

Abstract
We have found that activation of human adult T cell leukemia (Jurkat) cells with anti-Fas Ab leads, in a concentration-dependent manner, to an early burst of production of nitric oxide (NO), which inhibits cell respiration. This results in mitochondrial hyperpolarization, dependent on the hydrolysis of glycolytic ATP by the F1Fo-ATPase acting in reverse mode. During this early phase of activation, there is a transient release of superoxide anion. All these processes can be prevented by an inhibitor of NO synthase. Approximately 2 h after stimulation with anti-Fas Ab, a distinct second phase can be detected. This comprises a concentration-dependent collapse in mitochondrial membrane potential, a second wave of free radical production, and activation of caspase-8 leading to apoptosis. This second phase is abolished by an inhibitor of caspase activation. In contrast, inhibition of NO synthesis leads to an enhancement and acceleration of these latter processes, suggesting that the early NO-dependent phase represents a protective mechanism. The significance of the two phases in relation to cell survival and death remains to be studied.
We have previously shown in Jurkat cells that inhibition of respiration by exogenous nitric oxide (NO) leads to mitochondrial membrane hyperpolarization dependent on the utilization of glycolytic ATP by the F1Fo-ATPase and other transporters acting in reverse mode (1). This process also occurs in astrocytes, which are highly glycolytic cells, but not in neurones (2), which do not invoke glycolysis to maintain ATP concentrations. In addition, we have demonstrated that this hyperpolarization correlates with protection against apoptotic cell death (1). As a result of these observations, we speculated that endogenous NO released during cellular stress may trigger such a defense response (1).
Recently, several groups have reported that, after treatment of a variety of cells with different pro-apoptotic stimuli, there is an early phase of mitochondrial hyperpolarization (3–6). This has been shown to precede the generation of free radicals (3) and has been variously interpreted as being independent of (7) or part of the apoptotic process (3, 6). At present, no satisfactory explanation has been proposed to explain the mechanism of hyperpolarization, the reasons why free radicals are released from the mitochondrion, or the connection of these phenomena with apoptosis.
Our present results show that a pro-apoptotic stimulus, anti-Fas Ab, leads to release of endogenous NO from Jurkat cells in sufficient amounts to inhibit cell respiration and cause a hyperpolarization dependent on the reversal of the F1Fo-ATPase. Moreover, the reduction of the mitochondrial electron transport chain, after inhibition of cytochrome oxidase by NO, leads to generation of superoxide anion (O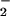 ). We suggest that this process is a cellular defense response that may be overcome by pro-apoptotic mechanisms that occur in parallel.
). We suggest that this process is a cellular defense response that may be overcome by pro-apoptotic mechanisms that occur in parallel.
Materials and Methods
Cell Culture and Preparation.
Human adult T cell leukemia (Jurkat) cells were grown in suspension using a biological stirring platform (Techne Laboratories, Princeton) in RPMI medium 1640 (GIBCO) supplemented with FBS (10% vol/vol), l-glutamine (4 mM), penicillin (100 units/ml), streptomycin (100 μg/ml), and gentamycin (5 μg/ml), at 37°C in a humidified atmosphere containing 5% CO2. Experiments were conducted using cells in log phase growth at a concentration of 0.3–0.6 × 106 per ml. Cells were centrifuged at 200 × g and resuspended in a specially produced RPMI medium 1640 lacking l-arginine and supplemented with Hepes (pH 7.2–7.5; 25 mM). The experiments described in this paper were all carried out in RPMI lacking l-arginine; this was supplemented with 10–15% FBS, which gave an l-arginine concentration of 30–45 μM. Under these conditions, a relatively low concentration (500 μM) of NG-nitro-l-arginine methyl ester (l-NAME) fully inhibited the generation of NO without altering the pH of the medium. Comparison to experiments carried out using RPMI containing l-arginine (1.4 mM) showed that this reduction of the concentration of l-arginine in the medium did not affect the results of any of the experiments.
For each experiment, the cells were divided into two batches, each of which was maintained in a 15-ml Falcon tube in a water-bath at 37°C with gentle agitation for a resting period of 1 h. Unless otherwise stated, drugs and fluorescent probes (see below) were administered to the treatment batch 15 min before administration of anti-Fas Ab (10–1,000 ng/ml). Control cells were treated with the corresponding vehicle. At various times after addition of anti-Fas Ab, 1-ml aliquots were removed and different parameters were measured as indicated.
Most experiments were carried out at a cell density of 106 per ml; however, in those experiments in which cell respiration was measured, oxygen consumption could not be satisfactorily registered at a density of <107 cells per ml.
Detection of NO.
The production of NO in cultured cells was assayed using the fluorescent probe 4,5-diaminofluorescein diacetate (DAF-2/DA) as described (8). The cells (106 per ml) were incubated in 10 μM DAF-2/DA for 25 min before addition of anti-Fas Ab (10 or 100 ng/ml). Green fluorescence of the dye was analyzed at 525 nm every 10 min for 2 h using an EPICS XL flow cytometer (Beckman Coulter). Data were acquired and analyzed using SYSTEM III Version 3.0. Dead cells and debris were excluded from the analysis by electronic gating of forward and side scatter measurements.
Measurement of Oxygen Consumption and NO.
Samples of cells (1 ml at 107 cells per ml) were analyzed at the time points indicated in a gas-tight vessel maintained at 37°C, equipped with a Clark-type oxygen electrode (Rank Brothers, Cambridge, U.K.) connected to a computer with the DUO.18 processing program (World Precision Instruments, Sarasota, FL).
The effect of anti-Fas Ab on cell respiration in the presence or absence of l-NAME (500 μM) was determined. Because the sensitivity of the oxygen electrode required these experiments to be carried out using 10 times more cells (107 per ml) than were used in the other experimental groups, higher concentrations of anti-Fas Ab (up to 1,000 ng/ml) were used to maintain the same ratio of Ab to cell as in the other experimental procedures.
All of the results given for oxygen consumption refer to mitochondrial oxygen consumption. To achieve this, each value was calculated as the percentage of the inhibition of oxygen consumption that was achieved by addition at that time point of 500 nM myxothiazol, the inhibitor of complex III. Initially >95% of oxygen consumption was found to be mitochondrial (and therefore inhibited by myxothiazol), but after ≈2 h some of the oxygen consumption was found to be due to an additional chemical, non-mitochondrial mechanism similar to the one we have described previously in cells treated with exogenous NO (9).
Detection of Mitochondrial Membrane Potential (Δψm).
The Δψm was measured by flow cytometry, using the J-aggregate-forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide (JC-1). In the cytosol and in the mitochondrion at low membrane potential, the monomeric form of the dye fluoresces green (emission at 525 nm), whereas within the mitochondrial matrix at high membrane potential, the concentrated JC-1 forms aggregates that fluoresce red (emission at 590 nm). Samples were incubated with JC-1 at a final concentration of 1 μM at 37°C and were maintained in the dark for 30 min after administration of anti-Fas Ab. Preliminary experiments demonstrated that, under the conditions described, the dye reached near equilibrium distribution and gave a maximal fluorescence response to a fall in Δψm induced by the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (5 μM). Flow cytometry was carried out using an EPICS XL flow cytometer. Results are expressed as either the mean aggregate fluorescence (red) alone or as the ratio of aggregate:monomer (red:green fluorescence).
In a group of experiments in which Δψm was measured concomitantly with the presence of NO, the lipophilic cation tetramethylrhodamine methyl ester (TMRM) (50 nM) was used to measure membrane potential differences. JC-1 could not be used in this particular experiment as it fluoresces in green and red and would interfere with DAF-2/DA fluorescence. TMRM equilibrates readily between compartments in response to potential differences and fluoresces only in orange, thus permitting the simultaneous use of the green fluorescent DAF-2/DA.
To study the involvement of the F1Fo-ATPase in the generation of Δψm, oligomycin (6 μM) was added in the presence or absence of anti-Fas Ab, and Δψm was assessed by flow cytometry.
Detection of Apoptosis.
The viability of cells was monitored for up to 8 h after treatment with anti-Fas Ab. Apoptotic cells were detected by flow cytometry after staining with fluorescein isothiocyanate-conjugated annexin V and propidium iodide (PI) using a commercially available kit (Annexin V-FLUOS, Roche Molecular Biochemicals). Cells were considered apoptotic when they were annexin V-positive and PI-negative. Staining of cells by PI was an indicator of the loss of plasma membrane integrity.
Production of Reactive Oxygen Species (ROS).
The production of ROS was estimated fluorometrically using hydroethidine (HE; Molecular Probes). This oxidation-sensitive fluorescent probe reacts predominantly with O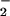 . At each time point studied, cells (106 per ml) were incubated in Hepes-buffered RPMI medium 1640 with 3 μM HE for 15 min, and samples were analyzed using an EPICS XL flow cytometer. Fluorescence emission from oxidized HE (ethidium) was detected at 605 nm. ROS levels, as determined by fluorescence of control cells, served as a baseline for assessment of increased ROS levels in response to stimulation by anti-Fas Ab (10 ng/ml). In some experiments, the complex I inhibitor rotenone (10 μM) was used to determine whether the ROS were generated by the electron transport chain.
. At each time point studied, cells (106 per ml) were incubated in Hepes-buffered RPMI medium 1640 with 3 μM HE for 15 min, and samples were analyzed using an EPICS XL flow cytometer. Fluorescence emission from oxidized HE (ethidium) was detected at 605 nm. ROS levels, as determined by fluorescence of control cells, served as a baseline for assessment of increased ROS levels in response to stimulation by anti-Fas Ab (10 ng/ml). In some experiments, the complex I inhibitor rotenone (10 μM) was used to determine whether the ROS were generated by the electron transport chain.
Determination of Caspase-8 Activity.
Jurkat cells (106 per ml) were treated with anti-Fas Ab (10 ng/ml) and incubated at 37°C. Samples were harvested 0, 30, 60, 90, 180, and 360 min after addition of Ab and lysates were evaluated for caspase-8 activity using a FLICE/Caspase-8 Fluorometric Protease Assay kit (Medical Biological Laboratories, Nagoya, Japan) according to the manufacturer's instructions. Levels of released 7-amino-4-trifluoromethylcoumarin were measured with a Victor2 spectrofluorometer (Wallac, Gaithersburg, MD) with excitation at 395 nm and emission at 505 nm.
Actions of the Caspase Inhibitor N-benzoylcarbonyl-Val-Ala-Aspfluoromethyl ketone (zVADfmk).
In some experiments, the caspase inhibitor zVADfmk (50 μM) was added to cell preparations 3 h before the addition of anti-Fas Ab (10 ng/ml), and its effects on the generation of NO, Δψm, ROS, and cell viability were measured as described above.
Materials and Drugs.
Culture media and FBS were purchased from GIBCO. JC-1, TMRM, and HE were from Molecular Probes. DAF-2/DA and zVADfmk were from Calbiochem. DETA-NO [(Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium 1,2-diolate] was from Alexis (Nottingham, U.K.). Anti-Fas Ab, clone CH11, was from Upstate Biotechnology. l-NAME, oligomycin, and all other chemicals were from Sigma.
Statistics.
Data were analyzed with a two-tailed Student's t test using the graphpad program (GraphPad, San Diego). Values are given as mean ± SEM. A P value <0.05 was considered to be statistically significant.
Results
Measurement of NO by DAF-2/DA.
Anti-Fas Ab (10–100 ng/ml) induced a concentration-dependent increase in intracellular NO (Fig. 1a). This was observed at the earliest reading (2 min) and progressed for up to 1.5 h, at which time it peaked. Thereafter, there was a decrease toward control values (Fig. 1b). Addition of l-NAME (500 μM) to control cells had no significant effect on the fluorescence detected but pretreatment with this concentration of l-NAME inhibited the increase in NO produced by anti-Fas Ab (Fig. 1c).
Figure 1.

Anti-Fas Ab-induced generation of intracellular NO. (a) Anti-Fas Ab (10–100 ng/ml) induced a concentration-dependent shift in fluorescence of DAF-2/DA. Measurements made 40 min after addition of the Ab. (b) Time dependence of the production of NO in response to anti-Fas Ab (10 ng/ml). (c) l-NAME (500 μM) alone had no effect on the DAF-2/DA fluorescent signal but caused a significant shift in the response to anti-Fas Ab (10 ng/ml) toward the baseline value. Measurements made 1.5 h after addition of Ab. Results shown are representative or the mean ± SEM of five experiments.
In an attempt to quantify the amount of NO generated, in some experiments DAF-2/DA-treated cells were incubated with a concentration of DETA-NO (100 μM) which constantly released 200 nM of NO, as measured by using an NO electrode (Iso-NO, World Precision Instruments, Sarasota, FL; lowest limit of detection equals 200 nM NO). The fluorescence generated by the cells was always less than that produced by 200 nM of NO.
Oxygen Consumption.
The rate of oxygen consumption of control cells was unchanged throughout the experiment (5 h, 12.8 ± 1.4 μM/min, data not shown). However, addition of anti-Fas Ab (10–1,000 ng/ml) inhibited cell respiration in a concentration-dependent manner so that 2 h after 100 ng/ml anti-Fas Ab the oxygen consumption was 27 ± 3% (n = 3) of control values (Fig. 2). Cell respiration also was inhibited by the lower concentration of anti-Fas Ab (10 ng/ml); however, this did not become significant until 2.5 h. l-NAME (500 μM) prevented the anti-Fas Ab-induced inhibition of respiration, demonstrating that it depended on the generation of NO (Fig. 2).
Figure 2.

Inhibition of mitochondrial respiration after administration of anti-Fas Ab. Anti-Fas Ab (10–1,000 ng/ml) caused a time-and concentration-dependent inhibition of mitochondrial respiration (see Materials and Methods), which was prevented by treatment with l-NAME (500 μM). n = 3; *, P < 0.05.
Mitochondrial Membrane Potential.
A concentration-dependent hyperpolarization was observed after addition of anti-Fas Ab (10–100 ng/ml, Fig. 3). After 10 ng/ml of anti-Fas Ab the increase in Δψm was detectable at 30 min and peaked at 1.5 h. Thereafter it decreased toward control levels, leading to a progressive depolarization after 3 h. The increase in Δψm was faster after 100 ng/ml of anti-Fas Ab, peaked at 30 min, and thereafter decreased rapidly, resulting in membrane depolarization (Fig. 3). l-NAME (500 μM) inhibited the hyperpolarization and increased the rate of depolarization for both concentrations of anti-Fas Ab (not shown).
Figure 3.

Effect of anti-Fas Ab on Δψm. Anti-Fas Ab (10 and 100 ng/ml) caused an initial hyperpolarization of the mitochondrial membrane, followed by a depolarization, the onset of each of which occurred earlier at the higher concentration. n = 4; *, P < 0.05.
To investigate whether reversal of F1Fo-ATPase was involved in the hyperpolarization of the mitochondria, some experiments were carried out in the presence of oligomycin (6 μM). No hyperpolarization was detected after administration of anti-Fas Ab to oligomycin-treated cells (fluorescence 102 ± 3% of control value in the presence of oligomycin compared to 137 ± 4% in the absence of oligomycin 1.5 h after treatment with 10 ng/ml anti-Fas Ab).
To study the generation of NO and changes in Δψm concomitantly, cells were co-incubated with DAF-2/DA and TMRM. After treatment with anti-Fas Ab (10 or 100 ng/ml), DAF-2/DA fluorescence increased 15–25 min earlier than that of TMRM, indicating that NO is generated before hyperpolarization. The production of NO seemed to be proportional to the degree of hyperpolarization. l-NAME prevented both the increase in NO and in Δψm (data not shown).
ROS.
Treatment of cells with anti-Fas Ab (10 ng/ml) resulted in an increase in HE fluorescence from 30 min onwards, reaching a maximum after 1 h and returning to control levels after 2 h (Fig. 4). The HE fluorescence remained at control levels until a second increase was detected, which became significant after 6 h. This second wave of O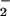 production continued rising until the end of the experimental period (fluorescence 187 ± 5% at 8 h, n = 5). l-NAME (500 μM) prevented the formation of the first peak of O
production continued rising until the end of the experimental period (fluorescence 187 ± 5% at 8 h, n = 5). l-NAME (500 μM) prevented the formation of the first peak of O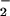 whereas the second wave was enhanced and detected earlier (Fig. 4). The early phase of ROS generation was abolished (inhibition of fluorescence >99%, n = 3) in the presence of rotenone (10 μM), indicating that it originated in the mitochondrion. The later phase of ROS generation was unaffected by treatment with rotenone (n = 3, data not shown).
whereas the second wave was enhanced and detected earlier (Fig. 4). The early phase of ROS generation was abolished (inhibition of fluorescence >99%, n = 3) in the presence of rotenone (10 μM), indicating that it originated in the mitochondrion. The later phase of ROS generation was unaffected by treatment with rotenone (n = 3, data not shown).
Figure 4.

Anti-Fas Ab-induced generation of ROS. After treatment with anti-Fas Ab (10 ng/ml), there was an initial burst of ROS reaching a maximum at 1 h; this was followed, after a delay of several hours, by a prolonged generation of ROS. The early phase of ROS production was inhibited by l-NAME (500 μM) but the second phase was enhanced. In contrast, zVADfmk (50 μM) had no effect on the early phase of ROS generation but inhibited the second phase. n = 5; *, P < 0.05.
Involvement of Caspase.
Caspase-8 activity was not detected until 60 min after administration of anti-Fas Ab, at which time the fluorescence was 124% ± 7% of control activity (n = 4). Its activity then increased to significant levels at 90 min (fluorescence 169% ± 11%) and reached a maximum at 180 min (230% ± 9%). This was maintained for the duration of the experiment (6 h). The caspase inhibitor zVADfmk had no effect on the generation of NO by anti-Fas Ab-treated cells, as detected by DAF-2/DA. Thus the production of NO after anti-Fas Ab stimulation is independent of caspase activation. zVADfmk had no effect on the first peak of O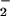 generation but completely abolished the second wave (Fig. 4). In the presence of zVADfmk, no apoptosis could be detected throughout the experimental period (8 h).
generation but completely abolished the second wave (Fig. 4). In the presence of zVADfmk, no apoptosis could be detected throughout the experimental period (8 h).
Cell Viability.
In control cells over a period of 8 h, there was no change in cell viability, measured as a lack of annexin V or PI staining. Anti-Fas Ab treatment (10–100 ng/ml) led to a concentration-dependent decrease in cell viability (Fig. 5a). These cells initially became annexin V positive and PI negative, indicating that apoptosis was occurring (Fig. 5b). A significant number of cells became necrotic in a concentration-dependent manner such that cells treated with a higher concentration of anti-Fas Ab became positive for PI sooner (Fig. 5c). Although l-NAME itself did not affect cell viability, pretreatment of anti-Fas Ab-treated cells with l-NAME resulted in an increase in apoptotic cell death (Fig. 5d).
Figure 5.

Effect of anti-Fas Ab on cell viability determined in cells treated with annexin V and PI. (a) Anti-Fas Ab caused a time- and concentration-dependent fall in cell viability. (b) Treatment with anti-Fas Ab led to apoptosis (cells annexin V positive, PI negative). (c) Treatment with anti-Fas Ab led to necrosis (cells annexin V positive, PI positive). (d) l-NAME (500 μM) alone had no effect on cell viability but significantly enhanced cell mortality 3 h after treatment with anti-Fas Ab (10 ng/ml). n = 5; *, P < 0.05.
Discussion
In recent years, many studies have demonstrated that the mitochondrion is implicated in the process of apoptosis initiated by a variety of stimuli (10–12). Mitochondrial depolarization marks an irreversible commitment of the cell to apoptosis (13). Recently, however, an earlier event, namely hyperpolarization of the mitochondrial membrane, has been observed after the administration to different cell types of several apoptotic stimuli (3–6). This hyperpolarization has been interpreted as being either independent or part of the apoptotic process (3, 7), but the mechanism by which it is generated and its significance remain unclear.
The cell surface receptor Fas is a member of the tumor necrosis factor receptor family expressed in a wide variety of tissues. Interaction between Fas receptor and its ligand, which is found predominantly on activated T cells and natural killer cells, has been implicated in several immune phenomena (14). Fas receptor can induce apoptosis when ligated by natural Fas ligand or by Fas agonist Ab. Fas activation elicits apoptosis by direct activation of caspases and by mitochondrial apoptotic signaling (15).
The role of NO in apoptosis has been widely investigated, and both pro- and anti-apoptotic actions have been reported (16). Furthermore, several pro-apoptotic stimuli, including tumor necrosis factor α (17) and IL-1β (18) have been shown to induce the release of NO. The action of NO in Fas-induced apoptosis also remains controversial (19, 20).
NO has been shown to inhibit the mitochondrial enzyme cytochrome oxidase (complex IV) in competition with oxygen (21, 22), and we have suggested that, under some circumstances, the concentrations of NO may rise so that inhibition of mitochondrial respiration becomes persistent (23, 24). We have shown that, after inhibition of complex IV by continuous exposure to NO, oxidative stress develops, with the subsequent inhibition of other mitochondrial (25) and cytosolic enzymes (23). Furthermore, during blockade of complex IV, cells respond with a defense mechanism that depends on the reversal of the F1Fo-ATPase. Glycolytic ATP is used to extrude protons from the mitochondrial matrix and thus maintain Δψm. This will protect the cell from entering into an apoptotic program (1).
We now show that treatment of Jurkat cells with anti-Fas Ab results in an almost immediate and concentration-dependent increase in the intracellular concentration of NO, which inhibits mitochondrial respiration. We did not establish which NO synthase was responsible for this production; however, mRNAs for endothelial NO synthase (26) and constitutively expressed inducible NO synthase (27) have been identified in Jurkat cells.
Our results also confirm that anti-Fas Ab induces mitochondrial membrane hyperpolarization (3), the extent and duration of which are also concentration-dependent. The hyperpolarization of the mitochondrial membrane is the result of inhibition of cell respiration by the endogenously released NO because treatment with l-NAME, which blocks NO synthesis, reverses both the inhibition of respiration and the hyperpolarization. Furthermore, the hyperpolarization depends on the reversal of the F1Fo-ATPase, as it is blocked by treatment with oligomycin. This is in keeping with our previous results with exogenous NO in Jurkat cells (1) and in astrocytes (2).
Caspase-8 activation was only observed after 1.5 h, confirming previous results in Jurkat cells (28). The caspase inhibitor zVADfmk did not affect the increase in NO, the subsequent inhibition of respiration or the hyperpolarization, indicating that these events precede caspase activation. The hyperpolarization was followed by a collapse in membrane potential. This was more pronounced with higher concentrations of anti-Fas Ab and occurred earlier and proceeded at a faster rate after treatment with l-NAME.
An increase in cellular ROS production has been observed in apoptotic processes triggered by various stimuli (3, 29, 30) and has been claimed to be responsible for the increase in Δψm (6) as well as for its collapse (3, 29, 31). Furthermore, some authors suggest that ROS generation precedes caspase activation (3, 6) whereas others claim that it is a consequence (29). Thus it remains unclear whether the release of ROS is part of a protective mechanism or contributes to apoptosis (see ref. 32). Although several possible sources of ROS have been proposed after activation of cells with apoptotic stimuli (33, 34), a clear mitochondrial source has been identified after activation with tumor necrosis factor α (35), Fas ligand (3), and p53 (6). Only one of these studies (3) has reported a detailed time course for the release of ROS; however, a recent report indicates an early release of mitochondrial ROS, most probably O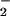 , following tumor necrosis factor stimulation (36).
, following tumor necrosis factor stimulation (36).
Our results show that O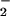 production develops in two sequential stages, which can be manipulated in different pharmacological ways, indicating different mechanisms of production. The early peak of ROS can be completely inhibited by l-NAME and by rotenone, demonstrating that it depends on NO production and is generated in mitochondria. In contrast, the later, more prolonged release of ROS is insensitive to rotenone, enhanced by l-NAME, and abolished by treatment with the general inhibitor of caspases, zVADfmk.
production develops in two sequential stages, which can be manipulated in different pharmacological ways, indicating different mechanisms of production. The early peak of ROS can be completely inhibited by l-NAME and by rotenone, demonstrating that it depends on NO production and is generated in mitochondria. In contrast, the later, more prolonged release of ROS is insensitive to rotenone, enhanced by l-NAME, and abolished by treatment with the general inhibitor of caspases, zVADfmk.
Anti-Fas Ab induces a concentration-dependent decrease in cell viability whose onset was always subsequent to depolarization of the mitochondrial membrane. The apoptotic action of anti-Fas Ab was significantly increased in the presence of l-NAME, indicating that NO and the early increase in ROS is part of a protective mechanism. Indeed, the release of ROS has been implicated as a signaling mechanism, which activates “protective” transcription factors such as NFκB and AP-1 (see refs. 34 and 37). The apoptotic action was completely abolished by zVADfmk, as was the later increase in ROS generation. These results not only confirm previous observations indicating the fundamental role of caspase activation in Fas-induced apoptosis but also strongly suggest that only the later generation of ROS is closely associated with caspase activation and thus with apoptosis.
In conclusion, we have shown that, following anti-Fas Ab in Jurkat cells, there is an increase in NO generation that inhibits cell respiration. This results in mitochondrial membrane hyperpolarization and the early release of free radicals. Both of these events are protective and occur before the activation of caspases. The mechanism by which NO is released and the significance of this defense response remain to be determined. However, it seems to occur in parallel to the activation of pro-apoptotic mechanisms, which also includes a component of ROS formation. It remains to be determined whether these two processes are linked so that the early protective events we have now described are just a negative feedback mechanism in the apoptotic process or whether they are independent, serving biological purposes other than cell death.
Acknowledgments
We thank Annie Higgs for valuable critical insight and Brian Normanly for help with writing the text. This study was financed by Grants SAF98-0118, SAF2001-0763, and 1F97-1029 from the Comisión Interministerial de Ciencia y Technología. E.G.-Z. and S.M. are the recipients of grants from the Ministerio de Ciencia y Tecnologia and Medical Research Council, respectively.
Abbreviations
- ROS
reactive oxygen species
- JC-1
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide
- DAF-2/DA
4,5-diaminofluorescein diacetate
- PI
propidium iodide
- HE
hydroethidine
- TMRM
tetramethylrhodamine methyl ester
- zVADfmk
N-benzoylcarbonyl-Val-Ala-Asp fluoromethyl ketone
- Δψm
mitochondrial membrane potential
- l-NAME
NG-nitro-l-arginine methyl ester
References
- 1.Beltrán B, Mathur A, Duchen M R, Erusalimsky J D, Moncada S. Proc Natl Acad Sci USA. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almeida A, Almeida J, Bolanos J P, Moncada S. Proc Natl Acad Sci USA. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banki K, Hutter E, Gonchoroff N J, Perl A. J Immunol. 1999;162:1466–1479. [PMC free article] [PubMed] [Google Scholar]
- 4.Scarlett J L, Sheard P W, Hughes G, Ledgerwood E C, Ku H H, Murphy M P. FEBS Lett. 2000;475:267–272. doi: 10.1016/s0014-5793(00)01681-1. [DOI] [PubMed] [Google Scholar]
- 5.Khaled A R, Reynolds D A, Young H A, Thompson C B, Muegge K, Durum S K. J Biol Chem. 2001;276:6453–6462. doi: 10.1074/jbc.M006391200. [DOI] [PubMed] [Google Scholar]
- 6.Li P F, Dietz R, von Harsdorf R. EMBO J. 1999;18:6027–6036. doi: 10.1093/emboj/18.21.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hortelano S, Alvarez A M, Bosca L. FASEB J. 1999;13:2311–2317. doi: 10.1096/fasebj.13.15.2311. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Figueroa M O, Day H E, Lee S, Rivier C, Akil H, Watson S J. Brain Res. 2000;852:239–246. doi: 10.1016/s0006-8993(99)02241-6. [DOI] [PubMed] [Google Scholar]
- 9.Orsi A, Beltran B, Clementi E, Hallen K, Feelisch M, Moncada S. Biochem J. 2000;346:407–412. [PMC free article] [PubMed] [Google Scholar]
- 10.Kluck R M, Martin S J, Hoffman B M, Zhou J S, Green D R, Newmeyer D D. EMBO J. 1997;16:4639–4649. doi: 10.1093/emboj/16.15.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P, Nijhawan D, Budihardjo I, Srinivasula S M, Ahmad M, Alnemri E S, Wang X. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 12.Kroemer G, Reed J C. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 13.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere J L, Petit P X, Kroemer G. J Exp Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Famularo G, Nucera E, Marcellini S, De Simone C. Med Hypotheses. 1999;53:50–62. doi: 10.1054/mehy.1997.0712. [DOI] [PubMed] [Google Scholar]
- 15.Nagata S. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 16.Kim P K, Zamora R, Petrosko P, Billiar T R. Int Immunopharmacol. 2001;1:1421–1441. doi: 10.1016/s1567-5769(01)00088-1. [DOI] [PubMed] [Google Scholar]
- 17.Binder C, Schulz M, Hiddemann W, Oellerich M. Anticancer Res. 1999;19:1715–1720. [PubMed] [Google Scholar]
- 18.Ehrlich L C, Peterson P K, Hu S. NeuroReport. 1999;10:1849–1852. doi: 10.1097/00001756-199906230-00009. [DOI] [PubMed] [Google Scholar]
- 19.Mannick J B, Miao X Q, Stamler J S. J Biol Chem. 1997;272:24125–24128. doi: 10.1074/jbc.272.39.24125. [DOI] [PubMed] [Google Scholar]
- 20.Hayden M A, Lange P A, Nakayama D K. J Surg Res. 2001;101:183–189. doi: 10.1006/jsre.2001.6257. [DOI] [PubMed] [Google Scholar]
- 21.Brown G C, Cooper C E. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 22.Cleeter M W, Cooper J M, Darley-Usmar V M, Moncada S, Schapira A H. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 23.Beltran B, Orsi A, Clementi E, Moncada S. Br J Pharmacol. 2000;129:953–960. doi: 10.1038/sj.bjp.0703147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moncada S, Erusalimsky J D. Nat Rev Mol Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. [DOI] [PubMed] [Google Scholar]
- 25.Clementi E, Brown G C, Feelisch M, Moncada S. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiling N, Kroncke R, Ulmer A J, Gerdes J, Flad H D, Hauschildt S. Eur J Immunol. 1996;26:511–516. doi: 10.1002/eji.1830260302. [DOI] [PubMed] [Google Scholar]
- 27.Amin A R, Attur M, Vyas P, Leszczynska-Piziak J, Levartovsky D, Rediske J, Clancy R M, Vora K A, Abramson S B. J Inflamm. 1995;47:190–205. [PubMed] [Google Scholar]
- 28.Hentze H, Schmitz I, Latta M, Krueger A, Krammer P H, Wendel A. J Biol Chem. 2002;277:5588–5595. doi: 10.1074/jbc.M110766200. [DOI] [PubMed] [Google Scholar]
- 29.Johnson T M, Yu Z X, Ferrans V J, Lowenstein R A, Finkel T. Proc Natl Acad Sci USA. 1996;93:11848–11852. doi: 10.1073/pnas.93.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Um H D, Orenstein J M, Wahl S M. J Immunol. 1996;156:3469–3477. [PubMed] [Google Scholar]
- 31.Vander Heiden M G, Chandel N S, Williamson E K, Schumacker P T, Thompson C B. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 32.Droge W. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 33.Friedl H P, Till G O, Ryan U S, Ward P A. FASEB J. 1989;3:2512–2518. doi: 10.1096/fasebj.3.13.2806779. [DOI] [PubMed] [Google Scholar]
- 34.Matthews N, Neale M L, Jackson S K, Stark J M. Immunology. 1987;62:153–155. [PMC free article] [PubMed] [Google Scholar]
- 35.Schulze-Osthoff K, Bakker A C, Vanhaesebroeck B, Beyaert R, Jacob W A, Fiers W. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- 36.Ko S, Kwok T T, Fung K P, Choy Y M, Lee C Y, Kong S K. Biol Signals Recept. 2001;10:326–335. doi: 10.1159/000046900. [DOI] [PubMed] [Google Scholar]
- 37.Chandel N S, Schumacker P T, Arch R H. J Biol Chem. 2001;276:42728–42736. doi: 10.1074/jbc.M103074200. [DOI] [PubMed] [Google Scholar]