

Abstract
Experimentally, protein engineering and φ-value analysis is the method of choice to characterize the structure in folding transition state ensemble (TSE) of any protein. Combining experimental φ values and computer simulations has led to a deeper understanding of how proteins fold. In this report, we construct the TSE of chymotrypsin inhibitor 2 from published φ values. Importantly, we verify, by means of multiple independent simulations, that the conformations in the TSE have a probability of ≈0.5 to reach the native state rapidly, so the TSE consists of true transition states. This finding validates the use of transition state theory underlying all φ-value analyses. Also, we present a method to dissect and study the TSE by generating conformations that have a disrupted α-helix (α-disrupted states) or disordered β-strands 3 and 4 (β-disrupted states). Surprisingly, the α-disrupted states have a stronger tendency to fold than the β-disrupted states, despite the higher φ values for the α-helix in the TSE. We give a plausible explanation for this result and discuss its implications on protein folding and design. Our study shows that, by using both experiments and computer simulations, we can gain many insights into protein folding.
Protein folding is one of the biggest challenges in structural biology (1). To understand how a protein goes from highly disordered unfolded states to its unique native state, the structure of the transition state ensemble (TSE) must be known. By definition, the TSE is at the top of the reaction barrier and has a probability to rapidly reach the native state (Pfold, see Methods for details) of ≈0.5. Experimentally, the most powerful way to determine the TSE structure is site-directed mutagenesis and φ-value analysis, first introduced by Fersht and coworkers (2). This analysis relies on the quantity φ, defined as ΔΔG‡/ΔΔGU-F, where ΔΔG‡ is the change in free energy between the unfolded states and TSE induced by the mutation, and ΔΔGU-F is the change in free energy between the unfolded and native state caused by the same mutation. φ-Value analysis on different proteins has greatly advanced our understanding of protein folding (2–16).
However, as φ values are calculated from the free energy of various states, they do not directly reveal the structure of the TSE. Furthermore, it is often assumed that residues with high φ values are kinetically more influential than residues whose φ values are lower, so there have been many recent efforts to predict φ values from theory (17–20). But in some cases, this assumption could be incorrect. For example, a recent study on bovine pancreatic trypsin inhibitor has shown that φ values may underestimate the degree of structure in the TSE when compared with NMR data (21). Hamill et al. (9) have convincingly argued that, in the third fibronectin type III domain of human tenascin, Ile-59 is not necessarily more nucleating than Ile-20, despite the higher φ value of I59A (0.6) vs. I20A (0.4). Similarly, Ladurner et al. (4) have demonstrated that Ile-57 in chymotrypsin inhibitor 2 (CI2) belongs to the folding nucleus despite its low φ value. In α-spectrin and src SH3, the distal loop has high φ values, but it is possible that these high φ values are just artifacts from topological constraints (6, 7). And residues with “abnormal” φ values (larger than unity or negative) seem to make non-native interactions in the TSE (6, 7) and may have been conserved in protein evolution (22). So, to better understand φ values and protein folding, it is vital to ask the following questions. First, can we reliably reconstruct the TSE from experimental φ values? Second, are residues with higher φ values kinetically more significant than those with lower φ values? Finally, and most importantly, if the answer to our second question is negative, why is it negative?
To answer these questions, computer simulation is ideal for several reasons. First, it can readily generate different structures and trajectories for the same system. So we can compare one trajectory with another, as well as combine all trajectories to obtain statistical analyses (23–28). Second, simulations can investigate protein conformations that are highly unstable and difficult to characterize by experiments, such as the TSE (29). Finally, computer simulations offers the freedom to create protein conformations that are not found in experiments. By studying these artificial conformations, we could gain insight into the naturally occurring folding nucleus.
We decided to look at the TS of CI2 by using an all-atom model with a Go potential (see Methods for details). CI2 is a 64-aa protein that folds into an α-helix packed against six β-strands (Fig. 1a). It is the first two-state protein to be found, with no detectable intermediate along its folding pathway (30). From extensive φ-value analyses (3), the CI2 TS is found to consist of the α-helix and β-strands 3 and 4 (see Fig. 1 legend for the residues in each secondary element). The residues Ala-16, Leu-49, and Ile-57 form the folding nucleus (3, 4). The α-helix has the highest average φ value, followed by β-strands 3 and 4 (3). To elucidate the folding of CI2, several simulation studies have been done. High-temperature unfolding with molecular dynamics (MD) have shed light on the putative TS (31), the unfolded state (32), and the unfolding pathways (33) of CI2. Unfortunately, given the current limits on computers, it is still impractical to repeatedly fold a protein from a random coil in a MD simulation, with all protein and solvent atoms represented (34). Clementi et al. (35) used a C-α model to investigate CI2 folding, but their model has no side chain. In contrast, our model includes all heavy atoms but a simplified energy function, so it lies between all-atom MD and the model in ref. 35 in complexity. Furthermore, we are able to obtain hundreds of trajectories by using our model with modest computer resources and time cost (36). This makes our study different from, and complementary to, the ones already done.
Figure 1.

Different states of CI2. (a) Native state of CI2 from x-ray crystallography (40). The secondary elements are as follows: β-strand 1 = residues 3–5, β-strand 2 = residues 10 and 11, α-helix = residues 12–24, β-strand 3 = residues 28–34, β-strand 4 = residues 45–51, β-strand 5 = residues 56–60, and β-strand 6 = residues 61–64. Ala-16, Leu-49, and Ile-57 are shown as space-filling spheres. To aid identification in all four structures, the α-helix is in red, and β-strands 3 and 4 are in blue. (b) A representative conformation in the TSE. The disordered β-strands 1 and 6 are in magenta. (c) An α-disrupted state. (d) A β-disrupted state.
In this paper we report three results. First, we constructed the TSE of CI2 from published φ values (Fig. 1b). Importantly, we have verified, from multiple simulations, that the TSE has Pfold ≈0.5 and therefore consists of true TSs. Second, we have studied two sets of alternative conformations: one set with a disrupted α-helix but intact β-strands 3 and 4 (Fig. 1c), and the other set with a well-formed α-helix but disordered β-strands 3 and 4 (Fig. 1d). The conformations in the first set are called α-disrupted and the ones in the second set β-disrupted. Surprisingly, the α-disrupted conformations have an average Pfold ≈0.3, whereas β-disrupted states show almost no tendency to fold (Pfold ≈0). This finding suggests that, compared with perturbing β-strands 3 and 4, disrupting the α-helix has a weaker effect on CI2 folding, despite the higher φ values in the α-helix. We give a plausible explanation for this result and discuss its implications on folding in other proteins. Finally, because the α-disrupted conformations have a Pfold significantly greater than 0, we propose them as possible candidates for a re-engineered folding nucleus, which may be realized by redesigning the amino acid sequence, as is recently attempted with protein G (37).
Methods
Model and Simulation Protocol.
The all-atom model and simulation protocol have been described in detail (36). Briefly, the model includes all nonhydrogen atoms in the backbone and side chains of the protein and uses a square-well potential between any two atoms. Each native contact has an attractive energy of −1, whereas each non-native contact is repulsive with an energy of +1. During a simulation, the program randomly moves up to three residues, calculates the energy before and after the move, and accepts or rejects the move according to the Metropolis criterion (38). This random movement and subsequent acceptance or rejection constitutes a Monte Carlo step (MCS). Chain connectivity and excluded volume is preserved at every MCS.
Construction and Verification of the TSE.
The putative TSE is constructed in a way similar to that in ref. 29. We define a quantity φsim = N‡/NNS, where N‡ and NNS are the number of native contacts in the putative TS and the native state, respectively. Then we unfold the protein from its x-ray structure (Fig. 1a) with the following energy function:
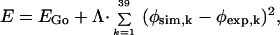 |
where EGo is the Go potential (−1 and +1 for native and non-native contact, respectively). φexp,k is the kth experimental φ value in the following list of mutations from ref. 3: K2 M, T3A, P6A, E7A, L8A, S12A, E14N, E15N, A16G, K17A, K18G, I20V, L21A, Q22G, K24G, P25A, E26A, I29A, I30A, L32A, V34G, T36V, V38A, T39A, E41A, Y42G, R43A, D45A, V47A, L49A, F50A, V51A, D52A, N56A, I57A, A58G, V60A, P61A, and V63A. So φexp,k=1 = experimental φ value for K2 M = 0.03 (3). We use the φexps at 0 M guanidinium hydrochloride (3). φexp for I57A has been set to 0.5 because this residue participates in the TS despite its low φ value (4). φsim,k is the φsim for the kth residue in the same list above (i.e., φsim,k=3 is the φsim for Pro-6). The value of the constant Λ is 1,000 and, as a result, all putative TSs have φsim ≈ φexp at most positions because the condition φsim ≈ φexp minimizes the energy. Typically, |φsim − φexp| is less than 0.1 (data not shown). The unfolding simulations have been performed at a high temperature of 2.3 to remove as much of the native structure as possible. The wild-type CI2 sequence is used throughout. We collected 40 random states from multiple unfolding trajectories after 2 × 106 MCSs. This ensemble of 40 conformations is the putative TSE.
To determine the Pfold of the putative TSE, we eliminate the second term of the energy function by setting Λ = 0 and perform 20 independent runs at T = 1.2 for each member of the putative TSE. So, in total, 800 simulations (i.e., 40 putative TSs × 20 runs per putative TS) have been conducted. The length of each simulation is 5 × 107 MCSs, which is less than 5% of the time required by a random coil to reach the native state. The protein is considered as native if its backbone rms deviation from the native state drops below 1 Å. Pfold is calculated as: (the number of simulations that folded within 5 × 107 MCSs)/800. T = 1.2 is chosen for the following reason. CI2 is thermally very stable, with a transition temperature (Tf) of 73.8°C at pH 3.5 (39), whereas the protein engineering experiments have been done at 25°C and pH 6.3 (3). The transition temperature of our CI2 model cannot be located exactly (1.7 < Tf < 1.9, data not shown), and we believe T = 1.2 is approximately the same as experimental conditions in ref. 3.
Construction and Folding of α- and β-Disrupted States.
The α- and β-disrupted conformations are generated in a similar way as the TSE. The same form for the energy function as above has been used. The only difference is that the φexps are not from ref. 3. Instead, the φexps have been chosen to give conformations with a disordered α-helix or disrupted β-strands 3 and 4. Among the α-disrupted states, the φexps are: 0 for residues 1–18 and 22–24; 0.1 for residue 59; 0.2 for residues 25, 29, 47, 49, and 55–57; 0.3 for residues 21 and 61–64; 0.4 for residues 20 and 27; 0.42 for residue 26; 0.44 for residue 45; and 0.5 for all remaining positions. For the β-disrupted structures, φexp = 0.5 for residues 2, 4, 7, 8, 11, 15, 18, 19, and 23 and φexp = 0.4 for residues 51 and 60–63. We have studied 20 α-disrupted and six β-disrupted conformations and determined their Pfold in an identical manner as for the TSE. The simulation temperature for folding is again 1.2, so it is meaningful to compare our results from the TSE, α- and β-disrupted states.
Results and Discussion
The TSE.
Although our putative TSs have been generated from experimental φ values, there is no a priori reason to expect that they are automatically true TSs with Pfold ≈ 0.5. Only repeated computer simulations can settle this issue. Here, we have demonstrated conclusively that the conformations in our putative TSE have an average Pfold of 0.59 (Table 1), so they are true TSs. Furthermore, because the TSE is generated from experimental φ values, a Pfold of ≈0.5 proves that protein engineering and φ-value analysis indeed probes the TS along the folding pathway of a protein, and that the use of TS theory in φ-value analysis is justified.
Table 1.
Structural and kinetic properties for various states of CI2
| State | 〈Rg〉* (Å) | 〈Etot〉† | 〈drms〉‡ (Å) | 〈SASA〉§ (Å2) | Pfold |
|---|---|---|---|---|---|
| TSE | 13.1 ± 0.4 | −96 ± 16 | 5.4 ± 0.8 | 6,500 ± 160 | 0.59 |
| α-disrupted | 16.9 ± 1.6 | −133 ± 11 | 11.6 ± 2.6 | 6,700 ± 120 | 0.32 |
| β-disrupted | 13.8 ± 0.4 | −130 ± 19 | 5.9 ± 0.3 | 7,100 ± 300 | 0.02 |
| Native | 11.0 | −804 | 0 | 4,500 | — |
Radius of gyration.
Total energy (native and non-native).
rms deviation of the backbone from the native state.
Solvent accessible surface area, calculated with a probe sphere of radius 1.4 Å using naccess (41).
In general agreement with previous studies (3, 31, 33, 35), the TSE resembles an expanded native state (Fig. 1b). The α-helix is the most structured, followed by β-strands 3 and 4, with β-strands 1 and 6 almost completely disordered (Fig. 2). We focus on Ala-16 in the α-helix, as this residue is found by experiments to be fully structured in the TS (3). Among the 40 conformations in our TSE, Ala-16 makes, on average, 28.5 native contacts with other amino acids. Fifty five percent of these contacts are with other residues in the α-helix; 29% are with Leu-8 in the type III reverse turn; and 13% are with Leu-49 or Ile-57. The high ratio of intrahelical contacts agrees with the results from molecular dynamics simulations (31, 33). The contacts with Leu-49 and Ile-57 are consistent with the three residues forming the folding nucleus (3, 4). Of particular interest is the significant fraction of contacts between Leu-8 and Ala-16. These contacts appear to maintain the reverse turn (residues 8–11) between β-strand 1 and the α-helix and prevent the N-terminal amino acids from moving too faraway from the α-helix (Fig. 3). This finding suggests that, in addition to forming the nucleus with Leu-49 and Ile-57, Ala-16 may stabilize the reverse turn in the TSE by interacting with Leu-8. Apparently, Leu-8 does not require many contacts to keep the reverse turn in position, because its φsim = 0.22 ± 0.03 in the TSE and its φ value from experiment is only 0.15 (3). So, Leu-8 may be more important to the TSE than its low φ value may indicate at first glance.
Figure 2.

The average degree of native structure for each secondary element in the TSE (black), the α-disrupted states (green), and the β-disrupted states (red). A high <φ> value in a particular element means that the element is well-formed. See Fig. 1 legend for definitions of secondary elements.
Figure 3.

The major contact partners of Ala-16 outside the α-helix in the TSE. Leu-8 is in magenta, Ala-16 is in blue, Leu-49 is in red, and Ile-57 is in cyan. The turn between β-strand 1 and the α-helix is in black.
The α- and β-Disrupted States.
To selectively investigate the kinetic importance of various parts of CI2, we generate states with a disordered α-helix (Figs. 1c and 2) and states with disordered β-strands 3 and 4 (Fig. 1d and 2). Table 1 shows that, by many macroscopic measures, the α- and β-disrupted states are similar to the TSE. The α-disrupted states have the highest radius of gyration and rms deviation from the native state (Table 1).
Protein engineering experiments indicate that β-strands 3 and 4 are partially formed in the CI2 TS, with Leu-49 on β-strand 4 participating strongly in the folding nucleus (3). So disrupting these two β-strands is expected to drastically reduce Pfold. This is indeed observed for the β-disrupted states, whose Pfold is close to 0 (Table 1). So our simulations confirm the importance of β-strands 3 and 4 for CI2 folding.
Both experiments (3) and our simulations (Fig. 2) show that the α-helix is the most structured element in the CI2 TSE. So it might be expected that disrupting the α-helix would abolish folding. Surprisingly, the α-disrupted states have a significant tendency to rapidly reach the native state (Pfold = 0.32, Table 1). However, instead of contradicting the results from protein engineering, we believe our simulations have improved our understanding of how CI2 folds. Disordering the α-helix has reduced Pfold from 0.59 (the value for TSE) to 0.32. This finding is consistent with the experimental conclusion that the α-helix, including Ala-16, is important for CI2 folding (3). However, as stated in our Introduction, experiments cannot unambiguously decide whether the α-helix is more or less influential than β-strands 3 and 4, because a residue with a higher φ value is not necessarily more nucleating than one with a lower φ value. One advantage of computer simulations is that we can readily generate the α- and β-disrupted conformations and study their folding without mutating or redesigning the protein. And our results suggest that, relative to β-strands 3 and 4, disordering the α-helix is less destructive to CI2 folding, despite the higher φ values in the α-helix (Fig. 2).
Furthermore, based on the simulation results, we can explain why disordering the α-helix is less destructive. The α-helix makes 93 local (intrahelical) contacts and 57 nonlocal contacts with β-strands 3 and 4 in the CI2 native state. Local contacts are more entropically favored than nonlocal contacts because they constrain fewer monomers. So it is likely that, as an α-disrupted state folds, the local contacts within the α-helix form easily. These local contacts could then facilitate the nonlocal contacts between the α-helix and β-strands 3 and 4. The cooperation between local and nonlocal contacts would help the protein reach the native state and lead to a significant Pfold for the α-disrupted states, as is observed (Table 1). The two types of contacts probably form simultaneously, in accordance with the nucleation-condensation mechanism of protein folding (1, 3, 24, 25).
On the other hand, the contacts made by the β-strands 3 and 4 are mostly nonlocal. In the native state of CI2, β-strand 3 has 25 contacts with the α-helix and 85 contacts with β-strand 4. For β-strand 4, there are 32 contacts with the α-helix, 33 with β-strand 5, and 38 with β-strand 6. Importantly, β-strands 3 and 4 make no intrastrand contact in the native state. So a β-disrupted state has no local contact to guide it to the native state. This probably explains why the β-disrupted conformations have a much lower Pfold than the α-disrupted states.
In summary, we find that, in comparison to β-strands 3 and 4, disrupting the α-helix is a weaker perturbation on CI2 folding. The disrupted α-helix can readily reform because of the local and nonlocal contacts made by the helix. In this sense, the α-helix is kinetically less important than β-strands 3 and 4. However, this does not mean that, relative to β-strands 3 and 4, the α-helix contributes less to the energetic stability of the TSE.
Implications for Protein Folding and Design.
We have provided a plausible reason why, in CI2, the α-helix is kinetically less important than β-strands 3 and 4, even when the α-helix is the most structured element in the TSE. Our explanation is based on the balance between local and nonlocal interactions, so it may apply generally to other α/β proteins (5, 8, 11–13, 15, 16). Therefore a φ value of 0.6 in an α-helix could, in general, be less important than the same value in a β-strand. In addition, to pinpoint the folding nucleus of a protein, it may be necessary to consider low φ values, especially in elements with long-range contacts such as β-strands. More experiments and computer simulations on other proteins should be done to test the above hypotheses. In this study, we have specifically perturbed different parts of the protein and looked at subsequent folding. This approach is a potentially powerful way to dissect the folding nucleus and could help us locate protein moieties that are essential to folding.
We also have demonstrated that the α-disrupted states, while structurally different from the experimental TSE of CI2, have a significant probability of reaching the native state rapidly. So, within a fixed protein topology, there may be diverse structures that can fold rapidly and repeatedly. This observation raises the exciting possibility of shifting the folding nucleus by protein design (8), and some results have already been reported for protein G (37). Given the significant Pfold for the α-disrupted states, it may be interesting to redesign the CI2 sequence so that the α-helix is destabilized but the β-strands are stabilized. The almost identical exposed surface area for the TSE and the α-disrupted states (Table 1) shows that the hydrophobic core of the protein is buried to a similar extent in the two ensembles. This finding gives us hope that the α-disrupted states could function as the TSE in a redesigned CI2. Such a redesigned protein will illuminate the relative effect of sequence and topology on the TS in protein folding (6, 7, 12, 13, 15–17, 27, 35).
Conclusions
First, we have validated protein engineering and φ-value analysis, by constructing the TSE from experimental φ values and verifying that the TSE has Pfold ≈0.5. Second, we have presented a method that dissects the folding TS and applied the method to CI2. Surprisingly, the α-helix seems to be kinetically less important than β-strands 3 and 4, despite its higher φ values. We have given a plausible explanation for this result, and our explanation may apply to other α/β proteins. Third, we have suggested the α-disrupted states (Fig. 1c) as possible candidates for a redesigned TS in CI2 folding. Finally, our study shows that, to locate the most nucleating region of a protein, both experimental φ values and exhaustive computer simulations are vital.
Acknowledgments
We thank Jun Shimada and Edo Kussell for the all-atom simulation program. L.L. is particularly grateful to Jun Shimada and Alexey Ishchenko for computer assistance throughout this project. Dr. N. V. Dokholyan is acknowledged for useful discussion. This research is supported by National Institutes of Health Grant GM52126.
Abbreviations
- TS
transition state
- TSE
TS ensemble
- CI2
chymotrypsin inhibitor 2
- MCS
Monte Carlo step
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Fersht A. Enzyme Structure, Mechanism, and Protein Folding. 3rd Ed. New York: Freeman; 1998. [Google Scholar]
- 2.Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 3.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 4.Ladurner A G, Itzhaki L S, Fersht A R. Folding Des. 1997;2:363–368. doi: 10.1016/S1359-0278(97)00050-3. [DOI] [PubMed] [Google Scholar]
- 5.Fulton K F, Main E R G, Daggett V, Jackson S E. J Mol Biol. 1999;291:445–461. doi: 10.1006/jmbi.1999.2942. [DOI] [PubMed] [Google Scholar]
- 6.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 7.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 8.Choe S E, Li L, Matsudaira P T, Wagner G, Shakhnovich E I. J Mol Biol. 2000;304:99–115. doi: 10.1006/jmbi.2000.4190. [DOI] [PubMed] [Google Scholar]
- 9.Hamill S J, Steward A, Clarke J. J Mol Biol. 2000;297:165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- 10.Burton R E, Huang G S, Daugherty M A, Calderone T L, Oas T. Nat Struct Biol. 1997;4:305–310. doi: 10.1038/nsb0497-305. [DOI] [PubMed] [Google Scholar]
- 11.López-Hernández E, Serrano L. Folding Des. 1996;1:43–55. [PubMed] [Google Scholar]
- 12.Villegas V, Martínez J C, Avilés F X, Serrano L. J Mol Biol. 1998;283:1027–1036. doi: 10.1006/jmbi.1998.2158. [DOI] [PubMed] [Google Scholar]
- 13.Chiti F, Taddei N, White P M, Bucciantini M, Magherini F, Stefani M, Dobson C M. Nat Struct Biol. 1999;6:1005–1009. doi: 10.1038/14890. [DOI] [PubMed] [Google Scholar]
- 14.Kragelund B B, Osmark P, Neergaard T B, Schiødt J, Kristiansen K, Knudsen J, Poulsen F M. Nat Struct Biol. 1999;6:594–601. doi: 10.1038/9384. [DOI] [PubMed] [Google Scholar]
- 15.Kim D E, Fisher C, Baker D. J Mol Biol. 2000;298:971–984. doi: 10.1006/jmbi.2000.3701. [DOI] [PubMed] [Google Scholar]
- 16.McCallister E L, Alm E, Baker D. Nat Struct Biol. 2000;7:669–673. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 17.Guerois R, Serrano L. J Mol Biol. 2000;304:967–982. doi: 10.1006/jmbi.2000.4234. [DOI] [PubMed] [Google Scholar]
- 18.Galzitskaya O V, Finkelstein A V. Proc Natl Acad Sci USA. 1999;96:11299–11304. doi: 10.1073/pnas.96.20.11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alm E, Baker D. Proc Natl Acad Sci USA. 1999;96:11305–11310. doi: 10.1073/pnas.96.20.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muñoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulaj G, Goldenberg D. Nat Struct Biol. 2001;8:326–330. doi: 10.1038/86200. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Mirny L A, Shakhnovich E I. Nat Struct Biol. 2000;7:336–341. doi: 10.1038/74111. [DOI] [PubMed] [Google Scholar]
- 23.Guo Z, Brooks C L., III Biopolymers. 1997;42:745–757. doi: 10.1002/(sici)1097-0282(199712)42:7<745::aid-bip1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 24.Abkevich V I, Gutin A M, Shakhnovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 25.Guo Z, Thirumalai D. Biopolymers. 1995;36:83–102. [Google Scholar]
- 26.Klimov D K, Thirumalai D. Protein Struct Funct Genet. 2001;43:465–475. doi: 10.1002/prot.1058. [DOI] [PubMed] [Google Scholar]
- 27.Ferrara P, Caflisch A. J Mol Biol. 2001;306:837–850. doi: 10.1006/jmbi.2000.4400. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y, Karplus M. Nature (London) 1999;401:400–403. doi: 10.1038/43937. [DOI] [PubMed] [Google Scholar]
- 29.Vendruscolo M, Paci E, Dobson C M, Karplus M. Nature (London) 2001;409:641–645. doi: 10.1038/35054591. [DOI] [PubMed] [Google Scholar]
- 30.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 31.Li A, Daggett V. J Mol Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 32.Kazmirski S L, Wong K-B, Freund S M V, Tan Y-J, Fersht A R, Daggett V. Proc Natl Acad Sci USA. 2001;98:4349–4354. doi: 10.1073/pnas.071054398. . (First Published March 27, 2001; 10.1073/pnas.071054398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 34.Duan Y, Kollman P A. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 35.Clementi C, Nymeyer H, Onuchic J N. J Mol Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 36.Shimada J, Kussell E, Shakhnovich E I. J Mol Biol. 2001;308:79–95. doi: 10.1006/jmbi.2001.4586. [DOI] [PubMed] [Google Scholar]
- 37.Nauli S, Kuhlman B, Baker D. Nat Struct Biol. 2001;8:602–605. doi: 10.1038/89638. [DOI] [PubMed] [Google Scholar]
- 38.Metropolis N, Rosenbluth A W, Rosenbluth M N, Teller A H, Teller E. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- 39.Jackson S E, Moracci M, elMasry N, Johnson C M, Fersht A R. Biochemistry. 1993;32:11259–11269. doi: 10.1021/bi00093a001. [DOI] [PubMed] [Google Scholar]
- 40.Harpaz Y, elMasry N, Fersht A R, Henrick K. Proc Natl Acad Sci USA. 1994;91:311–315. doi: 10.1073/pnas.91.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hubbard S J, Thornton J M. naccess. London: University College; 1993. [Google Scholar]