

Abstract
Background
Myocardial cells from failing human hearts are characterized by abnormal calcium handling, a negative force-frequency relationship, and decreased sarcoplasmic reticulum Ca2+ ATPase (SERCA2a) activity. In this study, we tested whether contractile function can be improved by decreasing the inhibitory effects of phospholamban on SERCA2a with adenoviral gene transfer of antisense phospholamban (asPL).
Methods and Results
Myocardial cells isolated from 9 patients with end-stage heart failure and 18 donor nonfailing hearts were infected with adenoviruses encoding for either the antisense of phospholamban (Ad.asPL), the SERCA2a gene (Ad.SERCA2a), or the reporter genes β-galactosidase and green fluorescent protein (Ad.βgal-GFP). Adenoviral gene transfer with Ad.asPL decreased phospholamban expression over 48 hours, increasing the velocity of both contraction and relaxation. Compared with cardiomyocytes infected with Ad.asPL (n = 13), human myocytes infected with Ad.βgal-GFP (n = 8) had enhanced contraction velocity (20.3 ± 3.9% versus 8.7 ± 2.6% shortening/second; P < 0.01) and relaxation velocity (26.0 ± 6.2% versus 8.6 ± 4.3% shortening/second; P < 0.01). The improvement in contraction and relaxation velocities was comparable to cardiomyocytes infected with Ad.SERCA2a. Failing human cardiomyocytes had decreased contraction and Ca2+ release with increasing frequency (0.1 to 2 Hz). Phospholamban ablation restored the frequency response in the failing cardiomyocytes to normal; increasing frequency resulted in enhanced sarcoplasmic reticulum Ca2+ release and contraction.
Conclusion
These results show that gene transfer of asPL can improve the contractile function in failing human myocardium. Targeting phospholamban may provide therapeutic benefits in human heart failure.
Keywords: calcium, heart failure, gene therapy
Congestive heart failure has been intimately associated with abnormalities in intracellular [Ca2+] handling.1 In the failing heart, resting [Ca2+] is elevated, the amplitude of the [Ca2+] transient is decreased, and its duration is prolonged.2 These changes are the direct result of an abnormal sarcoplasmic reticulum (SR) Ca2+ ATPase pump (SERCA2a), the activity of which is reduced in heart failure.3–5 SERCA2a is regulated by phospholamban, the phosphorylation of which relieves the inhibition of SERCA2a. The phospholamban/SERCA2a interaction controls the calcium content of the SR and ultimately controls cardiac contractility.6,7 A decrease in the phosphorylation of phospholamban, along with an increase in the phoshpholamban/SERCA2a ratio, contributes to contractile dysfunction in heart failure.3,5 Contractility, calcium handling, and the frequency response were restored in isolated failing human cardiomyocytes by restoring this ratio by means of a gene transfer that caused the overexpression of SERCA2a.8 Another approach to restoring the phospholamban/SERCA2a ratio in failing hearts to normal would be to decrease levels of phospholamban through the use of antisense strategies.
In the present study, we have used such a strategy by generating adenoviral vectors expressing antisense of phospholamban and examining their effects on phospholamban expression, SERCA2a activity, and myocyte function in failing human cardiomyocytes.
Methods
Failing human ventricular myocardial tissue was obtained from 9 explanted hearts (5 ischemic and 4 with dilated cardiomyopathy), and nonfailing tissue was obtained from 18 donor hearts. Myocytes were isolated from 1 g of myocardial tissue removed from the free wall of the left ventricle by enzymatic digestion, as described previously.8 The proportion of rod-shaped viable cells at the time of isolation was 30% to 50% (n = 9) for failing and 40% to 60% (n = 18) for nonfailing cardiomyocytes. After isolation, the cells were resuspended in M199, 50 U/mL penicillin, and 50 U/mL streptomycin; equilibrated to pH 7.4; and infected with the adenoviruses at a multiplicity of infection of 100.
The antisense cDNA of phospholamban was first cloned into a shuttle vector, pAdTrack-CMV.9 The resultant plasmid was linearized with restriction endonuclease PmeI and subsequently was cotransformed into E coli BJ5183 cells with an adenoviral backbone plasmid, pAdEasy-1. Recombinants were selected for kanamycin resistance, and recombination was confirmed by multiple-restriction endonuclease analyses. The linearized recombinant plasmid was transfected into adenovirus-packaging cell lines (293 cells). Ad.βgal-GFP, which contains both β-galactosidase and green fluorescent protein (GFP) controlled by separate cytomegalovirus promoters, was used as control. Ad.SERCA2a, which carries both the SERCA2a and GFP genes, was also used.
Forty-eight hours after infection, myocytes were placed in a flow chamber on the stage of an inverted microscope, superfused with oxygenated Krebs-Henseleit solution, and electrically stimulated with biphasic pulse (0.2 Hz, 50% above threshold). Contraction amplitude and rates of contraction and relaxation were recorded online using a video-edge-detection system and data acquisition software (IonOptix). The fluorescent Ca2+ indicator Fura-2 (Molecular Probes) was used to measure intracellular Ca2+ through the use of a dual excitation spectrofluorometer (IonOptix), as described previously.8 Cumulative concentration-response experiments to isoproterenol, ranging from 0.01 nmol/L to 1 μmol/L, were performed. Cell shortening was measured at each dose and also at the maximum shortening response reached.
We isolated SR membranes from ventricular myocytes, and SERCA2a activity assays were carried out on the basis of Pyruvate/NADH-coupled reactions at [Ca2+] of 10 μmol/L, as previously described.8 Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed on the isolated membranes under reducing conditions on a 7.5% to 12.5% separation gel with a 4% stacking gel, and membranes were immunoblotted with 1:2500 diluted monoclonal anti-phospholamban and anti-SERCA2 antibody (Affinity BioReagents, Golden, Colo). The blot was then incubated in a chemiluminescence system and exposed to an X-OMAT AR x-ray film (Fuji Films) for 1 minute. Data are presented as mean ± SEM and were analyzed using a 1-way ANOVA, with statistical differences identified at P < 0.05.
Results
The coexpression of GFP allowed us to identify the cells that were infected and expressing the transgene after 48 hours. Both nonfailing and failing cardiac cells infected with Ad.asPL showed a >50% decrease in the expression of phospholamban, as shown in Figure 1A. SERCA2a measured from failing cardiomyocytes showed a decrease compared with nonfailing cardiomyocytes. Both ablation of phospholamban and increased SERCA2a overexpression enhanced ATPase activities. However, the increase in ATPase activity was larger in cells overexpressing SERCA2a, as shown in Figure 1B. Figure 1C shows tracings from representative cardiomyocytes isolated from failing hearts, which are characterized by decreased shortening and prolonged relaxation when compared with the donor nonfailing cardiomyocytes. As shown in Table, ablation of phospholamban and overexpression of SERCA2a were associated with marked improvement in contraction, relaxation, and calcium handling. Ablation of phospholamban also restored the frequency response in failing cardiomyocytes (data not shown).
Figure 1.
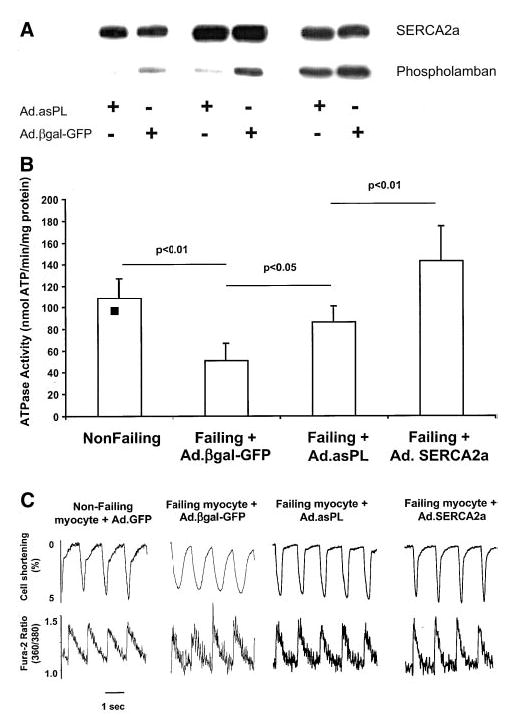
A, Phospholamban and SERCA2a expression from 3 different isolated cardiomyocyte preparations 48 hours after infection with Ad.asPL or Ad.βgal-GFP. B, Measurements of ATPase activity performed at [Ca2+] of 10 μmol/L in membrane preparations from nonfailing cardiomyocytes (n = 10) and failing cardiomyocytes infected for 48 hours with Ad.asPL (n = 6), Ad.SERCA2a (n = 8), or Ad.βgal-GFP (n = 8). C, Recordings from cardiomyocytes isolated from a donor nonfailing heart and from a failing heart infected with an adenovirus expressing either Ad.βgal-GFP, Ad.asPL, or Ad.SERCA2a, stimulated at 1 Hz at 37°C. The failing cells had a characteristic decrease in contraction and prolonged relaxation, along with a prolonged Ca2+ transient. Ablation of phospholamban in the failing cardiomyocytes normalized these parameters.
Phospholamban Ablation, SERCA2a Overexpression, and Improvements in Contraction, Relaxation, and Calcium Handling
| % Contraction | Contraction Velocity, %/s | Relaxation Velocity, %/s | τ, s | Cells, n | |
|---|---|---|---|---|---|
| Ad.βgal-GFP | 2.7 ± 1.0 | 8.7 ± 2.6 | 8.6 ± 4.3 | 0.6 ± 0.13 | 8 |
| Ad.asPL | 5.0 ± 0.9* | 20.3 ± 3.9* | 26.0 ± 6.2* | 0.29 ± 0.05* | 13 |
| Ad.SERCA2a | 4.9 ± 1.8* | 25.4 ± 8.8* | 27.4 ± 10.0* | 0.2 ± 0.04* | 9 |
τ indicates time course of relaxation as a function of increasing extracellular calcium.
P < 0.01 compared with Ad.βgal-GFP.
To examine whether ablation of phospholamban improves calcium handling in human cardiomyocytes, we increased extracellular calcium and measured contractile parameters. As shown in Figure 2A, ablation of phospholamban improved contraction and relaxation velocities to the same degree as did overexpression of SERCA2a at calcium levels up to 4 mmol/L. However, cells overexpressing SERCA2a were more tolerant to the higher calcium concentrations than were the cells infected with Ad.asPL.
Figure 2.
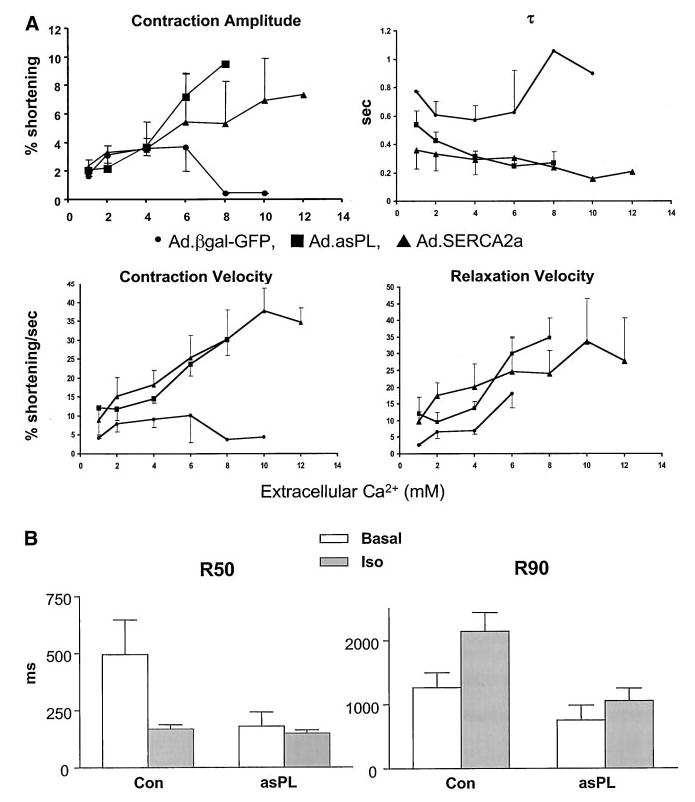
A, Contractile parameters, including percent shortening, velocity of contraction, velocity of relaxation, and τ(time course of relaxation as a function of increasing extracellular calcium), in human cardiomyocytes infected with Ad.asPL (n = 6), Ad.SERCA2a (n = 6) or Ad.βgal-GFP (n = 8). B, Tabulated effects of isoproterenol in human cardiomyocytes infected with either Ad.βgal-GFP or Ad.asPL (control, n = 5; +isoproterenol, n = 14). *P < 0.01 compared with Ad.βgal-GFP.
Phospholamban plays an important role in modulating the response to β-adrenergic stimulus because its phosphorylation leads to increased ATPase activity10 and acceleration of both contraction and relaxation. Addition of isoproterenol in nonfailing cardiomyocytes enhanced contraction but also was associated with aftercontractions (data not shown). Ablation of phospholamban increased contraction and significantly decreased the time of contraction, as depicted in Figure 2B. With the substantial decrease in the time course of contraction in failing human cardiomyocytes infected with Ad.asPL, the addition of isoproterenol still had additive effects on the time course of contractions, as shown in Figure 2B.
Discussion
The present study revealed important findings with regard to targeting calcium cycling proteins in failing human cardiomyocytes: (1) Decreasing phospholamban expression restores contractility in failing ventricular cells of different etiologies; and (2) ablation of phospholamban results in improvement in contractility similar to that of SERCA2a overexpression.
Studies by Kranias et al6,7 have shown clearly that murine models of phospholamban knockout have enhanced contractility. Furthermore, ablation of phospholamban prevented the development of heart failure and restored cardiac function in several murine models of heart failure, including muscle lim protein–knockout (MLP−/−) mice.11 Our results show that improving calcium cycling by decreasing phospholamban inhibition to SERCA2a restores contractility in failing human ventricular cardiomyocytes. These findings also extend previous results showing that overexpression of SERCA2a improves contractile function in human failing cardiac myocytes.
Even though the two strategies—phospholamban decrease and overexpression of SERCA2a—improved contractile function in the failing human cardiomyocytes to the same extent, there were certain differences. SERCA2a activity increased to a greater degree in cardiomyocytes overexpressing SERCA2a. In failing cardiomyocytes, relieving inhibition to SERCA2a pumps, which may be impaired because of oxidative stresses,12 may not restore ATPase activity to normal.
One concern about the strategy of increasing SERCA2a by diminishing phospholamban inhibition is the energy cost, which would be anticipated to increase ATP hydrolysis. Our group has recently shown that overexpression of SERCA2a in a rat model of heart failure enhances contractility without energetic compromise.13 In fact, in this model of heart failure, overexpression of SERCA2a restored the balance between ATP and creatine phosphate.
Another concern with the antisense approach is that it uncouples the β-receptors from one of their downstream targets, phospholamban, rendering them “spare receptors.” This would decrease the modulatory effects of isoproterenol. Interestingly, though, with phospholamban ablation, the contractile state of the cardiomyocyte at baseline is high. Furthermore, the human heart has only a small receptor reserve (if any) for β-adrenoceptors.14 ,15 It is unclear from our study what the long-term effects of functionally increasing these spare receptors will be. Also, the ratio of the Na/Ca exchanger to SERCA2a has been shown to be increased in failing hearts and to be predictive of diastolic function in these hearts.16 Even though ablation of phospholamban would not affect the ratio of SERCA2a to Na/Ca, the resultant enhancement of SERCA2a activity would contribute to restoring diastolic function.
Our results demonstrate that targeting calcium regulation by ablation of phospholamban improves contractile function in failing human cardiomyocytes. This study validates the feasibility of cardiac gene transfer in failing hearts as a therapeutic modality.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (HL 57623 to Dr Hajjar and HL 49574 to Dr Gwathmey) and the British Heart Foundation (Dr Harding) and by a Doris Duke Charitable Foundation Clinician Scientist Award (to Dr Hajjar). Dr Hajjar is a Beeson Scholar of the American Federation of Aging Research. The authors thank the cardiac surgeons at Massachusetts General Hospital and the National Disease Research Interchange.
References
- 1.Gwathmey JK, Copelas L, MacKinnon R, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 2.Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt U, Hajjar RJ, Kim CS, et al. Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am J Physiol. 1999;277:H474–H480. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- 4.Schwinger RH, Bohm M, Schmidt U, et al. Unchanged protein levels of SERCAII and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 5.Schwinger RH, Munch G, Bolck B, et al. Reduced Ca2+-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- 6.Koss KL, Kranias EG. Phospholamban: a prominent regulator of myocardial contractility. Circ Res. 1996;79:1059–1063. doi: 10.1161/01.res.79.6.1059. [DOI] [PubMed] [Google Scholar]
- 7.Luo W, Grupp IL, Harrer J, et al. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res. 1994;75:401–409. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 8.del Monte F, Harding SE, Schmidt U, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He TC, Zhou S, da Costa LT, et al. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tada M, Kirchberger MA, Katz AM. Regulation of calcium transport in cardiac sarcoplasmic reticulum by cyclic AMP-dependent protein kinase. Recent Adv Stud Cardiac Struct Metab. 1976;9:225–239. [PubMed] [Google Scholar]
- 11.Minamisawa S, Hoshijima M, Chu G, et al. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 12.Xu KY, Zweier JL, Becker LC. Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ Res. 1997;80:76–81. doi: 10.1161/01.res.80.1.76. [DOI] [PubMed] [Google Scholar]
- 13.del Monte F, Williams E, Lebeche D, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–1429. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwinger RH, Bohm M, Erdmann E. Evidence against spare or uncoupled beta-adrenoceptors in the human heart. Am Heart J. 1990;119:899–904. doi: 10.1016/s0002-8703(05)80329-1. [DOI] [PubMed] [Google Scholar]
- 15.Brown L, Deighton NM, Bals S, et al. Spare receptors for beta-adrenoceptor-mediated positive inotropic effects of catecholamines in the human heart. J Cardiovasc Pharmacol. 1992;19:222–232. doi: 10.1097/00005344-199202000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Hasenfuss G, Schillinger W, Lehnart SE, et al. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]