

Abstract
Nonketotic hyperglycinaemia (NKH) is an autosomal recessive neurometabolic disorder resulting from deficient glycine cleavage system activity, causing severe neurological impairment. While NKH is typically associated with pathogenic variants in glycine decarboxylase (GLDC) or aminomethyltransferase, the role of synonymous variants remains uncertain. To date, no cases of NKH caused by GLDC homozygous synonymous variants have been reported. Herein, a female infant born to consanguineous parents who developed refractory seizures, progressing to infantile epileptic spasms syndrome at 2 months is reported. Initial genetic testing identified a homozygous synonymous GLDC variant (c.1023G > A, p.Val341=), previously classified as “likely benign” in ClinVar (variation identification number: 1108119). Minigene splicing analysis revealed that the c.1023G > A variant caused a 38-base pair deletion in exon 7 (r.1021_1058del), Given the phenotypic characteristics of the child, we predict that this may resulting in a frameshift mutation (p.Val341ArgfsTer56) and a truncated protein. This functional evidence confirmed the pathogenicity of the variant.
Keywords: Nonketotic hyperglycinemia, GLDC, Synonymous variants, Minigene splicing assays
1. Introduction
The first mention of the term “nonketotic hyperglycinemia” is from 1968, is a rare neurometabolic disorder with an estimated incidence of 1 in 76,000 live births [1,2]. Its rarity complicates timely diagnosis and treatment. NKH results from defective glycine cleavage system (GCS) function, a mitochondrial enzyme complex responsible for glycine catabolism. Impaired GCS activity leads to glycine accumulation, causing severe neurological dysfunction. The GCS comprises four protein complexes (P, T, H, and L), encoded by glycine decarboxylase (GLDC), aminomethyltransferase (AMT), GCS H protein, and GCS L protein respectively. Classic NKH primarily results from pathogenic variants in GLDC or AMT [2].
Based on epilepsy severity and neurodevelopmental outcomes, Kuseyri et al. categorised classic NKH into severe and attenuated subtypes [3]. Severe NKH typically presents in the neonatal period with lethargy, apnoea, seizures, and coma, which often resolve within 1–3 weeks [4]. Long-term complications include intellectual disability, recurrent seizures, axial hypotonia, and progressive spastic paraplegia. Brain anomalies such as corpus callosum hypoplasia and cerebellar cysts with hydrocephalus are common in severe cases [5]. Conversely, attenuated NKH exhibits variable phenotypes, including mild-to-moderate intellectual disability, seizures, hyperactivity, choreoathetosis, and episodic somnolence and ataxia [4,6]. Atypical presentations occur in 15 % of neonatal and 50 % of infantile cases [6]. Severe NKH is strongly linked to biallelic pathogenic variants in GLDC or AMT, including GLDC exon deletions [7]. Diagnosis relies on biochemical markers, notably elevated glycine levels in plasma and cerebrospinal fluid (CSF). A CSF/plasma glycine ratio exceeding >0.08 is a key diagnostic criterion [8].
Unlike severe NKH, attenuated NKH is associated with lower CSF glycine levels and a reduced CSF/plasma glycine ratio [8]. Severe NKH presents earlier and is more readily diagnosed, particularly when genetic testing identifies a definite pathogenic variant. However, diagnosis is more challenging in attenuated cases, where glycine elevation might be borderline, clinical features are nonspecific, and genetic testing reveals variants of uncertain significance (VUS). Herein, a novel case of NKH caused by a homozygous synonymous GLDC variant, resulting in aberrant messenger ribonucleic acid (mRNA) splicing and the production of a truncated, non-functional protein is reported.
2. Methods
Whole-exome sequencing of the proband and her parents revealed a homozygous synonymous variant in GLDC (c.1023G > A, p.Val341=), classified as a VUS. Existing databases provided inconclusive clinical interpretations; ClinVar (variation identification number [ID]: 1108119; https://www.ncbi.nlm.nih.gov/clinvar/) previously annotated it as “likely benign”. In vitro minigene splicing experiments assessed the potential splice-altering effects of the c.1023G > A variant, the detailed experimental methods refer to our previous studies [9], Detailed experimental procedures were provided in Supplemental information(S1).
3. Results
3.1. Case presentation
The proband was a 4-month, 22-day-old female infant born to consanguineous parents. She was referred for evaluation of refractory epilepsy and neurodevelopmental delay. Family history was unremarkable for neurological disorders. She was delivered at full term via uncomplicated vaginal delivery and initially hospitalised for neonatal jaundice, which was resolved with phototherapy.
At 2 months of age, she developed focal seizures characterised by asymmetric tonic-clonic limb movements. Electroencephalography (EEG) revealed sporadic sharp waves localised to the bilateral central and temporal lobes during sleep, consistent with focal epilepsy. Initial treatment with valproic acid (20 mg/kg/day) and levetiracetam (30 mg/kg/day) failed to control seizures. By 3 months of age, she developed epileptic spasms with hypsarrhythmia on EEG, which showed a disorganised background, multifocal irregular spikes, and fast rhythms in the occipital, parietal, and posterior temporal regions. Despite dose adjustments of valproic acid and the addition of topiramate (5 mg/kg/day), seizures remained intractable.
Neurodevelopmental assessment revealed severe global developmental delay. The infant exhibited only occasional smiling and auditory/visual tracking but lacked head control, purposeful grasping, or hand-to-mouth coordination. Cranial magnetic resonance imaging shows focal gyral thickening in the left frontal lobe (Fig. 1), The corpus callosum appears thin(Fig. 1), We measured the child's corpus callosum according to the method described by Stence NV [10] (Fig. 2). She was treated with adrenocorticotropic hormone(2.5 U/kg/day); however, the spasms remained uncontrolled. We immediately discontinued sodium valproate and ACTH once the glycine test results were available.
Fig. 1.

Patient's brain MRI. At 2 months of age, (A) the T2 axial view shows enlarged and slightly thickened local gyrus of the left frontal lobe; (B) the T1 axial view shows enlarged and slightly thickened local gyrus of the left frontal lobe; (C) the T2 flair axial view shows enlarged and slightly thickened local gyrus of the left frontal lobe; (D) the T1 sagittal view shows thinning of the corpus callosum.
Fig. 2.
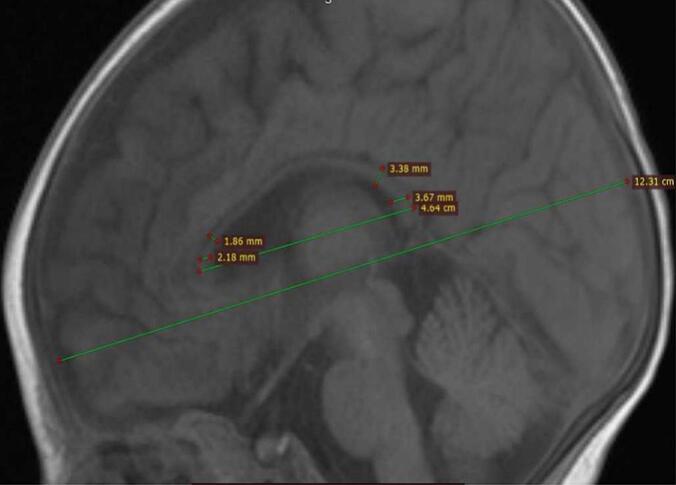
Patient's brain MRI. At 2 months of age, the T1 sagittal view shows thinning of the corpus callosum, and measurements of the corpus callosum were performed according to the method described by Stence NV.
3.2. Metabolic and genetic investigations
CSF analysis revealed elevated glycine levels (425 μmol/L) [11], with a CSF/serum glycine ratio of 0.076. Whole-exome sequencing identified a homozygous synonymous variant in GLDC (c.1023G > A, p.Val341=), According to ClinVar (variation ID: 1108119), this variant had previously been annotated as “likely benign”. We used a SpliceAI prediction score of 0.62, and this variant may affect normal splicing and generate abnormal splice sites. It has not been recorded in the gnomAD database. In accordance with the variant classification guidelines of ACMG and ClinGen, based on the available evidence, the c.1023G > A (p.Val341=) variant is classified as a variant of uncertain significance (VUS) (supported by PM3 + PM2 + PP3 + PP4). However, a VUS is insufficient for molecular diagnosis and genetic counseling; therefore, An in vitro minigene assay was performed to assess its splice-altering potential.
3.3. Functional validation
The minigene assay revealed aberrant exon 7 splicing, resulting in a 38-base pair deletion (NM_000170.3:r.1021_1058del); in the mature mRNA, This deletion introduced a frameshift mutation (p.Val341ArgfsTer56), Given the phenotypic characteristics of the child, we predict that this may resulting in a frameshift mutation (p.Val341ArgfsTer56) and a truncated protein (Fig. 3). These findings provided definitive evidence of loss of function, supporting the reclassification of c.1023G > A as a pathogenic variant. This molecular evidence correlated with the biochemical and clinical phenotype of NKH, reinforcing its causal role in disease pathology. We intended to conduct further functional verification, but the child's parents refused to undergo additional testing.
Fig. 3.

(A) shows an agarose gel electrophoresis result. On the left, there's a DNA ladder with size markers from 100 bp to 5000 bp. Lane 1 contains the GLDC Wild type fragment with a size of 513 bp, and Lane 2 has the Mutant (c.1023G > A) fragment with a size of 475 bp. On the right, the DL5000 DNA Marker is presented, indicating the amounts (50 ng or 100 ng) corresponding to each band size (100 bp to 5000 bp) in a 1.0 % TAE Agarose Gel. (B) illustrates the splicing effect of the c.1023G > A mutation. For the Wild type, Exon 6, Exon 7, and Exon 8 are spliced together normally. In the Mutant type, due to the c.1023G > A mutation, there is a deletion (r.1021_1058del) involving parts of Exon 7 during splicing, altering the normal splicing pattern.
4. Discussion
NKH is a rare genetic metabolic disorder characterised by a deficiency in GCS activity, leading to markedly elevated glycine levels in plasma and CSF. The clinical presentation of NKH is heterogeneous, ranging from severe neonatal-onset cases to milder, attenuated forms. The severe phenotype typically manifests in the neonatal period with profound developmental delay, lethargy, apnoea, intractable seizures, and coma. In contrast, the attenuated phenotype presents later in infancy or childhood and is associated with intellectual disability, seizures, lethargy, chorea, hyperactivity, and ataxia [4].
Glycine primarily functions as an inhibitory neurotransmitter in the brainstem and spinal cord. However, it also acts as a co-agonist of the N-methyl-d-aspartate (NMDA) receptor, exerting excitatory effects in the cerebral cortex, prefrontal cortex, and other brain regions. Pathologically elevated glycine levels result in NMDA receptor overstimulation, leading to neuronal and axonal injury, particularly in the hippocampus, cerebral cortex, olfactory bulb, and cerebellum. Additionally, excessive activation of glycine receptors in the spinal cord and brainstem induces central respiratory depression, leading to apnoea, hiccups, and hypotonia, which are hallmark features of early-onset NKH [4].
This report describes a case of early-onset epilepsy with profound motor and cognitive regression. At nearly 5 months, the patient had failed to achieve any developmental milestones, exhibiting an inability to grasp objects or roll over and laughing only occasionally. Based on previous studies, these clinical features align with severe NKH. To assess disease severity, the NKH severity prediction model developed by Swanson MA et al. [8], which assigns scores based on key clinical parameters was applied. Excluding genetic results, the patient received a total score of 1, calculated as follows: age of onset at 2 months: (−1 point), CSF glycine level (−1 point), CSF/plasma glycine ratio of 0.076 (2 points), and the absence of corpus callosum agenesis or thinning (1 point). This prediction model suggests that the patient harbours a severe variant that impacts protein function. Whole-exome sequencing (WES) in a trio-based analysis identified a homozygous GLDC gene variant (c.1023G > A, p.Val341=). This variant was previously classified as likely benign.
The GLDC gene, located on chromosome 9p24.1, comprises 25 exons spanning approximately 113.15 kb and encodes GLDC, a 1020-amino acid enzyme crucial for glycine metabolism. To date, more than 431 mutations in GLDC have been identified, accounting for approximately 80 % of NKH cases. The majority are missense mutations, with a smaller proportion involving exonic deletions [2,7].
Functional assay confirmed that the c.1023G > A variant induced an aberrant splicing event (r.1021_1058del), resulting in a frameshift mutation (p.Val341ArgfsTer56) that maybe produced a truncated, non-functional protein. This finding highlights the critical need for phenotype-driven analysis in variant classification, particularly for synonymous variants, which might be misclassified as benign despite potential functional consequences.
Yuan et al. [12] also confirmed that the synonymous mutation c.1023G > A (p.Val341=) resulted in abnormal splicing of the GLDC gene, causing a 38 bp deletion in exon 7. These findings highlight the pathogenic nature of a novel synonymous mutation c.1023G > A and expand the genetic spectrum of GLDC. Synonymous variants could disrupt normal splicing mechanisms [13]. Certain homologous variants exert functional effects by influencing transcription factor binding, mRNA binding sites, stability and splicing, and translation efficiency [14]. Contrary to the assumption that synonymous variants are selectively neutral or functionally silent [15], studies indicate that most synonymous variants in yeast genes exhibit non-neutral effects [16]. Liang et al. demonstrated that a synonymous variant in ATPase H+ transporting accessory protein 2 could contribute to neurodevelopmental disorders and epileptic encephalopathy by competitively reducing full-length transcript production [9]. Similarly, Gaysinskaya et al. identified a novel synonymous variant (c.942G > A.P.K314k) in dyskerin pseudouridine synthase 1, a telomere maintenance gene, as a causative factor in familial pulmonary fibrosis [17]. These findings emphasise the necessity of comprehensive functional assessment when evaluating the pathogenicity of synonymous variants, particularly in cases with strong phenotypic correlation. These findings highlight the need for comprehensive functional validation of VUS, particularly synonymous variants, which might disrupt normal splicing despite being previously classified as benign.
Our study underscores the phenotypic variability of NKH. While genetic testing offers valuable diagnostic insights, the interpretation of WES results requires careful consideration, particularly for synonymous variants. A thorough evaluation of clinical manifestations is essential, and functional validation might be necessary to confirm pathogenicity, thereby reducing the risk of misdiagnosis or missed diagnosis.
CRediT authorship contribution statement
Ping Pang: Writing – original draft. Lin Wan: Writing – review & editing. Yan Liang: Data curation. Xia Zhao: Investigation. Guang Yang: Supervision, Project administration, Funding acquisition, Conceptualization.
Consent for publication
All participants provided written informed consent before participation.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of The Second Affiliated Hospital of Guizhou University of Traditional Chinese Medicine (LW20250104), The study was conducted in compliance with local laws and institutional requirements. Written informed consent was obtained from all individual participants (or their legal guardians) included in the study for the publication of any potentially identifiable data or images.
Funding
This research was funded by the general project of National Key Research and Development Program of China (Reference: 2023YFC2706405, 2022YFC2705300), Capital's Funds for Health Improvement and Research (Reference: 2024–2-5082), Beijing Natural Science Foundation (Reference: 7222187), the key project of innovation cultivation fund of the Seventh Medical Center of Chinese PLA General Hospital (Reference: qzx-2023-1) and Innovation Talent Fund of Senior Department of Pediatrics, The Seventh Medical Center of PLA General Hospital (Reference: QZX-04-EKLHJH-3 and QZX-04-EKCLJH-2).
Declaration of competing interest
Ping Pang, Lin Wan, Yan Liang, Xia Zhao, Guang Yang declare that they have no competing interests. No financial or nonfinancial benefits have been received or will be received from any party directly or indirectly related to the subject of this study. We conform that we have read the journal's stance on ethical publication issues and affirm that this report adheres to these guidelines.
Acknowledgements
We sincerely appreciate all the children and their families who participated in this study, as well as the medical staff who helped in our experiment.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymgmr.2025.101268.
Appendix A. Supplementary data
Supplementary material
Data availability
To safeguard patient privacy, the Ethics Committee of the The Second Affiliated Hospital of Guizhou University of Traditional Chinese Medicine restricted access to the original quencing data used to support the conclusions of this study. Researchers who meet the criteria for access to confdential data may obtain data from the corresponding author, Guang Yang (E-mail: yangg301@126.com).
References
- 1.Ziter F.A., Bray P.F., Madsen J.A., et al. The clinical findings in a patient with nonketotic hyperglycinemia. Pediatr. Res. 1968;2(4):250–253. doi: 10.1203/00006450-196807000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Coughlin C.R., 2nd, Swanson M.A., Kronquist K., et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet. Med. 2018;20:1098. doi: 10.1038/gim.2017.232. [DOI] [PubMed] [Google Scholar]
- 3.Kuseyri Hübschmann O., Juliá-Palacios N.A., Olivella M., et al. Integrative approach to predict severity in nonketotic hyperglycinemia. Ann. Neurol. 2022;92:292–303. doi: 10.1002/ana.26423. [DOI] [PubMed] [Google Scholar]
- 4.Hennermann J.B., Berger J.M., Grieben U., et al. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J. Inherit. Metab. Dis. 2012;35:253–261. doi: 10.1007/s10545-011-9398-1. [DOI] [PubMed] [Google Scholar]
- 5.Van Hove J.L., Kishnani P.S., Demaerel P., et al. Acute hydrocephalus in nonketotic hyperglycinemia. Neurology. 2000;54:754–756. doi: 10.1212/wnl.54.3.754. [DOI] [PubMed] [Google Scholar]
- 6.Dinopoulos A., Matsubara Y., Kure S. Atypical variants of nonketotic hyperglycinemia. Mol. Genet. Metab. 2005;86:61–69. doi: 10.1016/j.ymgme.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Kanno J., Hutchin T., Kamada F., et al. Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J. Med. Genet. 2007;44 doi: 10.1136/jmg.2006.043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanson M.A., Coughlin C.R., Jr., Scharer G.H., et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann. Neurol. 2015;78:606–618. doi: 10.1002/ana.24485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang Y., Wan L., Yan H., et al. Synonymous variants in the ATP6AP2 gene may lead to developmental and epileptic encephalopathy. Front. Neurol. 2024;14:1320514. doi: 10.3389/fneur.2023.1320514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stence N.V., Fenton L.Z., Levek C., et al. Brain imaging in classic nonketotic hyperglycinemia: quantitative analysis and relation to phenotype. J. Inherit. Metab. Dis. 2019;42(3):438–450. doi: 10.1002/jimd.12072. [DOI] [PubMed] [Google Scholar]
- 11.Applegarth D.A., Toone J.R. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol. Genet. Metab. 2001;74:139–146. doi: 10.1006/mgme.2001.3224. [DOI] [PubMed] [Google Scholar]
- 12.Yuan F., Song X., Yin R., et al. The precise molecular diagnosis of novel compound heterozygous variants highlights the benefits for a Chinese family with nonketotic hyperglycinemia. Mol. Genet. Metab. Rep. 2025;43 doi: 10.1016/j.ymgmr.2025.101209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548. doi: 10.1016/j.cell.2018.12.015. e24. [DOI] [PubMed] [Google Scholar]
- 14.Hanson G., Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018;19:20–30. doi: 10.1038/nrm.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharp N. Mutations matter even if proteins stay the same. Nature. 2022;606:657–659. doi: 10.1038/d41586-022-01091-6. [DOI] [PubMed] [Google Scholar]
- 16.Shen X., Song S., Li C., et al. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature. 2022;606:725–731. doi: 10.1038/s41586-022-04823-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaysinskaya V., Stanley S.E., Adam S., et al. Synonymous mutation in DKC1 causes telomerase RNA insufficiency manifesting as familial pulmonary fibrosis. Chest. 2020;158:2449–2457. doi: 10.1016/j.chest.2020.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
To safeguard patient privacy, the Ethics Committee of the The Second Affiliated Hospital of Guizhou University of Traditional Chinese Medicine restricted access to the original quencing data used to support the conclusions of this study. Researchers who meet the criteria for access to confdential data may obtain data from the corresponding author, Guang Yang (E-mail: yangg301@126.com).