

Abstract
Molecular surveillance of pathogenic microbes works by genotyping isolates with DNA fingerprinting techniques and then using these genotypes to assign individuals to populations. Clonality is assumed in many fingerprinting studies, although this assumption has been shown to be false for many organisms. To accommodate recombining organisms into surveillance programs, methods using population allele frequencies in combination with individual multilocus genotypes are necessary. Here, we develop a statistical method appropriate for haploid recombining microbes that allows individuals to be assigned to populations. We illustrate the usefulness of this technique by inferring the source populations for Coccidioides isolates recovered from patients treated outside the endemic area of Coccidioides sp., the etiological agents of human coccidioidomycosis, but with a travel history including visits to one or more endemic areas.
Globalization has increased the rate at which organisms are being introduced into previously nonendemic areas. This traffic has spawned an increase in the number of emerging infectious diseases of animals and plants as a result of nonnative introductions (1). Central to developing policy aimed at controlling such immigration events is the implementation of surveillance systems that enable epidemiologists to identify sources of infection. An example of this type of surveillance is the nationally networked U.S. Pulsenet database, which uses DNA fingerprints for detecting outbreaks of bacterial foodborne disease (www.cdc.gov/ncidod/dbmd/pulsenet/pulsenet.htm). Although the need for such surveillance systems exists for other infectious organisms, differences in their basic biology require alternative theoretical approaches. This fact is true especially for eukaryotic microbes where, in contrast to many bacteria, meiotic recombination is a regular and obligatory feature of most life cycles. These microbes typically differ from higher eukaryotes, however, in having a haploid, rather than a diploid, genome; recently developed methods for assigning diploid individuals to populations (2) must therefore be modified for use with recombining pathogens.
Where population genetic structure is principally clonal, single DNA sequences of sufficient variability are all that is needed to identify source populations (or strains) because of the high degree of linkage among regions of the genome. However, in recombining organisms, each locus has a separate evolutionary history. Therefore, a number of independent loci are required to estimate the relatedness between isolates (3). In this article, we develop statistical methods for determining the source populations of haploid recombining microbes from multilocus data sets, by extending the method developed by Rannala and Mountain (2). We demonstrate the utility of this test by considering the worldwide population genetic structure of the closely related fungal pathogens Coccidioides immitis and Coccidioides posadasii (4). Coccidioides sp. are saprobic soil fungi adapted to growth in the highly arid Lower Sonoran Life Zone. When vertebrates inhale arthroconidia, the fungus undergoes a dimorphic transition to a yeast-like phase that can cause a severe disseminated disease, coccidioidomycosis (5). The incidence of human coccidioidomycosis in endemic areas increased during the 1990s (6), and a concomitant rise in nonendemic areas seems to have occurred (7). A recent study by Chaturvedi et al. (7) of mycology data in New York State showed 181 hospitalizations with coccidioidomycosis. A substudy of 16 of these cases from which patient isolates were available found that all had a history of travel to Coccidioides endemic areas, and multilocus genotyping demonstrated that all isolates were C. posadasii and most likely from Arizona.
Here, we formulate a Bayesian assignment test (BAT), using Bayesian estimation of allele frequencies that assigns unknown isolates of Coccidioides sp. to their population of origin. The method assumes that loci are statistically independent (i.e., in linkage equilibrium) at the intrapopulation level and that all potential source populations have been sampled. The performance of the method, and the effect of violating these assumptions, is assessed. We do this by simulating populations and varying a critical set of population genetic parameters, then evaluate the strength of assignment of random samples of known origin. We subsequently field-test our method by reanalyzing the 16 clinical isolates from patients in New York State and demonstrate a great increase in sensitivity for detecting their source population. These methods provide an important advance in the epidemiological tools necessary for molecular surveillance in this, and other, haploid recombining organisms.
Data and Analyses
Theory and Methodology.
The statistical method used to assign individuals of uncertain origin to populations is similar to that of Rannala and Mountain (2), except that the approach is modified to accommodate haploid, rather than diploid, genotypes. Another change is that the Bayesian posterior probability that an individual derives from any one of I potential source populations is used to assign the individual to a source population rather than basing the decision on a series of likelihood ratio tests. Assuming that individual m derives from a particular population, the probability of the individual's multilocus genotype, Ym = {Yjm }, where Yjm is the allele present at the jth locus in individual m, is used to assign the individual to a source population conditional on a preexisting sample of genotypes from all potential source populations and assuming random mating within populations.
Define x = {xhji} to be a matrix of the allele frequencies in each of the sampled populations, where xhji is the frequency of allele h at locus j in population i. The probability of allele h at locus j is
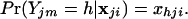 |
1 |
The allele frequencies are, in reality, unknown and therefore we instead consider
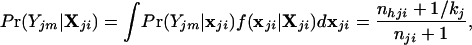 |
2 |
where X = {Xijm} is a matrix of multilocus genotypes among n individuals sampled from the I populations, and Xijm is the allele present at the jth locus of the mth individual sampled from the ith population. Note that nhji is the total number of h alleles at locus j in the sample from population i, and nji is the total number of alleles typed for locus j from population i (both are functions of Xji). We define kj to be the total number of alleles present at locus j (assumed to be identical in all populations and set equal to the observed number of alleles present in all populations). To generate the posterior density of xji, given Xji, we have assumed a Dirichlet prior density for the allele frequencies (2). The posterior probability density that individual m arose from population i is
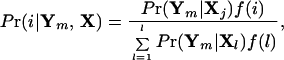 |
3 |
where
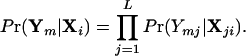 |
4 |
If it is assumed that all populations are equally likely sources for individual m, a priori, then f(i) = 1/I for all i. The potential source populations for an unknown isolate were considered to be those populations contained in the 95% credible set. The 95% credible set is obtained by ranking potential source populations according to their posterior probabilities and sequentially placing populations into the credible set until the posterior probabilities of the populations contained in the set sum to be greater than 0.95 (see Berger ref. 8).
Analysis with Population Simulations.
We studied the expected proportion of BATs that will correctly assign isolates by simulating haploid populations under a range of models using the software environment written by Balloux (9). For each run, 5 populations consisting of 1,000 fully recombining haploid isolates were allowed to evolve for 5,000 generations under an island model of population dispersal. Here, an isolate undergoing dispersal had an equal probability of migrating to any population, and the probability of migration per generation was varied between 0.1 and 0.0001 for each set of simulations. Loci evolved according to the stepwise (10) or infinite alleles (11) models of mutation, and the total genetic diversity was determined by setting the mutation rate to either 0.001, 0.0001, or 0 per generation. The impact of multilocus linkage disequilibrium on the population assignments was investigated by varying the per generation recombination rate between 0 and 1. For each simulation, reference samples of n = 30 genotypes were randomly picked from every population, except where the effects of sample size were being investigated in which case n varied between 5 and 45. BATs were then used to determine the source population of 50 genotypes. The resulting posterior probabilities were assessed as a function of Fst (12), here estimated between pairs of populations with the Weir and Cockerham estimator, θ (13), and calculated with the program fstat (14). The strength of multilocus linkage associations was assessed with the standardized index of association (IA; ref. 15).
Population Allele Frequencies for C. immitis and C. posadasii.
Here, we use data from nine microsatellite loci that were isolated from C. immitis (loci GAC, 621, GA37, GA1, and ACJ) and C. posadasii (loci KO3, KO7, KO1, and KO9). These loci are highly informative because of their extensive polymorphism (between 8 and 26 alleles per locus), making them useful for discriminating between genetically similar isolates (16, 19). Multilocus genotypes were determined for 161 clinical and environmental isolates from 8 populations (3 from C. immitis and 5 from C. posadasii) spanning the worldwide distribution of Coccidioides sp., as described (4).
Within both species, Fst is significant between most pairs of populations (Table 1), and isolates tend to cluster in phylogenetic analyses according to where they were found, showing that pronounced phylogeographical structure exists (4). Previous analyses have shown that levels of linkage disequilibria are low within these populations (barring South American isolates), indicating that recombination has caused local panmixia (17, 18). Within South America, the likelihood that this population results from a postpleistocene radiation from North America, and has therefore undergone a population bottleneck, means that levels of linkage disequilibria are high relative those observed in North America (4). However, pairwise comparisons between unlinked loci show that recombination is (or has) occurred and the IA for this population is low but significant (IA = 0.12, P < 0.01).
Table 1.
Genetic differentiation (Fst) between populations for C. posadasii and C. immitis
|
C. posadasii
|
|||||
|---|---|---|---|---|---|
| SA | SC | TX | AZ | MX | |
| SA | 0.000 | ||||
| SC | 0.2606** | 0.000 | |||
| TX | 0.1389** | 0.1486** | 0.000 | ||
| AZ | 0.3984** | 0.1203* | 0.2785** | 0.000 | |
| MX | 0.1966** | 0.0820* | 0.1191** | 0.2021** | 0.000 |
| C. immitis | |||||
| SC | CC | MX | |||
| SC | 0.000 | ||||
| CC | 0.1056** | 0.000 | |||
| MX | −0.0047 | 0.0662** | 0.000 | ||
SA = South America; SC = Southern California; TX = Texas; AZ = Arizona; MX = Mexico; CC = Central California.
Denotes significance at P = 0.99;
Denotes significant Fst at P = 0.95. Source of the data is from an online database at http://plantbio.berkeley.edu/∼taylor/ftp/genotypes.pdf (4).
Determining the Source Population for Isolates of Unknown Origin.
We applied BATs to determine where patients from nonendemic areas had acquired their infections. Hospitalization records from 1992–1997 were obtained from the state of New York and used to identify 161 persons who were diagnosed with coccidioidomycosis; of this set, 16 patients had positively identified Coccidioides sp. cultures that were available to us (7). All of these 16 patients had a clinical history that recorded travel to areas that are endemic for Coccidioides sp. The isolates were genotyped at all nine microsatellite loci and, based on two diagnostic loci (621 and GAC2; ref. 19), were identified as C. immitis or C. posadasii. An individual is unambiguously assigned to a population if the posterior probability that it belongs to that population is greater than 0.95, given its genotype and the sample of reference genotypes from all potential source populations. The accuracy of the test for determining the correct source population was estimated by simulating novel genotypes from each population with the observed allele frequencies. BATs were then used to assign each simulated genotype, and the numbers of individuals correctly assigned to their source population were determined.
Results
Assignment to Simulated Populations.
We analyzed the performance of the BATs by observing the posterior probabilities and measuring the proportion of individuals correctly assigned to their population as a function of Fst. Fig. 1A shows the relationship, approximated by logistic regression, between the numbers of isolates correctly assigned to the source population and Fst for high (an average of 10 alleles per locus) and low (an average of 4 alleles per locus) diversity populations. The relationships are also shown as a function of the numbers of loci used in the BATs. These data show that to achieve assignment of over 95% of isolates for the levels of genetic divergence seen in this study (where Fst between populations ranges from 0 to 0.4; Fig. 1A), then locus diversity needs to be high (10 alleles per locus). At this level of diversity, only 5 loci were needed to correctly assign 95% of isolates to populations of Fst between 0.3 and 0.4. However, for populations that are more closely related (Fst ≈ 0.2), then between 10 and 20 loci were needed to correctly assign isolates. Further, these data showed that the power of the BATs critically depended on the levels of diversity found at loci. Here, Fst between populations needed to be in the order of 0.5–0.8 to correctly assign isolates to populations for low diversity loci (4 alleles per locus).
Figure 1.

(A) The percentage of individuals correctly assigned to their source population as a function of Fst for loci with high allelic diversity (an average of 10 alleles locus−1, thick lines) and low allelic diversity (an average of 4 alleles locus−1, thin lines). The numbers of loci used in the BATs are shown for 20 (diamonds), 10 (triangles), and 5 loci (squares). Curves are approximated by using logistic regression (P < 0.001 for all curves). (B) Mean posterior probabilities for assignment to their original source population for 50 randomly selected isolates. Reference sample populations of size 5, 15, 30, and 45 are used, drawn from a simulated population of n = 5,000. Fst ≈ 0.16 between populations and assignments are shown for samples of 20, 10, and 5 loci (see A for the definitions of symbols).
In a separate series of analyses, we examined the effect of increasing sample size on the accuracy of assignment for a series of populations with values of Fst similar to those observed in this study (Fst 0.15–0.17; Fig. 1B). Here, posterior probabilities were seen to plateau at values above n = 15 showing that little statistical power was gained by increasing the size of sampled populations. The effect of the model of mutation on posterior probabilities was also examined. The BATs were found to be relatively impervious to how loci were evolving, although assignments for loci mutating under a stepwise mutation model were found to have slightly reduced power as a result of the occurrence of back mutations that lowered the overall genetic diversity of the simulated populations (data not shown). However, these data only affect the general conclusions drawn here in that they tend to the conservative.
The effect of interlocus associations was determined by setting Fst to ≈0.2 and then analyzing the magnitude and variance of posterior probabilities as the IA increased. The simulations (Fig. 2) showed a significant reduction in posterior probabilities and an associated rise in their variances, as the IA rose above 0.02. This change corresponded to a 60% loss in the power of the analysis. Further loss in power associated with increasing linkage disequilibria was not observed and increasing the IA until total clonality was observed demonstrated that 40% of isolates were still being correctly assigned to their source population with a probability of >0.95.
Figure 2.

Mean posterior probabilities for assignment to their original source population for isolates selected from simulated populations with varying recombination rates. Reference sample populations of 30 isolates and 20 loci are used with Fst set to ≈0.2. Posterior probabilities (line marked with ×) and their associated SDs (line marked with +) are marked, as well as a curve plotting the increase of the standardized IA across the simulations.
Assignment of Clinical Isolates to Populations.
Diagnostic loci showed that the isolates of Coccidioides sp. from New York clinical cases consisted of both C. posadasii (12 isolates, Table 2 C. posadasii) and C. immitis (4 isolates, Table 2 C. immitis). BATs assigned 12 of 16 isolates to a source population with high probability (Table 2). For four isolates, assignment was equivocal between two or more populations (isolates 269.97, 376.95, 639.97, and 131.96). We empirically determined the accuracy of the BATs for all populations and species by simulating novel genotypes and then determining the numbers that were correctly assigned to their source population (Table 3). These simulations showed that 97% of C. posadasii and 93% of C. immitis isolates were assigned to the correct source populations (76% and 66% of isolates were assigned with posterior probabilities of >0.95).
Table 2.
Results of assignment tests for nonendemic clinical cases of coccidioidomycosis found in New York State for C. posadasii and C. immitis
| Isolate | Travel | Species | Potential source | Posterior probabilities |
|---|---|---|---|---|
| 269.97* | Arizona | C. posadasii | SC | 0.2086 |
| TX | 0.6580 | |||
| AZ | 0.0324 | |||
| MX | 0.0998 | |||
| 376.95* | Arizona | C. posadasii | AZ | 0.6419 |
| MX | 0.3364 | |||
| 104.94 | Arizona | C. posadasii | AZ | 0.9884 |
| 639.97* | California/Mexico/Southwest U.S. | C. posadasii | AZ | 0.4911 |
| MX | 0.2939 | |||
| 815.97 | California/Arizona | C. posadasii | AZ | 0.9954 |
| 515.95 | California/Arizona | C. posadasii | AZ | 0.9705 |
| 588.97 | Arizona | C. posadasii | AZ | 0.9887 |
| 229.96 | Arizona | C. posadasii | AZ | 0.9500 |
| 686.97 | Southwest U.S. | C. posadasii | AZ | 0.9989 |
| 819.96 | California/Arizona | C. posadasii | AZ | 0.9508 |
| 371.97 | Arizona | C. posadasii | AZ | 0.9980 |
| 750.95 | Arizona | C. posadasii | AZ | 0.9992 |
| 779.97 | California | C. immitis | SC | 0.9995 |
| 131.96* | Arizona/Texas | C. immitis | SC | 0.1307 |
| CC | 0.8265 | |||
| 366.96 | Arizona | C. immitis | CC | 0.9898 |
| 474.97 | California | C. immitis | CC | 0.9750 |
SA = South America; CC = Central California; SC = Southern California; TX = Texas; AZ = Arizona; MX = Mexico.
Signifies isolates that have a significant posterior probability of having come from one or more source populations. Isolates marked in bold are the most likely source population taken from within the 95% credible set.
Table 3.
Results of assigning simulated genotypes to observed populations and species by using observed allele frequencies
| Fst* | Alleles per locus (range) | % Simulated loci correctly assigned (with P > 0.95) | |
|---|---|---|---|
| C. posadasii | 0.216 ± 0.07 | 9.25 (4–16) | 97 (76) |
| C. immitis | 0.081 ± 0.04 | 7.30 (4–12) | 93 (66) |
Average Fst between all geographic populations within species. The ±95% confidence interval is assessed by bootstrap.
In 11 of the 16 tests, the statistically determined source population matched the travel history of the patient and often differentiated among a number of potential regions where infection might have been acquired, e.g., the patient contributing isolate 686.97 had traveled throughout the Southwest U.S. (and therefore had four potential sources of infection), but the genotype showed that it was most likely to have been acquired in Arizona. In 2 of 16 tests, the source population did not match the known travel history of the patient (isolates 269.97 and 366.96). In these cases it is possible that either the case history of the patient is incomplete or the patient had acquired an isolate that had undergone long-distance dispersal.
Discussion
A number of assignment methods for diploid populations have been rigorously tested by using simulation approaches by Cornuet et al. (20), with the general conclusion that a Bayesian approach displayed uniformly higher probabilities of the correct assignment of an individual by comparison with the other methods. The study further concluded that knowledge of the Fst values among an array of populations provided a means of predicting the performance of assignment tests. This now enables genotyping experiments to be optimally designed with respect to the numbers and type of loci required for accurate assignment. The simulations described here bear out their conclusions. We show that there are considerable gains in statistical power resulting from increasing the numbers of loci and targeting highly polymorphic loci. This finding is illustrated by the observation that Fst would need to be on the order of 0.5–0.8 to accurately assign isolates for low-diversity loci. These levels of genetic differentiation are more representative of between-species levels of genetic differentiation (e.g., many alleles are fixed between populations). As a consequence, studies on intraspecific genetic differentiation should aim to maximize the numbers of alleles found at a locus, as well as increasing the numbers of loci themselves.
Our analyses of natural infective Coccidioides isolates show that genetic differentiation among spatially separated populations is sufficient to allow assignment of infections of unknown origin to a source population with high probability. For our clinical data, the expected fraction of misassigned isolates is low, as measured by the fraction of misassigned genotypes observed when simulating with the estimated population frequencies (between 3 and 7%). Moreover, simulated data sets suggest that typing a further 5–10 loci would achieve nearly perfect assignments given the levels of differentiation seen in our study populations. These data are in contrast to a previous study by Chaturvedi et al. (7) who, using these same 16 isolates, assigned them all to the same species (C. posadasii) and population (Arizona). This prior study compared the genotypes of five single nucleotide polymorphism-containing loci against their published allele frequencies (21). Problems inherent in that study were twofold: (i) the loci and alleles used had low discriminatory power between populations and species and (ii) statistical methods for gauging the significance of the associations were not available. The data presented here demonstrate the increased resolution and confidence that is possible when more, highly polymorphic, genetic markers are used in conjunction with the newly developed statistical tools.
Our clinical data show that there are two cases where clinical history and assignment are in conflict (isolates 269.97 and 366.96). That there are high levels of population subdivision for Coccidioides shows that effective long-distance migration between geographical areas has been low historically and suggests that effective wind dispersal of spores is rare. It is therefore unlikely that these two isolates represent cases where the patient has inhaled spores undergoing long-distance dispersal. Rather, incomplete clinical histories are a more likely explanation. One such instance has been recognized by Burt et al. (21), who showed that a Californian isolate with atypical genotype was the result of a patient having acquired an infection in Texas, then having moved to Central California where the infection was diagnosed.
In the case of isolate 269.97, the BATs show that four populations are the possible sources, including the region that the patient traveled through, suggesting that the Arizona population (AZ) is still a possible, albeit less likely, source of infection. In some cases, the set of alleles found within a population may be uninformative relative to the observed genotype. In this case, the power of the test will be low, resulting in posterior probabilities the are nearly uniform (each population having posterior probability of 1/I) as is seen in the case of 269.97. For such isolates, examination of additional loci might resolve this ambiguity by increasing the power of the assignment test.
Active surveillance of infectious diseases allows rapid comparison of DNA fingerprints with centralized, and standardized, databases. Matches made in this manner promise to revolutionize the early detection and management of common-source outbreaks as well as acts of bioterrorism. Moreover, once these databases achieve sufficient coverage of natural genetic and geographical diversity, matching cases to sources of infection becomes possible. However, these techniques are relevant for organisms that reproduce mainly clonally, and hence have relatively stable genomes. Because of the effect of genetic recombination shuffling alleles, the multilocus genotypes of pathogens undergoing random genetic exchange will typically not match those in fingerprint databases, unless by chance alone. Such considerations are paramount in the medically important fungi where estimates show that 75% are morphologically sexual, and where several of the morphologically asexual species (which include C. immitis and C. posadasii) have been shown to undergo cryptic recombination in nature (3, 22, 23). Within Coccidioides, the rate of recombination is high enough that in the two populations that have been extensively surveyed for single nucleotide polymorphisms, only three instances of genetically identical isolates have been found (of which two could have occurred by chance alone), and tests consistently confirm the occurrence of panmixia and linkage equilibrium among markers (17, 18). For such organisms where clonal reproduction has a low impact on the population genetic structure, DNA fingerprinting studies should take into account recombination by comparing the variation in allele frequencies between populations, as described here, rather than the simple identity of multilocus genotypes (24).
There are two main caveats associated with our method. First, our simulations show that as significant linkage associations occur as a result of a decrease in recombination rates, then posterior probabilities drop by about 60%. This finding confirms the necessity of limiting analyses to recombining populations, although loci in linkage disequilibrium can be accommodated at a significant cost to the power of the analysis. In cases where low levels of linkage occur (IA = 0 − 0.1), clone correcting data sets by removing identical genotypes may be applicable. Second, a wide sampling of genotypes are needed to include all potential source populations, and as it is likely that sampling will be focused on clinical isolates then some potential environmental source populations may be missed, initially at least. Unidentified population structure may also occur and further refinement of our method will focus on the development of clustering algorithms to define populations based on local genetic equilibria, as recently proposed by Dawson and Belkhir (25). However, once a scientific community has decided on a standardized set of marker loci on which to focus, multi-user submission of data then will generate data sets of useful depth and quality, as has been the case with the Multilocus Sequence Typing projects of bacteria (26). For recombining pathogens, adopting standardized markers will enable the development of high-resolution data sets that will allow the assignment of isolates to increasingly finer levels of population structure, ultimately providing a powerful tool for detecting the source of disease outbreaks.
Although this article has focused on haploid eukaryotes, the techniques described are potentially applicable to other systems. Multiclone infections are being observed in many bacterial and viral diseases (27), and within these infections chimeric genomes are often observed, implying recombination. Even for taxa lacking meiotic capability, recombination can be frequent, as the often high levels of segregation within populations of certain bacterial species show (28). This mixing illustrates the pressing need to develop the theory and methodology that is necessary to allow the incorporation of data from recombinant populations into molecular epidemiological studies.
Program Availability.
A computer program (bayesass) implementing these procedures was written by B.R. and used to perform the BATs carried out in this article. The program is available from www.rannala.org.
Acknowledgments
This work was supported by National Institutes of Health Grants PO1 AI37232 (to J.W.T.) and R01 HG01988 (to B.R.) and by a Wellcome Trust Biodiversity Fellowship (to M.C.F.).
Abbreviations
- BAT
Bayesian assignment test
- IA
standardized index of association
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Daszak P, Cunningham A A, Hyatt D. Science. 2000;287:443–449. doi: 10.1126/science.287.5452.443. [DOI] [PubMed] [Google Scholar]
- 2.Rannala B, Mountain J L. Proc Natl Acad Sci USA. 1997;94:9197–92013. doi: 10.1073/pnas.94.17.9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor J W, Geiser D M, Burt A, Koufopanou V. Clin Microbiol Rev. 1999;12:126–146. doi: 10.1128/cmr.12.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher M C, Koenig G L, White T J, San-Blas G, Negroni R, Alvarez I G, Wanke B, Taylor J W. Proc Natl Acad Sci USA. 2001;98:4558–4562. doi: 10.1073/pnas.071406098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rippon J W. Medical Mycology. Philadelphia: Saunders; 1988. [Google Scholar]
- 6.Kirkland T M, Fierer J. Emerg Infect Dis. 1996;2:192–199. doi: 10.3201/eid0203.960305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaturvedi V, Ramani R, Gromadzki S, Rodeghier B, Hwa-Ghan C, Morse D. Emerg Infect Dis. 2000;6:25–29. doi: 10.3201/eid0601.000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger J O. Statistical Decision Theory and Bayesian Analysis. 2nd Ed. New York: Springer; 1985. [Google Scholar]
- 9.Balloux F. J Hered. 2001;92:301–302. doi: 10.1093/jhered/92.3.301. [DOI] [PubMed] [Google Scholar]
- 10.Ohta T, Kimura M. Genet Res. 1973;22:201–204. doi: 10.1017/s0016672300012994. [DOI] [PubMed] [Google Scholar]
- 11.Kimura M, Crow J F. Genetics. 1964;49:725–738. doi: 10.1093/genetics/49.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright S. Ann Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 13.Weir B S, Cockerham C C. Evolution (Lawrence, Kans) 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 14.Goudet J. FSTAT 2.9.3. Lausanne, Switzerland: Institute of Ecology; 2001. [Google Scholar]
- 15.Haubold B, Hudson R R. Bioinformatics. 2000;16:847–848. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
- 16.Fisher M C, Koenig G L, White T J, Taylor J W. Mol Ecol. 1999;8:1082–1084. doi: 10.1046/j.1365-294x.1999.00655_5.x. [DOI] [PubMed] [Google Scholar]
- 17.Burt A, Carter D A, Koenig G L, White T J, Taylor J W. Proc Natl Acad Sci USA. 1996;93:770–773. doi: 10.1073/pnas.93.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher M C, Koenig G L, White T J, Taylor J W. J Clin Microbiol. 2000;38:807–813. doi: 10.1128/jcm.38.2.807-813.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher M C, Koenig G L, White T J, Taylor J W. Mycologia. 2001;94:73–84. [PubMed] [Google Scholar]
- 20.Cornuet J-M, Piry S, Luikart G, Estoup A, Solignac M. Genetics. 1999;153:1989–2000. doi: 10.1093/genetics/153.4.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burt A, Dechairo B M, Koenig G L, Carter D A, White T J, Taylor J W. Mol Ecol. 1997;6:781–786. doi: 10.1046/j.1365-294x.1997.00245.x. [DOI] [PubMed] [Google Scholar]
- 22.Gräser Y, Volovsek M, Arrington J, Schonian G, Presber W, Mitchell T G, Vilgalys R. Proc Natl Acad Sci USA. 1996;93:12473–12477. doi: 10.1073/pnas.93.22.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez E, De Meeus T, Mallie M, Renaud F, Symoens F, Mondon P, Piens M A, Lebeau B, Viviani M A, Grillot R, et al. J Clin Microbiol. 1996;3:2559–2568. doi: 10.1128/jcm.34.10.2559-2568.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soll D R. Clin Microbiol Rev. 2000;13:332–370. doi: 10.1128/cmr.13.2.332-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson K J, Belkhir K. Genet Res. 2001;78:59–77. doi: 10.1017/s001667230100502x. [DOI] [PubMed] [Google Scholar]
- 26.Maiden M C J, Bygraves J A, Feil E, Morelli G, Russell J E, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant D A, et al. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Read A F R, Taylor L H. Science. 2001;292:1099–1101. doi: 10.1126/science.1059410. [DOI] [PubMed] [Google Scholar]
- 28.Feil A J, Spratt B G. Annu Rev Microbiol. 2001;55:561–590. doi: 10.1146/annurev.micro.55.1.561. [DOI] [PubMed] [Google Scholar]