

Abstract
The evolutionary significance of introns remains a mystery. The current availability of several complete eukaryotic genomes permits new studies to probe the possible function of these peculiar genomic features. Here we investigate the degree to which gene structure (intron position, phase and length) is conserved between homologous protein domains. We find that for certain extracellular-signalling and nuclear domains, gene structures are similar even when pro tein sequence similarity is low or not significant and sequences can only be aligned with a knowledge of protein tertiary structure. In contrast, other domains, including most intracellular signalling modules, show little gene structure conservation. Intriguingly, many domains with conserved gene structures, such as cytokines, are involved in similar biological processes, such as the immune response. This suggests that gene structure conservation may be a record of key events in evolution, such as the origin of the vertebrate immune system or the duplication of nuclear receptors in nematodes. The results suggest ways to detect new and potentially very remote homologues, and to construct phylogenies for proteins with limited sequence similarity.
Keywords: evolution/exon/immune system/intron/protein structure
Introduction
A major feature of eukaryotic genomes is that the DNA coding for proteins is frequently interrupted by introns. Splicing machinery in cells removes introns from mRNA (thus joining together exons) before it is translated into protein. Why organisms have evolved such a seemingly convoluted system for transmitting information between the nucleus and the rest of the cell has been the subject of much debate during the past three decades. Most theories, including the ‘introns-early’ (Gilbert, 1986) and ‘introns-late’ (Orgel and Crick, 1980; Cavalier-Smith, 1985; Sharp, 1985; Stoltzfus et al., 1994) theories, argue for a role of exon shuffling in the evolution of proteins. For example, studies of intron positions within modular extracellular proteins have suggested that exon shuffling played a key role in the evolution of complex multi-domain proteins first used for cell–cell communication within multicellular animals (Patthy, 1994, 1999). However, until recently the relative paucity of genomic sequence information meant that most studies were limited to a handful of protein families.
The current abundance of genomic sequence data (The C.elegans sequencing consortium, 1998; Adams et al., 2000; The Arabidopsis Initiative, 2000; Lander et al., 2001) permits large-scale studies on the relationship between introns and their associated proteins. Here we assess the degree to which exon/intron (gene) structure, defined by the position, phase and length of introns, is conserved within diverse protein domains. Like previous studies on single proteins (e.g. Craik et al., 1981, 1983; Rokas et al., 1999; Venkatesh et al., 1999), we mapped genomic DNA sequence onto protein alignments. We took protein sequence alignment data from the SMART database (Schultz et al., 2000), which contains a diverse set of carefully aligned protein domains from all species and with diverse functions and cellular locations. We compared each protein sequence with genomic sequences and merged gene structure data with the aligned protein sequences. We devised a simple measure of gene structure similarity and sought groups of proteins with conserved gene structures at low sequence similarities. We discuss several examples where gene structures are conserved at low sequence similarity, and possible reasons for this strange phenomenon. We also describe how conserved gene structures could aid phylogenetic reconstruction and the detection of new members of diverse protein superfamilies.
Results
Figure 1 shows the distribution of gene structure similarities for all homologous pairs of protein domains with sequence identities of <40%. Although the majority of pairs show little similarity in gene structure, there are clearly peaks to the right of the figure indicating high gene structure similarities, particularly for nuclear and extracellular signalling domains (broad classifications from the SMART database; Schultz et al., 2000).

Fig. 1. Distribution of gene structure similarities for pairs of proteins with <40% sequence identity. Protein pairs are grouped according to the broad classifications for cellular location given in the SMART database: Extracellular signalling, Intracellular signalling, Nuclear, Others. Contributions to the frequencies were weighted according to the number of pairs from each family. This was to avoid large families (e.g. MATH domains with 4031 pairs) obscuring the plot.
Table I shows that several domains contain large clusters (sets of sequences from a larger alignment) with gene structure similarity despite low sequence identity, and for two (marked with +), gene structures are still similar even though protein sequences can only be accurately aligned with a knowledge of protein tertiary structure (see Materials and methods). These clusters come frequently from extracellular signalling and nuclear domains, though only rarely from intracellular signalling or other domains. Although the location and phase of introns is conserved for the clusters shown in Table I, intron length is highly variable (from <10 to >1000 nucleotides), even at comparatively high sequence similarities (results not shown).
Table I. Protein domains with similar gene structures.
| Domain | Np | Ng | Taxg | N2+ | Nc | %Imin | %Iave | Taxc | Introns | |
|---|---|---|---|---|---|---|---|---|---|---|
|
Extracellular signalling (162) |
10752 |
2396 |
|
964 |
|
|
|
|
|
|
| Meprin and traf homology domains | 116 | 98 | V F M | 93 | 77 | 6 | 26 | At Ce | 2. | • |
| 7 | 11 | 18 | At Ce | 21 | • | |||||
| Chemokines | 64 | 30 | Vert | 29 | 29 | 13 | 31 | Vert | 2 | • |
| Four-helical cytokines + | 110 | 38 | Vert | 36 | 18 | 4 | 43 | Vert | 000 | • |
| Trypsin-like proteases | 223 | 54 | F M | 43 | 13 | 26 | 42 | mammals | 20.0 | • |
| 10 | 27 | 37 | Vert | 2.10 | • | |||||
| Cystatins | 56 | 19 | V M | 18 | 12 | 7 | 27 | Vert | 00 | • |
| Lysozymes | 97 | 21 | M | 14 | 12 | 30 | 53 | Vert | 112 | • |
| β-trefoil cytokines + | 67 | 17 | M | 12 | 11 | 7 | 33 | M | 10 | • |
| Albumins | 84 | 14 | Vert | 12 | 10 | 16 | 28 | primates | 202 | • |
| Ly-6 like proteins | 30 | 15 | M | 13 | 9 | 14 | 22 | mammals | 1 | • |
| Granulins | 18 | 9 | V M | 7 | 7 | 32 | 45 | Mm | 0 | • |
| Hemopexin repeats | 124 | 27 | M | 16 | 7 | 4 | 20 | Ce Pl Hs | 1 | • |
| Phospholipases A2 |
142 |
11 |
Vert |
7 |
7 |
30 |
48 |
Vert |
21 |
• |
|
Nuclear (77) |
6143 |
1866 |
|
859 |
||||||
| Homeobox | 276 | 135 | V F M | 64 | 37 | 7 | 30 | V M | .0 | • |
| 7 | 19 | 29 | V F M | 00 | ||||||
| Nuclear receptors (ligand binding) | 311 | 128 | M | 120 | 28 | 8 | 22 | M | ..1 | |
| 11 | 9 | 29 | Ce | 02. | • | |||||
| 10 | 13 | 27 | Ce | 021 | • | |||||
| 10 | 9 | 29 | Ce | .21 | • | |||||
| 8 | 9 | 21 | Ce | 0.1 | ||||||
| Krueppel associated box | 58 | 23 | Hs Mm | 22 | 22 | 12 | 41 | Hs Mm | 1 | • |
| Nuclear receptors (Zn finger domain) | 215 | 126 | M | 115 | 20 | 27 | 38 | Ce | ....1. | |
| 16 | 28 | 39 | Ce | ...2.. | ||||||
| 14 | 34 | 46 | Ce | ...2.2 | • | |||||
| 13 | 26 | 39 | Ce | 0...1. | • | |||||
| 9 | 28 | 38 | Ce | .2.... | ||||||
| 8 | 28 | 42 | Ce | .2..1. | • | |||||
| 7 | 42 | 54 | M | ..0... | ||||||
| 6 | 40 | 45 | Ce | .....2 | ||||||
| 6 | 32 | 38 | Ce | 0..... | ||||||
| Leucine-rich region | 20 | 12 | Hs | 12 | 12 | 47 | 58 | Hs | 0 | • |
| B-box Zn finger | 61 | 23 | V M | 9 | 8 | 15 | 32 | At Ce | 0 | • |
| IPT domains |
103 |
32 |
F M |
21 |
6 |
13 |
19 |
Vert |
20 |
• |
|
Intracellular signalling (138) |
10773 |
2808 |
|
977 |
|
|
|
|
|
|
| C1 domains | 58 | 27 | V F M | 16 | 9 | 16 | 27 | F M | 1 | • |
The table is divided into three sections corresponding to extracellular, nuclear and signalling classes as defined by the SMART database. Domain names are taken from SMART; Np gives the number of protein sequences for each domain; Ng, the number of genomic DNA entries found; N2+, the number of domains containing two or more exons (or parts thereof); and Nc, the number (out of N2+) having pairwise SG ≥0.8. %Imin gives the lowest pairwise protein % sequence identity observed for the cluster; %Iave gives the average. Taxonomic data were extracted from DDBJ/EMBL/GenBank and the descriptions given for all genomic DNA sequences (Ng, Taxg) and for those in the clusters (Nc, Taxc). When more than three species were represented in the genomic DNA or the clusters, the taxonomic groups were abbreviated as: V, Viriplantae; F, Fungi; M, Metazoan; Vert, vertebrates or mammals; otherwise species are given: At, Arabidopsis thaliana; Ce, Caenorhabditis elegans; Mm, Mus musculus; Pl, Paracentrotus lividus (urchin); Hs, Homo sapiens. ‘Introns’ gives a short description of the conserved gene structure by giving the phases of the equivalent introns. Where more than one group is present in a single SMART domain, the intron patterns are aligned to show their relationships to one another (a ‘.’ denotes a gap). Groups are separated according to the four broad classes in SMART, with the number in parentheses giving the total number of domains in the class, and Np, Ng and N2+ in these rows giving totals for the class. No groups with similar gene structures were found for the ‘Others’ class (Np = 3168, Ng = 560, N2+ = 159). Domain names are taken from SMART: Meprin and traf homology, MATH; chemokines, SCY; trypsin-like proteases, Tryp_SPc; cystatins, CY; lysozymes, LYZ1; albumins, ALBUMIN; Ly-6 like, LU; granulins, GRAN; hemopexin repeats, HX; phospholipases A2, PA2; homeobox, HOX; nuclear receptors ligand binding, HOLI; Krueppel associated box, KRAB; nuclear receptors Zn finger, ZnF_C4; leucine-rich region, LRR; B-box Zn finger, BBOX; IPT domains, IPT; C1 domains, C1. Those domains marked with a ‘+’ symbol are those comprising multiple SMART alignments linked via structure based sequence alignment: four-helical cytokines, CSF2 IFabd IL10 IL2 IL4_13 IL6 LIF_OSM; β-trefoil cytokines, FGF IL1.
It is clear from the table that most clusters from extracellular signalling domains come from vertebrates, and that the domains themselves are often involved in the immune system. Four of these domains are effector or cytokine molecules: the chemokines, four-helical cytokines (the cluster includes interleukins 2, 4, 6, 10, 13 and colony stimulating factor), β-trefoil cytokines (fibroblast growth factors and interleukin 1), and granulins (GRAN). Other domains involved in the immune system include the lysozymes, one cluster of 13 trypsin-like proteases (including granzyme B, mast-cell proteases, cathepsin G and leukocyte elastases) and Ly-6-like proteins. Within these clusters there is only one sequence from the β-trefoil cytokines (an FGF homologue from Drosophila melanogaster) from outside the vertebrates. Vertebrate-specific clusters are also found in the cystatins and albumins, which can be loosely grouped into blood constituents, and in the phospholipases A2, which include several snake venom components.
Figure 2 shows an alignment of four-helical cytokines with the location, length and phase of introns, and a representative tertiary structure coloured according to the location of the core exons. For this diverse superfamily sequence identities are as low as 4% and homology can only be inferred from a similar protein structure and function. Nevertheless, many family members share a common gene structure comprising four core exons, with all introns having phase 0. Despite great sequence diversity this superfamily is very specific to the vertebrates; with the exception of viral homologues, no non-vertebrate members have been found (Lander et al., 2001), nor has any non-vertebrate protein to date been found to adopt the unusual up-up-down-down topology (Lo Conte et al., 2000).

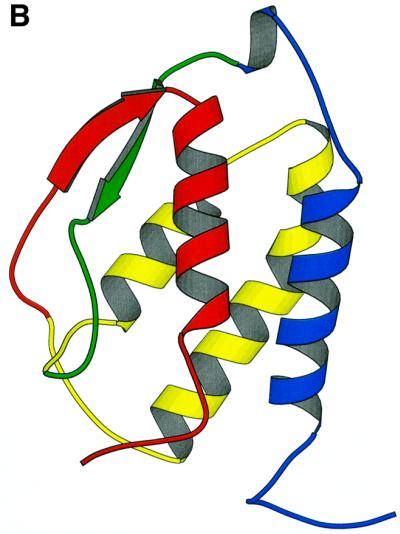
Fig. 2. Gene structure conservation within the four helical cytokines. (A) Alscript (Barton, 1993) figure showing a member of the four-helical cytokine superfamily (SMART domains: IL2, IL6, CSF2, IL4_13, LIM_OSF, IL10, IFabcd merged via the SCOP ‘four-helical cytokine’ superfamily). Aligned sequences for individual SMART entries were taken from the SMART database. Representative three-dimensional structures were aligned using the STAMP package (Russell and Barton, 1992), and this was used to merge the different SMART alignments in this superfamily. Residues are coloured according to conservation of hydrophobic properties: yellow background, conserved hydrophobic character; red characters, conserved hydrophobic. Structurally similar regions, defined by STAMP (Russell and Barton, 1992) are boxed. The location of introns within each sequence is shown within braces; the two numbers denote the phase and length of the intron. A ‘×’ denotes regions where unconserved sequence is not shown for clarity. The location of the four helices common to all proteins in this superfamily is shown below the alignment, coloured blue, yellow and red to agree with the order shown in (B). (B) Molscript (Kraulis, 1991) figure showing the structure of granulocyte macrophage colony stimulating factor (PDB code 2gmf). α-helices are shown as ribbons, β-strands as arrows and other structures as coil. Segments are coloured blue, green, yellow and red to show the order of core exons in the superfamily.
Gene structures for meprin and traf homology (MATH) domains from Arabidopsis thaliana and Caenorhabditis elegans are also surprisingly similar. A total of 87 out of 98 proteins with known gene structures contain a phase 2 intron at the same position, and for 86 the codon interrupted is TGG (tryptophan). Conserved or nearly conserved tryptophans like this are also found in the first (phase 1) and last (phase 2) introns of the lysozymes, and a thymidine is conserved in the second codon position within the phase 2 intron in the chemokines, which leads to conservation of hydrophobic character in the amino acid.
Several nuclear domains also show conservation of gene structure despite little sequence similarity. For example, several large clusters with similar gene structures are found in homeodomains and nuclear receptors. Here there is no obvious common theme uniting the functions of the domains, although the many C.elegans-specific clusters in the nuclear receptors are possibly associated with the diversification of this protein family within the nematodes (Sluder et al., 1999).
Table I also contains instances where domains duplicated in tandem show preservation of gene structure. Clusters from albumins, hemopexin repeats (Altruda et al., 1988) and granulins show a similarity in gene structure within tandem repeats despite limited sequence similarity. This parallels the situation seen previously for enzymes such as phosphoglycerate kinase (PGK) (Michelson, 1985).
The majority of domains containing introns show little conservation of gene structure. Figure 3 shows an alignment and introns for the GAP superfamily, a diverse group of intracellular signalling molecules. Modifications in the number, position and phase of introns are common, even with phase differences at the same approximate alignment positions. The lack of gene structure conservation within these and other intracellular signalling domains broadly supports suggestions that the spread of these modules may have preceded the rise of exon shuffling (Patthy, 1999).

Fig. 3. Gene structure differences within the GAP superfamily. Alscript (Barton, 1993) figure showing GAP domains (SMART domains RasGAP and RhoGAP linked by SCOP ‘GTPase activation domain, GAP’ superfamily). Details are as for Figure 2.
Discussion
Why can gene structure be conserved in certain domains despite little or no protein sequence similarity? For clusters specific to vertebrates or to C.elegans the most plausible explanation is that the proteins have diversified quickly via point mutations, and within too short a time to permit changes to the gene structure (e.g. such changes are rare in vertebrates; Guigo et al., unpublished results; Venkatesh et al., 1999). Rapid evolution has clearly lead to situations where the position of introns and the protein three-dimensional structure are apparently more conserved than the protein sequence. There could well be other examples where conservation of gene structure occurs within domains under evolutionary pressure, but there is currently not enough genomic DNA sequence from a sufficient diversity of organisms to see them.
There could also be functional reasons for the conservation of intron position. For example, conservation of phase 1 or 2 introns (where an amino acid is split across an intron) could be due to pressure to conserve the ‘split’ amino acid and/or the integrity of the splice site (Fichant, 1992). This could lead to a situation where an intron position becomes inexorably linked to protein structure and/or function (e.g. MATH domains). Another possible role for intron position conservation may be in alternative splicing, reflecting sites on protein structures that are useful for modulating function. An investigation of this possibility awaits large sets of carefully annotated splice variants.
The fact that intron positions (particularly phase 1 introns) sometimes correspond to domain boundaries in modular extracellular proteins led to the suggestion that exonic recombination played a key role in the ‘big bang’ of metazoan radiation (Patthy, 1994, 1999). During this period many new multi-domain, modular proteins evolved to function in the cell–cell communication processes first seen in the multicellular animals. The observation that phase and position of introns is conserved within certain diverse domain families similarly suggests that exon shuffling may have played key roles during other dramatic events in evolutionary history, such as the evolution of molecules involved in the immune system, or the diversification of nuclear receptors in C.elegans.
An intriguing possibility is that fixation of intron positions permitted the construction of chimeric domains through unequal crossing-over events between members of the same family. Shuffling equivalent parts of different proteins sharing the same fold could be a mechanism for rapid evolution. This phenomenon would suggest that one might obtain different phylogenies for the exons comprising the domains. We could not identify a clear example where this was the case, although the sequences are often too short, or have diverged beyond the point where phylogenetic reconstruction gives reliable trees. Other evidence for unequal crossing over comes from clustering of gene families on chromosomes. This is indeed the case for several of the cytokines (e.g. Kelleher et al., 1991) and a preliminary investigation with human proteins found that several of the clusters in Table I are within close proximity on the same chromosome. For example, two clusters of trypsin-like proteases on human chromosomes 14 and 19 correlate with the different gene structures given in Table I.
Genetic features beyond DNA and protein sequences, such as intron position and phase, can be excellent sources of phylogenetic information (e.g. Patthy, 1999; Rokas et al., 1999; Venkatesh et al., 1999; Rokas and Holland, 2000; Telford et al., 2000; Krem and Di Cera, 2001). Several of the groups shown in Table I contain similar but not identical gene structures. For example, the nuclear receptor ligand binding domains in Table I all appear to contain gene structures that are variations on a similar three intron (phase 0, 2 and 1) theme, with different clusters being formed possibly by different intron loss events. Differences in gene structure might thus provide clues as to the phylogeny of distantly related proteins.
Intron position could also be used for the detection and accurate alignment of new members of diverse protein families. Where gene structure conservation is seen in the absence of protein sequence identity it might be possible to detect additional, and very remote members of families simply by searching for genes with a similar set of intron positions and phases (Brown et al., 1995). Similarly, in regions of alignment ambiguity (e.g. due to low local sequence similarity) it may be fruitful to consider whether or not intron positions are equivalent.
Our results argue strongly neither for the ‘introns-early’ (Gilbert, 1986; Dorit et al., 1990; de Souza et al., 1998) nor the ‘introns-late’ (Orgel and Crick, 1980; Cavalier-Smith, 1985; Sharp, 1985; Stoltzfus et al., 1994) hypotheses. However, they do suggest that the conservation of intron location and phase within diverse protein domains might be explained by comparatively recent evolutionary events, rather than by an ancient correspondence between exons and local regions of structure or function.
We have found clear instances of gene structure conservation despite low protein sequence similarity. We anticipate that more examples will be uncovered as genomic sequences are determined for more organisms, giving further clues to events in evolution.
Materials and methods
We took 419 protein domain sequence alignments from SMART (Schultz et al., 2000) and information on superfamilies defined by analysis of protein structure from SCOP (Lo Conte et al., 2000), which groups proteins into homologous superfamilies based on similarities beyond sequence (e.g. structural features, common active sites, etc.). A total of 58 SCOP superfamilies were found to contain two or more SMART domains, and these were merged by structure alignment (Russell and Barton, 1992).
We extracted a database of eukaryotic genomic DNA sequences from DDBJ/EMBL/GenBank release 120 (rod, mam, pri, pln, inv, vrt). Each domain sequence was compared against this database to identify putative identical matches using WU-tblastn (W = 5 hitdist = 11 –filter xnu + seg T = 1000 –matrix identity –nogap S = 40 topcomboN = 2). Each pair of protein–genomic DNA sequences was aligned using genewise (Birney, unpublished) to refine the exon–intron boundaries and to obtain phase information. The SMART database contained a total of 27 168 protein sequences (including Eukarya, Archaea and Bacteria), of which 7701 gene sequences could be matched to a genomic DNA sequence. Ignoring the genes that lacked introns left 2987 gene sequences. Of the 419 protein domain alignments, 250 contained at least two gene sequences with one or more introns, and 151 contained six or more. We merged gene structures with their associated proteins and incorporated the gene structures into the alignment. For each pair of proteins in every alignment the associated sets of intron locations were matched by finding the optimal set of exon matches, fusions and insertions that could transform one gene structure into the other.
We constructed a simple gene structure similarity measure, which assesses the similarity between a pair of aligned proteins based solely on the location and phase of introns:
![]()
ai and bi refer to the ith equivalent intron positions in the two proteins (one on each side of an exon), Nmax is the largest number of introns found in either protein, Nequiv is the number of introns that have been assigned to be equivalent, di is the difference in position of the introns within the two proteins (in amino acids), and ϕ(ai,bi) is 1 if the phases are the same and 0 otherwise. γ and δ are constants (0.2, 30) in a sigmoid function (i.e. the first term above), and were chosen such that the function was insensitive to small changes in intron positions (i.e. ± 10 residues). We weighted the terms for position and phase equally, although modifications to weights do not change the results significantly. We found intron size varied considerably even in closely related proteins, and thus did not develop a scoring term for differences between intron lengths. SG ranges from 0 (no similarity) to 1 (identical), and we found that requiring values to be ≥0.8 gives pairs or groups of proteins with gene structures that we consider to be similar. This allows for short insertions/deletions or errors/ambiguities in the alignments.
For each domain we calculated all pairwise SG values and used these to cluster sequences by complete linkage. Only clusters with six or more sequences were considered further. We performed a simple χ2-test with 1 degree of freedom for each domain and each gene structure cluster, to test the hypothesis that the fraction of domains with that gene structure (considering only intron phases) was significantly more than the background frequency of the gene structure in the entire data set. Table I shows domains containing at least one significant cluster (p <0.001), which are indicated by black dots in the rightmost column.
All data used in this study are available from the authors.
Acknowledgments
Acknowledgements
We thank Max Telford (Dept of Zoology, Cambridge, UK), Rich Copley, José Castresana, Peer Bork (EMBL, Heidelberg, Germany), Chris Larmine and Rosemary Kelsell (GlaxoSmithKline) for helpful discussions, and Jim Fickett and David Searls (GlaxoSmithKline) for encouragement and support.
References
- Adams M.D. et al. (2000) The genome sequence of Drosophila melanogaster. Science, 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- Altruda F., Poli,V., Restagno,G. and Silengo,L. (1988) Structure of the human hemopexin gene and evidence for intron-mediated evolution. J. Mol. Evol., 27, 102–108. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796–815. [DOI] [PubMed] [Google Scholar]
- Barton G.J. (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng., 6, 37–40. [DOI] [PubMed] [Google Scholar]
- Brown N.P., Whittaker,A.J., Newell,W.R., Rawlings,C.J. and Beck,S. (1995) Identification and analysis of multigene families by comparison of exon fingerprints. J. Mol. Biol., 249, 342–359. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. (1985) Selfish DNA and the origin of introns. Nature, 315, 283–284. [DOI] [PubMed] [Google Scholar]
- C. elegans sequencing consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science, 282, 2012–2018. [DOI] [PubMed] [Google Scholar]
- Craik C.S., Buchman,S.R. and Beychok,S. (1981) O2 binding properties of the product of the central exon of β-globin gene. Nature, 291, 87–90. [DOI] [PubMed] [Google Scholar]
- Craik C.S., Rutter,W.J. and Fletterick,R. (1983) Splice junctions: association with variation in protein structure. Science, 220, 1125–1129. [DOI] [PubMed] [Google Scholar]
- de Souza S.J., Long,M., Klein,R.J., Roy,S., Lin,S. and Gilbert,W. (1998) Toward a resolution of the introns early/late debate: only phase zero introns are correlated with the structure of ancient proteins. Proc. Natl Acad. Sci. USA, 95, 5094–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorit R.L., Schoenbach,L. and Gilbert,W. (1990) How big is the universe of exons? Science, 250, 1377–1382. [DOI] [PubMed] [Google Scholar]
- Fichant G.A. (1992) Constraints acting on the exons positions of the splice site sequences and local amino acid composition of the protein. Hum. Mol. Genet., 1, 259–267. [DOI] [PubMed] [Google Scholar]
- Gilbert W. (1986) The RNA world. Nature, 319, 618. [Google Scholar]
- Kelleher K., Bean,K., Clark,S.C., Leung,W.Y., Yang-Feng,T.L., Chen,J.W., Lin,P.F., Luo,W. and Yang,Y.C. (1991) Human interleukin-9: genomic sequence, chromosomal location and sequences essential for its expression in human T-cell leukemia virus (HTLV)-I-transformed human T cells. Blood, 77, 1436–1441. [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst., 24, 946–950. [Google Scholar]
- Krem M.M. and Di Cera,E. (2001) Molecular markers of serine protease evolution. EMBO J., 20, 3036–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lo Conte L., Ailey,B., Hubbard,T.J., Brenner,S.E., Murzin,A.G. and Chothia,C. (2000) SCOP: a structural classification of proteins database. Nucleic Acids Res., 28, 257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson A.M., Blake,C.C., Evans,S.T., Orkin,S.H. (1985) Structure of the human phosphoglycerate kinase gene and the intron mediated evolution and dispersal of the nucleotide binding domain. Proc. Natl Acad. Sci. USA, 82, 6965–6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel L.E. and Crick,F.H. (1980) Selfish DNA: the ultimate parasite. Nature, 284, 604–607. [DOI] [PubMed] [Google Scholar]
- Patthy L. (1994) Exons and introns. Curr. Opin. Struct. Biol., 4, 383–392. [Google Scholar]
- Patthy L. (1999) Protein Evolution. Blackwell Science Ltd, Oxford, UK.
- Rokas A. and Holland,P.W. (2000) Rare genomic changes as a tool for phylogenetics. Trends Ecol. Evol., 15, 454–459. [DOI] [PubMed] [Google Scholar]
- Rokas A., Kathirithamby,J. and Holland,P.W. (1999) Intron insertion as a phylogenetic character: the engrailed homeobox of Strepsiptera does not indicate affinity with Diptera. Insect Mol. Biol., 8, 527–530. [DOI] [PubMed] [Google Scholar]
- Russell R.B. and Barton,G.J. (1992) Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins, 14, 309–323. [DOI] [PubMed] [Google Scholar]
- Schultz J., Copley,R.R., Doerks,T., Ponting,C.P. and Bork,P. (2000) SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res., 28, 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P.A. (1985) On the origin of RNA splicing and introns. Cell, 42, 397–400. [DOI] [PubMed] [Google Scholar]
- Sluder A.E., Mathews,S.W., Hough,D., Yin,V.P. and Maina,C.V. (1999) The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res., 9, 103–120. [PubMed] [Google Scholar]
- Stoltzfus A., Spencer,D.F., Zuker,M., Logsdon,J.M. and Doolittle,W.F. (1994) Testing the exon theory of genes: the evidence from protein structure. Science, 265, 202–207. [DOI] [PubMed] [Google Scholar]
- Telford M.J., Herniou,E.A., Russell,R.B. and Littlewood,D.T. (2000) Changes in mitochondrial genetic codes as phylogenetic characters: two examples from the flatworms. Proc. Natl Acad. Sci. USA, 97, 11359–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh B., Ning,Y. and Brenner,S. (1999) Late changes in spliceosomal introns define clades in vertebrate evolution. Proc. Natl Acad. Sci. USA, 96, 10267–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]