

Abstract
Dissociation of human β-2-microglobulin (β2m) from the heavy chain of the class I HLA complex is a critical first step in the formation of amyloid fibrils from this protein. As a consequence of renal failure, the concentration of circulating monomeric β2m increases, ultimately leading to deposition of the protein into amyloid fibrils and development of the disorder, dialysis-related amyloidosis. Here we present the crystal structure of a monomeric form of human β2m determined at 1.8-Å resolution that reveals remarkable structural changes relative to the HLA-bound protein. These involve the restructuring of a β bulge that separates two short β strands to form a new six-residue β strand at one edge of this β sandwich protein. These structural changes remove key features proposed to have evolved to protect β sheet proteins from aggregation [Richardson, J. & Richardson, D. (2002) Proc. Natl. Acad. Sci. USA 99, 2754–2759] and replaces them with an aggregation-competent surface. In combination with solution studies using 1H NMR, we show that the crystal structure presented here represents a rare species in solution that could provide important clues about the mechanism of amyloid formation from the normally highly soluble native protein.
Beta-2-microglobulin (β2m) constitutes the noncovalently bound light chain of the class I human leukocyte antigen (HLA class I). The protein is 99 residues in length and has a seven-stranded β sandwich fold typical of the Ig superfamily (1). In vivo, β2m is continuously shed from the surface of cells displaying HLA class 1 molecules into the serum, where it is transported to the kidneys to be degraded and excreted. As a consequence of renal failure, the concentration of β2m circulating in the serum increases up to 60-fold (2). Free β2m then associates to form amyloid fibrils that typically accumulate in the musculoskeletal system and result in the development of the disorder dialysis-related amyloidosis. The majority of ex vivo β2m amyloid is comprised of full-length wild-type β2m, although small amounts (<30%) of modified or truncated forms are found (2–4).
As for other amyloidogenic proteins (5–7), mild acidification has been shown to promote formation of amyloid fibrils in vitro from pure β2m, both de novo (8–10) and by extension of ex vivo material (11). Under acidic conditions, high yields of amyloid-like fibrils displaying a variety of morphologies form spontaneously in vitro (8–10). Incubation of β2m at pH 3.6 results in the formation of a partially unfolded species that assembles into short curly fibrils ≈10 nm wide and <200 nm in length. By contrast, fibrils with a long straight morphology, more reminiscent of ex vivo amyloid, are generated by incubation of acid-denatured forms of the protein at low ionic strengths at pH 2.5 and below, either alone (10) or in the presence of fibrillar seeds (11). Amyloid fibrils of β2m have also been generated in vitro at neutral pH by deletion of the N-terminal 6 residues (12); incubation of the full length protein in the presence of Cu2+ ions (13); dialysis of the protein at neutral pH into H2O followed by its concentration by drying onto the membrane surface (14); and by the extension of ex vivo material, believed to proceed by the assembly of a rarely populated intermediate onto the ends of existing fibrillar seeds (11, 15). The common theme to emerge from these studies is that conformational rearrangements of the normally highly soluble native protein are required for amyloid fibrils to form.
More than 80 crystal structures of human β2m bound to the heavy chain of the class I HLA complex (HLAβ2m) have been solved to date (16). These structures all display the classical seven-stranded β sandwich fold typical of the Ig superfamily (1). β strands A, B, D, and E comprise one β sheet, whereas β strands C, F, and G form the second β sheet (Fig. 1c). The protein is stabilized by a single disulphide bond between Cys-25 and Cys-80, which links the two β sheets (1, 9, 17). In addition, the protein contains a short (two-residue) C′ β strand that is located in the loop connecting strands C and D. The fourth β strand (D), which lies at one edge of the β sandwich, is divided into two short two-residue β strands (D1 and D2) by a two-residue β bulge. The distortion from regularity of a β strand at the edge of a β sandwich is a common feature of β sheet proteins which, together with conserved inwardly pointing charges, has recently been proposed to prevent edge-to-edge aggregation of normally soluble β sheet proteins (18).
Figure 1.

Ribbon diagram of the crystal structures of (a) MHβ2m and (c) HLAβ2m. Detailed views of the conformation of residues 49–68 are also shown for (b) MHβ2m and (d) HLAβ2m. The structure of HLAβ2m was taken from PDB ID code 1DUZ (25). Individual β strands are labeled A although G. a and c were drawn by using the program MOLSCRIPT (37) and RASTER 3D (38) and b and d, by using SPOCK (39).
Despite the wealth of structures of HLAβ2m, the only crystal structure of monomeric β2m solved to date is that of the closely related bovine protein (MBβ2m) (76% identity to human β2m) (19). In a recent study, the conformational properties of monomeric human β2m (MHβ2m) in solution were determined by using 1H NMR at pH 6.6, 33°C (20). These data showed that the protein has the expected seven-stranded β sandwich fold, consistent with earlier observations (1, 21), although residues 53–62 (the β bulge, strand D2, and its succeeding loop) are dynamic and not uniquely structured in solution (20, 21). In addition, changes in the structure of the loop linking strands A and B were observed.
Here, we report the crystal structure of MHβ2m that reveals structural changes relative to both HLAβ2m and MHβ2m in solution. Combined with additional solution studies using 1H NMR, we show that the conformation of MHβ2m in the crystal represents a rarely populated species in solution that could be important in the generation of amyloid fibrils.
Materials and Methods
Materials.
Escherichia coli strain BL21(DE3) was obtained from Promega; Q-Sepharose and all other reagents were purchased from Sigma; Butyl-Sepharose and Superdex 75 were purchased from Amersham Bioscience, Littie Chalfont, U.K.; and Carbenicillin was from Melford Chemicals, Ipswich, U.K.
β2m Overexpression and Purification.
Monomeric human β2m was expressed from the plasmid pINKwt and purified as previously described (8). The protein was shown to be >99% pure by SDS/PAGE and of the expected molecular weight by electrospray ionization MS (11,860 ± 0.72 Da).
Crystal Structure Determination.
Crystals of monomeric β2m were grown as thin plates at 16°C by the sitting-drop vapor diffusion method from a precipitant buffer solution containing 4–8% (wt/vol) polyethylene glycol 4000, 20% (vol/vol) isopropanol in 100 mM sodium citrate at pH 5.7. The inclusion of isopropanol is essential for crystal formation. Crystals belong to space group C2 with unit cell dimensions a = 77.4 Å, b = 29.1 Å, c = 54.6 Å and β = 121.6°. Data collection was carried out at 100 K by using 25% glycerol, 20% sucrose, and 10% xylitol as a cryoprotectant.
X-ray diffraction data were collected to 1.8-Å resolution on station 14.2 at Daresbury Synchrotron Radiation Source by using an Area Detector System Corporation (ADSC, Poway, CA) Quantum IV charge-coupled device detector. The data were integrated by using mosflm (22), then scaled and reduced by using scala and truncate from the CCP4 program suite (23).
The molecular replacement program amore (24) was used to solve the structure by using data from 8.0 to 3.0-Å resolution. The search model was based on the coordinates of β2m in the HLA class 1 complex (Protein Data Bank ID code 1DUZ) (25). The R factor and correlation coefficient for the best solution were 48.0 and 45.4%, respectively.
The model was built by using the program o (26) and refined by using cns (27, 28) with data to 1.8-Å resolution. Progress of the refinement was monitored by means of the free R factor (29). Water molecules were included in the model once the R factor reached 30%, but only where clear peaks were present in both the 2Fo − Fc and Fo − Fc maps, and where appropriate hydrogen bonds could be made to surrounding residues or other water molecules. Refinement was judged complete when the R factor had converged to 18.8% (Rfree = 23.4%), and no significant interpretable features remained in the Fo − Fc map. The geometry of the model was monitored by using the program procheck (30), the Ramachandran plot showing that 98% of the residues lie in the allowed region. A summary of the refinement statistics is given in Table 1. The coordinates and structure factor amplitudes have been deposited in the Protein Data Bank (ID code 1LDS).
Table 1.
Crystallographic data collection and refinement statistics
| Space group | C2 |
| Unit cell | a = 77.4 Å, b = 29.1 Å, c = 54.6 Å, β = 121.5° |
| Resolution (Å) | 26.7–1.8 |
| Number of observed reflections | 46,169 |
| Number of unique reflections | 9,565 |
| Completeness (%)* | 97.5 (90.6) |
| I/σ(I)* | 15.8 (9.6) |
| Rsym (%)* | 3.5 (6.2) |
| Rfactor (%) | 18.7 |
| Rfree (%) | 23.3 |
| Number of protein atoms | 809 |
| Number of water molecules | 106 |
| Average overall B factor (Å2) | 14.6 |
| rms deviation ideal stereochemistry | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.5 |
Values given in parentheses correspond to those in the outermost shell of the resolution range.
NMR.
For homonuclear experiments, samples of 0.8 mM β2m were prepared at pH 5.7 in 90% H2O/10% D2O or 99.9% D2O. Additional experiments were carried out by using buffer solutions containing 172 mM NaCl (the ionic strength of the crystal growth buffer) and 20% (vol/vol) isopropanol-d8. No significant differences in interresidue nuclear Overhauser effect (NOE) patterns were observed under the different conditions examined. 1H-15N heteronuclear single quantum correlation (HSQC) experiments were recorded as a function of pH on a sample of 0.4 mM protein that was uniformly labeled with 15N (31). Analytical sedimentation velocity ultracentrifugation experiments confirmed that in D2O or H2O the protein is monodisperse and monomeric. In 20% (vol/vol) isopropanol and 172 mM NaCl, however, ≈20% of the protein forms dimers or higher oligomers. These species did not perturb the spectra obtained. All NMR experiments were performed on a Varian Inova spectrometer operating at a 1H frequency of 500 MHz. Gradient-enhanced HSQC spectra were acquired with 128 complex points and 8 scans per increment. Spectral windows were 1,800 Hz (15N) and 3,597 Hz (1H). Mixing times were 50 ms for the total correlation without NOESY (TOWNY) spinlock sequence (32) in the total correlation spectroscopy and 100 ms (D2O) or 150 ms (H2O) in the NOE spectroscopy (NOESY). Water suppression in the spectra acquired in H2O was achieved by adding the excitation sculpting gradient double echo to the end of the sequences (33). Complex points (256 × 1,024) were acquired with a spectral window of 6,400 Hz, and 80–96 scans per increment were collected with a relaxation delay of 1 sec. All NMR data were processed by using nmrpipe, Ver. 97.027.12.56 (34). The data were apodized by using a cosine bell function, followed by zero filling and Fourier transformation. The two-dimensional spectra were analyzed in nmrview (34). The spectra were referenced by using 2,2-dimethyl-2-silapentane-5-sulfonic acid.
Results and Discussion
The Crystal Structure of MHβ2m,
Crystals of MHβ2m grown from pure recombinant material (see Materials and Methods) reached 170 × 80 × 40 μm and diffract to a resolution of 1.8 Å. Data were collected from a single crystal at 100 K and the structure determined by molecular replacement by using the structure of its HLA-bound counterpart (PDB ID code 1DUZ) as a starting model (see Materials and Methods). The final electron-density map is of high quality for the entire main chain, except for the three C-terminal residues that are disordered. As expected, the overall structure of MHβ2m determined here is very similar to that of its HLA-bound counterpart (Fig. 1). Both structures comprise a seven-stranded antiparallel β sandwich, typical of the Ig CH3 domain (1). Residues involved in the β strands include A6–12, B22–30, C36–41, D51–56, E62–69, F78–83 and G91–94 for MHβ2m and A6–11, B21–30, C36–41, D150–51, D255–56, E62–70, F78–83 and G91–94 for HLAβ2m (Fig. 2 a and b). In addition, a short β strand connecting strands C and D is observed (residues 44–45) in both structures and labeled C′, in accord with the β strand assignments by Saper et al. (1). Superposition of all Cα atoms in the crystal structures of HLAβ2m and MHβ2m gives a rms deviation (rmsd) of 3.1 Å. The largest deviations involve residues 12–20, 49–60, and 71–77 (maximum deviation up to 13.5, 5.7, and 3.4 Å, respectively). Omitting these regions from the alignment leads to an rmsd of 0.5 Å. Interestingly, residues 13–18, which lie in the loop linking strands A and B, have been shown to be dynamic in solution (20, 21), whereas 71–77 involves a smaller movement of the loop between strands E and F.
Figure 2.

Topology diagram of the structures of (a) MHβ2m and (b) HLAβ2m showing the location of individual β strands in each structure together with their hydrogen-bonding networks. Solid straight line depicts residues in which both the amide nitrogen and carbonyl oxygen form main-chain–main-chain hydrogen bonds; dotted line, residues that make a main-chain–main-chain hydrogen bond involving either the amide nitrogen or the carbonyl oxygen. Data for b were taken from PDB ID code 1DUZ (25). Strands and hydrogen bonds were determined by using the criteria of Kabsch and Sander (40).
The most significant difference in the crystal structures of MHβ2m and HLAβ2m involves residues in β strand D (50–56) and the succeeding loop. When complexed with the HLA heavy chain, residues 50–56 of HLAβ2m form two short (two-residue) β strands that are separated by a two residue β bulge. These strands (depicted as D1 and D2 in Fig. 1c) each form three main-chain–main-chain hydrogen bonds to the adjacent β strand E (Figs. 1d and 2b). The β bulge in HLAβ2m effectively twists the edge strand, which facilitates its binding to the surface of the heavy chain. The interface with the HLA heavy chain is stabilized by numerous interactions involving residues 1, 6–12, 24–33, 53–56, 60–65, and 98–99 of HLAβ2m. The most significant of these for the structure of MHβ2m presented here is Asp-53, which is located in the β bulge of HLAβ2m and hydrogen bonds to Gln-32, Arg-35, and Arg-48 in the α1 domain of the HLA heavy chain.
The β bulge found in strand D is present in all of the structures of HLAβ2m determined to date as well as in the crystal structure of MBβ2m (19). The crystal structure of MHβ2m determined here is striking in that this feature is no longer present. Instead, residues 51–56 form a continuous six-residue β strand (Fig. 1 a and b) that involves regular backbone φ/ψ angles stabilized by six main-chain–main-chain hydrogen bonds with β strand E, typical of formation of a regular antiparallel β sheet. Loss of the β bulge, together with changes in the positions of three residues in the loop linking strands C and D, causes structural rearrangements of the N-terminal section of strand D. These result in a slip of i + 1 in the hydrogen-bonding partners of residues 50–53 relative to their positions in HLAβ2m, with concomitant reorganization of their side chains (Figs. 1 and 2). Thus, residues 50 and 52 in HLAβ2m form hydrogen bonds with residues 67 and 65, respectively, in strand E. By contrast, loss of the β bulge in the crystal structure of MHβ2m results in residues 51 and 53 forming new hydrogen bonds with residues 67 and 65, respectively (Figs. 1 and 2). The hydrogen-bonding pattern and orientation of side chains at the C-terminal end of the strand (residues 55–57) remain largely invariant in the two crystal structures.
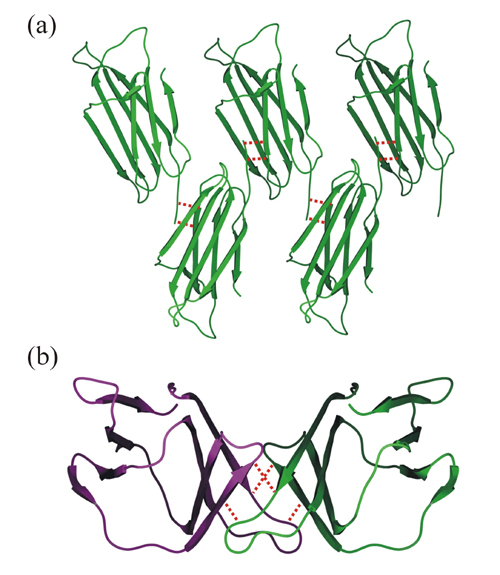
Analysis of crystal contacts revealed a number of interactions between neighboring molecules within the lattice. One of the two major contacting regions is formed between residues 1–4 (N-terminal region) of one molecule and 34–37 (loop B/C and strand C) of a neighboring molecule, forming a continuous array throughout the lattice. This contact involves the formation of a number of main-chain–main-chain hydrogen bonds between adjacent monomers and buries 621 Å2 of the total 1,375 Å2 involved in the crystal contacts. The second region involves residues 12–15 of one molecule (loop A/B) and 54–57 (strand D) of an adjacent molecule (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). These interactions also involve the formation of intermolecular main-chain hydrogen bonds, burying 754 Å2 and resulting in the formation of head-to-tail dimers. In both contacts, the arrangement is similar to adjacent strands in an antiparallel β sheet. Although these contacts are in regions that undergo structural changes in the free and bound forms, it is unlikely that they result from the process of crystallization; rather, the changes are required to initiate formation of the lattice.
Conformational Properties of MHβ2m in Solution.
Previous studies using two-dimensional 1H NMR have suggested that the conformation of MHβ2m in solution resembles closely that of its HLA-bound counterpart (20, 21). Most intriguingly, however, whereas the region encompassing residues 50–57 is highly ordered in the crystal structure of MHβ2m described here, this region appears to be dynamic in solution (20, 21). To determine whether the β bulge and its subsequent β strand (D2) persist in MHβ2m in solution under the crystallization conditions, the protein was also analyzed by using two-dimensional 1H NMR at pH 5.7 in the presence and absence of isopropanol-d8 [20% (vol/vol)] and/or 172 mM NaCl to match the conditions used for crystallization (see Materials and Methods). Backbone assignments for the main-chain hydrogens in strands D and E at pH 5.7 at both 37 and 25°C were determined from those previously obtained at pH 7.0 and 37°C (21) by carefully tracking the resonances of 15N-labeled protein as a function of temperature and pH. The CαΗ chemical shifts were then confirmed directly by NOESY and total correlation spectroscopy experiments. By this approach, 17 of the CαH and 17 of the NH resonances from the 20 residues between residues 50 and 69 (encompassing strands D and E) were assigned unambiguously (the remaining resonances including those of residues 54 and 57 showed line broadening effects or were absent in some of the spectra). The pattern of interstrand NOEs involving CαH and NH hydrogens in strands D and E were then used to determine the secondary structural properties of residues 51–56 in solution. If the β bulge comprising residues 53 and 54 is retained in solution in a major population of molecules at pH 5.7, NOEs between the amide hydrogens of residues 52/65 and 50/67, together with an NOE between the CαH hydrogens of residues 51/66 should be observed (Fig. 3b). However, if the conformation of MHβ2m in solution is identical to that of MHβ2m in the crystal (i.e., the β bulge is absent), then crosspeaks between the amide hydrogens of residues 53/65 and 51/67, and between the CαH hydrogens of residues 54/64 and 52/66 should result (Fig. 3a). Alternatively, if this region is dynamic, the spectra could be more complex. The CαH-CαH and NH-NH regions of 1H-1H NOESY spectra of MHβ2m obtained at pH 5.7, 37°C in D2O and H2O, respectively, are shown in Fig. 3 c and d. The crosspeak pattern observed matches precisely that predicted for retention of the β bulge in solution. All three NOEs that are uniquely characteristic of the presence of the β bulge in solution are clearly observed in the spectrum, whereas those characteristic of the crystal structure presented here are absent. Notably, the peak observed for 51/66 is of similar intensity to those from residues in other regions of the protein that are known to be highly structured in solution (20). The data indicate, therefore, that the major proportion of MHβ2m in solution under the conditions used to crystallize the protein adopts a conformation in which the β bulge involving residues 53 and 54 persists.
Figure 3.

Schematic diagram showing the pattern of NH-NH and CαH-CαH NOEs predicted for conformations of MHβ2m (a) lacking and (b) containing the β bulge involving residues 53 and 54. CαH-CαH region (c) and NH-NH region (d) of a 1H-1H NOESY spectrum of MHβ2m acquired at pH 5.7, 37°C, in D2O and H2O, respectively. Crosspeaks consistent with the presence of the β bulge in solution are marked in bold and underlined. Crosspeaks expected were MHβ2m to adopt a conformation in solution identical to that in a are shown as solid circles.
Resonances from residues 54, 57, and 58 are missing from the two-dimensional total correlation spectroscopy spectrum of MHβ2m at pH 5.7. In addition, the 1H-15N HSQC spectrum of MHβ2m at pH 5.7 shows significant line broadening for residues 55 and 56 (data not shown). Consistent with previous observations (20, 21), our data suggest that these residues, which lie in strand D2 in the crystal structure of HLAβ2m and in the C-terminal region of strand D in MHβ2m, are in intermediate exchange between different conformations in solution, one of which presumably includes the continuous β strand involving residues 51–56 observed here by crystallography. The NMR data described here indicate, therefore, that at least the vast majority of molecules of MHβ2m in solution adopt a structure distinct from that observed in the crystal structure presented here, indicating that a rare conformation of monomeric β2m in solution can form a stable crystal lattice that displaces the equilibrium toward this state.
Implications for Amyloid Formation.
Previous data on a number of proteins, including MHβ2m, have suggested that partial unfolding is a prerequisite for the assembly of native proteins into amyloid fibrils (5–8). Our previous experiments on MHβ2m have suggested that the amyloid precursor state formed at pH 3.6 retains a native-like core involving strands C, D, E, and F that comprise the central Greek key domain in the native protein, whereas the precursor formed at lower pH (pH 2.5) is more highly unfolded (8, 10, 31). Interestingly, the N- and C-terminal regions of the polypeptide, which form β strands A and G in the native protein, are highly destabilized in the amyloid precursor conformation at pH 3.6 and are also relatively weakly protected from hydrogen exchange in the amyloid fibril (31, 35). Despite the rapid increase in our knowledge of the conformation of the amyloid precursors of MHβ2m formed at low pH (8, 10, 31), little is known about the manner by which these precursor states assemble into amyloid. Moreover, the structural characteristics and assembly mechanism of the amyloid precursor populated from the intact protein at physiological pH are also relatively poorly understood (13, 15). The data presented here provide important new clues into these questions. Ordered assembly is most likely to occur by the association of preformed complementary surfaces (18). Our data suggest that the long straight conformation of strand D in the crystal structure of MHβ2m could provide such a surface. Although an increase in conformational dynamics in this region, caused by dissociation of β2m from the heavy chain of the HLA, could be involved in the initiation of amyloid fibril formation (20, 21), we propose that the ordering of strand D to the conformation observed here by crystallography could provide a template for the propagation of ordered assembly. This rarely populated conformation of β2m offers a rationale for the observation that at pH 5 and above, fibrils of full length β2m are formed only at very high concentrations (14), or in the presence of copper ions (13), and involve a rarely populated state that is capable of extending preexisting fibrillar material (15).
How could the structural rearrangements in strand D facilitate assembly? In a recent study, Richardson and Richardson (18) have identified features common to β sheet proteins that may have evolved to prevent aggregation. One such feature, often found in the edge strands of β sheet-containing proteins, is the presence of a twisted or bulged edge strand. Such features are proposed to disfavor edge strand docking (see Table 2, which is published as supporting information on the PNAS web site). By contrast, long (>6 residues) regular β strands at the edges of β sheets are proposed to favor edge-to-edge strand association between adjacent monomers, so that their hydrogen-bonding potential can be satisfied. A second common feature, often observed in β sandwich proteins involves protection of one or more of the edge β strands from intermolecular self association by the possession of an inwardly pointing charged side chain, which would be buried by dimerization. Indeed, the addition of only a single lysine to the edge of de novo designed β sheet proteins has been shown to inhibit polymerization into amyloid fibrils (36).
Monomeric β2m possesses four edge strands; strands A and G form one edge pair, whereas strands C and D form the second edge strand pair (Fig. 1). Strands A and G are protected in both HLAβ2m and MHβ2m by an inwardly pointing charge contributed by Lys-91 in strand G. The short β strands D1 and D2 separated by a β bulge provide an excellent mechanism for prohibiting self assembly at the other edge of the β sandwich. However, the conformation of β strand D observed here in the crystal structure of MHβ2m provides an ideal assembly surface, making this edge-strand pair vulnerable to aggregation. For the crystal structure shown here, the hydrogen-bonding potential of strand D is satisfied by the formation of intermolecular interactions with adjacent molecules within the lattice, demonstrating the potential for this region to propagate assembly through edge-strand interactions. In addition, the changes observed in strand D result in different orientations of the side chains of residues 50–54. This arises because of the i to i + 1 slippage in the hydrogen-bond pairings in this strand. As a result, His-51 (which points inwards in the structure of HLAβ2m) rotates by approximately 180°, such that it now points away from the hydrophobic core of the protein. This would remove the second protective feature from the edge strand, facilitating further interaction in this region (His-51 would be at least partially charged at pH 7.0 and below) (Fig. 4).
Figure 4.

Ribbon diagram showing the position of His-51 in the crystal structures of (a) MHβ2m and (b) HLAβ2m. Note that in a, His-51 is outwardly pointing, whereas in b, this residue points inward. Drawn by using SPOCK (39).
Whatever the origin of intermolecular associations, it is clear that further conformational changes are required to promote assembly of adjacent monomers into amyloid fibrils. Such changes are likely to include the fraying of the N- and C-terminal strands, which would expose new assembly-competent sites (12, 31, 35). The structure of MHβ2m identified here by crystallization suggests that the reorganization of strand D and the resulting loss of the protective features in this region could provide one such site. Ongoing studies aim to provide a quantitative estimate of the populations and interconversions between the observed conformational states of human β2m and to test the hypothesis that specific rare conformations are involved in amyloid assembly under physiological conditions. The data presented here support the suggestion of Richardson and Richardson (18) that alteration of β sheet edge strands could be a critical first step in the formation of amyloid.
Supplementary Material
Acknowledgments
We thank Jane and David Richardson for sending us a preprint of their paper (18) and for inspiring this work. We gratefully acknowledge Neil Kad and Victoria McParland for acquiring the 1H-15N HSQC spectra and for help with their processing. We thank Victoria McParland, Neil Kad, Fabrizio Chiti, Steve Homans, and members of the S.E.R. group for helpful discussions. We acknowledge the University of Leeds, The Wellcome Trust, Biotechnology and Biological Sciences Research Council (BBSRC), and Engineering and Physical Sciences Research Council (EPSRC) for financial support. C.H.T., D.P.S., and A.P.K. are supported by the BBSRC, EPSRC, and The Wellcome Trust. S.E.R. is a BBSRC Professorial Fellow. This paper is a contribution from the Astbury Centre for Structural Molecular Biology, which is part of the North of England Structural Biology Centre and is supported by the BBSRC.
Abbreviations
- β2m
β-2-microgloblin
- MHβ2m
monomeric human β2m
- MBβ2m
monomeric bovine β2m
- HLAβ2m
human β2m bound to the HLA class 1 complex
- HSQC
heteronuclear single quantum correlation
- NOE
nuclear Overhauser effect
- NOESY
NOE spectroscopy
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1LDS).
References
- 1.Saper M A, Bjorkman P J, Wiley D C. J Mol Biol. 1991;219:277–319. doi: 10.1016/0022-2836(91)90567-p. [DOI] [PubMed] [Google Scholar]
- 2.Floege J, Ehlerding G. Nephron. 1996;72:9–26. doi: 10.1159/000188801. [DOI] [PubMed] [Google Scholar]
- 3.Linke R P, Bommer J, Ritz E, Waldherr R, Eulitz M. Biochem Biophys Res Commun. 1986;136:665–671. doi: 10.1016/0006-291x(86)90492-4. [DOI] [PubMed] [Google Scholar]
- 4.Bellotti V, Stoppini M, Mangione P, Sunde M, Robinson C, Asti L, Brancaccio D, Ferri G. Eur J Biochem. 1998;258:61–67. doi: 10.1046/j.1432-1327.1998.2580061.x. [DOI] [PubMed] [Google Scholar]
- 5.Lai Z H, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 6.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C F, Pepys M B. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 7.Khurana R, Gillespie J R, Talapatra A, Minert L J, Ionescu-Zanetti C, Millett I, Fink A L. Biochemistry. 2001;40:3525–3535. doi: 10.1021/bi001782b. [DOI] [PubMed] [Google Scholar]
- 8.McParland V J, Kad N M, Kalverda A P, Brown A, Kirwin-Jones P, Hunter M G, Sunde M, Radford S E. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 9.Smith D P, Radford S E. Protein Sci. 2001;10:1775–1784. doi: 10.1110/ps.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kad N M, Thomson N H, Smith D P, Smith D A, Radford S E. J Mol Biol. 2001;313:559–571. doi: 10.1006/jmbi.2001.5071. [DOI] [PubMed] [Google Scholar]
- 11.Naiki H, Hashimoto N, Suzuki S, Kimura H, Nakakuki K, Gejyo F. Amyloid Int J Exp Clin Invest. 1997;4:223–232. [Google Scholar]
- 12.Esposito G, Michelutti R, Verdone G, Viglino P, Hernandez H, Robinson C V, Amoresano A, Dal Piaz F, Monti M, Pucci P, et al. Protein Sci. 2000;9:831–845. doi: 10.1110/ps.9.5.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan C J, Gelfand M, Atreya C, Miranker A D. J Mol Biol. 2001;309:339–345. doi: 10.1006/jmbi.2001.4661. [DOI] [PubMed] [Google Scholar]
- 14.Connors L H, Shirahama T, Skinner M, Fenves A, Cohen A S. Biochem Biophys Res Commun. 1985;131:1063–1068. doi: 10.1016/0006-291x(85)90198-6. [DOI] [PubMed] [Google Scholar]
- 15.Chiti F, De Lorenzi E, Grossi S, Mangione P, Giorgetti S, Caccialanza G, Dobson C M, Merlini G, Ramponi G, Bellotti V. J Biol Chem. 2001;276:46714–46721. doi: 10.1074/jbc.M107040200. [DOI] [PubMed] [Google Scholar]
- 16.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohhashi Y, Hagihara Y, Kozhukh G, Hoshino M, Hasegawa K, Yamaguchi I, Naiki H, Goto Y. J Biochem (Tokyo) 2002;131:45–52. doi: 10.1093/oxfordjournals.jbchem.a003076. [DOI] [PubMed] [Google Scholar]
- 18.Richardson J S, Richardson D C. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker J W, Reeke G N. Proc Natl Acad Sci USA. 1985;82:4225–4229. doi: 10.1073/pnas.82.12.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verdone G, Corazza A, Viglino P, Pettirossi F, Giorgetti S, Mangione P, Andreola A, Stoppini M, Bellotti V, Esposito G. Protein Sci. 2002;11:487–499. doi: 10.1110/ps.29002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okon M, Bray P, Vucelic D. Biochemistry. 1992;31:8906–8915. doi: 10.1021/bi00152a030. [DOI] [PubMed] [Google Scholar]
- 22.Leslie A G W. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. Vol. 26. Warrington, U.K.: Science and Engineering Research Council Daresbury Lab.; 1992. [Google Scholar]
- 23.Collaborative Computational Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 24.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 25.Khan A R, Baker B M, Ghosh P, Biddison W E, Wiley D C. J Immunol. 2000;164:6398–6405. doi: 10.4049/jimmunol.164.12.6398. [DOI] [PubMed] [Google Scholar]
- 26.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 27.Adams P D, Pannu N S, Read R J, Brünger A T. Proc Natl Acad Sci USA. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pannu N S, Read R J. Acta Crystallogr A. 1996;52:659–668. [Google Scholar]
- 29.Brunger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski R A, Rullmannn J A, MacArthur M W, Kaptein R, Thornton J M. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 31.McParland V J, Kalverda A P, Homans S W, Radford S E. Nat Struct Biol. 2002;9:326–331. doi: 10.1038/nsb791. [DOI] [PubMed] [Google Scholar]
- 32.Kadkhodaei M, Hwang T L, Tang J, Shaka A J. J Magn Reson A. 1993;105:104–107. [Google Scholar]
- 33.Hwang T L, Shaka A J. J Magn Reson A. 1995;112:275–279. [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister G W, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Hoshino M, Katou H, Nakasima Y, Hagihama Y, Hasegawa K, Naiki H, Goto Y. Nat Struct Biol. 2002;9:332–336. doi: 10.1038/nsb792. [DOI] [PubMed] [Google Scholar]
- 36.Wang W, Hecht M H. Proc Natl Acad Sci USA. 2002;99:2760–2765. doi: 10.1073/pnas.052706199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 38.Merritt E A, Murphy M. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 39.Christopher J A. SPOCK: The Structural Properties Observation and Calculation Kit. College Station, TX: The Center for Macromolecular Design, Texas A&M Univ.; 1998. [Google Scholar]
- 40.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}