

Abstract
Alcohol is a teratogen that induces a variety of abnormalities including brain and facial defects [Jones, K. & Smith, D. (1973) Lancet 2, 999-1001], with the exact nature of the deficit depending on the time and magnitude of the dose of ethanol to which developing fetuses are exposed. In addition to abnormal facial structures, ethanol-treated embryos exhibit a highly characteristic pattern of cell death. Dying cells are observed in the premigratory and migratory neural crest cells that normally populate most facial structures. The observation that blocking Sonic hedgehog (Shh) signaling results in similar craniofacial abnormalities prompted us to examine whether there was a link between this aspect of fetal alcohol syndrome and loss of Shh. We demonstrate that administration of ethanol to chick embryos results in a dramatic loss of Shh, as well as a loss of transcripts involved in Shh signaling pathways. In contrast, other signaling molecules examined do not demonstrate such dramatic changes. Furthermore, we demonstrate that both the ethanol-induced cranial neural crest cell death and the associated craniofacial growth defect can be rescued by application of Shh. These data suggest that craniofacial anomalies resulting from fetal alcohol exposure are caused at least partially by loss of Shh and subsequent neural crest cell death.
The secreted protein Sonic hedgehog (Shh) is essential for multiple events during embryogenesis, including proper craniofacial development (1–4). Of the vertebrate homologues of the Drosophilia segment polarity gene Hedgehog, Shh has the widest expression pattern in the developing head, including the presence of transcripts in the developing branchial arches during the time of neural crest migration (5). SHH mutations in both mice and humans cause significant defects in head development, including distinctive facial and brain abnormalities (3, 4). Analogously, inhibition of Shh-signaling during facial development causes neural crest cell death and a subsequent reduction in head size (1).
There are several intriguing similarities in the craniofacial phenotypes observed in embryos treated with ethanol and those treated with antibodies that block Shh signaling. Both treatments result in a reduction in head size and selective apoptosis of cranial neural crest cells. The neural crest cell death observed after blocking Shh signaling in the chick (1) is similar to that observed after ethanol exposure in a number of species including mouse, chick, and quail, both in vivo and in vitro (6–10). However, the mechanisms by which alcohol affects selective target tissues remain unknown.
In this study, we investigate the impact of embryonic exposure to ethanol on Shh signaling during the critical period of early cranial neural crest migration. After ethanol exposure, we noted a reduction in the mRNA levels of Shh both by in situ hybridization and reverse transcriptase (RT)-PCR, as well as a reduction in the mRNA for a number of genes in the Shh signaling cascade. We further demonstrate that cranial neural crest cells can be rescued from ethanol-induced cell death, and that the associated decrease in size of the frontonasal mass can be prevented by application of exogenous Shh. We have thus identified a molecular deficit associated with embryonic exposure to ethanol that in turn represents a link to the etiology of craniofacial defects.
Materials and Methods
Embryos.
Chicken eggs were obtained from local sources. Eggs were incubated at 37°C with constant humidity. In preliminary experiments, unopened eggs were injected with 200 μl of a solution of 10% ethanol in Ringer's solution, or with Ringer's solution alone, at 26 h of incubation. This procedure has been previously demonstrated to result in craniofacial abnormalities (6). We then modified this procedure, such that eggs were opened at stage 9–10, and 20 μl of a 1% ethanol solution (in Ringers) was placed between the vitelline membrane and the embryo. Both procedures resulted in similar cranial neural crest cell death and loss of frontonasal tissues. Embryos were collected at the desired time, and either harvested in TRIzol (Life Technologies, Grand Island, NY) for RNA isolation or fixed in 4% paraformaldehyde (PFA) for either cell death analysis or in situ hybridization. Whole-mount in situ hybridizations were performed as described (11). Whole mount HNK-1 staining was performed by soaking fixed embryos in HNK-1 supernatant (Development Studies Hybridoma Bank, Iowa City), followed by overnight incubation in 1:100 goat anti-mouse IgM–AP (Zymed), using BCIP (5-bromo-4-chloro-3-indolyl phosphate)/NBT (nitroblue tetrazolium chloride) to visualize.
Cranial Neural Crest Cell Death.
Fixed embryos were cryoprotected in sucrose, then embedded in gelatin and sectioned on a cryostat at 10 μm. Sections were briefly heated to 42°C to remove the gelatin, then blocked in 20% heat-inactivated goat serum in a solution of PBS+BSA + 0.1% Triton, with all further dilutions also being made in this solution. The sections were incubated overnight at 4°C in HNK-1 supernatant (1:10), followed by anti-mouse IgM–FITC (1:500; Zymed). Finally, the sections were incubated for 10 min in 10 μg/ml of the nuclear marker 4′,6-diamidino-2-phenylindole (DAPI) made in water, then coverslips were applied. The percentage of dead cranial crest cells was determined as described (1). All statistics were performed using statview software.
Size Measurements.
Embryos were photographed at fixed magnifications, which changed at different ages and with different head positions. The digital photographs were analyzed using the measuring tool available in photoshop software (Adobe Systems, Mountain View, CA).
Hybridoma Treatments.
Stage 9–10 chick embryos were injected with a function-blocking anti-Shh antibody (Development Studies Hybridoma Bank) as described (1).
RT-PCR.
RNA was pooled from 4–6 embryos per experiment and treated to remove genomic DNA, then cDNA was synthesized using SuperscriptII. Alternatively, RNA was collected from the heads of 6–7 embryos per condition. The relative amounts of cDNA in each matched set was normalized for the presence of the ubiquitous gene GAPDH (12), using a Molecular Dynamics PhosphorImager and imagequant software (Molecular Dynamics). GAPDH was used as a loading control for each PCR reaction. By the amounts of GAPDH and by visual assessment of 18S RNA, there did not appear to be a significant alteration in the amount of RNA that was obtained from ethanol-treated versus control embryos. The optimal cycle number for linear amplification for each set was determined by titration using whole-embryo cDNA from matched stages. The PCR program used was: 94°C for 45 s, 55°C for 45 s, 72°C for 2 min. In addition to GAPDH, the primer pairs for cPtc (13), cSmo (13), and BMP7 (14) were previously published. New primers designed for this study were (sense primer first): cGli2/4, 5′-GTCTATCTCCCCGCTGTCTG-3′ and 5′-CAGGATCTGCTGGTACGTCA-3′ (202 bp); cShh, 5′-TGCTAGGATCGGTGGATAG-3′ and 5′-ACAAGTCAGCCCAGAGGAGA-3′ (197 bp); cGli3, 5′-GCAGGCTAGAGCATTTGGAC-3′ and 5′-CTGCTGACGCTTTCACCATA-3′ (207 bp); and cGli1, 5′-GAAACCTGAACTGGAGACCA-3′ and 5′-GTGCACCACCAGCATGTACT-3′.
Viral Preparations.
Concentrated virus stocks were obtained by infecting DF-1 cells (American Type Culture Collection) with plasmids carrying a retroviral construct capable of expressing cShh, cFgf-8, or control constructs. The resulting supernatant was concentrated in an ultra-centrifuge (15).
Viral Rescue Experiments.
Chick embryos were incubated to the three somite stage [HH stage 8+ (16)]. They were opened after removal of 2 ml of albumin, and a small amount of contrast agent (dilute India Ink) was placed under the embryo. Retrovirus encoding chick Shh was placed over the neural tube and ectoderm using a Picospritzer (General Valve, Fairfield, NJ). The embryos were re-incubated for an additional 12 h, then reopened, and either 20 μl of a 1% ethanol solution or the same amount of Ringer's was placed directly on the embryo surface, below the vitelline membrane. Embryos were collected 12–14 or 20–24 h later, fixed in 4% PFA, and analyzed for cranial neural crest cell death as described above.
Protein and Cell Rescue Experiments.
Chick embryos were incubated to the 8–11 somite stage (HH stage 9–10). They were opened and exposed to ethanol as described above. Using a pressure-injection system we injected ≈3 μl of a 8 ng/μl solution of recombinant mouse Shh amino-terminal terminal peptide (R & D Systems) into the lateral head mesenchyme. Such bacterially expressed recombinant Shh has been used successfully to activate Shh signaling by previous investigators (17, 18). To maintain Shh protein levels for rescue of frontonasal mass size, we performed a second injection between 12–14 h after the first injection and collected the embryos 24 h after exposure to ethanol. Alternatively, DF-1 cells (chicken embryo fibroblasts) engineered to express Shh were injected into the lateral head mesenchyme at the same time as ethanol exposure (stage 9–10), and embryos were collected 24 h later to determine head size.
Results
There are several similarities in the craniofacial phenotypes between embryos treated with ethanol and those treated with antibodies that block Shh signaling. Both display reduction of the frontonasal process, hypoplastic branchial arches, and apoptotic death of cranial neural crest cells (Figs. 1 and 2, Table 1; refs. 1 and 6). Cranial neural crest cells are more susceptible to ethanol treatment than are trunk neural crest cells (Fig. 1 c–f; ref. 20). Adapting a previously established protocol as a model for fetal alcohol syndrome in the chick (6, 7, 19), eggs were injected at stage 9–10 with sufficient ethanol to achieve an 0.15% alcohol level to total egg volume (20). Our results confirm that ethanol treatment results in a loss of frontonasal mass tissue and death of cranial neural crest cells (Figs. 1 and 2, Table 1), which are predominantly responsible for the growth of cranial tissues during early development (1). Measurements of the developing eye did not reveal any reduction in eye growth at this stage (Fig. 1, Table 1). Total growth inhibition did not reach statistical significance at early stages (i.e., 3 days post treatment; Table 1). This finding suggests that the ethanol-induced reduction in head size may be distinct from the general growth inhibition effects of ethanol (21). Although a number of developmental abnormalities were noted in addition to neural crest anomalies, we focused this study on the molecular mechanisms underlying craniofacial growth reduction and cranial neural crest cell death.
Fig 1.
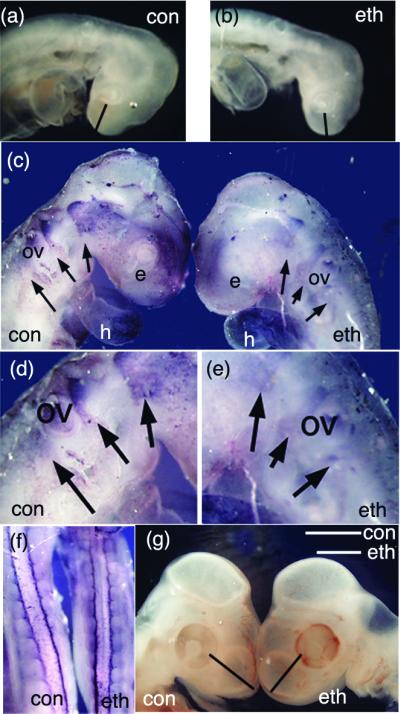
Ethanol treatment results in a reduction in the size of craniofacial tissues. (a and b) Embryos collected approximately 24 h after ethanol treatment (stage 9–10) show distinct differences in craniofacial growth. The measurements for the frontonasal mass are indicated by the bar in each figure; control, 0.42 mm; ethanol, 0.33 mm (see Table 1). (c–f) HNK-1 whole-mount analysis of embryos ≈1 day after ethanol treatment demonstrates a reduction in the HNK-1-positive cranial neural crest. Streams of neural crest are indicated with arrows, and the region around the otic vesicle (ov) is enlarged in d and e. Although crest is clearly present in all appropriate regions, the intensity of staining appears reduced in the whole-mount. HNK-1 staining of non-neural tissue, including the heart (h) appears unchanged. (f) The trunk region of ethanol-treated embryos did not display any neural crest abnormalities when stained with HNK-1. (g) Ethanol-treated (stage 9–10) embryos collected 3 days after treatment show distinct size differences of the frontonasal mass (Table 1).
Fig 2.

There is increased cranial neural crest cell death after ethanol treatment. Embryos were treated with ethanol at stage 9–10, collected 1 day later, fixed, sectioned, and the sections stained for the neural crest marker HNK-1 (in green) and the nuclear marker DAPI (in blue). (a and b) A section taken from the optic cup (oc) region of a control embryo contains HNK-1-positive neural crest cells (nc). (c and d) A section taken from the same region of an ethanol-treated embryo also contains HNK-1-positive neural crest cells. ec, ectoderm; di, diencephalon. (e–e′′). Magnified views of the area outlined in a. A single pyknotic nucleus is present (arrow). This nucleus has an HNK-1-positive membrane (e′ and e′′). (f–f′′). Magnified views of the area outlined in c. Arrows point to three examples of pyknotic nuclei in f, all of which have HNK-1-positive membranes (f′). (g) Analysis of the pyknotic cells demonstrates that exposure to ethanol resulted in a dramatic increase in the percentage of the cranial neural crest cells (HNK-1-positive) that appear pyknotic, whereas no change in the survival of the non-neural crest mesenchyme (HNK-1-negative) was observed. HNK-1-positive: control n = 44; ethanol n = 48. *, Unpaired t test, P < 0.0001. HNK-1-negative: control n = 44; ethanol n = 39, P not significant. (h) Merged image of DAPI and HNK-1 from the region marked “h” in c, a region that contains almost no neural crest cells, and demonstrates no pyknotic nuclei.
Table 1.
Frontonasal mass, eye, and body measurements respond differently to ethanol treatment
| Measurement | Control, mm | Ethanol-treated, mm | P value |
|---|---|---|---|
| 1 day embryo length | 7.08 ± 0.21 (n = 13) | 6.51 ± 0.25 (n = 9) | 0.10 |
| 1 day FNM | 0.44 ± 0.03 (n = 9) | 0.30 ± 0.04 (n = 11) | <0.05 |
| 1 day eye diameter | 0.25 ± 0.012 (n = 14) | 0.25 ± 0.18 (n = 11) | 0.63 |
| 3 day embryo length | 12.20 ± 0.31 (n = 13) | 11.6 ± 0.25 (n = 7) | 0.07 |
| 3 day FNM | 0.78 ± 0.06 (n = 13) | 0.53 ± 0.07 (n = 7) | <0.05 |
| 3 day eye diameter | 2.2 ± 0.08 (n = 13) | 2.0 ± 0.14 (n = 7) | 0.23 |
| 5 day FNM | 1.13 ± 0.06 (n = 4) | 0.81 ± 0.07 (n = 13) | <0.05 |
| 5 day eye diameter | 3.06 ± 0.5 (n = 4) | 2.69 ± 0.15 (n = 13) | 0.32 |
The reduction in head size and the loss of neural crest cells was previously attributed to apoptotic death of cranial neural crest cells (6). To confirm neural crest cell death after ethanol exposure, we measured the percentage of pyknotic nuclei in the cranial neural crest population rostral to the otic vesicle (Fig. 2). HNK-1 immunoreactivity was used to identify cranial neural crest cells and DAPI was used to label all of the nuclei in the section. We found a significant enhancement of cell death in the HNK-1-positive cranial neural crest cells after ethanol treatment compared with control embryos (Fig. 2). In contrast, no increase in cell death was observed in the non-neural crest regions of cranial mesenchyme (Fig. 2 g and h).
We next examined whether exposure to alcohol altered the expression of regulatory genes involved in craniofacial patterning. Fgf-8 and Wnt-1 are both diffusible factors that have been implicated in cranial neural crest development. Although Wnt1 has previously been shown to be involved in the proliferation of neural crest precursors (22), its expression appears unchanged after exposure to ethanol (Fig. 3a). Fgf-8 is a known survival factor for a subset of cranial neural crest cells (23). After ethanol exposure, Fgf-8 expression appears normal in the brain and frontonasal mass, but appears to be slightly reduced in the branchial arches. In transverse sections, Fgf-8 expression is present in the branchial arches (Fig. 3d). Examination of the amount of Fgf-8 by RT-PCR in whole embryos failed to detect a reproducible decrease in transcript levels. However, RT-PCR analysis of isolated heads demonstrated a low, 10% loss of Fgf-8 in ethanol compared with control embryos (data not shown), consistent with a slight reduction that may be confined to the branchial arches.
Fig 3.

In situ hybridizations demonstrate changes in gene expression after ethanol exposure. In some instances, embryos were implanted with a blue agarose bead postfixation to allow mixing during the in situ process. These can be seen in b, e, and f (white arrowheads). (a) There is no apparent change in the Wnt-1 mRNA expression 27 h after alcohol exposure (exposure at stage 10). The staining for Wnt-1 is primarily on the dorsal neural tube, but is also present in the isthmus (i). ov, otic vesicle; h, heart. (b) Fgf-8 mRNA levels remain high 27 h after alcohol exposure (treatment at stage 10). The isthmus is stained for Fgf-8, as is the tail bud and forebrain region (fb) at this stage. A close-up of the branchial arch region of the ethanol-treated embryos demonstrates the Fgf-8 expression is present but reduced ventral to the otic vesicle (ov) (Inset). (c and d) Sections immediately anterior to the otic vesicle through the branchial arch demonstrate that expression of Fgf-8 in this region. nt, neural tube; nc, notochord. (e–h) There are apparent reductions in the mRNA levels of genes in the Shh signaling pathway. (e) Ventral view of embryos collected 24 h after ethanol treatment (at stage 9–10), with in situ hybridization for Shh. Similar results were seen for embryos collected 27–30 h after ethanol treatment. There is a prominent reduction in head Shh (arrow). The reaction product is also decreased in the notochord (nc). (f–h) There is a reduction in the extent and intensity of Gli mRNA in embryos collected 25 h after ethanol treatment. (g and h) Sections through the developing eye cup demonstrate a reduction in Gli message after ethanol treatment. Arrows point to regions demonstrating loss of Gli, compared with control sections. fb, forebrain; h, heart; i, isthmus; nc, notochord; ov, otic vesicle; nt, neural tube.
In contrast to Wnt-1 and Fgf-8, expression of a number of genes in the Shh signaling pathway are drastically reduced after ethanol treatment. The expression of Shh (Fig. 3e) and Ptc transcripts (data not shown) are markedly down-regulated after ethanol exposure. This effect is seen primarily in the developing head (arrow in Fig. 3e); however, Shh also appears reduced in more posterior notochord tissues. The expression of the transcription factors Gli (Fig. 3 f–h) and Gli2 (data not shown) are also reduced by ethanol exposure.
We next examined changes in mRNA levels after ethanol exposure by using RT-PCR (Fig. 4). RNA was collected from embryos 24 h after treatment. The samples were normalized for the content of GAPDH. Phosphorimaging analysis (imagequant software) of matched samples demonstrates a 2–4-fold decrease in Shh transcripts in embryos exposed to ethanol. An even greater reduction was noted in the transcripts for Ptc (from 5–8-fold reduction) and all three Gli genes (at least 6-fold decrease; Fig. 4 Left). Similar findings were seen when the RNA was obtained only from the heads of the embryos (Fig. 4 Center). In contrast, the expression of smoothened (Smo), which is also part of the transmembrane receptor complex for Shh, was not altered by ethanol exposure, as was true for a number of other transcription factors and signaling molecules tested, such as BMP-7 (Fig. 4). The effects of ethanol exposure mirror those observed after treatment with Shh-blocking antibody, which results in significant reduction of mRNAs of genes involved in Shh signaling with the exception of Shh message itself (Fig. 4 Right). This finding suggests that ethanol treatment, unlike anti-Shh antibody exposure, alters regulation of the expression of Shh mRNA.
Fig 4.

Expression of certain genes is altered by exposure to ethanol. RNA was collected from embryos one day after treatment, with Ringer's-treated embryos as controls. RT-PCR was used to determine the relative amounts of the genes of interest in each set of samples. Lane 1, control whole embryo; lane 2, ethanol whole embryo; lane 3, −RT whole embryo; lane 4, control head only; lane 5, ethanol head only; lane 6, −RT head only; lane 7, control hybridoma head only; lane 8, anti-Shh head only; lane 9, −RT head only. The enzyme GAPDH was used as a loading control, and showed no significant variability between samples (less than 0.2-fold difference). In contrast, a number of genes were examined that did show significant decreases in embryos exposed to ethanol (Left). The fold differences in these examples were (control over experimental): GADPH, 1.2× (no difference); Shh, 4× decrease; Smo, 0.9× (no difference); Ptc, 6.8× decrease; Gli1, 100× decrease; Gli2/4, 6× decrease; Gli3, 5.8× decrease; BMP7, 1.0× (no difference). The fold differences when the samples were limited to head RNA were (control over experimental): GADPH, 1.0× (no difference); Shh, 2.5× decrease; Smo, 1.2× (no difference); Ptc, 5× decrease; Gli1, 100× decrease; Gli2/4, 7.5× decrease; Gli3, 6× decrease; BMP7, 1.0× (no difference). This pattern of changes was also seen in embryos where Shh was directly inhibited using anti-Shh hybridoma cells (Right). The fold differences in these examples were (control over experimental): GADPH, 0.8× (no difference); Shh, 1.3× (no difference); Smo, 1.0× (no difference); Ptc, 14.5× decrease; Gli1, 38× decrease; Gli2/4, 10.6× decrease; Gli3, 6.5× decrease.
The results of the in situ hybridization and RT-PCR comparisons suggest that ethanol significantly reduces the amount of Shh made, and thus down-regulates genes involved in Shh signaling. Because blocking Shh signaling results in cranial neural crest cell death (1), we tested whether addition of exogenous Shh could rescue the neural crest cell death resulting from ethanol exposure. To this end, Shh was overexpressed in the head region by retrovirally mediated gene transfer. As shown above, ethanol exposure results in significant death of cranial neural crest cells (Fig. 5 a and a′′). Importantly, the ethanol-induced cell death was completely prevented by concomitant overexpression of Shh (Fig. 5 b–b′′). In control embryos, we observed a low but significant level of cranial neural crest cell death, as has been described (1). Although this level of cell death might be due to limiting amounts of Shh, addition of exogenous Shh by retroviral infection only slightly reduced the level of cell death in controls (Fig. 5c). This finding suggests that Shh may not be a limiting factor during normal development. As noted above, Fgf-8 was slightly reduced by ethanol exposure. Therefore, we used retrovirally mediated gene transfer to overexpress Fgf-8 (24) in the identical manner done for Shh. In contrast to Shh-retrovirus, the Fgf-8 virus was unable to rescue neural crest cell death induced by ethanol exposure (Table 2).
Fig 5.

Sonic hedgehog rescues cranial neural crest cell death and frontonasal growth defects resulting from ethanol exposure. (a and b) Representative sections through the frontonasal cranial mesenchyme, stained with DAPI (a and b) to show nuclear phenotype or with HNK-1 (a′ and b′) to stain neural crest cells; a′′ and b′′ show merged images. (a) DAPI staining reveals numerous pyknotic nuclei (arrows) in the cranial mesenchyme of embryos exposed to ethanol when infected with a control retrovirus. (a′ and a′′) These cells express the neural crest marker HNK-1. (b–b′′) Few pyknotic nuclei are seen in the cranial mesenchyme of embryos that are infected with a retrovirus that expresses cShh and exposed to ethanol, including cells that express the neural crest marker HNK-1. (c) Analysis of the cranial mesenchyme demonstrates that exposure to ethanol resulted in a dramatic increase in the percentage of the cranial neural crest cells that were pyknotic (first bar), an effect that is completely lost when the embryo is treated with a virus expressing cShh (second bar). The cranial neural crest cell death in embryos that were infected with cShh containing virus but not exposed to alcohol was not different from that seen under control conditions (third and fourth bars). The number of sections analyzed is noted above each bar. (d and e) Shh overexpression rescues the frontonasal growth defect induced by ethanol. (d) embryos treated with Shh were fixed and examined as for Table 1. The number of embryos examined is noted above each bar. The asterisk indicates that ethanol causes a significant decrease in frontonasal mass (P < 0.01), which is no longer true when Shh is present. (e) examples of embryos treated with ethanol and BSA (Left) or Shh (Right).
Table 2.
Fgf-8 viral overexpression does not reduce the percentage of apoptotic cranial neural crest cell death after ethanol treatment
| Control, % | Ethanol, % | P value | |
|---|---|---|---|
| Control virus | 4.0 ± 1.0 (n = 42) | 11.1 ± 2.5 (n = 23) | <0.003 |
| Fgf-8 virus | 2.5 ± 0.5 (n = 40) | 9.6 ± 1.7 (n = 46) | <0.003 |
To control for the possible complication of secondary infection by Shh-producing retroviruses, we tested the ability of purified Shh protein to rescue cranial neural crest cell death. We injected approximately 50 ng of recombinant Shh into the lateral mesenchyme on both sides of the heads of stage 9–10 embryos, then exposed them to ethanol or control carrier. We found that Shh protein reduced ethanol-induced cranial neural crest cell death 12–14 h after treatment (Table 3).
Table 3.
Shh protein reduces the percentage of cranial neural crest cells that are apoptotic after ethanol treatment
| Control, % | Ethanol, % | P value | |
|---|---|---|---|
| No protein 12 h | 3.0 ± 0.54 (n = 45) | 16.2 ± 2.4 (n = 38) | <0.0001 |
| BSA 12 h | 3.1 ± 0.54 (n = 47) | 10.27 ± 1.8 (n = 43) | <0.0002 |
| SHH 12 h | 3.67 ± 0.72 (n = 52) | 2.20 ± 0.42 (n = 48) | n.s. |
n.s., not significant.
In addition to causing neural crest cell death, ethanol administration resulted in significant reductions in the size of the frontonasal mass (Fig. 1, Table 1). We examined whether the rescue of cranial neural crest cell death by Shh was sufficient to reverse this reduction in head growth. Because the Shh protein degrades with time (data not shown), we treated embryos with two doses of Shh protein with the second dose administered 12–14 h after the first, or implanted Shh-expressing cells to have continuous presence of protein; both methods yielded similar results. Such continuous administration of Shh resulted in frontonasal size that was not significantly different from control embryos (Fig. 5 d and e). Embryos exposed to excess Shh in the absence of ethanol demonstrated a distinct set of abnormalities (data not shown), as has been reported (18). Thus, the combination of Shh and ethanol exposure prevents or reverses the craniofacial growth defect observed in the embryos exposed to ethanol alone.
Discussion
The data presented here demonstrate a link between loss of Shh signaling and the craniofacial growth defects resulting from fetal alcohol exposure. Ethanol exposure causes a marked down-regulation of Shh and other molecular components of the Shh signaling pathway, Patched, Gli, Gli2, and Gli3, with no detected change in the levels of other signaling molecules like Wnt1 or BMP7 and only minor effects on Fgf-8. The specificity of the effect is confirmed by the rescue of the cranial neural crest cell death and size of the frontonasal mass by administration of Shh, but not Fgf-8. Embryos treated with antibodies to Shh demonstrate a similar down-regulation of Shh signaling components and also have similar craniofacial phenotypes.
In a previous study in zebrafish, exposure to ethanol were reported to down-regulate Shh levels (25). However, the doses used were sufficiently high to cause failure of prechordal mesoderm to migrate anteriorly. This alone can lead to loss of cranial Shh. Therefore, the apparent down-regulation may have been secondary to mesodermal defects. In the present study, we see significant effects on Shh message and cranial neural crest survival even in embryos treated at stage 10, well after prechordal mesoderm migration has occurred. In addition, we do not see any sign of cyclopia in our embryos, which would indicate a loss of prechordal mesoderm. Finally, the levels of BMP7, another marker of prechordal mesoderm (26), appears unchanged. Therefore, we conclude that our effects are not due to loss of prechordal mesoderm.
Although we do not know whether the actions of Shh on cranial neural crest cells are direct or indirect, we previously showed that cranial neural crest cells migrating toward the branchial arches express the Ptc receptor (1), as well as the transcription factor Gli2 (data not shown), consistent with the possibility that they directly respond to Shh. Additional evidence for a direct effect of Shh on cranial neural crest cells comes from studies that drive excess Ptc by using a first arch neural crest specific promoter, and in which a decrease in the size of the first arch was noted, suggesting the size difference is due to a direct effect of Shh on cranial neural crest (27). It is interesting to note that the presence of excess Shh protein does not reduce the cell death in the cranial neural crest, and does not appear to be a limiting survival factor. Trunk neural crest cells in culture, although not requiring Shh for survival or proliferation, respond to Shh by altering their migratory behavior, and do so independently of the canonical Shh signaling pathway (28).
Shh may represents a pivotal “checkpoint” in craniofacial development on which many environmental and growth factors act. For example, the reduction of Shh shown here is reminiscent of that seen when retinoid levels are perturbed (29), consistent with the suggestion that one action of ethanol during development is to perturb retinoic acid levels (30, 31). Perturbing retinoic acid appears to affect both Shh and Fgf-8 genes equally and rescue requires expression of both genes (29), unlike the effects seen with ethanol treatment. The relationship between the molecular changes demonstrated here and those seen when retinoid signaling is perturbed need to be further studied. Fetal exposure to alcohol can now be added to the list of environmental insults that result in craniofacial birth defects via an interruption of Shh signaling or message levels. Other such environmental insults include retinoic acid exposure (29), cholesterol biosynthesis inhibition (32), and some plant teratogens (27, 33). Taken together, these data establish a firm link between fetal alcohol syndrome and decreased Shh signaling, leading to subsequent cranial neural crest cell death, and thus reinforcing the importance of Shh in craniofacial development.
Acknowledgments
We thank Sarah Mahoney for technical assistance, Laura Gammill and Tanya Moreno for advice, and Clare Baker and Martin Garcia-Castro for reading the manuscript. We extend many thanks to Drs. Cliff Tabin, Juan Carlos Izpisua-Belmonte, and Mary Dickinson for materials. S.C.A. was supported by an American Heart Association fellowship, Award 1168-F11, as well as by National Institutes of Health Grant USPHS NS36585, awarded to M.B.-F.
Abbreviations
Shh, Sonic hedgehog
RT, reverse transcriptase
DAPI, 4′,6-diamidino-2-phenylindole
References
- 1.Ahlgren S. C. & Bronner-Fraser, M. (1999) Curr. Biol. 9, 1304-1314. [DOI] [PubMed] [Google Scholar]
- 2.Hu D. & Helms, J. A. (1999) Development (Cambridge, U.K.) 126, 4873-4884. [DOI] [PubMed] [Google Scholar]
- 3.Chiang C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H. & Beachy, P. A. (1996) Nature (London) 383, 407-413. [DOI] [PubMed] [Google Scholar]
- 4.Roessler E., Belloni, E., Gaudenz, K., Jay, P., Scherer, S. W., Tsui, L.-C. & Muenke, M. (1996) Nat. Genet. 14, 357-360. [DOI] [PubMed] [Google Scholar]
- 5.Echelard Y., Epstein, D. J., St.-Jacques, B., Shen, L., Mohler, J., McMahon, J. A. & McMahon, A. P. (1993) Cell 75, 1417-1430. [DOI] [PubMed] [Google Scholar]
- 6.Cartwright M. M. & Smith, S. M. (1995) Alcohol Clin. Exp. Res. 19, 1454-1462. [DOI] [PubMed] [Google Scholar]
- 7.Cartwright M. M. & Smith, S. M. (1995) Alcohol Clin. Exp. Res. 19, 378-386. [DOI] [PubMed] [Google Scholar]
- 8.Chen S. Y., Periasamy, A., Yang, B., Herman, B., Jacobson, K. & Sulik, K. K. (2000) Alcohol 20, 75-81. [DOI] [PubMed] [Google Scholar]
- 9.Rovasio R. A. & Battiato, N. L. (1995) Int. J. Dev. Biol. 39, 421-422. [PubMed] [Google Scholar]
- 10.Sulik K. K., Johnston, M. C. & Webb, M. A. (1981) Science 214, 936-938. [DOI] [PubMed] [Google Scholar]
- 11.Nieto M. A., Patel, K., Wilkinson, D. G. & Cooke, J. (1994) in Methods in Avian Embryology, ed. Bronner-Fraser, M. (Academic, San Diego), pp. 220–235.
- 12.Munsterberg A. E., Kitajewski, J., Bumcrot, D. A., McMahon, A. P. & Lassar, A. B. (1995) Genes Dev. 9, 2911-2922. [DOI] [PubMed] [Google Scholar]
- 13.Lewis K. E., Drossopoulou, G., Paton, I. R., Morrice, D. R., Robertson, K. E., Burt, D. W., Ingham, P. W. & Tickle, C. (1999) Development (Cambridge, U.K.) 126, 2397-2407. [DOI] [PubMed] [Google Scholar]
- 14.Wilson S. I., Graziano, E., Harland, R., Jessell, T. M. & Edlund, T. (2000) Curr. Biol. 10, 421-429. [DOI] [PubMed] [Google Scholar]
- 15.Marcelle C., Ahlgren, S. & Bronner-Fraser, M. (1999) Dev. Biol. 214, 277-287. [DOI] [PubMed] [Google Scholar]
- 16.Hamburger V. & Hamilton, H. L. (1951) J. Morphol. 88, 49-67. [PubMed] [Google Scholar]
- 17.Carpenter D., Stone, D. M., Brush, J., Ryan, A., Armanini, M., Frantz, G., Rosenthal, A. & de Sauvage, F. J. (1998) Proc. Natl. Acad. Sci. USA 95, 13630-13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasrallah I. & Golden, J. A. (2001) Teratology 64, 107-113. [DOI] [PubMed] [Google Scholar]
- 19.Cartwright M. M., Tessmer, L. L. & Smith, S. M. (1998) Alcohol Clin. Exp. Res. 22, 142-149. [PubMed] [Google Scholar]
- 20.Smith S. M. (1997) Alcohol Health Res. World 21, 287-295. [PMC free article] [PubMed] [Google Scholar]
- 21.Pennington S. N. (1988) Alcohol 5, 91-94. [DOI] [PubMed] [Google Scholar]
- 22.Ikeya M., Lee, S. M., Johnson, J. E., McMahon, A. P. & Takada, S. (1997) Nature (London) 389, 966-970. [DOI] [PubMed] [Google Scholar]
- 23.Crossley P. H. & Martin, G. R. (1995) Development (Cambridge, U.K.) 121, 439-451. [DOI] [PubMed] [Google Scholar]
- 24.Vogel A., Rodriguez, C. & Izpisua-Belmonte, J.-C. (1996) Development (Cambridge, U.K.) 122, 1737-1750. [DOI] [PubMed] [Google Scholar]
- 25.Blader P. & Strahle, U. (1998) Dev. Biol. 201, 185-201. [DOI] [PubMed] [Google Scholar]
- 26.Dale J. K., Vesque, C., Lints, T. J., Sampath, T. K., Furley, A., Dodd, J. & Placzek, M. (1997) Cell 90, 257-269. [DOI] [PubMed] [Google Scholar]
- 27.ten Berge D., Brouwer, A., Korving, J., Reijnen, M. J., van Raaij, E. J., Verbeek, F., Gaffield, W. & Meijlink, F. (2001) Development (Cambridge, U.K.) 128, 2929-2938. [DOI] [PubMed] [Google Scholar]
- 28.Testaz S., Jarov, A., Williams, K. P., Ling, L. E., Koteliansky, V. E., Fournier-Thibault, C. & Duband, J. L. (2001) Proc. Natl. Acad. Sci. USA 98, 12521-12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider R. A., Hu, D., Rubenstein, J. L. R., Maden, M. & Helms, J. A. (2001) Development (Cambridge, U.K.) 128, 2755-2767. [DOI] [PubMed] [Google Scholar]
- 30.Duester G. (1991) Alcohol Clin. Exp. Res. 15, 568-572. [DOI] [PubMed] [Google Scholar]
- 31.Zachman R. D. & Grummer, M. A. (1998) Alcohol Clin. Exp. Res. 22, 1544-1556. [PubMed] [Google Scholar]
- 32.Gofflot F., Gaoua, W., Bourguignon, L., Roux, C. & Picard, J. J. (2001) Dev. Dyn. 220, 99-111. [DOI] [PubMed] [Google Scholar]
- 33.Cooper M. K., Porter, J. A., Young, K. E. & Beachy, P. A. (1998) Science 280, 1603-1607. [DOI] [PubMed] [Google Scholar]