

Abstract
Most studies of microsatellite evolution utilize long, highly mutable loci, which are unrepresentative of the majority of simple repeats in the human genome. Here we use an unbiased sample of 2,467 microsatellite loci derived from alignments of 5.1 Mb of genomic sequence from human and chimpanzee to investigate the mutation process of tandemly repetitive DNA. The results indicate that the process of microsatellite evolution is highly heterogeneous, exhibiting differences between loci of different lengths and motif sizes and between species. We find a highly significant tendency for human dinucleotide repeats to be longer than their orthologues in chimpanzees, whereas the opposite trend is observed in mononucleotide repeat arrays. Furthermore, the rate of divergence between orthologues is significantly higher at longer loci, which also show significantly greater mutability per repeat number. These observations have important consequences for understanding the molecular mechanisms of microsatellite mutation and for the development of improved measures of genetic distance.
The human genome is composed of 40–50% repetitive DNA, an important class being simple tandem repeats or microsatellite DNA sequences (1). Microsatellites are iterations of short (1–6 bp) sequence motifs, repeat numbers generally being less than 30 (2). They are spread over the genome with an estimated average density of one locus per 2–30 kb, the frequency being dependent on the criteria used for defining a microsatellite locus (1, 3). Similar microsatellite densities have also been documented for other eukaryotic genomes (4). Microsatellites are instrumental as genetic linkage markers in genome mapping projects (5) and have also found widespread use for evolutionary and population genetics studies in many species (6, 7), including humans (8).
Microsatellites differ from most other DNA sequences in their high degree of polymorphism, with heterozygosities commonly exceeding 70%. As they generally seem to be free of selective constraints, it is evident that the extensive degree of genetic variability requires a high underlying mutation rate. Estimates of the human genomic mutation rate are in the range of 10−4 to 10−2 per meiosis (9), several orders of magnitude higher than that of unique DNA sequences (10, 11). It is commonly assumed that microsatellite mutations arise from replication slippage (slipped strand mispairing), a process thought to result in the gain or loss of one or a few repeat units (12–14). A microsatellite locus may initially evolve from the random occurrence of a few repeat units embedded within unique sequence and subsequently, after several steps of repeat expansion, reach a stage of an appreciable number of repeats. In theory, such expansion can proceed indefinitely in the absence of selection unless there are mechanisms that direct mutations toward contractions (15–17) or that make loci collapse (e.g., point mutations or large deletions; ref. 18).
Based on the assumption that replication slippage is the main cause of mutation, a stepwise mutation model (SMM; refs. 19 and 20), or derivatives thereof, has been suggested to be applicable to microsatellite data and for deriving genetic distance measures (21–24). In its simplest form, the model assumes that the mutation process is characterized by constant rates of single step changes per locus with equal likelihood of expansion and contraction. However, recent observations suggest that the process of microsatellite mutation and evolution is more complex. A number of mutation biases have been indicated, such as an excess of gains over losses (9, 15, 25, 26), a propensity for long alleles to contract upon mutation (9, 15–17), and an increase in mutation rate with allele size (4, 9, 15, 27–30). In addition, it has been suggested that the mutation process may differ even between closely related species, leading to interspecific differences in microsatellite length. One such example is the report of human microsatellites generally being longer than orthologous loci in chimpanzee (ref. 31; but see ref. 32). Furthermore, there are several lines of evidence suggesting that locus-specific mutation rates may vary both within and between species (18, 33, 34), as well as between different types of repeat motifs (33, 35, 36). Together, these observations point at microsatellite evolution being a highly dynamic and variable process. Understanding the character and cause of this variation is pivotal for the proper implementation of microsatellite data in evolutionary studies, and for explaining their widespread occurrence in eukaryotic genomes.
The information currently available on biases in the microsatellite mutation process suffers from the common problem of itself being based on data from biased selections of loci. First, loci where it has been possible to characterize de novo mutations by pedigree analysis clearly represent the upper extreme end of the distribution of mutation rates. Mutations at loci that might better represent the mean genomic rate are not sufficiently common to make scoring and characterization of reasonable numbers feasible by pedigree analysis. Second, comparisons of microsatellite length and structure at orthologous loci in related species are generally based on sets of loci selected on the basis of polymorphism in the species from which they were isolated (screening and development methods thus select for long microsatellites). For this reason, such interspecific comparisons are likely to be flawed because of ascertainment bias (32, 37–39).
We have adopted an unbiased empirical approach to the study of microsatellite evolution by analyzing long genomic sequences, containing numerous microsatellite loci, from humans and chimpanzees. We aligned 5.1 Mb of orthologous genomic sequence and utilized this data set to address fundamental questions regarding the microsatellite mutation process. Using this approach, we assayed differences in repeat lengths of orthologous microsatellites in humans and chimpanzees and characterized the relationships among mutability, allele length, and repeat motif.
Materials and Methods
Human–Chimpanzee DNA Alignments.
We constructed a data set composed of 5.1 Mb of human and chimpanzee genomic DNA alignments in a number of stages. First, we extracted chimpanzee (Pan troglodytes) bacterial artificial chromosome (BAC) clone sequences from the National Center for Biotechnology Information (NCBI) Entrez (http://www.ncbi.nlm.nih.gov/Entrez/). The majority of the retrieved sequence data were generated by the NIH Intramural Sequencing Center (NISC) comparative vertebrate sequencing project, which aims to sequence in several vertebrate species genomic DNA sequence orthologous to five large regions of human chromosome 7 (see http://www.nisc.nih.gov/). The sequences reported as “working draft sequence” were broken up into their constituent unordered pieces. Corresponding orthologous human sequences were obtained by performing blast searches against the human genome (http://www.ncbi.nlm.nih.gov/BLAST/). The position of each blast match on a human genome contig enabled removal of any overlapping portions of DNA sequence. Alignments were then generated by using the default values of clustalw (40) as implemented on the Multiple Alignment General Interface (MAGI) server at the Human Genome Mapping Project (http://www.hgmp.mrc.ac.uk/). The resulting alignments were checked by eye to remove poorly aligned regions, such as are often found at the ends of alignment, thereby ensuring high-quality genomic alignments. In total, we obtained 80 human–chimpanzee genomic alignments by using this method. Details of the positions of all human–chimpanzee alignments within human and chimpanzee sequences in GenBank are provided in Table 5, which is published as supporting information on the PNAS web site, www.pnas.org.
Microsatellite Analyses.
Microsatellites within the genomic DNA sequences were identified by using the program sputnik (http://abajian.net/sputnik/), which uses a recursive algorithm to search for repeated patterns of nucleotides of length between 1 and 5. Sputnik does not search against a “library” of known microsatellites, but instead applies simple scoring rules to identify those simple repeats for which the maximum score is above a certain threshold. Imperfect repeats, those with insertions, deletions and mismatches, are allowed by sputnik, although such imperfections reduce the score of the microsatellite. We altered the “error_match_points” parameter to −6 to allow microsatellites with small proportion of imperfect repeats to be detected. The results of this study are insensitive to this parameter: no qualitative changes were observed when only perfect repeats were considered (results not shown).
To perform orthologous comparisons of microsatellite loci, we ran the Sputnik analysis separately on each sequence in the alignments. A second algorithm was then implemented to identify orthologous microsatellites (i.e., those that occur in the same position of an alignment in both of the constituent sequences) on the condition that none of the orthologues were overlapped by any other microsatellites in the aligned sequence. All microsatellites where both orthologues were above a certain length threshold were recovered. This length threshold was set at 9 bp (9 repeat units) for mononucleotides, 10 bp (5 repeat units) for dinucleotides, 9 bp (3 repeat units) for trinucleotides, 12 bp (3 repeat units) for tetranucleotides, and 10 bp (2 repeat units) for pentanucleotides. All microsatellites found in coding sequences, as identified by using the annotation data of the human sequences, were removed from the analysis.
Microsatellites detected by the above methods were then checked for a number of possible anomalies. A number of different errors in measuring allele length are possible due to the combined effects of misaligned flanking sequences, single-base substitutions, mutations in nearby microsatellites, and actual length changes. For example, point substitutions close to the end of repeat arrays, which have been shown to be more common than expected (41), could cause the array to appear shorter because of a length change, when no actual change has occurred. In addition, the presence of repetitive DNA can cause errors to occur in the alignment because of the higher incidence of length mutations. For these reasons, microsatellites not flanked by conserved matching nonrepetitive sequence where length changes could be unambiguously detected were omitted from the analysis. The locations, motifs, and lengths of all microsatellites included in the analysis are provided in Table 6, which is published as supporting information on the PNAS web site.
To determine the mutability of different length classes of microsatellites, pairs of orthologous loci of motif size 1–4 were placed in 6-bp bins according to allele length (pentanucleotides were excluded from the analysis as few long alleles were observed). This procedure was done twice, first on the basis of human length, assuming that this was the ancestral state, and then assuming that allele lengths in chimpanzees were ancestral. An estimate of mutability, based on the average squared divergence between orthologues (22), which is affected by both mutation rate and the variance in mutation step size, was then calculated for each bin. The same method was also used to divide microsatellites into categories above and below a length threshold of 18 bp by both human and chimpanzee length to test the significance of changes in mutability with repeat length.
As both human and chimpanzee orthologues of a microsatellite had to be above a particular length threshold for a microsatellite locus to be detected by our methods, contraction mutations that cause the length of one orthologue to sink below the threshold would not be detected. Without correction, this would cause an underestimation of mutability, mainly affecting microsatellites whose lengths are close to the minimum threshold. Therefore, when examining the effect of allele length on mutability, we corrected for this bias by adding to our results a number of hypothetical loci representing the reciprocal contractions of inferred expansions, assuming an equal likelihood of expansions and contractions in the microsatellite mutation process. Whether the assumption of equal proportions of expansion and contraction mutations is realistic across all loci is unclear, although a recent study found that it was adequate to explain length distributions of human dinucleotide loci (42).
When orthologues at a locus have different lengths, we assume that either an expansion from the shorter allele length or a contraction from the longer one has occurred, depending on which orthologue is considered ancestral. In cases where an expansion was inferred and a contraction of the same magnitude could not have been observed (because one allele would lie below the length threshold), a correction was made by counting the observed change twice. When this correction is taken into account an estimate of mutability for each length class was obtained by using the following expression:
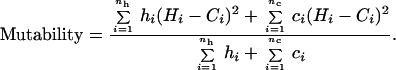 |
Hi and Ci are, respectively, the human and chimpanzee allele lengths of all of the nh pairs of orthologues sorted by human length and nc pairs of orthologues sorted by chimpanzee length in a given length bin. hi and ci are weighting parameters. hi = 2 when a correction for reverse mutation is required when human length is considered ancestral [i.e., when Hi − (Ci − Hi) is less than the detection threshold] and otherwise hi = 1. ci = 2 when a correction for reverse mutation is required when chimpanzee length is considered ancestral [i.e., when Ci − (Hi − Ci) is less than the detection threshold] and otherwise ci = 1.
Simulations of Microsatellite Evolution.
The properties of a null model of no change in mutation rate with allele length under conditions of high and low mutation rates were investigated by simulation; 105 microsatellite alleles of random initial lengths up to 200 repeat units were evolved along two separate lineages for 1,000 generations starting from a common ancestor. All mutations were single-step changes, and expansions and contractions were treated as equally likely. The likelihood of a mutation occurring in each generation was 0.0005 for the lower mutation rate simulation and 0.05 for the higher rate simulation. The resulting distributions of allele lengths were analyzed by dividing orthologues into categories based on final length in one of the lineages, assuming that the simulated microsatellites below a threshold of 9 repeat units could not be identified. A correction was made to account for the reciprocal contractions of observed expansions that were missed because of sampling above the threshold in the same manner as for the empirical data, assuming that the lineage used to divide lengths represented the ancestral state, and the estimate of average mutability was calculated for each bin. Standard errors were estimated by repeatedly running each simulation 100 times and calculating the standard deviation.
Human–Chimpanzee–Baboon DNA Alignments.
To examine the possibility of different mutation rates along the human and chimpanzee lineages we constructed three-species alignments including baboon sequences as an outgroup, using a method similar to the human-chimpanzee alignments. Baboon (Papio cynocephalus anubis) bacterial artificial chromosome clone sequences generated by the NISC comparative vertebrate sequencing project were extracted from NCBI Entrez and, if necessary, separated into their constituent fragments. Standalone blast searches (43) were performed between all chimpanzee and baboon sequences to identify orthologous regions greater than 10 kb in length. The corresponding human sequences were then obtained by performing blast searches against the human genome and overlaps were removed. Three-species alignments were then created by using clustalw and checked for poorly aligned regions as described above. We obtained 43 human–chimpanzee–baboon genomic alignments, with a total length of 1.8 Mb.
Results and Discussion
Microsatellite Length in Humans and Chimpanzees.
Previous studies have suggested that human microsatellites are longer than their chimpanzee orthologues (31, 44). Here we test this observation by using an unbiased sample of orthologous noncoding microsatellites of repeat unit lengths 1–5 from 5.1 Mb of nonoverlapping human–chimpanzee genomic DNA sequence alignments. We observed 1,154 orthologous mononucleotide loci, 451 dinucleotide loci, 206 trinucleotide loci, 409 tetranucleotide loci, and 247 pentanucleotide repeat loci (Table 1).
Table 1.
Length differences between human and chimpanzee orthologous microsatellites
| Type of repeat | Minimum no. of repeat units | No. of loci | Total bp
|
Average length, bp
|
Human = chimp | Human > chimp | Human < chimp | χ2 difference from 1:1 | P | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | Chimp | Human | Chimp | ||||||||
| Mononucleotide | 9 | 1,154 | 17,922 | 17,946 | 15.53 | 15.55 | 288 | 407 | 459 | 3.12 | NS |
| Dinucleotide | 5 | 451 | 10,252 | 9,168 | 22.73 | 20.33 | 210 | 150 | 91 | 14.44 | <0.001 |
| Trinucleotide | 3 | 206 | 3,102 | 3,018 | 15.06 | 14.65 | 163 | 26 | 17 | 1.88 | NS |
| Tetranucleotide | 3 | 409 | 7,792 | 7,512 | 19.05 | 18.37 | 320 | 53 | 36 | 3.25 | NS |
| Pentanucleotide | 2 | 247 | 3,700 | 3,620 | 14.98 | 14.66 | 220 | 13 | 14 | 0.04 | NS |
NS, not significant.
The data reveal a highly significant tendency for human dinucleotide repeat arrays to be longer than their chimpanzee orthologues (Table 1; P < 0.001). None of the other repeat types exhibit significant differences, although tri- and tetranucleotide repeats both also have a larger proportion of orthologues that are longer in humans than chimpanzees. Conversely, mononucleotide repeats tend to be longer in chimpanzee than in human and, although the ratio is not significantly different from 1:1, show significantly different behavior compared with both dinucleotide (χ2 = 18.1; P < 0.001) and tetranucleotide (χ2 = 5.6; P < 0.025) repeats.
It has been suggested that microsatellite mutations are more common in heterozygotes (25) and, assuming that this observation is valid, it is argued that populations with higher heterozygosities could experience elevated mutation rates because of microsatellite loci being found in the heterozygous state more often. If directional biases in the mutation process are also assumed, such differences in microsatellite mutation rates could lead to length differences between humans and chimpanzees. Humans are believed to harbor lower levels of DNA sequence variation than chimpanzees and other primates, an observation that can be explained by a period of reduced population size followed by an expansion in the human population (45, 46). If heterozygosity increases mutation rate and chimpanzee microsatellites exhibit greater variation than humans, we would expect more of these loci to be longer in chimpanzees than humans. As the opposite trend is observed, it is unlikely that such a mechanism is operating.
Another possible explanation for the observed between-species differences in microsatellite lengths could be that general neutral mutation rates—including length and point mutations—are different along the lineages leading to humans and chimpanzees. For instance, this could result from human males having a higher average age of reproduction than other primates (31), as direct observations of germ-line transfers demonstrate that fathers passing on microsatellite mutations are older on average than those that do not (28, 30). In the presence of preexisting directional biases in the microsatellite mutation process this age difference could lead to average allele length differences between species. Alternatively, changes in the point substitution rate could directly affect microsatellite mutation rates by increasing or decreasing the rate of introduction of imperfections in repeat arrays (18, 47). However, there is no significant difference between the number of nucleotide substitutions in the human and chimpanzee lineages observed in 1.8 Mb of human–chimpanzee–baboon alignments, inferring lineage-specific substitutions by parsimony and using baboon as an outgroup (results not shown). It therefore appears that the observed differences in microsatellite allele lengths are not connected to changes in point mutation rates since the common ancestor of humans and chimpanzees.
A remaining possibility is that at least one fundamental change in a molecular mechanism that is specifically involved in the generation of microsatellite mutations has occurred on the lineage leading to humans or chimpanzees (or both). For example, this could be manifested as an alteration in the structure of an enzyme involved in DNA replication that specifically alters microsatellite mutation rate or the extent of directional biases in the mutational mechanism. However, any such changes appear to have affected arrays of different repeat motif sizes with different severity and bias, as mononucleotide arrays show significantly different behavior from di- and tetranucleotides. This observation suggests that the dynamics of the microsatellite mutation process are heterogeneous, and a change in the mutation process may have had different effects depending on repeat motif.
To determine the lineage in which the mutational processes might have changed, we examined mean allele lengths of microsatellites in the 1.8-Mb human–chimpanzee–baboon DNA sequence alignments (Table 2). For dinucleotide repeat arrays, the only motif type to show significant differences between humans and chimpanzees, average length in chimpanzees and baboons is similar, but is about 2 bp longer in humans. This difference could mean that, if a change in the mutation process governing dinucleotide repeats has occurred, it is more likely to have happened along the lineage leading to humans, leading to an increase in repeat size, rather than a decrease in size along the chimpanzee lineage.
Table 2.
Average length of human, chimpanzee, and baboon microsatellites in aligned genomic sequence
| Type of repeat | Human
|
Chimpanzee
|
Baboon
|
|||
|---|---|---|---|---|---|---|
| No. of loci | Average length, bp | No. of loci | Average length, bp | No. of loci | Average length, bp | |
| Mononucleotide | 508 | 14.59 | 473 | 14.61 | 522 | 15.72 |
| Dinucleotide | 219 | 20.09 | 213 | 18.39 | 224 | 17.73 |
| Trinucleotide | 80 | 15.75 | 83 | 14.71 | 105 | 14.54 |
| Tetranucleotide | 185 | 18.51 | 188 | 17.94 | 189 | 19.41 |
| Pentanucleotide | 97 | 15.46 | 101 | 14.55 | 101 | 13.81 |
Ascertainment Bias.
The ascertainment bias hypothesis suggests that, assuming a link between microsatellite length and polymorphism, microsatellites chosen because they are highly polymorphic in one species are likely to be longer than their orthologues in closely related species (32). This possibility was cited to account for previous findings of length differences between human and chimpanzee microsatellites (31). To illustrate the extent of the ascertainment bias we compared all microsatellites longer than 20 repeat units (≈10% of loci) in humans with their chimpanzee orthologues and then performed the reciprocal test by selecting chimpanzee loci on the same criteria (Table 3). Both selections of loci show highly significant tendencies toward longer alleles in the species in which loci were selected (P < 0.001 in both cases). Conversely, when loci are selected on the basis of being less than 20 repeat units, alleles are in both cases significantly shorter in the species in which loci were selected.
Table 3.
Illustration of ascertainment bias with complete set of orthologous human and chimpanzee microsatellites
| Selection | No. of repeat units | Human = chimp | Human > chimp | Human < chimp | χ2 difference from 1:1 | P |
|---|---|---|---|---|---|---|
| By human length | <20 | 1,179 | 449 | 523 | 5.63 | <0.025 |
| ≥20 | 22 | 200 | 94 | 38.22 | <0.001 | |
| By chimp length | <20 | 1,179 | 581 | 421 | 25.55 | <0.001 |
| ≥20 | 22 | 68 | 196 | 62.06 | <0.001 |
These results suggest that a strong ascertainment bias is likely to operate when loci cloned in one species are compared with their orthologues in a closely related species, and such comparisons should be therefore be stringently evaluated. However, although the earlier observation of human microsatellites being longer than their chimpanzee orthologues must have been affected by a strong ascertainment bias (31), our unbiased analysis confirms that such a difference exists, at least for dinucleotide repeats.
Effect of Repeat Length on Mutability.
The dependency of the mutation process on repeat length is an important issue in microsatellite evolution (48). Under the stepwise mutation model, the mutation rate of a locus is independent of repeat number (19). However, it is likely that the microsatellite mutation process exhibits some form of length dependency (9, 27, 28), although it is not known whether mutation rate increases linearly or exponentially with allele length. To characterize the relationship between mutability and allele length, arrays of repeat motif size 1–4 bp were binned according to both human and chimpanzee length. An estimate of mutability per locus per generation based on the average squared divergence between orthologues was calculated, assuming a 20-year generation time and a 5 million-year time of divergence of the human and chimpanzee lineages. This measure is affected by both mutation rate and the variance in step size. A consistent increase in mutability with allele length is shown for both mono- and dinucleotide loci (Fig. 1 Upper). Furthermore, and importantly, mutability per repeat unit also steadily increases for these motif sizes (Fig. 1 Lower), suggesting that the effect of allele length on mutability is greater than a linear increase. For all motif sizes, repeats in the shortest length category have the lowest average mutability both per locus and per repeat unit.
Figure 1.

Effect of allele length on mutability per generation measured per locus (Upper) and per repeat unit (Lower) for microsatellites of repeat motif sizes 1–4 bp. Loci with allele lengths on the boundary between classes were placed in the higher category.
To examine the significance of the increase in mutability with locus length, we divided the entire data set into loci longer or shorter than 18 bp (Table 4) on the basis of both human and chimpanzee length. There is a highly significant increase in mutability in the longer allele length class for all motif sizes. Additionally, mutability per repeat unit is also significantly higher in the longer length class for all motif sizes, indicating that mutability increases more than linearly with repeat number between these length categories.
Table 4.
Estimates of microsatellite mutability
| Type of repeat | Length class, bp | No. of loci (after correction)* | Divergent orthologues/no. of loci | P | Average mutability per locus × 10−5 | P | Mutability per repeat unit × 10−6 | P |
|---|---|---|---|---|---|---|---|---|
| Mononucleotide | 9–18 | 982 | 0.73 | <0.025 | 3.20 | <0.001 | 2.52 | <0.001 |
| >18 | 289 | 0.92 | 9.63 | 4.06 | ||||
| Dinucleotide | 10–18 | 296 | 0.40 | <0.001 | 2.29 | <0.001 | 3.58 | <0.001 |
| >18 | 199 | 0.84 | 10.22 | 6.14 | ||||
| Trinucleotide | 9–18 | 180 | 0.15 | <0.001 | 0.09 | <0.001 | 0.22 | <0.001 |
| >18 | 32 | 0.66 | 2.44 | 2.68 | ||||
| Tetranucleotide | 12–18 | 308 | 0.12 | <0.001 | 0.17 | <0.001 | 0.53 | <0.001 |
| >18 | 119 | 0.59 | 1.72 | 2.09 |
Average number of loci in length category when orthologues are sorted by human and chimpanzee length.
We ran simulations to compare the finding of an increase in mutability with allele length with the predictions of a null model of a constant single-step mutation rate across allele lengths. The purpose of these simulations was to check that, after correction for undetected contractions, there was no artificial increase in the mutability estimate with allele length caused by our sampling process. A high mutation rate (0.05 per generation) and a low mutation rate (0.0005 per generation) simulation were performed, leading to expected mutability estimates of 100 and 1, respectively. The expected mutability parameter from the high mutation rate simulation is larger than any observed in the data, whereas the lower parameter is consistent with values observed at the shorter tri- and tetranucleotide loci. After the correction for reverse mutations had been performed, none of the mutability values were significantly different from their expected values (Fig. 2), indicating that it is highly unlikely that observed increases in mutability with allele length in the empirical data are an artifact of the method of microsatellite detection and analysis.
Figure 2.

Results of simulations of microsatellite evolution in two species with constant mutation rates independent of allele length. Alleles were sampled from the final distribution on the basis of length in one species and a correction for unobserved reverse mutations was performed. Average mutability is plotted relative to its expected value given the simulated mutation rate. Both high (0.05 per generation) and low (0.0005 per generation) mutation rates were simulated, leading to expected average squared differences of 1 and 100, respectively.
Significant differences in the estimate of mutability are also exhibited between loci with different repeat motif sizes. Seventeen of the 24 possible pairwise comparisons of loci of motif sizes 1–4 taken from each of the four length categories shown in Fig. 1 Upper reveal highly significant differences (P < 0.001) in mutability. In general, the mutability of microsatellites in a given length class appears to be inversely related to motif size (Fig. 1 Upper), in concordance with the findings of ref. 36. Such differences in mutability between motif sizes could help to explain their relative abundance in the human genome (49).
As we have found a tendency toward length differences between human and chimpanzee orthologues, which are highly significant in dinucleotide repeats but may also exist in tri- and tetranucleotide repeats, our estimates of mutability based on average squared divergence between orthologues are likely to be biased. Consistent between-species differences invalidate the underlying assumptions of the distance measure and inflate the net magnitude of microsatellite length differences, leading to an overestimation of mutation rate. However, there are no significant differences between the respective proportions of loci that are longer in humans and chimpanzees when dinucleotide repeats are divided by average allele length into the longer (>18 bp; 113 longer in humans, 69 longer in chimpanzees) and shorter (18 bp or less; 37 longer in humans, 22 longer in chimpanzees) categories used to compare mutability. The finding of a significant increase in mutability with locus length is therefore unlikely to be affected by the significant length differences between humans and chimpanzees. A further potential bias could be caused by longer microsatellites being more highly mutable, and hence likely to have undergone more drastic changes, involving accumulation of point mutations and length differences, and leading to difficulties in the assignment of orthology. This scenario would result in their exclusion from the data set and an underestimate of mutability in the longer length categories and therefore could not account for an increase in mutability with locus length.
Because of the limited availability of long microsatellites in random genomic sequences, our analysis is restricted to repeat lengths shorter than microsatellites typically used as genetic markers. Data from large-scale human genome mapping and paternity testing based on many different loci generally suggest higher mutation rates (10−4 to 10−2) than our estimates for microsatellites longer than 18 bp (≈10−4 for mono- and dinucleotide and 10−5 for tri- and tetranucleotide repeat loci, assuming the stepwise mutation model). As loci used in genome mapping and paternity analysis are often selected on the basis of polymorphism and are longer on average than the repeats analyzed here, the discrepancies in mutation rate estimates are not surprising, given the significant increase in mutability at longer repeat lengths.
Conclusions
Our demonstration that microsatellite mutability per repeat unit is significantly higher at longer allele lengths may be suggestive of the process of microsatellite evolution. Assuming that microsatellites mutate by replication slippage, this observation indicates that the mutation mechanism is not solely dependent on single-step slippage of the nascent strand at the exact site of nucleotide incorporation, but rather the process of strand synthesis is additionally destabilized by the presence of runs of repeat elements surrounding the site of incorporation, leading to a greater occurrence of mutations in longer alleles or greater length changes in single events. The strong length dependence of microsatellite mutability also suggests an explanation for the large ascertainment bias effect (32): the greater the effect of repeat length on mutability, the greater the bias caused by selecting microsatellites on the basis of levels of polymorphism, which under neutrality will be directly linked to mutation rate. It has also been shown that estimates of genealogical depth based on distance measures are greatly affected by assumptions of length dependency of the mutation process (50). The data presented here should therefore be important for improving such evolutionary distance measures.
Another important conclusion of this study is that the microsatellite mutation process is highly heterogeneous. There are significant differences in mutability between both different allele lengths and different motif sizes. We have also found significant between-species differences in orthologous allele lengths, and significant differences in the magnitude and direction of this disparity depending on motif size. Together, these results indicate that distinct mechanisms operate to produce length mutations in loci of different motif sizes, and that one or more changes in these mechanisms have occurred on the lineages leading to humans and chimpanzees.
Supplementary Material
Acknowledgments
We thank Kristin Eliasson for help with sequence alignments. Financial support was obtained from the Swedish Research Council and from the Swedish Research Council for Agriculture and Forestry. H.E. is a Royal Swedish Academy of Sciences Research Fellow supported by a grant from the Knut and Alice Wallenberg Foundation.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lander E S, Linton L M, Birren B, Nusbaum C, Zody M C, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Nature (London) 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein D B, Schlötterer C, editors. Microsatellites: Evolution and Applications. Oxford: Oxford Univ. Press; 1999. [Google Scholar]
- 3.Stallings R L, Ford A F, Nelson D, Torney D C, Hildebrand C E, Moyzis R K. Genomics. 1991;10:807–815. doi: 10.1016/0888-7543(91)90467-s. [DOI] [PubMed] [Google Scholar]
- 4.Neff B D, Gross M R. Evolution (Lawrence, Kans) 2001;55:1717–1733. doi: 10.1111/j.0014-3820.2001.tb00822.x. [DOI] [PubMed] [Google Scholar]
- 5.Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. Nature (London) 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 6.Slatkin M. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarne P, Lagoda P J L. Trends Ecol Evol. 1996;11:424–429. doi: 10.1016/0169-5347(96)10049-5. [DOI] [PubMed] [Google Scholar]
- 8.Bowcock A M, Ruiz Linares A, Tomfohrde J, Minch E, Kidd J R, Cavalli-Sforza L L. Nature (London) 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- 9.Ellegren H. Nat Genet. 2000;24:400–402. doi: 10.1038/74249. [DOI] [PubMed] [Google Scholar]
- 10.Crow J F. Environ Mol Mutagen. 1993;21:122–129. doi: 10.1002/em.2850210205. [DOI] [PubMed] [Google Scholar]
- 11.Li W H. Molecular Evolution. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- 12.Levinson G, Gutman G A. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 13.Schlötterer C, Tautz D. Nucleic Acids Res. 1992;20:211–215. doi: 10.1093/nar/20.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisen J A. In: Microsatellites: Evolution and Applications. Goldstein D B, Schlötterer C, editors. Oxford: Oxford Univ. Press; 1999. pp. 34–48. [Google Scholar]
- 15.Primmer C R, Saino N, Møller A P, Ellegren H. Mol Biol Evol. 1998;15:1047–1054. doi: 10.1093/oxfordjournals.molbev.a026015. [DOI] [PubMed] [Google Scholar]
- 16.Harr B, Schlötterer C. Genetics. 2000;155:1213–1220. doi: 10.1093/genetics/155.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Peng M, Fang Z, Xu X P. Nat Genet. 2000;24:396–399. doi: 10.1038/74238. [DOI] [PubMed] [Google Scholar]
- 18.Kruglyak S, Durrett R T, Schug M D, Aquadro C F. Proc Natl Acad Sci USA. 1998;95:10774–10778. doi: 10.1073/pnas.95.18.10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura M, Ohta T. Proc Natl Acad Sci USA. 1978;75:2868–2872. doi: 10.1073/pnas.75.6.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimmel M, Chakraborty R. Theor Popul Biol. 1996;50:345–367. doi: 10.1006/tpbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 21.Valdes A M, Slatkin M, Freimer N B. Genetics. 1993;133:737–749. doi: 10.1093/genetics/133.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldstein D B, Linares A R, Cavalli-Sforza L L, Feldman M W. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shriver M D, Jin L, Boerwinkle E, Deka R, Ferrell R E, Chakraborty R. Mol Biol Evol. 1995;12:914–920. doi: 10.1093/oxfordjournals.molbev.a040268. [DOI] [PubMed] [Google Scholar]
- 24.Calabrese P P, Durrett R T, Aquadro C F. Genetics. 2001;159:839–852. doi: 10.1093/genetics/159.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amos W, Sawcer S J, Feakes R W, Rubinsztein D C. Nat Genet. 1996;13:390–391. doi: 10.1038/ng0896-390. [DOI] [PubMed] [Google Scholar]
- 26.Primmer C R, Ellegren H, Saino N, Møller A P. Nat Genet. 1996;13:391–393. doi: 10.1038/ng0896-391. [DOI] [PubMed] [Google Scholar]
- 27.Weber J L. Genomics. 1990;7:524–530. doi: 10.1016/0888-7543(90)90195-z. [DOI] [PubMed] [Google Scholar]
- 28.Brinkmann B, Klintschar M, Neuhuber F, Huhne J, Rolf B. Am J Hum Genet. 1998;62:1408–1415. doi: 10.1086/301869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlötterer C, Ritter R, Harr B, Brem G. Mol Biol Evol. 1998;15:1269–1274. doi: 10.1093/oxfordjournals.molbev.a025855. [DOI] [PubMed] [Google Scholar]
- 30.Kayser M, Roewer L, Hedman M, Henke L, Henke J, Brauer S, Kruger C, Krawczak M, Nagy M, Dobosz T, et al. Am J Hum Genet. 2000;66:1580–1588. doi: 10.1086/302905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubinsztein D C, Amos W, Leggo J, Goodburn S, Jain S, Li S H, Margolis R L, Ross C A, Ferguson-Smith M A. Nat Genet. 1995;10:337–343. doi: 10.1038/ng0795-337. [DOI] [PubMed] [Google Scholar]
- 32.Ellegren H, Primmer C R, Sheldon B C. Nat Genet. 1995;11:360–362. doi: 10.1038/ng1295-360. [DOI] [PubMed] [Google Scholar]
- 33.Schug M D, Hutter C M, Wetterstrand K A, Gaudette M S, Mackay T F C, Aquadro C F. Mol Biol Evol. 1998;15:1751–1760. doi: 10.1093/oxfordjournals.molbev.a025901. [DOI] [PubMed] [Google Scholar]
- 34.Di Rienzo A, Donnelly P, Toomajian C, Sisk B, Hill A, Petzl-Erler M L, Haines G K, Barch D H. Genetics. 1998;148:1269–1284. doi: 10.1093/genetics/148.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber J L, Wong C. Hum Mol Genet. 1993;2:1123–1128. doi: 10.1093/hmg/2.8.1123. [DOI] [PubMed] [Google Scholar]
- 36.Chakraborty R, Kimmel M, Stivers D N, Davison L J, Deka R. Proc Natl Acad Sci USA. 1997;94:1041–1046. doi: 10.1073/pnas.94.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellegren H, Moore S, Robinson N, Byrne K, Ward W, Sheldon B C. Mol Biol Evol. 1997;14:854–860. doi: 10.1093/oxfordjournals.molbev.a025826. [DOI] [PubMed] [Google Scholar]
- 38.Fitzsimmons N N, Moritz C, Moore S S. Mol Biol Evol. 1995;12:432–440. doi: 10.1093/oxfordjournals.molbev.a040218. [DOI] [PubMed] [Google Scholar]
- 39.Forbes S H, Hogg J T, Buchanan F C, Crawford A M, Allendorf F W. Mol Biol Evol. 1995;12:1106–1113. doi: 10.1093/oxfordjournals.molbev.a040284. [DOI] [PubMed] [Google Scholar]
- 40.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brohede J, Ellegren H. Proc R Soc London Ser B. 1999;266:825–833. doi: 10.1098/rspb.1999.0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renwick A, Davison L, Spratt H, King J P, Kimmel M. Genetics. 2001;159:737–747. doi: 10.1093/genetics/159.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul S F, Madden T L, Schaffer A A, Zhang J H, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubinsztein D C, Amos W, Leggo J, Goodburn S, Jain S, Li S H, Margolis R L, Rose C A, Ferguson-Smith M A. Am J Hum Genet. 1995;57:214–214. doi: 10.1038/ng0795-337. [DOI] [PubMed] [Google Scholar]
- 45.Kaessmann H, Wiebe V, Pääbo S. Science. 1999;286:1159–1162. doi: 10.1126/science.286.5442.1159. [DOI] [PubMed] [Google Scholar]
- 46.Kaessmann H, Wiebe V, Weiss G, Pääbo S. Nat Genet. 2001;27:155–156. doi: 10.1038/84773. [DOI] [PubMed] [Google Scholar]
- 47.Santibanez-Koref M F, Gangeswaran R, Hancock J M. Mol Biol Evol. 2001;18:2119–2123. doi: 10.1093/oxfordjournals.molbev.a003753. [DOI] [PubMed] [Google Scholar]
- 48.Ellegren H. Trends Genet. 2000;16:551–558. doi: 10.1016/s0168-9525(00)02139-9. [DOI] [PubMed] [Google Scholar]
- 49.Katti M V, Ranjekar P K, Gupta V S. Mol Biol Evol. 2001;18:1161–1167. doi: 10.1093/oxfordjournals.molbev.a003903. [DOI] [PubMed] [Google Scholar]
- 50.Stumpf M P H, Goldstein D B. Science. 2001;291:1738–1742. doi: 10.1126/science.291.5509.1738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.