

Abstract
The tumor suppressor protein p53 localizes to microtubules (MT) and, in response to DNA damage, is transported to the nucleus via the MT minus-end-directed motor protein dynein. Dynein is also responsible for MT-mediated nuclear targeting of adenovirus type 2 (Ad2). Here we show that treatment with low concentrations of MT-targeting compounds (MTCs) that do not disrupt the MT network but are known to suppress MT dynamics enhanced p53 nuclear accumulation, and the activation of the p53-downstream target genes. p53 nuclear accumulation required binding of MTCs to MTs and enhanced the induction of p53-up-regulated modulator of apoptosis (PUMA) mRNA and apoptosis on challenging cells with the DNA-damaging drug adriamycin. Low concentrations of MTCs enhanced the rate of movement of fluorescent Ad2 to the nucleus and increased the nuclear targeting efficiency of Ad2. We propose that suppression of MT dynamics by low concentrations of MTCs enhances MT-dependent trafficking toward the minus ends of MTs and facilitates nuclear targeting.
Microtubules (MTs) are regulated dynamic cytoskeletal polymers, crucial for many important cellular functions, including the spatial organization of the interphase cytoplasm, cell signaling, and chromosome segregation in mitosis (1–3). In most cells, MTs are organized in a single array with their minus ends associated with the MT organizing center located near the nucleus and their plus ends toward the cell periphery. Therefore, MTs are uniquely positioned to transmit signals to and from the nucleus and may play a central role in intracellular transport and signal transduction (1). A wide variety of natural products, including paclitaxel (PTX) and the vinca alkaloids, target MTs and are widely used in cancer chemotherapy (4, 5). At high concentrations, these MT-targeting compounds (MTCs) disrupt normal MT function by either stabilizing or destabilizing MTs. It has been shown that low doses of both MT-stabilizing or -destabilizing drugs potently suppress MT dynamics without any alterations in the MT polymer mass (6, 7). Suppression of MT dynamics is important for the antimitotic action of these drugs, because inhibition of MT dynamics results in kinetic stabilization of the mitotic spindle leading to mitotic arrest (8, 9). However, whether inhibition of MT dynamics without changes in the MT polymer mass affects MT functions in interphase is currently unknown.
We have recently shown that the tumor suppressor protein p53 associates with MTs and uses the MT-dependent motor complex dynein/dynactin for nuclear targeting, e.g., after DNA damage (10). Disruption of the MT network by polymerization with high concentrations of PTX or depolymerization with vincristine (VCR) impedes p53 translocation to the nucleus and in turn inhibits activation of downstream targets by p53. Although an intact MT network is required for p53 trafficking, the role of a dynamic MT network for p53 nuclear accumulation is not known.
Using low concentrations of PTX or VCR, we investigate herein the effects of suppressing MT dynamics on the translocation of p53 to the nucleus. We show that after treatment with concentrations of PTX or VCR lower than those required to affect polymerization, p53 nuclear accumulation is enhanced. This accumulation was accompanied by induction of downstream targets of p53. In a cell line harboring wild-type (wt) p53 but a mutant β tubulin insensitive to PTX, nuclear targeting of p53 by low concentrations of PTX did not occur. In addition, low concentrations of PTX or nocodazole (Noc) enhanced the nuclear targeting and the rate of movement of the human adenovirus type 2 (Ad2), a nonenveloped virus that replicates within the nucleus of an infected cell and uses MTs to traffic from the cytoplasm to the nucleus (11). Our data demonstrate that MT-mediated trafficking in interphase cells can be regulated and enhanced above physiological levels. It is possible that the manipulation of MT dynamics by MT-interacting compounds can be exploited to enhance cell death in human cancer cells.
Materials and Methods
Cell Lines and Antibodies.
Human lung cancer A549 cells were obtained from the National Cancer Institute Drug Anticancer Drug Screen and the human cervical cancer HeLa cells from American Type Culture Collection. A2780 (1A9) human ovarian carcinoma cells and their epothilone (Epo)-resistant subline, A2780/Epo B, were a generous gift from Craig Fairchild (Bristol-Myers Squibb). Mouse monoclonal antibodies against p53 (Ab 6: Mab Do1) and mdm2 (Ab-2) were purchased from Oncogene Science. The sheep polyclonal antibody against p53 (Ab-7) was from Oncogene Science. The mouse monoclonal anti-α-tubulin (DM1α) antibody and the rabbit polyclonal antiactin antibody were from Sigma. The antibody against the cleaved epitope of poly(ADP-ribose)polymerase (PARP) (p85) was from Upstate Biotechnology (Lake Placid, NY). PTX was purchased from Bristol-Myers Squibb, VCR from Eli Lilly, and colchicine, Noc, and adriamycin (ADR) were from Sigma.
Western and Northern Blot Analysis.
Western blot analysis was performed as previously described (10). Briefly, 30 μg of total cell protein was resolved by 10% SDS/PAGE and immunoblotted with the indicated antibodies. Northern blot analysis for the p53-up-regulated modulator of apoptosis (PUMA) mRNA was performed as previously described (12).
Immunofluorescence and Microscopy.
Exponentially growing cells were plated on 12-mm glass coverslips (A. Daigger, Vernon Hills, IL) and incubated overnight. The next day, after drug treatment, cells were fixed and processed for double immunofluorescence labeling as previously described (10). The primary antibodies used were the mouse monoclonal anti-α-tubulin (DM1α) antibody for tubulin/MT staining and the sheep polyclonal (Ab-7) antibody for p53 staining. The coverslips were examined on a Zeiss axioplasm microscope by using a Zeiss 100 × 1.3 oil immersion objective. Confocal images were obtained on an LSM 510 laser scanning (Zeiss) microscope (Zeiss axioplasm).
Time-Lapse Fluorescence Microscopy and Quantitative Subcellular Analyses of Ad2-TR.
Ad2 was isolated and labeled with the fluorophore Texas red (Ad2-TR) (13). Infections of A549 human lung epithelial carcinoma cells that were pretreated or not with the MT active drugs Noc (Sigma) and PTX (Sigma) at low concentrations were carried out as described (14), except that cold synchronization was omitted, and virus was added directly to the cells in RPMI-BSA at 37°C for 10 min (about 2,000 particles per cell). Cells were washed with warm DMEM-BSA and further incubated at 37°C as indicated. Under these conditions, 50–100 particles were bound per cell, independent of the pretreatments with Noc or Taxol. Virus motility was analyzed by live-mode video microscopy and the elementary motion steps as well as motility frequencies determined exactly as described (11). Population velocities were calculated from the net distance traveled to and from the nucleus divided by the total time of sampling of the analyzed particles (n). Population motilities represented the mean number of elementary motion steps (ES) larger than 0.1 μm⋅s−1 divided by the total number of ES of each particle. Motilities c and p were defined as the average fractions of the vectorial ES components larger than 0.1 μm⋅s−1 directed toward the center or the periphery. P values were determined by using one-sided t tests. Quantifications of Ad2-TR in subcellular regions of fixed cells were as described (13), and one-sided t tests were applied as indicated.
Results
Low Concentrations of PTX or VCR Enhance p53 Nuclear Accumulation and Result in Transcriptional Activation of mdm2.
To assess the effects of the MT-stabilizing drug PTX and the MT-destabilizing drug VCR on p53 cellular localization, A549 human lung carcinoma cells (with wt p53) were treated with low (6 nM) and high (100 nM) concentrations of each drug for 18 hr. At low concentrations, both the MT-stabilizing drug PTX and the MT-destabilizing drug VCR cause suppression of MT dynamics (4, 8, 9, 15–17). The subcellular localization of p53 was monitored by indirect immunofluorescence followed by confocal laser scanning microscopy (CLSM; Fig. 1A). Cells treated with the DNA-damaging drug ADR readily accumulated p53 in the nucleus, as expected (18). Treatment with 6 nM PTX or 6 nM VCR (without ADR) resulted in enhanced p53 nuclear accumulation (red p53 staining), similar to the ADR-treated cells. In agreement with previous reports (17, 19), these low concentrations of PTX and VCR had no apparent effects on MTs or the organization of the MT network (Fig. 1A and Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). In contrast, treatment with higher PTX concentrations (100 nM) resulted in extensive stabilization of MTs, as evidenced by tubulin bundling, whereas treatment with 100 nM VCR resulted in MT depolymerization (Fig. 1A). In agreement with our previous findings, interference with the MT network by either hyperstabilization with PTX (100 nM) or depolymerization with VCR (100 nM) impaired p53 nuclear accumulation. Consistent with this finding, low concentrations of PTX (3–25 nM) enhanced p53 nuclear accumulation and induced mdm2, a surrogate marker of p53 nuclear accumulation (Fig. 1B). Higher doses of PTX (50 and 100 nM), however, failed to induce mdm2, consistent with a lack of p53 in the nucleus. Similar to the PTX treatment, 3–12 nM VCR resulted in induction of mdm2, whereas higher concentrations had no effect on mdm2 expression levels, consistent with the loss of the MT network and a lack of p53 nuclear accumulation. To investigate whether the above findings were specific to the A549 human lung carcinoma cells, we performed a similar experiment by using the A2780 (1A9) human ovarian carcinoma cells containing wt p53 (20). The results confirmed the enhanced p53 nuclear accumulation seen after treatment with low concentrations of either PTX or VCR (Fig. 2 A and B).
Fig 1.

(A) Low concentrations of PTX or VCR enhance p53 nuclear accumulation in A549 cells. A549 cells were treated with the indicated drugs for 18 hr and processed for double immunofluorescence labeling with antibodies against p53 (red, labeled with Rhodamine red X) or α-tubulin (green, labeled with FITC). Untreated A549 cells were included as control (CTRL). Staining was analyzed by CLSM. (Bar = 10 μm.) (B) Low concentrations of PTX or VCR induce mdm2 in A549 cells. Untreated A549 cells (0) or cells treated for 18 hr were processed for Western blotting. Cells treated with 400 ng/ml of ADR for 18 hr are included as a positive control for p53 and mdm2 increased levels. Thirty micrograms of total cellular protein from each sample were resolved in a 10% SDS/PAGE, transferred, and immunoblotted with antibodies against p53 and mdm2. Please notice that in the Westerns blots shown here, the control lane (CTRL) has been moved from the first position to the second to facilitate the direct comparison of the control lane with the ADR lane to the right and the PTX 3 lane to the left.
Fig 2.

Low concentrations of PTX or VCR enhance p53 nuclear accumulation, and induce mdm2 in A2780 (1A9) cells in a tubulin-dependent manner. 1A9 cells were plated in duplicate, subjected to the indicated drug treatments, and processed either for CLSM (A) or Western blotting (B). (A) Cells are stained with antibodies against p53 (red, labeled with Rhodamine red X) or α-tubulin (green, labeled with FITC). (Bar = 10 μm.) (B) Thirty micrograms of total cell protein from each sample were resolved in a 10% SDS/PAGE, transferred, and immunoblotted with antibodies against p53 and mdm2. Induction of mdm2 is consistent with enhanced p53 nuclear accumulation (A). (C) The β tubulin mutant A2780/Epo B cells were treated with the indicated drugs for 18 hr and processed for double immunofluorescence labeling with antibodies against p53 (red, labeled with Rhodamine red X) or α-tubulin (green, labeled with FITC). Untreated A2780/Epo B cells (CTRL) and cells treated with ADR are included as controls. Staining was analyzed by CLSM. (Bar = 10 μm.)
P53 Nuclear Accumulation by MTCs Requires Drug/Tubulin Interaction.
We next tested whether the effects of low concentrations of PTX and VCR on p53 nuclear accumulation were MT dependent. We used a subclone of A2780 cells (A2780/Epo B) that harbors an acquired β tubulin mutation at residue Arg-282 to Gln (R282Q). This mutation is located near the taxane-binding site on tubulin (21). It impairs the PTX/tubulin interaction, thus conferring 10- to 15-fold crossresistance to PTX, but has no effect on VCR's interaction with tubulin (22). Treatment of A2780/Epo B cells with 3 nM PTX had no effect on p53 nuclear accumulation, whereas treatment with 3 nM VCR resulted in increased p53 nuclear accumulation (Fig. 2C). As expected, 100 nM VCR resulted in the complete depolymerization of the MT network and did not enhance p53 nuclear accumulation. Interestingly, treatment with 100 nM PTX slightly increased p53 nuclear staining in A2780/Epo B cells. This observation is in agreement with the impaired PTX/tubulin interaction. As a consequence, much higher concentrations of PTX are required to mimic the effect of low PTX concentrations in a wt cell line. Together, these results suggest that suppression of MT dynamics by low concentrations of MTCs enhances the nuclear accumulation of p53 and activates the p53 target gene, mdm2.
Low Concentrations of Other MT-Interacting Compounds Enhance p53 Nuclear Accumulation.
To further test the above hypothesis, the effects of other MT-stabilizing compounds, Epo A and B, or destabilizing compounds, Noc and colchicine, on p53 nuclear accumulation were examined. The results shown in Fig. 3 A and B reveal that treatment of A549 cells with a low concentration (3 nM) of each of these compounds enhanced p53 nuclear accumulation (Fig. 3A, red p53 staining) and increased the transactivation of mdm2 (Fig. 3B).
Fig 3.

Low concentrations of other MT-active agents enhance p53 nuclear accumulation and induce mdm2 in A549 cells. A549 cells were plated in duplicate, subjected to the indicated drug treatments, and processed either for CLSM (A) or Western blotting (B) (see Fig. 2). Arrows in A point to either bundled MTs (Epo A, Epo B) or depolymerized tubulin network (colchicine, Noc). (Bar = 10 μm.)
MT-Mediated Trafficking of Ad2 Is Enhanced by Low Concentrations of PTX or Noc.
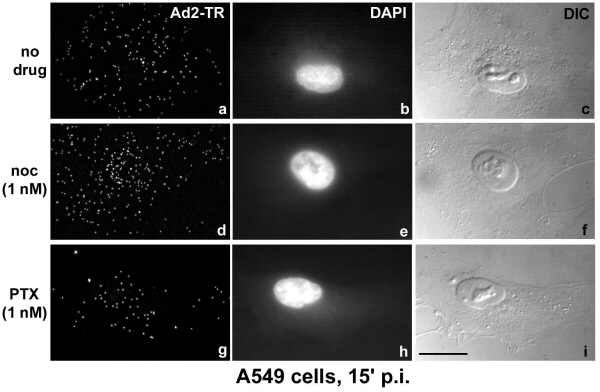
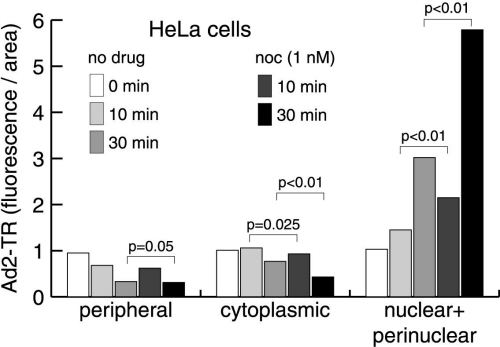
We next sought to investigate the effects of low concentrations of MTCs on the intracellular trafficking of the human Ad2. Like other viral pathogens (23), this virus replicates in the nucleus and utilizes the MT-dependent dynein/dynactin motor complex for targeting to the MT minus ends, typically located near the nuclear membrane (11). The motility of incoming virus is biased to the nucleus because of virus-induced cell signaling (24). The activation of an integrin–PKA pathway peaks at 15 min postinfection (p.i.), and the induction of the MKK6-p38/MAKP-MK2 module peaks at 30 min p.i. Minus-end-directed Ad2 transport is more efficient in cells expressing the MT-binding domain of the MT-associated protein MAP4 (11), which confers enhanced MT stability (25). We therefore tested whether the suppression of MT dynamics with low concentrations of MTCs facilitated MT-dependent Ad2 trafficking. A549 cells were treated with 1 nM PTX or Noc for 18 hr before infection with Ad2-TR. The intracellular localization of the labeled virus was quantified at 15 and 40 min p.i., i.e., before and after the onset of Ad-induced signaling affecting the MT-dependent transport rates (Fig. 4A). Enhanced Ad2-TR nuclear and perinuclear accumulation was observed in the presence of 1 nM PTX or 1 nM Noc at 15 min p.i., compared with the nontreated cells (P < 0.01 for PTX and 0.05 for Noc). This enhanced nuclear targeting of Ad2-TR was accompanied by a more rapid clearance of the periphery in drug-treated cells compared with nontreated cells (P < 0.01 for both drugs; Fig. 4A and Fig. 7, which is published as supporting information on the PNAS web site). Enhanced nuclear targeting of Ad2-TR by 1 nM PTX or 1 nM Noc was also measured by video microscopy (Fig. 4B). Until 20 min p.i., the population motility of Ad2 toward the nucleus was enhanced by both PTX and Noc by a factor of 3 compared with nontreated cells and reached 0.09 μm/min (Fig. 4B Upper, and Movies 1–6, which are published as supporting information on the PNAS web site). This enhanced motility was largely because of augmented minus-end-directed velocities of individual elementary motion steps. Interestingly, the frequencies of both minus-end- and plus-end-directed motilities were both reduced by PTX and Noc, consistent with the notion that low concentrations of PTX and Noc reduce, e.g., the motility of peripheral phagosomes (26). After the onset of MT-directed signaling by Ad2 (i.e., 20–50 min p.i.), we did not observe any significant differences in the population speeds of Ad2-TR in PTX- or Noc-treated cells compared with control cells (Fig. 4B Lower). This observation was consistent with the subcellular localizations of Ad2-TR at 40 min p.i. (Fig. 4A Lower). In addition, enhanced nuclear targeting of Ad2-TR early in infection was also obtained with a different cell line (HeLa) treated with 1 nM Noc (Fig. 8, which is published as supporting information on the PNAS web site). These data demonstrate that low concentrations of MTCs enhanced MT-mediated minus-end-directed trafficking of Ad2.
Fig 4.

Low concentrations of PTX or Noc enhance the nuclear targeting and the motility of Ad2 in A549 cells. (A) Untreated A549 cells (mock) or cells treated with 1 nM Noc or 1 nM PTX (Tax) for 18 hr were infected with Ad2-TR in RPMI-BSA at 37°C. In randomly selected untreated or drug-treated cells, the nuclear, perinuclear, cytoplasmic, and peripheral fluorescence of Ad2-TR was quantitated 15 min (Upper) or 40 min (Lower) p.i. The bars represent the average of fluorescence ± SEM of 20–40 independent measurements. (B) Untreated A549 cells (no drug) or cells treated with 1 nM of either PTX (Tax) or Noc for 18 hr were infected with Ad2-TR in RPMI-BSA at 37°C for 15 min followed by further incubation in RPMI-BSA in the presence of PTX or Noc but without the virus until imaging of Ad2-TR was completed. Ad2-TR was imaged in time-lapse mode, and ES were analyzed for each viral particle at 0–20 min (Upper) or 20–50 min (Lower) p.i. Population motilities (pop mot) represent the mean number of ES larger than 0.1 μm/s divided by the total of ES of each particle. Mot c and mot p were defined as the average fractions of the vectorial ES components larger than 0.1 μm/s directed toward the cell center (mot c) and toward the cell periphery (mot p), including the SEM and P values. For all measurements, (−) indicates motility toward the cell's center.
Potentiation of Apoptotic Cell Death in Cells Treated with Low Concentrations of PTX or VCR Followed by ADR.
Because p53 exerts many of its effects by transcriptional regulation (27), its translocation from cytoplasmic sites of synthesis to nuclear target genes is critical to elicit biological responses. To further investigate the biological effects of enhanced p53 nuclear targeting by low doses of MTCs, we performed the experiment shown in Fig. 5. A549 cells were first treated with 3, 6, or 100 nM of either PTX (Fig. 5A) or VCR (Fig. 5B) for 18 hr and then exposed to ADR (200 ng/ml) for another 6 hr. When 3 or 6 nM of each drug was combined with ADR (PTX3→ADR, PTX6→ADR, VCR3→ADR, and VCR6→ADR), apoptosis was observed. As a marker of apoptosis, the p85 cleaved epitope of poly(ADP-ribose)polymerase (PARP) (exposed only after it is cleaved in cells undergoing apoptosis) was detected (28). The results with PARP cleavage were confirmed with caspase-3 activation assay and the sulforhodamine-B cell survival assay (Fig. 9, which is published as supporting information on the PNAS web site). Although lower concentrations of MTCs did not induce caspase-3 activation and were only marginally cytotoxic, they rendered cells sensitive to posttreatment with ADR (Fig. 9). In the samples where PARP cleavage was observed (PTX3→ADR, PTX6→ADR, VCR3→ADR, and VCR6→ADR), induction of p53 and mdm2 was also observed. This finding is consistent with enhanced p53 nuclear accumulation and activation of apoptotic responses (29). Although treatment with higher concentrations of PTX (100 nM) alone or in combination with ADR induced PARP cleavage, it did not increase p53 or mdm2 levels. Similar results were observed with high concentrations of VCR. These results suggest that, at higher drug concentrations, MTCs induce cell death independently of p53-nuclear targeting. To further tighten the correlation between enhanced p53 nuclear accumulation and p53-mediated apoptosis, we performed a Northern blot analysis for a recently identified p53 target gene, the PUMA (Fig. 5C). PUMA is induced by both high levels of exogenous p53 and elevated endogenous p53 and results in apoptosis (12, 30). Increased PUMA levels were observed in cells treated with PTX3→ADR or PTX6→ADR but not with PTX100 or PTX100→ADR, suggesting that the apoptosis induced by high concentrations of PTX is p53-independent. These data confirm and extend the notion that low concentrations of MTCs enhance p53 nuclear accumulation and lead to apoptosis on short exposure to ADR.
Fig 5.

Treatment with low concentrations of PTX or VCR followed by ADR enhances apoptosis in A549 cells. (A) A549 cells were treated with 3, 6, or 100 nM of PTX or VCR for 18 hr followed by treatment with 200 ng/ml of ADR for an additional 6 hr (PTX3→ADR, PTX6→ADR, PTX100→ADR, and VCR3→ADR, VCR6→ADR, VCR100→ADR). ADR was added to the medium containing PTX or VCR at the indicated concentrations. Untreated cells (CTRL) or cells treated with 200 ng/ml of ADR alone (ADR), PTX alone, or VCR alone for 24 hr (PTX3, PTX6, PTX100; VCR3, VCR6, VCR100) were included as controls. Western blots were probed with antibodies against the cleaved (p85) form of PARP, p53, mdm2, and actin as a loading control. (B) A549 cells were treated with 3, 6, or 100 nM PTX for 18 hr, followed by treatment with 200 ng/ml of ADR for an additional 6 hr (PTX3→ADR, PTX6→ADR, PTX100→ADR). Untreated cells (CTRL) or cells treated with PTX alone for 24 hr (PTX3, PTX6, PTX100) are included as controls. Ten micrograms of total RNA from each sample were analyzed by Northern blotting. Ethidium bromide staining of 28S RNA is shown as loading control.
Discussion
We have previously shown that the tumor suppressor protein p53 is associated with the MT cytoskeleton and, in response to DNA damage, is transported toward the nucleus via the minus-end MT-associated motor protein dynein (10).
MT dynamics are essential for many MT-dependent cellular processes and in particular for mitosis, which is very sensitive to alteration of MT dynamics (4, 31–33). Both MT-stabilizing and -destabilizing agents at very low concentrations potently suppress MT dynamics, leading to kinetic stabilization of the mitotic spindle (8, 9, 15, 17). In addition, MTs provide tracks or scaffolds for the directional vesicle transport as well as organelle positioning.
We propose that suppressing the dynamics of interphase MTs may actually enhance their functions in transporting molecules. We demonstrate here that low concentrations of PTX or VCR (3 and 6 nM) enhance the nuclear accumulation of p53 and induce activation of the p53-dependent gene, mdm2. Furthermore, we showed that p53 nuclear accumulation depended on the ability of MTCs to bind tubulin, as evidenced by the lack of response of a cell line with a mutant β tubulin (R282Q mutation located at the PTX-binding pocket) to low doses of PTX. These findings favor the notion that suppression of MT dynamics leads to enhanced p53 nuclear targeting and is in concert with published reports showing that very low concentration of MTCs, well within the range of concentrations we used in this report, suppress MT dynamics both in vitro and in cells (4, 8, 9, 15, 17). More specifically, a recent study reported that treatment of A549 cells with as little as 2 nM PTX overnight was sufficient to suppress the overall dynamicity of MTs by 30% (34).
If suppression of MT dynamics affects MT-dependent cellular transport, it should not be limited to p53 but should apply to other entities that use MT-dependent transport. One such entity is the human common Ad2. The MT-dependent nature of Ad2 movement within the cell made it an ideal system to test the effects of suppressing MT dynamics by low concentrations of MTCs on Ad2 transport along MTs. Enhanced Ad2 nuclear and perinuclear accumulation was observed in A549 cells treated with 1 nM PTX or Noc compared with untreated cells (Fig. 4 and Figs. 7 and 8). Treated cells also demonstrated increased minus-end-directed population motilities of Ad2 (Fig. 4 and Movies 1–3). These observations from both the static and the dynamic analyses of Ad movement were limited to the early stages of infection (0–20 min p.i.). At later stages, no significant differences were observed between treated and untreated cells. The positive effect of MTCs on Ad2 nuclear targeting early in infection correlates with the absence of Ad-induced signaling known to stimulate nuclear targeting at 20 min p.i. (24). It also correlates with the enhanced nuclear targeting of Ad2 in cells where MTs are stabilized by overexpression of the MT-binding domain of MAP4 (MT-associated protein 4) (11). This correlation raises the possibility that the MT-stabilizing effects of MAPs and the suppression of MT dynamics by low concentrations of MTCs could elicit similar cellular effects, such as enhanced MT-dependent transport. At later stages of infection, we see no difference in the nuclear targeting of Ad2 between treated and untreated cells, possibly because Ad-induced signals boosting the perinuclear and nuclear accumulation of the virus mask the effects of suppressing MT dynamics in intracellular trafficking.
The involvement of MTs in signal transduction and the relationship between MTs and transcription factors in human cancer have only recently started to attract attention (for reviews, see refs. 1 and 35). For example, p53 function depends on its nuclear localization, and both p53 nuclear import and export are cellular processes tightly regulated (36). We showed here that enhanced p53 nuclear accumulation with low concentrations of PTX or VCR before short exposure to low concentrations of ADR resulted in apoptotic cell death in A549 cells, as evidenced by the induction of PARP cleavage and caspase-3 activation. This finding may be important in a clinical setting, as the taxanes are often combined with ADR or radiation for the treatment of different types of cancer (37, 38). Our findings demonstrate that pretreatment with low concentrations of MTCs (which do not induce either mitotic arrest or change tubulin polymerization) sensitizes cells to the effects of ADR, including induction of PUMA and apoptosis. This effect may be the result of additive phosphorylation of p53, as MTCs and DNA damage have been shown to induce p53 phosphorylation at distinct residues (39). In addition, p21 induction by the enhanced p53 nuclear accumulation has been shown to inhibit topoisomerase genes (38), which might cooperate with ADR-mediated inhibition of topoisomerase.
Our results demonstrate that drugs like PTX alter p53 nuclear trafficking at therapeutically relevant drug exposure and that combination therapies with known p53-dependent agents such as ADR can be potentiated by p53-enhancing concentrations of PTX. This conclusion supports the notion that lower doses of MTC can be therapeutically more attractive than higher doses (39). Finally, although disruption of MT functions by MTCs is well known and is the basis of mitosis arrest and of inhibition of MT-mediated trafficking, very low concentrations of MTCs can increase some functions of MTs in interphase cells. This is the first demonstration, to our knowledge, that MT-mediated trafficking can be pharmacologically increased.
Supplementary Material
Acknowledgments
We thank Garver Moore and Yvona Ward for help with confocal microscopy. This work was supported by a grant from the Swiss National Science Foundation (to U.F.G.), the Kanton of Zurich, and the Avon Foundation (A.O.).
Abbreviations
MT, microtubule
MTC, MT-targeting compound
PTX, paclitaxel
VCR, vincristine
ADR, adriamycin
Epo, epothilone
Noc, nocodazole
Ad2, adenovirus type 2
Ad2-TR, Ad2 labeled with Texas red
PARP, poly(ADP-ribose)polymerase
PUMA, p53- up-regulated modulator of apoptosis
CLSM, confocal laser scanning microscopy
ES, elementary motion steps
wt, wild type
p.i., postinfection
References
- 1.Gundersen G. G. & Cook, T. A. (1999) Curr. Opin. Cell Biol. 11, 81-94. [DOI] [PubMed] [Google Scholar]
- 2.Nogales E. (2000) Annu. Rev. Biochem. 69, 277-302. [DOI] [PubMed] [Google Scholar]
- 3.Brunet S. & Vernos, I. (2001) EMBO Rep. 2, 669-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan M. A. & Wilson, L. (1998) Curr. Opin. Cell Biol. 10, 123-130. [DOI] [PubMed] [Google Scholar]
- 5.Fojo T. & Giannakakou, P. (2000) Curr. Opin. Oncol. Endocrinol. Metab. Invest. Drugs 2, 293-304. [Google Scholar]
- 6.Toso R. J., Jordan, M. A., Farrell, K. W., Matsumoto, B. & Wilson, L. (1993) Biochemistry 32, 1285-1293. [DOI] [PubMed] [Google Scholar]
- 7.Dhamodharan R., Jordan, M. A., Thrower, D., Wilson, L. & Wadsworth, P. (1995) Mol. Biol. Cell 6, 1215-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derry W. B., Wilson, L. & Jordan, M. A. (1998) Cancer Res. 58, 1177-1184. [PubMed] [Google Scholar]
- 9.Ngan V. K., Bellman, K., Panda, D., Hill, B. T., Jordan, M. A. & Wilson, L. (2000) Cancer Res. 60, 5045-5051. [PubMed] [Google Scholar]
- 10.Giannakakou P., Sackett, D. L., Ward, Y., Webster, K. R., Blagosklonny, M. V. & Fojo, T. (2000) Nat. Cell Biol. 2, 709-717. [DOI] [PubMed] [Google Scholar]
- 11.Suomalainen M., Nakano, M. Y., Keller, S., Boucke, K., Stidwill, R. P. & Greber, U. F. (1999) J. Cell Biol. 144, 657-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu J., Zhang, L., Hwang, P. M., Kinzler, K. W. & Vogelstein, B. (2001) Mol. Cell 7, 673-682. [DOI] [PubMed] [Google Scholar]
- 13.Nakano M. Y. & Greber, U. F. (2000) J. Struct. Biol. 129, 57-68. [DOI] [PubMed] [Google Scholar]
- 14.Greber U. F., Suomalainen, M., Stidwill, R. P., Boucke, K., Ebersold, M. W. & Helenius, A. (1997) EMBO J. 16, 5998-6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jordan M. A., Toso, R. J., Thrower, D. & Wilson, L. (1993) Proc. Natl. Acad. Sci. USA 90, 9552-9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neumann T., Kirschstein, S. O., Camacho Gomez, J. A., Kittler, L. & Unger, E. (2001) Biol. Chem. 382, 387-391. [DOI] [PubMed] [Google Scholar]
- 17.Yvon A. M., Wadsworth, P. & Jordan, M. A. (1999) Mol. Biol. Cell 10, 947-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jimenez G. S., Khan, S. H., Stommel, J. M. & Wahl, G. M. (1999) Oncogene 18, 7656-7665. [DOI] [PubMed] [Google Scholar]
- 19.Torres K. & Horwitz, S. B. (1998) Cancer Res. 58, 3620-3626. [PubMed] [Google Scholar]
- 20.Giannakakou P., Poy, G., Zhan, Z., Knutsen, T., Blagosklonny, M. V. & Fojo, T. (2000) Oncogene 19, 3078-3085. [DOI] [PubMed] [Google Scholar]
- 21.Nogales E., Wolf, S. G. & Downing, K. H. (1998) Nature (London) 391, 199-203. [DOI] [PubMed] [Google Scholar]
- 22.Giannakakou P., Gussio, R., Nogales, E., Downing, K. H., Zaharevitz, D., Bollbuck, B., Poy, G., Sackett, D., Nicolaou, K. C. & Fojo, T. (2000) Proc. Natl. Acad. Sci. USA 97, 2904-2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ploubidou A. & Way, M. (2001) Curr. Opin. Cell Biol. 13, 97-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suomalainen M., Nakano, M. Y., Boucke, K., Keller, S. & Greber, U. F. (2001) EMBO J. 20, 1310-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen H. L., Chari, S., Gruber, D., Lue, C. M., Chapin, S. J. & Bulinski, J. C. (1997) J. Cell Sci. 110, 281-294. [DOI] [PubMed] [Google Scholar]
- 26.Blocker A., Severin, F. F., Burkhardt, J. K., Bingham, J. B., Yu, H., Olivo, J. C., Schroer, T. A., Hyman, A. A. & Griffiths, G. (1997) J. Cell Biol. 137, 113-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woods D. B. & Vousden, K. H. (2001) Exp. Cell Res. 264, 56-66. [DOI] [PubMed] [Google Scholar]
- 28.White M. K. & McCubrey, J. A. (2001) Leukemia 15, 1011-1021. [DOI] [PubMed] [Google Scholar]
- 29.Vousden K. H. (2000) Cell 103, 691-694. [DOI] [PubMed] [Google Scholar]
- 30.Nakano K. & Wousden, K. H. (2001) Mol. Cell 7, 683-694. [DOI] [PubMed] [Google Scholar]
- 31.Margolis R. L. & Wilson, L. (1998) BioEssays 20, 830-836. [DOI] [PubMed] [Google Scholar]
- 32.Mitchison T. & Kirschner, M. (1984) Nature (London) 312, 237-242. [DOI] [PubMed] [Google Scholar]
- 33.Rodionov V., Nadezhdina, E. & Borisy, G. (1999) Proc. Natl. Acad. Sci. USA 96, 115-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goncalves A., Braguer, D., Kamath, K., Martello, L., Briand, C., Horwitz, S., Wilson, L. & Jordan, M. A. (2001) Proc. Natl. Acad. Sci. USA 98, 11737-11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mimori-Kiyosue Y. & Tsukita, S. (2001) J. Cell Biol. 154, 1105-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan K. M., Phillips, A. C. & Vousden, K. H. (2001) Curr. Opin. Cell Biol. 13, 332-337. [DOI] [PubMed] [Google Scholar]
- 37.Crown J. & O'Leary, M. (2000) Lancet 355, 1176-1178. [DOI] [PubMed] [Google Scholar]
- 38.Zhu H., Chang, B. D., Uchiumi, T. & Roninson, I. B. (2002) Cell Cycle 1, 59-66. [PubMed] [Google Scholar]
- 39.Stewart Z. A., Tang, L. J. & Pietenpol, J. A. (2001) Oncogene 20, 113-124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}