

Abstract
Previous structural studies on native T5 5′ nuclease, a member of the flap endonuclease family of structure-specific nucleases, demonstrated that this enzyme possesses an unusual helical arch mounted on the enzyme's active site. Based on this structure, the protein's surface charge distribution, and biochemical analyses, a model of DNA binding was proposed in which single-stranded DNA threads through the archway. We investigated the kinetic and substrate-binding characteristics of wild-type and mutant nucleases in relation to the proposed model. Five basic residues R33, K215, K241, R172, and R216, are all implicated in binding branched DNA substrates. All these residues except R172 are involved in binding to duplex DNA carrying a 5′ overhang. Replacement of either K215 or R216 with a neutral amino acid did not alter kcat appreciably. However, these mutant nucleases displayed significantly increased values for Kd and Km. A comparison of flap endonuclease binding to pseudoY substrates and duplexes with a single-stranded 5′ overhang suggests a better model for 5′ nuclease-DNA binding. We propose a major revision to the binding model consistent with these biophysical data.
The flap endonucleases, or 5′ nucleases, are structure-specific endonucleases that also possess 5′–3′ exonucleolytic activity. They are involved in processing substrates with 5′ single-stranded tails such as those that arise during nick translation and replication and in some DNA damage-repair pathways (1, 2). These enzymes bind substrates containing a single-stranded 5′ end and a duplex region such as 5′ overhangs (5OVHs), 5′ flaps, and pseudoY (Ps-Y) substrates (Fig. 1; refs. 3–5). It has been suggested that the single-stranded 5′ tail of such substrates threads through the 5′ nuclease (4). Several prokaryotic and archaeal flap endonuclease structures have been solved (6–10). Because none of the reported structures contained bound DNA substrates, the precise mode of nucleic acid binding is unclear. These nucleases share significant primary sequence conservation as well as extensive structural similarities (11–14). A central β-sheet carrying many of the core metal-binding ligands is a striking feature of all 5′-nuclease structures reported to date. This core region contains acidic residues responsible for binding the two divalent metal ions required for nuclease activity in vitro. A series of helices and loops comprise the enzyme's core. The most variable region seems to be that comprising a helical arch in T5 5′ nuclease, the observation of which led us to develop a model of substrate binding (ref. 8; Fig. 2). In T5 5′ nuclease, the archway delineates a hole able to accommodate a threaded single-stranded nucleic acid. Although a similar hole is present in the Methanococcus jannaschii homologue (9), this region often appears disordered (6, 7) or as a folded loop in the structure of the Pyrococcus furiosus homologue (10). The conceptual model for flap endonuclease-DNA binding has been widely accepted (1, 7, 9, 10, 15, 16). Nevertheless, this model suffers from uncertainty with regard to the orientation of the flap substrate duplex because of a lack of experimental data identifying key protein–DNA interactions.
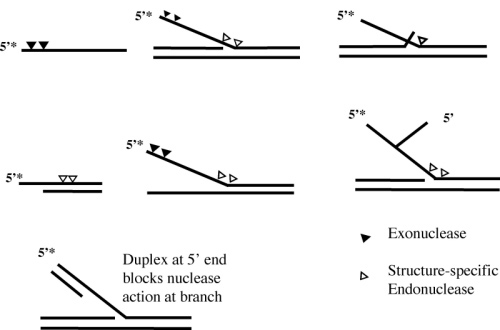
Figure 1.

Diagrammatic representation of the flap, Ps-Y, 5OVH, and HP1 oligonucleotide substrates referred to in this study. For each substrate the 5′-32P end-labeled oligonucleotide is shown as 5′-P. Exonucleolytic and structure-specific endonucleolytic cleavage sites are identified by filled and open triangles, respectively. I and II indicate duplex regions I and II, respectively.
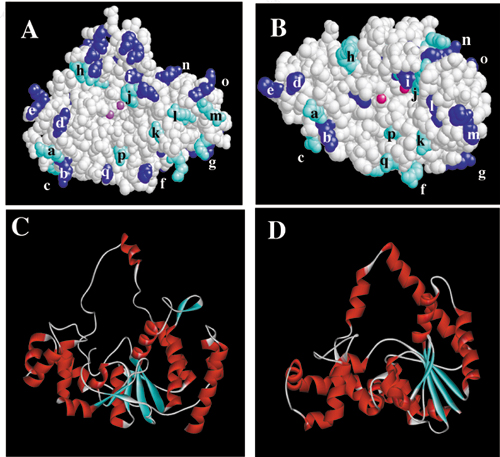
Figure 2.

(A) The structure (PDB ID code 1EXN) determined for T5 5′ nuclease showing the helical arch (red backbone), divalent metal ions (gray spheres), and space-filling representations of selected lysine (blue) and arginine (cyan) residues. (B) Original DNA-binding model proposed by Ceska et al. (8). The duplex parts (I and II) of the substrate lie across a slightly concave and positively charged region of the protein. In this model residues K241, K215, and R216 contact duplex I, and R172 contacts duplex II.
Binding studies with Ps-Y or flap substrates have been performed on mutant 5′ nucleases lacking conserved basic residues near their active sites (15, 17). Mutagenesis of the K83 in the T5 nuclease, a conserved residue at the base of the helical arch, severely compromises both exonuclease activity and the DNA-binding ability of the enzyme. However, the K83A substitution retained reduced but significant flap endonuclease activity. These observations support a role for K83 in catalysis and substrate binding and are compatible with the threading model proposed previously (4). However, the threading model does not readily account for some reactions catalyzed by the 5′ nucleases. Single-stranded circular DNA (18, 19) and double-stranded plasmid DNA can be cleaved under forcing conditions (20), implying that a 5′ end is not an absolute requirement for activity. Bambara and coworkers (21) showed that flap endonuclease I (FEN1, a eukaryotic 5′ nuclease) can effect endonucleolytic cleavage of substrates with 5′ tails containing 2′–5′ branch points (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). The same group also produced evidence to support a threading model, showing that a short duplex region at the extreme 5′ end of single-stranded tail inhibits the nuclease (see Fig. 6; ref. 22).
In this study we turned our attention to the roles of basic residues found near the C-terminal region of the T5 5′ nuclease on a helix–loop–helix/helix–3-turn–helix motif (10, 13) and those predicted to contact the DNA substrate in the model initially proposed. In this model, lysine and arginine residues (R33, R172, K215, R216, and K241) contact duplex regions of the DNA substrate (Fig. 2). Site-directed mutagenesis was used to modify the T5 5′-nuclease gene so as to substitute these basic amino acids with neutral residues, and their involvement in substrate binding was determined. Comparison of the results obtained with Ps-Y and 5OVH substrates sheds light on the likely structure of the protein–DNA complex. The original DNA-binding model presented for T5 5′ nuclease (8) is not fully compatible with the results of the quantitative binding assays presented herein. We propose a model for flap endonuclease–DNA interaction.
Materials and Methods
Site-Directed Mutagenesis and Enzyme Preparation.
Mutagenesis was carried out on an M13 derivative carrying the T5 D15 exonuclease gene (23) as described (24) in conjunction with the following oligonucleotides: R33A, d(TTACTATTGTTATGTTTGAAGGCAAAGCCTAAGTTAGTTCCAT); R172A, d(TATCACGAAGATGATACTCACGAGCTGTTGTGAAAGAAAAACG); K215A/R216S, d(AATATTATATCCGCTTGCTGCTCCTATTCC); K215R/R216K, d(CGAATAATATTATATCCTTTTCGTGCTCCTATTCC); R216A, d(CGAATAATATTATATCCGGCTTTTGCTCCTAT); and K241A, d(ATTCAGGTTCTGTATATATGCCTGCTTTCCAGGCAGTGGAA). Mutations resulting in codon changes are underlined. Dideoxy sequencing determined that only the desired sequence changes had been introduced. Mutated genes were subcloned into the expression vector pJONEX4 as described for the wild-type exonuclease gene (18). Mutant proteins were expressed and purified by ion-exchange chromatography until free of any detectable contaminating 3′–5′ exonuclease or endonuclease activity as described (23) except that heparin chromatography of the DNA-binding mutants required a pH of 6.5 (instead of pH 7.5) for efficient binding. T5 5′ nuclease carrying the K215A substitution was prepared as described (17).
Structure-Specific DNA-Cleavage Profiles.
The Ps-Y was prepared by annealing the 5′-32P end-labeled F1 oligonucleotide (Fig. 1), 3 nM, with the B1 oligonucleotide, 100 nM, heating to 80°C for 5 min, and then leaving at room temperature for 1 h in 25 mM potassium glycinate (pH 9.3)/100 mM KCl (3). Assays were diluted to contain 4.5 nM unlabeled oligonucleotide, 150 pM labeled oligonucleotide in 25 mM potassium glycinate (pH 9.3), 100 mM KCl, 10 mM MgCl2, 5% glycerol, 1 mM DTT, and 0.1 mg/ml acetylated BSA. The 5OVH substrate was prepared similarly by using 5′-32P end-labeled B1 annealed to an excess of unlabeled oligonucleotide In1, which produced a substrate with a duplex region of 16 bp and a 5′ end-labeled 14-nt single-stranded 5OVH. Reactions were terminated by the addition of an equal volume of 95% formamide/15 mM EDTA stop mix. Products were separated on a denaturing 7 M urea/15% polyacrylamide gel (50 cm), run in 1× TBE [90 mM Tris/64.6 mM boric acid/2.5 mM EDTA (pH 8.3)] at 50 W for 2 h, and visualized on a BioRad Molecular Imager phosphorimager. Quantification of the image was carried out by using Molecular Dynamics software.
Determination of Dissociation Constants.
Electrophoretic mobility-shift assays were carried out on 32P end-labeled flap, Ps-Y, and 5OVH substrates in the absence of divalent metal ions as described (17). Reactions contained 150 pM labeled oligonucleotide in 25 mM potassium glycinate/100 mM KCl/1 mM EDTA/5% glycerol/1 mM DTT/0.1 mg/ml acetylated BSA and enzyme at the appropriate concentration. Reactions (10 μl) were incubated on ice for 10 min and analyzed on a 17% native acrylamide gel in 50 mM Tris-Bicine (pH 8.3)/1 mM EDTA/1 mM DTT at 4°C for 2 h at 15 V/cm. The gel was analyzed by using a Molecular Imager (BioRad) and Molecular Dynamics software.
Kinetic Analysis.
Comparative specific activities were determined by measuring the rate of release of acid-soluble nucleotides from high molecular weight DNA (herring sperm type XIV, Sigma) in a UV spectrophotometric assay essentially as described (23). Assays (600 μl) contained 2 mM substrate DNA (in nucleotides), 25 mM potassium glycinate (pH 9.3), 1 μg of enzyme, and 10 mM MgCl2. Curves were plotted from the data obtained (from at least duplicate experiments), and estimates of the initial velocity were calculated.
Catalytic parameters of wild-type and R216A mutant proteins were determined by using HP1 substrate (Fig. 1) as described (25). HP1 concentrations were 0.005–1 μM for wild type and 0.1–20 μM for R216A with enzyme concentrations in the ranges of 5–30 pM for wild type and 0.12–1.8 nM for R216A.
Results
Structure-Specific Cleavage of DNA.
Cleavage specificity of mutant proteins was characterized by using Ps-Y and 5OVH substrates (Fig. 3). All mutant enzymes tested (R172A, K215A, R216A, K215R/R216K, K215I/R216S, and K241A) gave qualitatively similar patterns of hydrolysis by using either the Ps-Y (representative results presented in Fig. 3A) or flap (data not shown) substrates. However, some differences in the ratios of products generated by the wild type and mutants were observed, e.g., more of the 22-nt-long product is produced by the mutant nucleases when compared with the wild-type enzyme. In the latter case most of the radiolabeled products run as trimers or pentamers, whereas reactions catalyzed by the modified proteins produce predominantly endonucleolytic products (19, 21, and 22 mers). The major cleavage product obtained with the 5′ end-labeled 5OVH substrate arose from hydrolysis of the second base-paired nucleotide in the duplex region yielding a product 16 nt in length. Both wild-type and mutant enzymes gave very similar patterns of 5OVH digestion (Fig. 3B).
Figure 3.

Structure-specific cleavage of Ps-Y (A) and 5OVH substrates (B) by wild-type (WT) and mutant enzymes. Enzymes, at ≈1 nM wild-type and 10 nM for the mutants, were incubated at 37°C with substrate DNA [strand F1 labeled in Ps-Y (A) or B1 in 5OVH (B), 150 pM] in the presence of 10 mM MgCl2 for 0.5, 2, 5, 10, and 20 min (indicated by the filled triangles). Reaction products were separated by denaturing PAGE and visualized by autoradiography. Controls (0) that lacked enzyme but contained all other reactants were incubated for 30 min. Single nucleotide ladders (M) were produced by digestion of labeled F1 (A) or B1 (B) oligonucleotide with snake venom phosphodiesterase. Mutant exonucleases R33A, R172A, K241A, and K215A gave very similar results to those above (data not shown).
Quantitative Electrophoretic Mobility-Shift Assays.
The ability of the mutant proteins to bind Ps-Y and 5OVH DNA substrates was assayed by using an electrophoretic mobility-shift assay. All the modified nucleases bound the Ps-Y substrate less tightly than did the parent enzyme. Substitutions R216A and K241A produced the most dramatic impact on binding for single mutations, increasing the dissociation constant from 5–10 nM to over 500 and 850 nM, respectively. Binding of the double substitution K215A/R216S to DNA could not be detected (Kd > 10 μM). Exchanging two adjacent residues K215R/R216K with one another had less impact on Ps-Y DNA-binding affinity, resulting in a Kd of 61 nM.
The 5OVH substrate was bound more weakly than Ps-Y by all the enzymes. The dissociation constant for wild-type enzyme binding to the 5OVH substrate (Kd = 85 nM, range of 66–93 nM) increases 10–20-fold relative to the Kd for Ps-Y, implying that the 3′ arm (present in the Ps-Y substrate) makes substantial contacts with the protein. The R172A mutant bound the 5OVH substrate with the same affinity as wild type within the limits of experimental error (Kd = 93 nM). The R33A substitution caused only a modest increase in dissociation constant (230 nM). In contrast, the K241A mutant bound 5OVH weakly (Kd = 852 nM), and all the other mutant enzymes tested had vastly decreased affinity (Kd > 10,000 nM) for this substrate, which implies that residues R33, K215, R216, and K241 are involved in binding to the 5OVH substrate and R172 is not. These results are presented in Fig. 4 and Table 1. The wild-type protein bound the full flap structure and Ps-Y substrate with similar affinity, 10 vs. 4 nM, respectively (data not shown).
Figure 4.

Substrate binding by T5 5′ nucleases in the absence of cofactor. (A) Enzymes were incubated with Ps-Y substrate on ice, and then the enzyme–substrate complex was separated from unbound substrate by electrophoresis on a native gel. The enzyme concentrations were varied while the substrate concentration was kept at 150 pM for each experiment. Lanes marked “+” contained labeled 3 μM wild-type enzyme as a positive control. (B) Data from each experiment (n = 3 or more) were plotted as the percentage of unbound substrate against log[enzyme]. The error bars represent maximum and minimum values obtained at each protein concentration, and regression coefficients were >0.96. Dissociation constants (Kd) were determined for each enzyme as the enzyme concentration required to bind 50% of the substrate. Filled squares, filled triangles, filled circles, and open triangles denote wild type, R172A, K241A, and R216A, respectively. (C and D) As described for A and B but with labeled 5OVH substrate. Binding curves are shown for wild-type, K241A, and R172A enzymes. Table 1 summarizes these data.
Table 1.
Relative activity and substrate binding
| Nuclease | Relative activity* | Dissociation constant, nM
|
|
|---|---|---|---|
| Ps-Y | 5OVH | ||
| Wild type | 1.00 | 4 ± 1 | 85 ± 6 |
| R33A | 0.5 | 51 ± 20 | 230 ± 31 |
| R172A | 1.12 | 42 ± 14 | 93 ± 5 |
| K215A | 0.78 | 48† | >10,000 |
| R216A | 0.66 | 504 ± 150 | >10,000 |
| 215R/216K | 0.93 | 62 ± 6 | >10,000 |
| 215A/216S | 0.40 | >10,000 | >10,000 |
| K241A | 1.13 | 56 ± 5 | 852 ± 83 |
Relative activity is defined as the specific activity of each enzyme determined by using a spectrophotometric assay divided by the specific activity of the wild-type enzyme. The apparent increase in specific activity observed for R172A and K241A is insignificant within the limits of experimental error.
Reported previously (17).
Rates of Reaction and Specific Activity of Mutant Enzymes.
All the enzymes showed levels of specific activity approaching that of the wild-type enzyme under the conditions of high substrate concentrations used to determine specific activity on high molecular weight DNA. The K215A/R216S protein was the worst affected, but even it retained 40% of wild-type specific activity, followed by R33A (50%), R216A (66%), and K215A (78%) relative to that obtained for the wild-type protein (54–58 nmol per minute per μg of protein). The lysine/arginine swap (K215R/R216K) and the R172A and K241A substitutions showed no significant reduction in specific activity within the limits of experimental error (Table 1).
Steady-state kinetic analysis was performed by using the 5′-overhanging hairpin substrate HP1 (results shown in Table 2). This substrate undergoes a single endonucleolytic cleavage reaction to generate products of 8 and 21 nt in length (25). The catalytic parameters reported here for wild-type protein (kcat = 101 min−1, Km = 70 nM) agree with those reported earlier (25). Replacement of R216 with alanine resulted in no detectable decrease in kcat. However, Km was increased over 30-fold by this substitution (Km = 1.8 μM). As reported previously, the K215A substitution resulted in a modest decrease in kcat (38 min−1), and Km increased to 0.8 μM (25). These results suggest that the residues K215 and R216 play major roles in DNA binding but are not crucial for catalysis.
Table 2.
Kinetics of mutant and wild-type nucleases
| Nuclease | Catalytic parameters
|
|||
|---|---|---|---|---|
| kcat, min−1 | Km, μM | kcat/Km, μM−1⋅min−1 | ΔΔGapp, kJ·mol−1 | |
| Wild type | 101 ± 10 | 0.07 ± 0.01 | 1,400 | — |
| R216A | 97 ± 4 | 1.8 ± 0.3 | 54 | 8.4 |
| K215A* | 38 | 0.8 | 48 | 10 |
Result was reported previously (25).
Discussion
Our analysis of substrate binding using a 5OVH substrate allows us to interrogate the original model of flap endonuclease-DNA binding (Fig. 2B). In this model the 5′ end of the single-stranded substrate is threaded through a hole in the protein. In the present study, while developing simplified substrates to study flap endonuclease–DNA interactions, we found that substrates with a 5OVH were bound and processed efficiently by the wild-type enzyme. By extension of the original model proposed for binding to a flap structure, an analogous model can be proposed for binding of a 5OVH (i.e., a substrate lacking duplex region I), which is presented in Fig. 5A. In this model, there is no interaction of the substrate with the basic residues K241, K215, or R216. We tested this model by comparing the interaction of Ps-Y and 5OVH substrates (Fig. 1) with wild-type and mutant T5 5′ nucleases lacking one or more positively charged residue. The five positively charged residues span the length of a slightly concave surface, which is the only region of the protein with overall positive electrostatic potential (8) and thus is the most likely surface to interact with a polyanionic substrate.
Figure 5.

DNA-binding models. (A) The original model predicts no interaction of a 5OVH DNA substrate with residues K241, K215, or R216 but does suggest close contact with R172. (B) Such interactions are predicted in the revised model. (C) Stereoview of revised conceptual binding model (PDB ID code 1J5F). The model was energy-minimized by using X-PLOR and obvious steric clashes are absent (32). The position of the single-stranded tail (red) behind the nuclease is depicted arbitrarily.
Choice of Substrates.
The Ps-Y substrate was used in preference to the flap structure for these comparisons, because both are bound by wild-type enzyme with similar affinities. This result implies that, at least in the case of the T5 enzyme, duplex I plays no great role in substrate binding. Similarly, the enzyme cleaves flap and Ps-Y substrates to produce the same distribution of products at similar rates (17). The 5OVH structure is also a good substrate for the enzyme. We previously determined a kcat of 100 min−1 by using the same hairpin 5OVH substrate (25), which compares favorably with kinetic analysis of full flap structures, e.g., kcat values of 47 min−1 for Escherichia coli DNA polymerase I and 6 min−1 for human FEN1 (16, 26).
DNA-Binding and Kinetic Studies.
The 5OVH substrate was bound more weakly than the Ps-Y substrate (85 vs. 4 nM). This interaction with 5OVH is significant when compared with reported dissociation constants for flap structures that range from 9 to 4,700 nM for bacterial 5′ nucleases (16, 27).
Substitution of basic residues K215, R216, or K241 with a neutral amino acid affected 5OVH binding profoundly, increasing the Kd above practical assay limits (i.e., Kd > 10 μM). Not even the K215R/R216K double mutation was able to restore detectable binding to a 5OVH substrate. This result strongly suggests that the original model for flap (and 5OVH) binding is incorrect, because it predicts no contact between such a substrate and residues 215, 216, or 241. Furthermore, this model predicts that R172 should contact the duplex region of a 5OVH substrate. However, the R172A substitution's binding affinity for this substrate was similar to that of wild type.
Kinetic studies were performed on the R216A nuclease, because this substitution had the biggest impact on binding of any of the single mutations studied here as measured by the Ps-Y electrophoretic mobility-shift assay. The single-turnover substrate HP1, a hairpin duplex oligonucleotide possessing a single-stranded 7-nt 5OVH (Fig. 1), was used in this reaction. Both K215A and R216A produce minimal effects on kcat but impair substrate binding as evidenced by increased Km values. Thus, these basic residues play no role in chemical catalysis but are important for binding even for a 5OVH substrate such as HP1. The energetic penalties paid on substitution of R216 (ΔΔGapp, Table 2) or K215 (25) are consistent with the loss of either a strong hydrogen bonding interaction with an uncharged partner or a weak hydrogen bond to a charged partner (28). We previously reported kcat and Km values of 38 min−1 and 0.8 μM, respectively, for K215A with ΔΔGapp = 10 kJ·mol−1 by using the HP1 substrate (25). All the mutant nucleases displayed appreciable levels of activity in the spectrophotometric assay confirming that none of the residues is crucial for catalysis. All the proteins tested gave qualitatively indistinguishable patterns of digestion with either Ps-Y or 5OVH substrates, further providing confirmation that these residues are not crucial for catalysis.
New Binding Model.
Our results lead us to propose a revised model that is consistent with the results of these quantitative studies. They show that all residues interact with the Ps-Y, a result that would be expected from the original model. In contrast, results obtained by using the 5OVH substrate are not consistent with the original model, i.e., residues K215, R216, and K241 are important for binding to the 5OVH substrate. The observation that substitution of R172 with alanine results in a protein with essentially wild-type affinity for the 5OVH substrate further undermines the validity of the model originally proposed in which duplex I interacts with residues 215, 216, and 241. A new conceptual model for binding to the 5OVH substrate is presented (Fig. 5 B and C). In this revised model the duplex region of the 5OVH substrate (II) is in close contact with residues K215, R216, and K241, shown here to be required for substrate binding.
Joyce and coworkers (16) provided evidence to support the original model, suggesting that residue R20 in DNA polymerase I (R33 in T5 5′ nuclease) contacts the cleavable strand to the 3′ side of the cleavage site. They also showed that phosphodiesters in duplex region II on the uncleaved strand (B1 in the Ps-Y and flap structures, strand In1 on the 5OVH substrate) are protected from alkylation after binding to a flap endonuclease. Our conceptual model is consistent with these observations. The relevant phosphodiesters would be in intimate contact with the protein surface, thus blocking the approach of an alkylating agent. In the native T5 5′-nuclease structure, R33 points away from the active site but could rotate so as to come into close contact with a phosphodiester 3′ of the cleavage site. This theory is supported by the observation that in Taq polymerase, the equivalent arginine residue (R25) is nearer to the active site than in the T5 nuclease structure (6). The equivalent residue in our model (R33) is positioned so as to contact a phosphodiester residue 3′ to the cleavage site (assuming that hydrolysis must occur at one or the other of the bound metal ions), consistent with the observations of Xu et al. (16). Loss of this residue in the T5 nuclease R33A mutant resulted in only a 2-fold increase in dissociation constant for the 5′-overhanging substrate compared with the wild-type protein.
Broadly similar crystal structures of several 5′ nucleases have been solved in the last few years. The charge distribution around the surface of T5 5′ nuclease precludes binding of negatively charged DNA to most of the enzyme's surface apart from the active site and a patch of basic residues including all those substitutions studied here. Most conserved residues in the flap endonucleases reside within the active site (Fig. 2A). Recent studies have reinforced the notion that basic residues within the helical arch region (residues K83 and R86 in T5 5′ nuclease) are involved in DNA binding (15, 16). Positioned further away from the active site, residues K215 and R216 form part of a helix–loop–helix feature in the T5 nuclease. They form part of a conserved hexapeptide motif, GIGAKR. These residues also are well conserved in the 5′ nucleases, being present in those of the archaebacteria and eukaryotes (11). This helix–loop–helix feature in T5 nuclease was predicted to be a helix–hairpin–helix (HhH) on the basis of earlier work (29). The HhH motif consists of two antiparallel α helices connected by a single four-residue hairpin turn (30). However, the experimentally determined structures of T5 nuclease and Taq polymerase revealed two helices separated by a longer loop (6, 8, 13) than that in the HhH motif. Tainer and coworkers (10) identified a very similar H3TH motif in the Pyrococcus homologue. The distribution of positively charged residues on the surfaces of the 5′ nucleases from phage T5 and an archaeal homologue from M. jannaschii (PDB ID code 1A76) is remarkably similar despite the relatively low primary sequence homology shared by these two polypeptides. This charge distribution and the obvious structural similarities between these enzymes (14) suggest that the mode of DNA binding is likely to be similar in all 5′ nucleases (see Fig. 7, which is published as supporting information on the PNAS web site).
Our current study was designed to look at the complex and not to examine how such a complex might come to form. Lyamichev et al. (4) proposed a threading model to explain the action of 5′ nucleases on bifurcated DNA substrates. Essentially, they suggested that the free 5′ arm of a flap structure becomes threaded through the enzyme, which then tracks along the DNA until halted by the duplex region. This hypothesis was supported by observations from Bambara and coworkers (22) on the action of mammalian FEN1 on various oligonucleotide substrates. The presence of a hole in the structure of T5 5′ nuclease suggested a possible physical explanation of the threading models that had been proposed on the basis of biochemical studies (8).
However, recent studies are hard to reconcile with a threading model. Bambara and coworkers (21) found that FEN1 is able to process substrates containing bulky adducts (even branched DNA structures) in the 5′ tail. Our laboratory reported that T5 5′ nuclease is able to digest double-stranded closed-circular DNA under conditions that promote duplex melting (20). Lyamichev et al. (19) showed that the Thermus aquaticus 5′-nuclease homologue was able to process single-stranded circles. However, these purely endonucleolytic reactions seem to require forcing conditions such as high enzyme concentrations or the use of unusual cofactors. It is possible that such activity on closed-circular substrates is the result of low levels of contaminating endonucleases, but this explanation seems unlikely given that such activity is absent in catalytically inactive mutants and SDS/PAGE-purified protein (20).
Joyce and coworkers (16) suggested that the enzyme first might bind to the junction between duplex and single-stranded DNA followed by threading of the single-stranded region through the hole defined by the arch in T5 5′ nuclease. This “bind-then-thread” mechanism was likened to the process of threading a needle. It would explain the observations of Bambara and coworkers (22) that even a short oligonucleotide targeted to the 5′ end of the single-stranded arm is able to suppress the structure-specific endonuclease action of FEN1. A threading mechanism could even operate on circular substrates (19, 20) if extensive uncoiling of the helical arch were to occur, which could yield a loop able to thread large branched or fully circular substrates. A relatively large loop is visible in the M. jannaschii flap endonuclease structure, supporting such a hypothesis (9). However, it has been shown that an increase in helical content occurs after the binding of FEN1 to DNA (31), suggesting that the loop is likely to become more rather than less ordered. However, it may be that alternative mechanisms are used by different 5′ nucleases when faced with varying substrates. We proposed that the arch could fold backwards (or become disordered), allowing duplex DNA direct access to the active site (20). This region is disordered in the Taq and T4 flap endonuclease structures and also in the T5 K83A 5′-nuclease mutant (6, 7, 17), suggesting that it is far from rigid and could adopt alternative conformations.
Although the proposed model is consistent with the biochemical and biophysical properties displayed by these enzymes, the fine details of the mechanisms by which flap endonucleases interact with their substrates remain unclear. A DNA–protein cocrystal structure may shed light on these intriguing enzymes. In the meantime, we propose a revised model of protein–DNA interaction for the 5′ nucleases.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the Biotechnology and Biological Sciences Research Council (Studentship to J.J.D. and Advanced Fellowship to J.A.G.), The Wellcome Trust (ref. no. 052123), and The White Rose Consortium (Studentship to M.F.).
Abbreviations
- 5OVH
5′ overhang
- Ps-Y
pseudoY
- FEN1
flap endonuclease I
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1J5F).
References
- 1.Lieber M R. BioEssays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 2.Henricksen L A, Bambara R A. Leuk Res. 1998;22:1–5. doi: 10.1016/s0145-2126(97)00113-6. [DOI] [PubMed] [Google Scholar]
- 3.Garforth S J, Sayers J R. Nucleic Acids Res. 1997;25:3801–3807. doi: 10.1093/nar/25.19.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyamichev V, Brow M A D, Dahlberg J E. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 5.Harrington J J, Lieber M R. J Biol Chem. 1995;270:4503–4508. doi: 10.1074/jbc.270.9.4503. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y, Eom S H, Wang J M, Lee D S, Suh S W, Steitz T A. Nature (London) 1995;376:612–616. doi: 10.1038/376612a0. [DOI] [PubMed] [Google Scholar]
- 7.Mueser T C, Nossal N G, Hyde C C. Cell. 1996;85:1101–1112. doi: 10.1016/s0092-8674(00)81310-0. [DOI] [PubMed] [Google Scholar]
- 8.Ceska T A, Sayers J R, Stier G, Suck D. Nature (London) 1996;382:90–93. doi: 10.1038/382090a0. [DOI] [PubMed] [Google Scholar]
- 9.Hwang K Y, Baek K, Kim H Y, Cho Y. Nat Struct Biol. 1998;5:707–713. doi: 10.1038/1406. [DOI] [PubMed] [Google Scholar]
- 10.Hosfield D J, Mol C D, Shen B H, Tainer J A. Cell. 1998;95:135–146. doi: 10.1016/s0092-8674(00)81789-4. [DOI] [PubMed] [Google Scholar]
- 11.Shen B H, Qiu J Z, Hosfield D, Tainer J A. Trends Biochem Sci. 1998;23:171–173. doi: 10.1016/s0968-0004(98)01199-2. [DOI] [PubMed] [Google Scholar]
- 12.Gutman P D, Minton K W. Nucleic Acids Res. 1993;21:4406–4407. doi: 10.1093/nar/21.18.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Artymiuk P J, Ceska T A, Suck D, Sayers J R. Nucleic Acids Res. 1997;25:4224–4229. doi: 10.1093/nar/25.21.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sayers J R, Artymiuk P J. Nat Struct Biol. 1998;5:668–670. doi: 10.1038/1358. [DOI] [PubMed] [Google Scholar]
- 15.Bhagwat M, Meara D, Nossal N G. J Biol Chem. 1997;272:28531–28538. doi: 10.1074/jbc.272.45.28531. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Potapova O, Leschziner A E, Sun X C, Grindley N D F, Joyce C M. J Biol Chem. 2001;276:30167–30177. doi: 10.1074/jbc.M100985200. [DOI] [PubMed] [Google Scholar]
- 17.Garforth S J, Ceska T A, Suck D, Sayers J R. Proc Natl Acad Sci USA. 1999;96:38–43. doi: 10.1073/pnas.96.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sayers J R, Eckstein F. Nucleic Acids Res. 1991;19:4127–4132. doi: 10.1093/nar/19.15.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyamichev V, Brow M A D, Varvel V E, Dahlberg J E. Proc Natl Acad Sci USA. 1999;96:6143–6148. doi: 10.1073/pnas.96.11.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garforth S J, Patel D, Feng M, Sayers J R. Nucleic Acids Res. 2001;29:2772–2779. doi: 10.1093/nar/29.13.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bornarth C J, Ranalli T A, Henricksen L A, Wahl A F, Bambara R A. Biochemistry. 1999;38:13347–13354. doi: 10.1021/bi991321u. [DOI] [PubMed] [Google Scholar]
- 22.Murante R S, Rust L, Bambara R A. J Biol Chem. 1995;270:30377–30383. doi: 10.1074/jbc.270.51.30377. [DOI] [PubMed] [Google Scholar]
- 23.Sayers J R, Eckstein F. J Biol Chem. 1990;265:18311–18317. [PubMed] [Google Scholar]
- 24.Sayers J R, Krekel C, Eckstein F. BioTechniques. 1992;13:592–596. [PubMed] [Google Scholar]
- 25.Pickering T J, Garforth S J, Thorpe S J, Sayers J R, Grasby J A. Nucleic Acids Res. 1999;27:730–773. doi: 10.1093/nar/27.3.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen B, Nolan J P, Sklar L A, Park M S. Nucleic Acids Res. 1997;25:3332–3338. doi: 10.1093/nar/25.16.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amblar M, de Lacoba M G, Corrales M A, Lopez P. J Biol Chem. 2001;276:19172–19181. doi: 10.1074/jbc.M008678200. [DOI] [PubMed] [Google Scholar]
- 28.Fersht A R. Biochemistry. 1988;27:1577–1580. doi: 10.1021/bi00405a027. [DOI] [PubMed] [Google Scholar]
- 29.Doherty A J, Serpell L C, Ponting C P. Nucleic Acids Res. 1996;24:2488–2497. doi: 10.1093/nar/24.13.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thayer M M, Ahern H, Xing D, Cunningham R P, Tainer J A. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim C Y, Park M S, Dyer R B. Biochemistry. 2001;40:3208–3214. doi: 10.1021/bi002100n. [DOI] [PubMed] [Google Scholar]
- 32.Brunger A T, Karplus M, Petsko G A. Acta Crystallogr A. 1989;45:50–61. doi: 10.1107/s0108767389000437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}