bacillus subtilis RNase P RNA folds without a kinetic trap, thereby providing an ideal system to elucidate these steps. We analyzed the folding kinetics by using fluorescence and absorbance spectroscopies, catalytic activity, and synchrotron small-angle x-ray scattering. Folding begins with the rapid formation of early intermediates wherein the majority of conformational search occurs, followed by the slower formation of subsequent intermediates. Before the rate-limiting step, more than 98% of the total structure has formed. The rate-limiting step is a small-scale structural rearrangement involving prebound metal ions.
Understanding the nature of the rate-limiting step in tertiary RNA folding is crucial to our ability to design, modify, and elucidate the evolutionary relationship of functional RNA structures. Early in vitro studies showed that tRNA often folds slowly because of the need to undo nonnative structures (1–3). Recent studies on RNA folding have focused on large ribozymes (4–24), such as the group I and II ribozymes and the bacterial RNase P RNA. The rate-limiting step in the folding of the Tetrahymena group I ribozyme is the disruption of an alternate helix, AltP3, which takes minutes at 37°C. Under certain conditions, AltP3 does not form, and the Tetrahymena group I ribozyme folds to the native state (N) in seconds or less (12). Folding of the ai5γ group II ribozyme is limited by need to form key long-range tertiary interactions (24). Folding of the Bacillus subtilis RNase P RNA is limited by the need to undo a nonnative structure that forms between its two domains (14).
The transitory, nonnative structure in the folding of the full-length B. subtilis P RNA is eliminated when both domains are separated (15). The catalytic domain (C domain, Fig. 1A) folds in less than 0.2 s at 37°C and 30 s at 10°C, more than 100 times faster than the folding of the full-length P RNA. Furthermore, the presence of the denaturant urea can decrease the folding rate of the C domain, whereas urea increases the folding rate of RNAs known to be limited by error-correction steps. In conjunction with a detailed analysis of the metal ion dependence of the folding kinetics, we concluded previously that the folding of C-domain is not limited by misfolding events.
Figure 1.

(A) The B. subtilis C-domain (47). A fluorescein is covalently attached to the 5′ end that allows monitoring the folding kinetics by fluorescence spectroscopy. (B) Folding kinetics at 10°C starting from either the Uaq state (20 mM Tris⋅HCl, no Me2+) or the Uurea state (20 mM Tris⋅HCl, 8 M urea, 1 mM EDTA, no Me2+). Folding is monitored by absorbance from 0.002 to 0.2 s, and the dead-time change in signal is indicated with dashed arrows. Folding is monitored by fluorescence between 0.2 to 360 s. The fluorescence signal is fit with a two-exponential function. The slow phase has the same rate as that obtained by catalytic activity (open circles, right axis) and directly reports the rate-limiting step. The fast phase corresponds to the signal change of the Ieq-to-I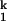 transition and is ≈6 times faster than the slow phase.
transition and is ≈6 times faster than the slow phase.
In the absence of misfold/reorganization barriers, what process is rate-limiting? Local secondary structures such as hairpin-loops can rapidly form in microseconds in the absence of tertiary structure. The formation of long-range helices and loop–loop interactions may require an extensive conformational search and is likely to be much slower (21, 24). Alternatively, the limiting process may involve interactions between the RNA and the divalent metal ions required for the formation of the native structure. Metal ion binding sites are crucial for maintaining the tertiary architecture of large RNAs (25–30) and their formation is an event that cannot be avoided.
In this work, we demonstrate that the limiting step in C-domain folding occurs after nearly all conformational search. This conclusion is derived from chevron analysis of the metal ion series Mg2+, Ca2+, Sr2+, and Ba2+ and the temperature dependence of the folding rate on these metal ions. Furthermore, this step involves small-scale consolidation of RNA structure around one or more prebound metal ions, even though the limiting step does not involve uptake of additional metal ions.
Materials and Methods
RNA Synthesis.
The 5′-fluorescein labeled C-domain was prepared by in vitro transcription using standard methods in 14 mM MgCl2, with 1 mM each ATP, CTP, and UTP, 0.5 mM GTP, and 0.75 mM 5′ fluorescein-G, and purified by denaturing gel electrophoresis. The unfolded state (Uaq) was obtained by heating the previously denatured C-domain in Me2+-free Tris⋅HCl buffer (pH 8.1) at 85°C for 2 min, followed by incubation at room temperature for 3 min. The completely unfolded state (Uurea) was obtained by the addition of urea and EDTA to the unfolded state at a final concentration of 8 M and 1 mM, respectively.
Folding Kinetics Monitored by Fluorescence Spectroscopy (0.2–400 s).
Folding was initiated on injecting concentrated MeCl2 by a computer-interfaced Hamilton titrator into 1 ml of a solution of 0.02–0.03 μM C domain in Tris⋅HCl, pH 8.1. For unfolding kinetics, prefolded C domain was injected into 1 ml of buffer containing the appropriate amount of MeCl2. Folding and unfolding kinetics were monitored by using λexcitation = 490 ± 10 nm and λemission > 500 nm. The dead-time of the experiment was 0.2 s.
Folding Kinetics Monitored by Absorbance Spectroscopy (0.002–1 s).
Rapid mixing absorbance experiments used a Biologic SFM4 stopped-flow apparatus. C domain (2 μM) in 20 mM Tris⋅HCl (pH 8.1) with or without 8 M urea and 1 mM EDTA was diluted 10-fold with 20 mM Tris⋅HCl (pH 8.1) containing the appropriate amount of MgCl2. Folding was monitored by the absorbance change at 267 nm.
Folding Kinetics Monitored by Synchrotron Small-Angle X-Ray Scattering (SAXS).
SAXS experiments were carried out at the BESSRC ID-12 beam line of the Argonne National Laboratory (31). Folding of 0.3 mg/ml C-domain (3.8 μM) at 20 mM Tris⋅HCl, pH 8.1/0.3 mM MgCl2 (Ieq state) was initiated on the addition of MgCl2 to a final concentration of 10 mM. In comparison to the spectroscopic measurements, higher Mg2+ concentration was needed to fully populate Ieq for the SAXS studies because of the higher RNA concentration used (31). Data were acquired for 1 s each at 3, 6, 10, 25, 35, and 75 s after the initiation of folding.
Chevron Analysis.
The dependence of relaxation rates of the Ieq-to-N transition on ion concentration (15) was fit assuming two sequentially populated kinetic intermediates, I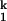 and I
and I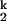 , according to
, according to
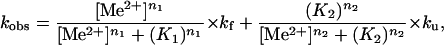 |
1 |
where K1, n1 and K2, n2 are the Me2+ concentration at the midpoint and the Hill constant of the Ieq-to-I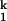 and I
and I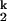 -to-N transition, respectively; kf and ku are the forward and backward rates of the I
-to-N transition, respectively; kf and ku are the forward and backward rates of the I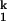 -to-I
-to-I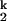 transition. Both the Ieq-to-I
transition. Both the Ieq-to-I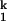 and I
and I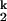 -to-N transitions are fast enough to be in equilibrium relative to the limiting I
-to-N transitions are fast enough to be in equilibrium relative to the limiting I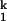 -to-I
-to-I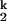 transition.
transition.
Results
Folding to the Equilibrium Intermediate, Ieq.
As described (31, 32), the equilibrium folding pathway of the C domain consists of two transitions: U-to-Ieq and Ieq-to-N. The unfolded state contains varying amount of structure, depending on the solution condition and temperature. Previous SAXS measurements at 37°C show that the radius of gyration (Rg) of the unfolded state is 80 Å in the aqueous solvent (Uaq), whereas in the presence of 8 M urea and 1 mM EDTA (Uurea), Rg increases to 180 Å (31). The formation of Ieq results in a dramatic collapse as indicated by the Rg change from 80–180 Å to 46 Å (native value is 38 Å). The intermediate is highly structured, and forms at ≈5 times lower Mg2+ concentration needed to fold the native structure (15, 31–33). The Ieq species contains the native amount of secondary structure as well as some tertiary structure, according to absorbance measurements, an analysis of urea sensitivity, and a comparative folding study with a thermophilic homologue (34). Regardless of the solution condition, however, Ieq has the same size, shape, and structure.
Folding to Ieq starting from Uaq and Uurea at 10°C (Fig. 1B) and 37°C (data not shown) is investigated by using stopped-flow absorbance spectroscopy. The UV absorbance significantly increases on the addition of urea (different starting level of absorbance of unfolded state at zero time in Fig. 1B), consistent with a loss of residual structure. In all measurements, Ieq is formed in less than 1 ms. Essentially all of the change in UV absorbance for the entire folding reaction to the native state occurs in this early step. Hence, an extensive conformational search occurs very quickly and results in the formation of this highly structured and compact intermediate.
Folding from Ieq to the Native Structure.
The Ieq-to-N transition is at least four orders of magnitude slower than the U-to-Ieq transition (Fig. 1B). Our previous study showed that C domain folds at the same rate whether folding was initiated from either Uaq or Ieq, indicating that the Ieq-to-N transition is always rate-limiting. We also demonstrated that the Ieq-to-N transition includes the transient formation of two kinetic intermediates, I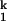 and I
and I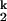 , and the rate-limiting step is the conversion between these two species:
, and the rate-limiting step is the conversion between these two species:
In this scheme, K1, K2 and n1, n2 are the Mg2+ midpoints and the Hill coefficients that define the relative populations of the Mg2+-dependent Ieq-to-I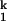 and the I
and the I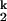 -to-N transitions, respectively; kf and ku are the folding and unfolding rates, respectively, of the Mg2+-independent I
-to-N transitions, respectively; kf and ku are the folding and unfolding rates, respectively, of the Mg2+-independent I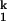 -to-I
-to-I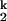 transition. Both the Ieq-to-I
transition. Both the Ieq-to-I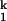 and I
and I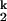 -to-N transitions are in fast equilibrium relative to the I
-to-N transitions are in fast equilibrium relative to the I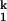 -to-I
-to-I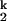 transition.
transition.
This kinetic folding pathway was deduced from the “Mg2+ chevron” (15), which reports the dependence of the folding and unfolding rates on the Mg2+ concentration, as measured by fluorescence spectroscopy (Fig. 2A). For C-domain folding, the Mg2+ dependence of the observed rate, kobs, produces a V or “chevron” shape. The vertex, where kobs is the slowest, generally represents the condition when the folding rate equals to the unfolding rate. Folding is faster than unfolding when Mg2+ concentration is above KMg (right side of chevron), whereas unfolding is faster than folding when Mg2+ concentration is below KMg (left side of chevron).
Figure 2.

(A) Metal-ion chevron analysis using fluorescence spectroscopy (Upper). In the chevron analysis, rate saturation at the high or low [Me2+] indicates the formation of a folding and unfolding intermediate, I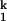 and I
and I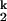 , respectively. [Me2+] at the vertex of the chevron is approximately the same as the midpoint of the equilibrium Ieq-to-N transition measured independently (Lower). (B) Folding kinetics of C-domain from Ieq at 10 mM MgCl2, 10°C, monitored by SAXS. The percentage of compaction is calculated from radius gyration. This result indicates that I
, respectively. [Me2+] at the vertex of the chevron is approximately the same as the midpoint of the equilibrium Ieq-to-N transition measured independently (Lower). (B) Folding kinetics of C-domain from Ieq at 10 mM MgCl2, 10°C, monitored by SAXS. The percentage of compaction is calculated from radius gyration. This result indicates that I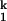 is highly compact and forms quickly.
is highly compact and forms quickly.
On the folding side, kobs becomes saturated when the Mg2+ concentration is above 3 mM (Fig. 2A), indicating a Mg2+-independent rate-limiting process in the Ieq-to-N transition (Scheme S1). This “rollover” in a chevron plot occurs because at high [Mg2+], the kinetic intermediate I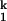 is fully populated, as opposed to folding at lower [Mg2+] where the kobs is the product of kf and the population fraction of I
is fully populated, as opposed to folding at lower [Mg2+] where the kobs is the product of kf and the population fraction of I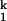 . On the unfolding side, kobs becomes saturated at [Mg2+] below 0.005 mM because the kinetic intermediate I
. On the unfolding side, kobs becomes saturated at [Mg2+] below 0.005 mM because the kinetic intermediate I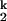 is fully populated at very low [Mg2+], as opposed to higher [Mg2+] where the kobs is the product of ku and the population fraction of I
is fully populated at very low [Mg2+], as opposed to higher [Mg2+] where the kobs is the product of ku and the population fraction of I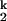 .
.
Scheme 1.

Because kf and ku, the interconversion rates of the I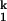 -to-I
-to-I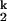 transition, do not depend on the Mg2+ concentration, the rate-limiting step in C-domain folding does not involve a change in the net number of metal ions bound. Furthermore, the interconversion between these two intermediates is accompanied by minimal surface burial, as indicated in our previous study by the identical folding rate at high Mg2+ concentration in the absence and presence of urea (15).
transition, do not depend on the Mg2+ concentration, the rate-limiting step in C-domain folding does not involve a change in the net number of metal ions bound. Furthermore, the interconversion between these two intermediates is accompanied by minimal surface burial, as indicated in our previous study by the identical folding rate at high Mg2+ concentration in the absence and presence of urea (15).
SAXS provides direct evidence on the transient accumulation of I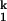 (Fig. 2B). Beginning from Ieq, folding occurs in two discrete steps according to SAXS. On the addition of 10 mM Mg2+, C domain collapses rapidly to I
(Fig. 2B). Beginning from Ieq, folding occurs in two discrete steps according to SAXS. On the addition of 10 mM Mg2+, C domain collapses rapidly to I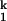 . Subsequent acquisition of the native state occurs >5-fold slower. The Ieq-to-I
. Subsequent acquisition of the native state occurs >5-fold slower. The Ieq-to-I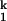 transition accounts for ≈85% of the total compaction between Ieq and N.
transition accounts for ≈85% of the total compaction between Ieq and N.
Combining the current results with our previous studies, we conclude that before the limiting step, C domain forms nearly all of the native structure. The high degree of compaction and structure formation is reflected in the SAXS data as well as in the surface burial measured previously by urea dependence (15). The urea m value, a valuable diagnostic of surface burial, for the I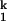 -to-N transition is only ≈0.2 kcal⋅mol−1⋅M−1, whereas the total m value for the Uurea-to-N transition is estimated to be ≈9 kcal⋅mol−1⋅M−1 (35). Similarly, the difference in Rg between I
-to-N transition is only ≈0.2 kcal⋅mol−1⋅M−1, whereas the total m value for the Uurea-to-N transition is estimated to be ≈9 kcal⋅mol−1⋅M−1 (35). Similarly, the difference in Rg between I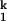 and N is only ≈1 Å, whereas the difference between Uurea and N is ≈140 Å (31). Therefore, more than 98% of the total surface burial and compaction has taken place on the formation of I
and N is only ≈1 Å, whereas the difference between Uurea and N is ≈140 Å (31). Therefore, more than 98% of the total surface burial and compaction has taken place on the formation of I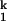 , relative to the fully unfolded state, Uurea. Furthermore, the folding rate of the limiting I
, relative to the fully unfolded state, Uurea. Furthermore, the folding rate of the limiting I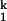 -to-I
-to-I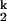 transition is insensitive to urea, indicating that most of the remaining 2% of the structure forms after the limiting step, in the I
transition is insensitive to urea, indicating that most of the remaining 2% of the structure forms after the limiting step, in the I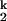 -to-N transition (15). Therefore, the rate-limiting step is a very small-scale event that involves only a minimal change in the RNA structure.
-to-N transition (15). Therefore, the rate-limiting step is a very small-scale event that involves only a minimal change in the RNA structure.
Metal Ion Dependence of the Limiting Step.
To further reveal the molecular origin of the rate-limiting step, we carried out metal ion chevron analysis using four group IIA ions, Mg2+, Ca2+, Sr2+, and Ba2+ (Figs. 2A and 3, Table 1). C domain folds to its native structure in the presence of each ion as confirmed by catalytic activity (data not shown). The chevron plots for all four metal ions have a similar characteristic shape and can be analyzed according to the same kinetic scheme, Ieq-to-I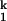 -to-I
-to-I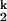 -to-N. The chevron analysis for all metal ions yields the same Hill constant for the Ieq-to-I
-to-N. The chevron analysis for all metal ions yields the same Hill constant for the Ieq-to-I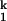 transition (n1 ≈ 2, folding side) and for the I
transition (n1 ≈ 2, folding side) and for the I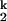 -to-N transition (n2 ≈ 1, unfolding side). Folding and unfolding rates (kf and ku) saturate at high and low metal ion concentration, respectively. This behavior demonstrates that folding induced by all four metal ions goes through the same two kinetic intermediates (I
-to-N transition (n2 ≈ 1, unfolding side). Folding and unfolding rates (kf and ku) saturate at high and low metal ion concentration, respectively. This behavior demonstrates that folding induced by all four metal ions goes through the same two kinetic intermediates (I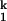 and I
and I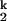 ) and has the same rate-limiting I
) and has the same rate-limiting I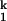 -to-I
-to-I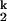 transition.
transition.
Figure 3.

(A) Temperature dependence of the folding rate. (B) Correlation between the activation enthalpy for folding and the dehydration of the metal ions measured by NMR (36).
Table 1.
Folding parameters of C-domain
| Metal ion | Mg2+ | Ca2+ | Sr2+ | Ba2+ |
|---|---|---|---|---|
| kf, s−1 | 0.021 ± 0.002 | 0.027 ± 0.003 | 0.071 ± 0.006 | 0.12 ± 0.01 |
| ku, s−1 | 0.55 ± 0.01 | 0.36 ± 0.02 | 0.14 ± 0.02 | 0.11 ± 0.01 |
| −RT ln (kf/ku), kcal/mol | 1.85 ± 0.05 | 1.47 ± 0.07 | 0.38 ± 0.09 | −0.05 ± 0.08 |
| K1, mM | 0.5 ± 0.1 | 0.5 ± 0.1 | 1.4 ± 0.2 | 2.4 ± 0.2 |
| n1 | 2.1 ± 0.3 | 2.3 ± 1.2 | 2.2 ± 0.5 | 2.2 ± 0.3 |
| K2, mM | 0.011 ± 0.004 | 0.028 ± 0.002 | 0.23 ± 0.04 | 0.27 ± 0.05 |
| n2 | 1.3 ± 0.2 | 1.3 ± 0.1 | 1.4 ± 0.3 | 1.3 ± 0.3 |
| KMe, mM* | 0.40 ± 0.02 | 0.41 ± 0.03 | 0.79 ± 0.03 | 0.72 ± 0.03 |
| ΔH‡, kcal/mol | 23.6 ± 1.0 | 21.2 ± 1.8 | 18.4 ± 1.3 | 11.4 ± 1.1 |
All measurements are in 20 mM Tris⋅HCl (pH 8.1), 10°C, except for ΔH‡ measurements, which are conducted over a temperature range of 2–30°C.
This value is the midpoint of the equilibrium Ieq-to-N transition and is also approximately the vertex of the chevron trace. R, gas constant; T, temperature.
The Hill constant and the midpoint of the equilibrium Ieq-to-N transition can be determined from the kinetic parameters obtained from the chevron analysis. These kinetically determined values agree well with the independently measured equilibrium parameters (Fig. 2A Lower, Table 1). Hence, the free energy and the metal-ion binding of the equilibrium Ieq-to-N transition can be accounted for in the same, minimal kinetic scheme for each of the four metal ions.
For all metal ions, both kf and ku saturate at high and low ion concentrations, respectively. This result confirms that the rate-limiting I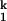 -to-I
-to-I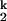 step does not involve bimolecular binding of a free metal ion to the RNA. Rather, the limiting step is a unimolecular event. On the other hand, kf and ku do depend on the particular divalent metal ion used to fold the RNA. Therefore, the rate-limiting step does involve at least one specific metal ion. These metal ions are already bound in the I
step does not involve bimolecular binding of a free metal ion to the RNA. Rather, the limiting step is a unimolecular event. On the other hand, kf and ku do depend on the particular divalent metal ion used to fold the RNA. Therefore, the rate-limiting step does involve at least one specific metal ion. These metal ions are already bound in the I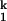 intermediate. Because the rate-limiting step involves a metal ion, but is accompanied by little change in the RNA structure, we conclude that the rate-limiting step is a small-scale conformational change, or structural consolidation involving prebound metal ions.
intermediate. Because the rate-limiting step involves a metal ion, but is accompanied by little change in the RNA structure, we conclude that the rate-limiting step is a small-scale conformational change, or structural consolidation involving prebound metal ions.
The metal ion dependence of kf and ku has a linear dependence on the reciprocal of the ionic radius. As the ionic radius increases (Mg2+ < Ca2+ < Sr2+ < Ba2+), the folding rate increases, whereas the unfolding rate decreases by almost equal measures. Therefore, the consolidation step is more destabilizing for the smaller metal ion [ΔG = −RT ln (kf/ku), Table 1]. For the smallest ion, Mg2+, the consolidation requires 1.9 kcal⋅mol−1 of free energy, whereas for the largest ion, Ba2+, the consolidation is energetically neutral.
The binding affinity of a metal ion to the RNA (K1, K2, and KMe, Table 1) is stronger for the ion with smaller radius. In the initial bimolecular binding event, smaller ions are preferred. But, in the subsequent unimolecular rate-limiting step, larger ions are favored. In short, the stronger the metal ion binds to the RNA, the slower is the folding rate and the faster is the unfolding rate.
Further indication that prebound metal ions are involved in the rate-limiting step is derived from the temperature dependence of the folding rate, kf (Fig. 3A). At a first glance, the activation enthalpy appears to correlate with the binding affinity of the metal ion to the RNA—the stronger the binding affinity, the higher is the activation enthalpy. A correlation also exists between the activation enthalpies and those of dehydration for these metal ions (ref. 36, Fig. 3B), potentially indicative of a change in the hydration state of the metal ions at the limiting step.
Discussion
How a large RNA folds into its biologically active structure has been intensely studied in the last decade. Treiber and Williamson (37) have proposed that the general RNA-folding mechanisms can be summarized into three classes. The first class includes fast folding RNAs with sequentially forming and productive intermediates. The second class includes slow folding RNAs with kinetic traps. The third class includes slow folding RNAs that result in unstable tertiary structures, which are subsequently captured by another molecule, e.g., a protein (21). Recently, Pyle and coworkers (24) proposed a fourth class wherein folding is trap-free, but still slow because of a conformational search for a key tertiary interaction early in the pathway. The B. subtilis C domain belongs to the first class of RNAs that fold with sequentially populated and productive intermediates. Because the C domain folds without kinetic traps, several important aspects of the RNA folding problem can be directly approached. In this work, we examined the speed and the extent of conformational search, the role of the productive intermediates, and the nature of the rate-limiting step (Fig. 4).
Figure 4.

Summary of C-domain folding. The rate-limiting step is shown in red. The folding half-lives at 10°C are shown, in seconds, above the arrows. The starting point is the Uurea state (C domain in 8 M urea and 1 mM EDTA), which has little RNA structure. Ieq contains the native amount of secondary structure and some tertiary structure. I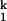 contains >98% of the native structure. In less than 1 ms, the majority of the conformational search occurs with the formation of Ieq. All folding half-lives are measured in this work. Percent compaction is calculated from the data shown in Fig. 2B and from our previous work (31). Percent m-value change is calculated from published data (15).
contains >98% of the native structure. In less than 1 ms, the majority of the conformational search occurs with the formation of Ieq. All folding half-lives are measured in this work. Percent compaction is calculated from the data shown in Fig. 2B and from our previous work (31). Percent m-value change is calculated from published data (15).
In C-domain folding, nearly all conformational search occurs before the rate-limiting step. Regardless of the residual structure, the addition of sufficient amount of Mg2+ allows the C-domain to fold into the first well-defined intermediate, Ieq, in less than 1 ms. When starting from the fully denatured state, Uurea, formation of Ieq compacts the RNA to ≈90% of the native dimension. The subsequent formation of the second intermediate, I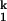 , further compacts the RNA to ≈98% of the native dimension. In each step, the large-scale compaction is accompanied by an extensive conformational search.
, further compacts the RNA to ≈98% of the native dimension. In each step, the large-scale compaction is accompanied by an extensive conformational search.
The present results, along with those from the group I ribozyme (38), indicate that when the RNA is permitted to overcome the electrostatic repulsion of its negatively charged backbone, it can compact extremely rapidly. Folding of the full-length RNase P RNA also involves very rapid compaction from the unfolded state to an intermediate state at the same time scale as in the folding of C domain (8). In the case of the full-length P RNA, however, a nonnative structure forms in the intermediate state and this significant nonnative structure has to be disrupted for the folding to proceed. The primary reason for the rapid, trap-free folding of the C domain to the native state is likely the early conformational search resulting in the formation of correct structures in Ieq and in I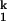 .
.
Three intermediates can be clearly identified in the folding kinetics of C-domain carried out in the four different metal ions, Mg2+, Ca2+, Sr2+, and Ba2+. The first intermediate, Ieq, is the same as the thermodynamic intermediate and it significantly populates at low Me2+ concentrations. The second and third intermediates, I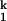 and I
and I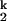 , only populate transiently, with a dwell time of ≈30 s for I
, only populate transiently, with a dwell time of ≈30 s for I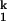 and ≈1 s for I
and ≈1 s for I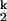 when folding in Mg2+.
when folding in Mg2+.
All three intermediates appear to be productive in folding. We have not been able to find conditions that permit the C domain to bypass any of them, and an increase in their stability enhances relaxation rates. Because the Ieq and the native state have the same UV absorbance, we suggested previously that all secondary structures of the C domain are already present in Ieq (32). On the basis of comparative studies with a thermophilic homologue (34), we also suggested that some tertiary structures are present in Ieq as well. Compared with Ieq, I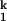 clearly has more structure and binds more metal ions (the Hill coefficient of the Ieq-to-I
clearly has more structure and binds more metal ions (the Hill coefficient of the Ieq-to-I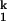 transition is ≈2). At this time, we have little information on the precise structure of these intermediates.
transition is ≈2). At this time, we have little information on the precise structure of these intermediates.
The rate-limiting step in C-domain folding is the conversion of two intermediates, from I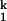 to I
to I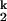 . We have shown that this step has no dependence on the Mg2+ and urea concentration (15). We demonstrate here that this step is always limiting when folding is carried out in other divalent metal ions. More importantly, the metal-ion chevron analysis demonstrates that the limiting step still involves metal ions that are prebound in I
. We have shown that this step has no dependence on the Mg2+ and urea concentration (15). We demonstrate here that this step is always limiting when folding is carried out in other divalent metal ions. More importantly, the metal-ion chevron analysis demonstrates that the limiting step still involves metal ions that are prebound in I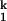 . The hydration properties of these metal ions may play a role as well. The simplest interpretation we can envision is that the limiting step represents a small-scale consolidation of RNA structure around prebound metal ions. Specifically bound divalent metal ions are present in the structure of all large RNAs. These metal ions are located in sites of high electrostatic potential, which stabilize interactions between different structural motifs, and hence, are crucial for maintaining the tertiary architecture (39–41). It is likely that the formation of one or more of these binding sites is rate-limiting, although concrete experimental evidence is still lacking.
. The hydration properties of these metal ions may play a role as well. The simplest interpretation we can envision is that the limiting step represents a small-scale consolidation of RNA structure around prebound metal ions. Specifically bound divalent metal ions are present in the structure of all large RNAs. These metal ions are located in sites of high electrostatic potential, which stabilize interactions between different structural motifs, and hence, are crucial for maintaining the tertiary architecture (39–41). It is likely that the formation of one or more of these binding sites is rate-limiting, although concrete experimental evidence is still lacking.
Models for Consolidation.
The dependence of the rate-limiting step on the size of the prebound metal ion suggests that structural changes occur in the rate-limiting step. There is a trend in the folding and unfolding rate regarding the four group IIA metal ions—the higher is the binding affinity of the metal ion to the RNA, the slower is the folding rate and the faster is the unfolding rate. Several models involving specific metal ion binding can be proposed to explain this trend.
(i) An interaction between an RNA ligand and metal ion is broken to allow a nucleotide base to flip, for example, and then the same or a different metal ion–RNA interaction forms. Allowing the same interaction to form would predict an increase in folding and unfolding rates with larger metal ions. Although increased folding rates are observed, the opposite trend is observed for the unfolding rate. Hence, the metal ion–RNA interaction formed after the rate-limiting step has to be different from that formed before the rate-limiting step, and be weaker for the smaller ions.
(ii) An RNA-RNA structural rearrangement, e.g., switching of a sugar pucker, occurs without the loss of RNA-metal ion interactions. This structural rearrangement would be retarded because of the increased stabilization of the I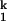 state through tighter binding with smaller ions. Again, for this model to be consistent with the unfolding rates, the RNA structure formed after the rate-limiting step has to have lower affinity for smaller metal ions.
state through tighter binding with smaller ions. Again, for this model to be consistent with the unfolding rates, the RNA structure formed after the rate-limiting step has to have lower affinity for smaller metal ions.
(iii) The rate-limiting step involves the release of a prebound metal ion at a high affinity site in I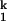 and rebinding at a low affinity site in I
and rebinding at a low affinity site in I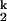 . This model can rationalize the observed trend for the folding rate, as a higher affinity ion would be released more slowly from the high-affinity site. As before, in order for this model to be consistent with the unfolding rates, the RNA structure formed after the rate-limiting step has to exhibit a lower affinity for smaller metal ions.
. This model can rationalize the observed trend for the folding rate, as a higher affinity ion would be released more slowly from the high-affinity site. As before, in order for this model to be consistent with the unfolding rates, the RNA structure formed after the rate-limiting step has to exhibit a lower affinity for smaller metal ions.
(iv) Consolidation of the RNA around smaller ions is more difficult because of steric and/or electrostatic crowding of the negatively charged RNA backbone. A more detailed model, based on the crystallographic observation of multiple inner-sphere metal-phosphate coordination, envisions that the I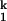 to I
to I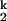 transition involves an increase in the number of RNA ligands around the prebound divalent metal ions. This model rationalizes the slower kf and the faster ku, as well as the free energy difference between I
transition involves an increase in the number of RNA ligands around the prebound divalent metal ions. This model rationalizes the slower kf and the faster ku, as well as the free energy difference between I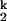 and I
and I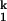 . Within an extremely constrained environment, a smaller ion shuns the bulkier and charged RNA ligand. The overall binding affinity for the particular metal ions in the rate-limiting step, however, may still be higher for the smaller ions. This increase in affinity is caused by the more favorable metal ion binding for the smaller ions on the formation of I
. Within an extremely constrained environment, a smaller ion shuns the bulkier and charged RNA ligand. The overall binding affinity for the particular metal ions in the rate-limiting step, however, may still be higher for the smaller ions. This increase in affinity is caused by the more favorable metal ion binding for the smaller ions on the formation of I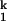 (K1 values in Table 1). The overall binding affinity would represent the combined free energy of the favorable binding step and the unfavorable, increased electrostatic and steric crowding after the rate-limiting step.
(K1 values in Table 1). The overall binding affinity would represent the combined free energy of the favorable binding step and the unfavorable, increased electrostatic and steric crowding after the rate-limiting step.
C-domain folding occurs many orders of magnitude slower than what one would expect for a simple ligand-exchange reaction. This discrepancy may be explained in part by the state of the metal ion that is already bound to the negatively charged RNA ligand, as well as a term related to RNA conformational change. The prebound metal ion probably is much less electrophilic. Thus, the rate of the ligand exchange on the C domain should be significantly slower than the rate of ligand exchange on a metal-ion free in solution. In the folding of our RNA, the conformational component must be consistent with the negligible amount of surface burial and compaction in the rate-limiting step. This component could represent the frequency of productive contacts in a small-scale conformational search.
Conformational Search in Protein and RNA Folding.
The folding of protein and RNA poses a considerable conformational search problem. However, the mode of search and the limiting steps have significant differences. In protein folding, bigger search units (e.g., α-helices) may form before the transition state, but these units are not stable, and the same stretch of amino acid residues may transiently adopt different conformations during the search. Such structural flexibility of protein secondary structure entails that the intrinsic rate-limiting step is a global conformational search involving all residues. The key element is the formation of the native topology of the backbone (42, 43), which often is accompanied by the formation of a hydrophobic core with long-range contacts along with the hydrogen-bond network needed to support this association (44).
In RNA folding, secondary structure often is independently stable and can form on the microsecond time-scale. A tertiary structure involving a three-way helical junction can form in a millisecond (45). Error-free folding can occur if the formation of stable nonnative structure is avoided, which depends on the sequence (21, 24, 37, 46) and connectivity (14). The rapid formation of stable secondary structure allows the search process to be conducted among preformed structural units whose number is significantly smaller than the total number of nucleotide residues. These units in turn assemble into a tertiary structure through nonlocal contacts mediated by loops, packed helices, and bridging metal ions. Two scenarios have been observed in tertiary RNA folding. In some cases, conformational search is rate-limiting because of the difficulty of forming key tertiary interactions (24). For the C domain, structural consolidation around prebound metal ions is rate-limiting. This event occurs after nearly the entire conformational search, and serves to lock down the tertiary structure at a late stage in folding.
Acknowledgments
We thank Drs. P. Cluzel, J. Piccirilli, J. R. Williamson, D. Herschlag, S. Chu, and J. Staley and the reviewers for helpful discussions and comments on the manuscript.This work was supported by National Institutes of Health Grant GM57880 (to T.P. and T.R.S.), and the U. S. Department of Energy, BES-Materials Science under Contract W-31-109-ENG-38.
Abbreviations
- C domain
catalytic domain
- N
native state
- Rg
radius of gyration
- SAXS
small-angle X-ray scattering
- U
unfolded state
References
- 1.Cole P E, Crothers D M. Biochemistry. 1972;11:4368–4374. doi: 10.1021/bi00773a025. [DOI] [PubMed] [Google Scholar]
- 2.Webb P K, Fresco J R. J Mol Biol. 1973;74:387–402. doi: 10.1016/0022-2836(73)90379-3. [DOI] [PubMed] [Google Scholar]
- 3.Lynch D C, Schimmel P R. Biochemistry. 1974;13:1841–1852. doi: 10.1021/bi00706a012. [DOI] [PubMed] [Google Scholar]
- 4.Zarrinkar P P, Williamson J R. Science. 1994;265:918–924. doi: 10.1126/science.8052848. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee A R, Turner D H. Biochemistry. 1995;34:6504–6512. doi: 10.1021/bi00019a031. [DOI] [PubMed] [Google Scholar]
- 6.Zarrinkar P P, Williamson J R. Nat Struct Biol. 1996;3:432–438. doi: 10.1038/nsb0596-432. [DOI] [PubMed] [Google Scholar]
- 7.Zarrinkar P P, Wang J, Williamson J R. RNA. 1996;2:564–573. [PMC free article] [PubMed] [Google Scholar]
- 8.Pan T, Sosnick T R. Nat Struct Biol. 1997;4:931–938. doi: 10.1038/nsb1197-931. [DOI] [PubMed] [Google Scholar]
- 9.Pan J, Thirumalai D, Woodson S A. J Mol Biol. 1997;273:7–13. doi: 10.1006/jmbi.1997.1311. [DOI] [PubMed] [Google Scholar]
- 10.Treiber D K, Rook M S, Zarrinkar P P, Williamson J R. Science. 1998;279:1943–1946. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- 11.Sclavi B, Sullivan M, Chance M R, Brenowitz M, Woodson S A. Science. 1998;279:1940–1943. doi: 10.1126/science.279.5358.1940. [DOI] [PubMed] [Google Scholar]
- 12.Pan J, Woodson S A. J Mol Biol. 1998;280:597–609. doi: 10.1006/jmbi.1998.1901. [DOI] [PubMed] [Google Scholar]
- 13.Rook M S, Treiber D K, Williamson J R. Proc Natl Acad Sci USA. 1999;96:12471–12476. doi: 10.1073/pnas.96.22.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan T, Fang X, Sosnick T R. J Mol Biol. 1999;286:721–731. doi: 10.1006/jmbi.1998.2516. [DOI] [PubMed] [Google Scholar]
- 15.Fang X, Pan T, Sosnick T R. Nat Struct Biol. 1999;6:1091–1095. doi: 10.1038/70016. [DOI] [PubMed] [Google Scholar]
- 16.Silverman S K, Deras M L, Woodson S A, Scaringe S A, Cech T R. Biochemistry. 2000;39:12465–12475. doi: 10.1021/bi000828y. [DOI] [PubMed] [Google Scholar]
- 17.Deras M L, Brenowitz M, Ralston C Y, Chance M R, Woodson S A. Biochemistry. 2000;39:10975–10985. doi: 10.1021/bi0010118. [DOI] [PubMed] [Google Scholar]
- 18.Pan J, Deras M L, Woodson S A. J Mol Biol. 2000;296:133–144. doi: 10.1006/jmbi.1999.3439. [DOI] [PubMed] [Google Scholar]
- 19.Zhuang X, Bartley L E, Babcock H P, Russell R, Ha T, Herschlag D, Chu S. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 20.Kent O, Chaulk S G, MacMillan A M. J Mol Biol. 2000;304:699–705. doi: 10.1006/jmbi.2000.4263. [DOI] [PubMed] [Google Scholar]
- 21.Buchmueller K L, Webb A E, Richardson D A, Weeks K M. Nat Struct Biol. 2000;7:362–366. doi: 10.1038/75125. [DOI] [PubMed] [Google Scholar]
- 22.Heilman-Miller S L, Pan J, Thirumalai D, Woodson S A. J Mol Biol. 2001;309:57–68. doi: 10.1006/jmbi.2001.4660. [DOI] [PubMed] [Google Scholar]
- 23.Russell R, Herschlag D. J Mol Biol. 2001;308:839–851. doi: 10.1006/jmbi.2001.4751. [DOI] [PubMed] [Google Scholar]
- 24.Swisher J F, Su L J, Brenowitz M, Anderson V E, Pyle A M. J Mol Biol. 2002;315:297–310. doi: 10.1006/jmbi.2001.5233. [DOI] [PubMed] [Google Scholar]
- 25.Cate J H, Hanna R L, Doudna J A. Nat Struct Biol. 1997;4:553–558. doi: 10.1038/nsb0797-553. [DOI] [PubMed] [Google Scholar]
- 26.Ferre-D'Amare A R, Zhou K, Doudna J A. Nature (London) 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 27.Shi H, Moore P B. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jovine L, Djordjevic S, Rhodes D. J Mol Biol. 2000;301:401–414. doi: 10.1006/jmbi.2000.3950. [DOI] [PubMed] [Google Scholar]
- 29.Ban N, Nissen P, Hansen J, Moore P B, Steitz T A. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 30.Rupert P B, Ferre-D'Amare A R. Nature (London) 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 31.Fang X, Littrell K, Yang X, Henderson S J, Siefert S, Thiyagarajan P, Pan T, Sosnick T R. Biochemistry. 2000;39:11107–11113. doi: 10.1021/bi000724n. [DOI] [PubMed] [Google Scholar]
- 32.Fang X, Pan T, Sosnick T R. Biochemistry. 1999;38:16840–16846. doi: 10.1021/bi991700n. [DOI] [PubMed] [Google Scholar]
- 33.Seifert S, Winans R E, Tiede D M, Thiyagarajan P. J Appl Crystallogr. 2000;33:782–784. [Google Scholar]
- 34.Fang X W, Golden B L, Littrell K, Shelton V, Thiyagarajan P, Pan T, Sosnick T R. Proc Natl Acad Sci USA. 2001;98:4355–4360. doi: 10.1073/pnas.071050698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shelton V M, Sosnick T R, Pan T. Biochemistry. 1999;38:16831–16839. doi: 10.1021/bi991699s. [DOI] [PubMed] [Google Scholar]
- 36.Helm L, Hertz H G. Z Physik Chem. 1981;127:23–44. [Google Scholar]
- 37.Treiber D K, Williamson J R. Curr Opin Struct Biol. 2001;11:309–314. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 38.Russell R, Millett I S, Tate M W, Kwok L W, Nakatani B, Gruner S M, Mochrie S G, Pande V, Doniach S, Herschlag D, Pollack L. Proc Natl Acad Sci USA. 2002;99:4266–4271. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermann T, Westhof E. Structure (London) 1998;6:1303–1314. doi: 10.1016/s0969-2126(98)00130-0. [DOI] [PubMed] [Google Scholar]
- 40.Chin K, Sharp K A, Honig B, Pyle A M. Nat Struct Biol. 1999;6:1055–1061. doi: 10.1038/14940. [DOI] [PubMed] [Google Scholar]
- 41.Misra V K, Draper D E. J Mol Biol. 2000;299:813–825. doi: 10.1006/jmbi.2000.3769. [DOI] [PubMed] [Google Scholar]
- 42.Sosnick T R, Mayne L, Englander S W. Proteins. 1996;24:413–426. doi: 10.1002/(SICI)1097-0134(199604)24:4<413::AID-PROT1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 43.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 44. Krantz, B. A., Srivastava, A. K., Nauli, S., Baker, D., Sauer, R. T. & Sosnick, T. R. (2002) Nat. Struct. Biol., in press. [DOI] [PubMed]
- 45.Kim H D, Nienhaus G U, Ha T, Orr J W, Williamson J R, Chu S. Proc Natl Acad Sci USA. 2002;99:4284–4289. doi: 10.1073/pnas.032077799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodson S A. Cell Mol Life Sci. 2000;57:796–808. doi: 10.1007/s000180050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown J W, Nolan J M, Haas E S, Rubio M A, Major F, Pace N R. Proc Natl Acad Sci USA. 1996;93:3001–3006. doi: 10.1073/pnas.93.7.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]