

Abstract
The folding of many small proteins is kinetically a two-state process that represents overcoming the major free-energy barrier. A kinetic characteristic of a conformation, its probability to descend to the native state domain in the amount of time that represents a small fraction of total folding time, has been introduced to determine to which side of the free-energy barrier a conformation belongs. However, which features make a protein conformation on the folding pathway become committed to rapidly descending to the native state has been a mystery. Using two small, well characterized proteins, CI2 and C-Src SH3, we show how topological properties of protein conformations determine their kinetic ability to fold. We use a macroscopic measure of the protein contact network topology, the average graph connectivity, by constructing graphs that are based on the geometry of protein conformations. We find that the average connectivity is higher for conformations with a high folding probability than for those with a high probability to unfold. Other macroscopic measures of protein structural and energetic properties such as radius of gyration, rms distance, solvent-accessible surface area, contact order, and potential energy fail to serve as predictors of the probability of a given conformation to fold.
The concept of the protein transition state ensemble (TSE) (1, 2) is the foundation of modern views on protein folding. Conformations of proteins belonging to the TSE are unstable and by definition have a 50% probability to fold to the protein native state and a 50% probability to unfold or misfold. The TSE conformations belong to the free-energy barrier separating native and unfolded or misfolded domains for two-state proteins. To understand the structure of the TSE conformations we must determine the difference between “pretransition” states (conformations that are en route to the native domain from the unfolded state but the transition barrier has not been crossed) and “posttransition” states (conformations that are en route to the unfolded domain from the native state but the transition barrier has not been crossed). The distinguishing kinetic feature between pre- and posttransition conformations is their probability to reach the native state domain, pFOLD (3). Because in the posttransition conformations the nucleus (4) is not disrupted, these conformations are more probable to fold than pretransition conformations in which the nucleus is not formed. If both pre- and posttransition states are structurally and energetically close to the TSE, the question then is what global properties distinguish these states from each other?
To answer this question, we selected pre- and posttransition states (see Methods) of two proteins: chymotrypsin inhibitor 2 (CI2) and C-Src SH3 domain, by using two different approaches (ref. 5 and F.D., N.V.D., S. Buldyrev, H. E. Stanley, and E.I.S., unpublished data) and two different simulation techniques (6, 7). Both the CI2 and C-Src SH3 domain proteins have been extensively studied experimentally (8–10), and both are known to be two-state proteins. We verify that the pFOLD of the selected pre- and posttransition conformations is ≈0 and 1 correspondingly for both proteins (Table 1).
Table 1.
The structural [RG, RMSD, solvent-accessible surface area (SASA), contact order, and number of contacts], energetic (E), and topological properties (L) of pre- and posttransition states of CI2 and C-Src SH3 domain proteins
| Protein | Relation to TSE | Number of conf. | pFOLD | RG, Å | RMSD, Å | SASA, × 103 Å2 | Contact order, % | No. of contacts | E | L |
|---|---|---|---|---|---|---|---|---|---|---|
| CI2 | post- | 20 | 0.89 ± 0.07 | 13.0 ± 0.5 | 5.2 ± 0.9 | 6.5 ± 0.2 | 19 ± 1 | 183 ± 5 | −102 ± 13 | 3.5 ± 0.1 |
| pre- | 6 | 0.02 ± 0.04 | 13.0 ± 0.2 | 5.9 ± 0.3 | 7.1 ± 0.3 | 19 ± 2 | 171 ± 4 | −130 ± 19 | 4.4 ± 0.4 | |
| C-Src | post- | 10 | 0.96 ± 0.01 | 11.2 ± 0.3 | 4.9 ± 0.3 | 4.5 ± 0.1 | 22 ± 3 | 110 ± 4 | −85 ± 2 | 2.73 ± 0.03 |
| SH3 | pre- | 10 | 0.26 ± 0.08 | 11.8 ± 0.3 | 4.7 ± 0.1 | 4.4 ± 0.1 | 16 ± 2 | 102 ± 7 | −84 ± 3 | 3.31 ± 0.06 |
The values of pFOLD correlate only with L values; the posttransition states are characterized by pFOLD ≈ 1, and their L values are smaller than that for the pretransition states, which are characterized by pFOLD ≈ 0. conf., conformations.
We find that such structural properties of protein conformations as radius of gyration (RG), rms displacement (RMSD) from the native state, solvent-accessible surface area, and contact order (11) cannot distinguish the pre- and posttransition conformations (Table 1). Correspondingly, the entropy of the pre- and posttransition conformations cannot account for the difference between these conformations. We also find that the potential energies (E) and the total number of contacts between amino acids are within error bars from each other in the pre- and posttransition conformations. If the pre- and posttransition conformations are similar to each other structurally, we hypothesize that there may be a difference in the topology of the network of amino acid interactions in these conformations.
To study the topology of pre- and posttransition conformations, we construct graphs corresponding to these conformations in which nodes represent amino acids and edges represent those pairs of amino acids that are geometrically located within interaction distance from each other. Vendruscolo et al. (12) have shown recently that the “small-world” feature (13–15) of proteins can be used to identify the key residues that stabilize the structure of the transition state. Our hypothesis is that the network of amino acid interactions in posttransition conformations is more small world-like (13–15) than that in pretransition conformations. The small-world graphs are a special class of random graphs that are connected as strongly as regular graphs (the clusters have a similar structure to regular graphs), but the average path that spans two nodes via a minimal set of graph edges is as low as that for random graphs (refs. 12 and 16 and figure 2 of ref. 13). The difference between regular, small-world, and random graphs is the “wiring” of these graphs: regular graphs are connected strongly locally, with no long-range edges; random graphs are disconnected locally but have many long-range edges; and small-world graphs are the blend of the high local connectivity with a number of the long-range contacts. Small-world graphs are characterized by small separation of nodes from each other, which for proteins means a higher degree of interaction cooperativity. Thus, we hypothesize that the wiring of the posttransitional conformation graphs is “tighter” than that of the pretransition conformation graphs, resulting in a cooperative folding to the native state domain.
To measure the wiring properties of pre- and posttransition conformation graphs, we compute the average minimal distance L between any pair of nodes of a graph by counting the minimal set of edges that connect these nodes (13). We find that the L values for posttransition conformation graphs are distinctly smaller than those for the pretransition conformation graphs, thus fully supporting our hypothesis (Table 1). We also observe that the posttransition conformation graphs have more edges that are of intermediate and long range than pretransition ones (Fig. 1), which shortens the minimal path for each node k, L(k) (Fig. 2), thus creating a more cooperative network for the former graphs. A similar mechanism was observed by Watts and Strogatz (13), who, by rewiring circular graphs by removing local edges and creating a few long-range edges, were changing the graph properties from the regular to the small world. Interestingly, some CI2 pretransition conformations have N and C termini in contact, in contrast to posttransition conformations (Figs. 1 and 2). Although the contact between the N and C termini is of the longest range, the lack of intermediate-range contacts nevertheless makes pretransition conformation networks less “cooperative” than posttransition ones. The difference between the numbers of long-range contacts in pre- and posttransition conformations is not statistically significant, thus the average contact orders for both conformation ensembles cannot discriminate between pre- and posttransition ensembles.
Figure 1.

The three-dimensional structure of the CI2 protein in post- (a) and pretransition (b) states. The protein graphs are constructed based on the structure of post- (c) and pretransition (d) states. Each node of protein graphs corresponds to an amino acid, whereas each edge between a pair of nodes corresponds to that pair of amino acids that are geometrically in contact with each other. For both CI2 and C-Src SH3 domain proteins' graph constructions, the contact between two amino acids is considered to be present if the distance between corresponding Cα atoms is less than 8.5 Å. In a and b, residues A16, L49, and I57 belonging to the specific nucleus of CI2 (8) are denoted by red spheres. A16, L49, and I57 form a triad of contacts in posttransition conformations (a), whereas such contacts are missing in the pretransition conformations. In both pre- and posttransition states the number of edges (contacts) are approximately the same.
Figure 2.

The dependence of the average minimal distance L(k) between a node k and the rest of the nodes on CI2 (a) and C-Src SH3 domain (b) proteins' graphs for post- (●) and pretransition (■) states. The error bars represent the standard deviation from the average values of L(k) over all post- and pretransition states. In a, by the open circles (○) we denote amino acids M40 and E41 that do not affect the protein three-dimensional structure after cleavage of the 40–41 bond (19), and by the open boxes (□) we denote the folding nucleus of CI2 (8), A16, L49, and I57.
An important property of the L values of protein conformations is that they can serve as a structurally reliable determinant of the pre- (pFOLD ≈ 0) and posttransition (pFOLD ≈ 1) states. The principal difficulty in selecting TSE conformations, the basis of the protein-engineering experiments, is the identification of the reaction coordinate for protein folding. The reaction coordinate for folding is not well defined (3, 17, 18) and has yet to be identified. The fact that average graph connectivity distinguishes the protein pre- and posttransition states, which can be close along the reaction coordinate to the TSE, tells us that any future constructions of the reaction coordinate should strongly depend on the structure of protein interaction networks.
Interestingly, in experimental studies of CI2, the cleavage between amino acids M40 and E41 is the only one that does not destroy the protein's three-dimensional structure (19). Neira et al. (19) cut CI2 at M40–E41 (without circular permutation) to separate fragments 1–40 and 41–64 and found that these fragments reassociate into CI2. We find that amino acids M40 and E41 have the largest values of L(k) in the pretransition states and among the largest values in the posttransition states (Fig. 2a), indicating that these amino acids are the most separated on interaction network from the rest of the amino acids. Weak participation of amino acids in the protein interaction network in pre- and posttransition states means that these amino acids have weak impact on protein folding kinetics and on the final native state of the protein (because the folding pathway is not altered). Thus, our findings are in agreement with ref. 19.
A crucial factor that distinguishes pre- and posttransition states is the protein folding nucleus, the formation of which in the TSE results in the rapid folding transition to the native state, and the disruption of which results in the global unfolding (4). Pretransition states lack the folding nucleus, whereas posttransition states have it intact (Fig. 1 a and b). Thus, the difference of L(k) between the pre- and posttransition states, ΔL(k), is most pronounced for those amino acids that are part of the protein folding nucleus. We find that for CI2 (Fig. 2a), the experimentally identified folding nucleus (8), A16, L49, and I57, has one of the largest ΔL(k) values.
We also find that for C-Src SH3 domain (Fig. 2b) ΔL(k) is most pronounced for two fragments, RT–loop (16–26) and β4 (54–61), suggesting a crucial role of the connectivity between these fragments in the TSE. This observation is in agreement with the finding of F.D., N.V.D., S. Buldyrev, H. E. Stanley, and E.I.S., (unpublished data), in which the nucleus of C-Src SH3 domain is identified on the RT–loop and β4.
The evolution of the protein graphs from pre- to posttransition states may be the key to better understanding the protein folding dynamics. It is possible that the formation of a specific nucleus (17) is the consequence of “specific rewiring” of protein graphs when a protein crosses the free-energy barrier en route to its folded state. Further studies are necessary to shed light on the relation of protein graph properties to the formation of a specific nucleus. The evolution of various networks has been extensively studied recently. Barabasi and Albert (14) recently proposed a model to explain scale-free networks, in which the distribution of the number of edges per node (node degree) scales as a power law. Their model is based on the idea of “preferential attachments”: the most connected nodes are more probable to acquire new edges with other graphs' nodes in the course of graph evolution. Although because of the finite size of protein graphs there is no evidence that our graphs are scale-free, we test whether evolution of the protein graphs during protein folding follows the preferential attachment scenario. We find (Fig. 3) that most connected residues actually decrease the number of edges rather than increase them (e.g., A16). In addition, we find that some residues that were less connected in the pretransition states become more connected (e.g., refs. 47–51). The correlation between change in the node degrees between post- and pretransition states and posttransition states is 0.61. We believe that when protein crosses its folding transition barrier, the network topology changes toward a specific one. Such rewiring herds the topology into a less random conformation. Thus, we observe specific rewiring rather than a preferential attachment.
Figure 3.

Plot of the degrees of each node versus the residue number for post- (thick solid line) and pretransition (thick broken line) states for CI2. The thin line represents the difference in node degrees between pre- and posttransition states.
In this work we presented a new structure-based topological criterion that seems to be a good predictor of kinetic ability to fold for a given conformation. The fact that this criterion performed equally well for two different proteins, simulated within different models by using different techniques, suggests its generality. Moreover, our recent all-atom Monte Carlo analysis of TSE of protein G (J. Shimada and E.I.S., unpublished data) also shows consistency with the proposed criterion. Further theoretical understanding of the deep connection between topological properties of protein conformations and their kinetic ability to fold is a challenging task for future studies.
Acknowledgments
We thank J. Shimada for helpful discussion of the folding kinetics of protein G. We acknowledge S. V. Buldyrev for help with simulations of C-Src SH3 and M. Karplus and M. Vendruscolo for critical reading of the manuscript. This work is supported by National Institutes of Health Grant GM52126 (to E.I.S.) and National Institutes of Health National Research Service Award Fellowship GM20251 (to N.V.D.).
Abbreviations
- TSE
transition state ensemble
- RMSD
rms displacement
Methods
CI2.
We perform Monte Carlo simulations of the all-atom model of CI2. The all-atom Monte Carlo has been described in detail elsewhere (7). We identify our putative TSE by using the method of Vendruscolo et al. (5). In our simulations, we approximate the experimentally determined φ values of amino acids ψm by the ratio of the number of contacts each amino acid makes in the TSE, N‡, to that in the native state of CI2, NNS,
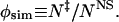 |
1 |
To generate the putative TSE, we perform unfolding simulations from the native structure of CI2 at T = 2.3 by using the form of the potential energy of amino acid interactions
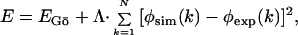 |
2 |
where EGō is the Gō potential energy (20–22), where the attractive potential (EGō = −1) between residues is assigned to the pairs that are in contact in the native state, and the repulsive potential (EGō = +1) is assigned to the pairs that are not in contact in the native state. The parameter Λ = 103 is set to a large value compared with the Gō potential to enforce the environment of each amino acid, k, in our simulations to be close to that observed experimentally. The φexp are the experimental φ values at 0 M GdHCl from Itzhaki et al. (8) for the following 39 mutations: K2M, T3A, P6A, E7A, L8A, S12A, E14N, E15Q, A16G, K17A, K18G, I20V, L21A, Q22G, K24G, P25A, E26A, I29A, I30A, L32A, V34T, T36V, V38A, T39A, E41A, Y42G, R43G, D45A, V47A, L49A, F50A, V51A, D52A, N56D, I57A, A58G, V60G, P61A, and V63A. ψm for I57A has been set to 0.5, because this residue is part of the nucleus despite its low φ value (23). The summation is over the n = 39 positions stated above.
For each putative TSE conformation (40 in total) we compute the probability to fold to its native state by performing 20 independent simulations at T = 1.2 for 5 × 107 Monte Carlo steps, which is less than 5% of the folding time from a random coil (data not shown; ref. 24). We consider the system folded when the RMSD for the backbone of the protein is less than 1Å. The average probability of folding pFOLD is 0.59, which confirms that the selected conformations do belong to the TSE. In Table 1 we present the data for the 20 conformations with pFOLD ≥ 0.8, which represent posttransition states.
We construct the pretransition conformations in a similar way to the putative TSE conformations except that the φexp values are set artificially and are not taken from experiments (24). We choose φexp to create structures with approximately the same energy and RMSD as the TSE conformations (Table 1). For the construction of the pretransition conformations we choose the φexp to be 0.5 for K2, E4, E7, L8, K11, E15, K18, V19, and D23 and 0.4 for V60, P61, R62, and V63. To create pretransition conformations that differ from each other we alternate the φexp values between these conformations. In Table 1 we present the data for the six conformations with pFOLD ≈ 0, which represent pretransition states.
C-Src SH3 Domain.
We model the C-Src SH3 domain by beads representing Cα and Cβ atoms. To mimic the flexibility of real proteins, we apply additional constraints (F.D., N.V.D., S. Buldyrev, H. E. Stanley, and E.I.S., unpublished data): (i) “covalent” bonds between Cαi and Cβi, (ii) “peptide” bonds between Cαi and Cα(i±1), (iii) effective bonds between Cβi and Cα(i±1), (iv) effective bonds between Cαi and Cα(i±2), where the subscript i denotes the amino acid sequence number. We use the Gō potential (20–22) to model interactions between Cβ atoms of the C-Src SH3 domain. It has been shown (F.D., N.V.D., S. Buldyrev, H. E. Stanley, and E.I.S., unpublished data) that our model of the C-Src SH3 domain can reproduce faithfully the thermodynamic and kinetic properties observed in experiments (9, 10).
To identify the TSE and then the pre- and posttransition states, we follow the method developed in ref. 32 (F.D., N.V.D., S. Buldyrev, H. E. Stanley, and E.I.S., unpublished data). We perform the discrete molecular dynamics simulation of the C-Src SH3 domain at the folding transition temperature, Tf ≈ 0.92. We select conformations that belong to the putative TSE from those with the potential energy in the range {ETS}, corresponding to the minimum of the probability of the potential energy histogram at T = Tf (figure 1a in ref. 4). We distinguish four types of fluctuations during the simulation of the C-Src SH3 domain that pass through the unstable states within energies in the range {ETS}: (i) FF, when the folded protein unfolds to {ETS} and then refolds rapidly to its native state, (ii) UU, when the unfolded protein partly folds into {ETS} and then unfolds rapidly, (iii) FU, when the folded protein unfolds to {ETS}, and then proceeds unfolding further, and (iv) UF, when the unfolded protein traverses the energy range {ETS} on its way to folded conformations.
Next, we compute pFOLD for each selected conformation by performing 102 independent simulations for 2 × 103 time units at Tf. The average time the C-Src SH3 domain spends in the unfolded state is 105 time units, whereas the average and maximum times of C-Src SH3 domain folding from an unfolded state at which C-Src SH3 domain is already committed to the folding transition is 102 and 103 time units correspondingly.
We find that the pFOLD of the most representative UF and FU conformations are ≈0.5, which suggests that these conformations belong to the TSE. For UU and FF conformations, we find that the pFOLD values are ≈0 and ≈1, respectively. Thus, we select the pre- and posttransition conformations from the UU and FF ensembles of conformations correspondingly.
The Protein Graphs.
The protein graphs are constructed on the basis of the Cα representation of proteins. Each graph node represents an amino acid. Each graph edge connects pairs of nodes that correspond to pairs of amino acids that are located geometrically within an interaction threshold radius, which we set to Rc = 8.5 Å. We test graph connectivity properties for various definitions of contacts and find that these properties are qualitatively invariant under contact definitions.
The average minimal distance L(k), also known as the chemical distance (25), between a node k and the rest of the graph nodes is defined as
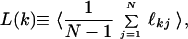 |
3 |
where N is the number of nodes in the graph or number of amino acids in proteins, ℓkj is the minimal number of edges one must transverse to reach a node j from a node k ≠ j. The averaging is done over all pre- or posttransition states. Analogously, we define the average minimal distance L between all pairs of nodes of the protein graph
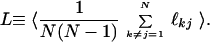 |
4 |
For example, the minimal distance between nodes N1 and V20 in the graph of Fig. 1c is 3, the path corresponding to the minimal distance passes through nodes E5 and P7: N1 → E5 → P7 → V20.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Fersht A R. Curr Opin Struct Biol. 1995;5:79–84. doi: 10.1016/0959-440x(95)80012-p. [DOI] [PubMed] [Google Scholar]
- 2.Mirny L A, Shakhnovich E I. Annu Rev Biophys Biomol Struct. 2001;30:361–396. doi: 10.1146/annurev.biophys.30.1.361. [DOI] [PubMed] [Google Scholar]
- 3.Du R, Pande V S, Grosberg A Y, Tanaka T, Shakhnovich E I. J Chem Phys. 1998;108:334–350. [Google Scholar]
- 4.Dokholyan N V, Buldyrev S V, Stanley H E, Shakhnovich E I. J Mol Biol. 2000;296:1183–1188. doi: 10.1006/jmbi.1999.3534. [DOI] [PubMed] [Google Scholar]
- 5.Vendruscolo M, Paci E, Dobson C, Karplus M. Nature (London) 2001;409:641–645. doi: 10.1038/35054591. [DOI] [PubMed] [Google Scholar]
- 6.Dokholyan N V, Buldyrev S V, Stanley H E, Shakhnovich E I. Folding Des. 1998;3:577–587. doi: 10.1016/S1359-0278(98)00072-8. [DOI] [PubMed] [Google Scholar]
- 7.Shimada J, Kussell E L, Shakhnovich E I. J Mol Biol. 2001;308:79–95. doi: 10.1006/jmbi.2001.4586. [DOI] [PubMed] [Google Scholar]
- 8.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 9.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 10.Riddle DS, Grantcharova V P, Santiago J V, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 11.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 12. Vendruscolo, M., Dokholyan, N. V., Paci, E. & Karplus, M. (2002) Phys. Rev. E, in press. [DOI] [PubMed]
- 13.Watts D J, Strogatz S H. Nature (London) 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 14.Barabasi A-L, Albert R. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 15.Jeong H, Tombor B, Albert R, Oltval Z N, Barabasi A L. Nature (London) 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 16.Bollobás B. Random Graphs. London: Academic; 1985. [Google Scholar]
- 17.Abkevich V I, Gutin A M, Shakhnovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 18.Klimov D K, Thirumalai D. Proteins Struct Funct Genet. 2001;43:465–475. doi: 10.1002/prot.1058. [DOI] [PubMed] [Google Scholar]
- 19.Neira J L, Davis B, Ladorner A G, Buckle A M, Gay G D, Fersht A R. Folding Des. 1996;1:189–208. doi: 10.1016/s1359-0278(96)00031-4. [DOI] [PubMed] [Google Scholar]
- 20.Gō N. Annu Rev Biophys Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Karplus M. Proc Natl Acad Sci USA. 1997;94:14429–14432. doi: 10.1073/pnas.94.26.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clementi C, Nymeyer H, Onuchic J N. J Mol Biol. 2000;278:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 23.Ladurner A G, Itzhaki L S, Fersht A R. Folding Des. 1997;2:363–368. doi: 10.1016/S1359-0278(97)00050-3. [DOI] [PubMed] [Google Scholar]
- 24.Li L, Shakhnovich E I. Proc Natl Acad Sci USA. 2001;98:13014–13018. doi: 10.1073/pnas.241378398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havlin S, Ben-Avraham D. Adv Phys. 1987;36:695–798. [Google Scholar]