

Abstract
The generation of >30 different HIV-1 mRNAs is achieved by alternative splicing of one primary transcript. The removal of the second tat intron is regulated by a combination of a suboptimal 3′ splice site and cis-acting splicing enhancers and silencers. Here we show that hnRNP A1 inhibits splicing of this intron via a novel heterogeneous nuclear ribonucleoprotein (hnRNP) A1-responsive intron splicing silencer (ISS) that can function independently of the previously characterized exon splicing silencer (ESS3). Surprisingly, depletion of hnRNP A1 from the nuclear extract (NE) enables splicing to proceed in NE that contains 100-fold reduced concentrations of U2AF and normal levels of SR proteins, conditions that do not support processing of other efficiently spliced pre-mRNAs. Reconstituting the extract with recombinant hnRNP A1 protein restores splicing inhibition at a step subsequent to U2AF binding, mainly at the time of U2 snRNP association. hnRNP A1 interacts specifically with the ISS sequence, which overlaps with one of three alternative branch point sequences, pointing to a model where the entry of U2 snRNP is physically blocked by hnRNP A1 binding.
Keywords: ASF/hnRNP A1/SF2/splicing silencer/U2AF
Introduction
Alternative splicing is a common mechanism for regulating gene expression in higher eukaryotes and it is controlled mainly at early stages of spliceosome assembly. Spliceosomes are assembled on pre-mRNA by stepwise association of the small nuclear ribonucleoprotein particles (snRNPs) U1, U2 and U5·U4/U6. Early steps in this process include the recognition of the 5′ splice site and the branch point sequence by the U1 snRNP and the U2 snRNP, respectively (Burge et al., 1999). Other splicing factors, several of which contain a domain that is rich in arginines and serines, known as an RS domain, assist these steps. One of these factors, U2AF, consists of two subunits, U2AF65 and U2AF35, which interact with the polypyrimidine tract and the 3′ splice site, respectively (Moore, 2000), and mediates the association of U2 snRNP with the branch point sequence (Zamore and Green, 1989; Valcárcel et al., 1996; Gozani et al., 1998).
Other important splicing factors that contain an RS domain belong to the family of SR proteins, of which 10 are known to date. SR proteins are essential for virtually every step of spliceosome assembly, including early recognition of splice sites, recruitment of basic splicing factors to the pre-mRNA, and formation of bridging contacts with other RS domain-containing splicing factors (Graveley, 2000).
Prior to formation of spliceosomes, the pre-mRNA is packed with proteins of the large and diverse family of heterogeneous nuclear ribonucleoproteins (hnRNPs) (Krecic and Swanson, 1999). Although the binding of hnRNPs appears to have an unspecific nature, different pre-mRNAs bind different subsets of hnRNPs, implying some degree of sequence specificity. A direct role of hnRNPs in constitutive splicing has not been observed, but hnRNPs may play important functions in modelling pre-mRNA structure and initial recognition of splice sites (Smith and Valcárcel, 2000).
Alternatively spliced cellular and viral pre-mRNAs often contain short cis-acting sequence elements that regulate splicing in either a positive or a negative way. These sequences generally bind trans-acting factors that interact with the basal splicing machinery. The majority of trans-acting factors that have been characterized so far belong to either the SR protein family or the hnRNP family, and proteins from both families have been implicated in positive as well as negative regulation. However, the emerging picture is that some of the factors from these two families have antagonistic effects, and that SR proteins mainly participate in positive regulation while some hnRNPs are more often involved in negative regulation (Graveley, 2000; Smith and Valcárcel, 2000).
Positively acting elements are generally known as exon splicing enhancers (ESEs) or intron splicing enhancers (ISEs). The best-characterized elements are the purine-rich ESEs that are prevalent in higher eukaryotes. Different subsets of SR proteins have been shown to interact with many of these ESEs in a sequence-specific manner (Sun et al., 1993; Ramchatesingh et al., 1995; Liu et al., 1998; Schaal and Maniatis, 1999). One commonly proposed model for ESE function suggests that ESE-bound SR proteins stabilize the binding of splicing factors to nearby splice sites, most often a weak upstream 3′ splice site. In particular, the U2AF factor has been suggested to be the target for the action of ESE-bound SR proteins (Wu and Maniatis, 1993; Wang et al., 1995; Zuo and Maniatis, 1996). Alternative ESE mechanisms that do not involve U2AF as primary target have recently been proposed (Kan and Green, 1999; Blencowe, 2000).
Exon splicing silencers (ESSs) and intron splicing silencers (ISSs) have been found to inhibit splicing of various cellular and viral pre-mRNAs, but the mechanisms are less well understood (McNally and Beemon, 1992; Valcárcel et al., 1993; Amendt et al., 1994; Del Gatto and Breathnach, 1995; Kanopka et al., 1996; Gooding et al., 1998; Blanchette and Chabot, 1999; Chen et al., 1999). Both inhibition of spliceosome assembly (Amendt et al., 1995; Kanopka et al., 1996) and formation of pseudo spliceosomes (Gontarek et al., 1993; Kan and Green, 1999) have been suggested as inhibitory mechanisms. In several cases, the nucleo-cytoplasmic shuttle protein hnRNP A1 has been shown to be implicated in this regulation (Fu et al., 1992; Mayeda et al., 1993; Blanchette and Chabot, 1999; Caputi et al., 1999; Del Gatto-Konczak et al., 1999; Eperon et al., 2000).
In order to express its full coding capacity, HIV-1 utilizes the host cell splicing machinery to produce >30 different mRNAs by alternative splicing of its primary transcript. These processes are controlled by suboptimal splice site sequences (Staffa and Cochrane, 1994; Dyhr-Mikkelsen and Kjems, 1995; O’Reilly et al., 1995; Si et al., 1997) and splicing regulatory elements (Amendt et al., 1994, 1995; Staffa and Cochrane, 1995; Wentz et al., 1997). One of the best-characterized splicing events in HIV-1 is the splicing of the first tat intron (Figure 1A, the joining of splice sites SD1 and SA3), which is downregulated by an ESS (ESS2) located downstream of SA3 in the second tat exon (Amendt et al., 1994, 1995; Si et al., 1997; Jacquenet et al., 2001). hnRNP A1 as well as other members of the hnRNP A/B family have been shown to interact with ESS2 and mediate the inhibition of splicing (Caputi et al., 1999; Del Gatto-Konczak et al., 1999).

Fig. 1. RNA substrates used in this study. (A) The primary transcript with indications of the tat splice sites. Boxes and thin lines denote exons and introns, respectively. The tat pre-mRNA contains SD4 and SA7. (B) The names and the schematic drawings of the various splicing substrates are shown to the left. Deletions are indicated by dotted lines and point mutations or scrambling by a cross. The 3′ exon was divided into regions 1–4 for visualization. The mutagenized regions, up- or downstream from SA7, are shown to the right. Lowercase letters indicate point mutations or scrambling, and the nucleotides are numbered relative to SA7. (C) The sequences of UVtat and UVPIP that were used for UV-crosslinking and gel shift analysis derive from tat and PIP7.A, respectively. The 3′ splice sites in tat and PIP7.A are marked SA7 and SA, respectively, and the experimentally verified alternative branch points at nucleotides –48, –26 and –16 relative to SA7 in tat and the branch point in PIP7.A are indicated with bold letters (Dyhr-Mikkelsen and Kjems, 1995; T.Ø.Tange, C.K.Damgaard and J.Kjems, in preparation). The ISS, ESE3 and ESS3 sequences are underlined, and putative hnRNP A1 binding sites that match the hnRNP A1 consensus binding site, UAGGG(U/A) (Burd and Dreyfuss, 1994), at five out of six positions are indicated by brackets above the sequence.
The focus of the present report is the splicing of the second tat intron (Figure 1A, the joining of splice sites SD4 and SA7), which in this study is referred to as the tat intron. Splicing of the tat intron is regulated by ESE and ESS elements in the third tat exon, named ESE3 and ESS3, respectively (Amendt et al., 1995; Staffa and Cochrane, 1995; Mayeda et al., 1999; Tange and Kjems, 2001) as well as a purine-rich ESE sequence (ESE2) located upstream of SD4 in the second tat exon (Kammler et al., 2001) (Figure 1A). The SR protein SF2/ASF is a trans-acting factor for the ESE3 sequence (Krainer et al., 1990; Fu, 1993; Mayeda et al., 1999; Tange and Kjems, 2001) and presumably also for the ESE2 sequence upstream of SD4 (Kammler et al., 2001).
We have reported elsewhere that ESE3 and ESS3 regulate the efficiency of SA7 utilization by modulating the level of U2AF65 that is associated with the polypyrimidine tract (Tange and Kjems, 2001). To study the molecular basis for this observation in further detail, we studied the splicing of the tat intron in a nuclear extract that was chromatographically depleted for U2AF activity. Here we report that hnRNP A1 was unexpectedly co-depleted by this procedure and that this led to a strong increase in tat intron splicing. Reconstitution experiments demonstrated that hnRNP A1 mainly acts through a novel ISS element inhibiting U2 snRNP association.
Results
Splicing of tat pre-mRNA is activated in U2AF depleted extract
Splicing of the HIV-1 tat pre-mRNA (Figure 1A and B) was performed in HeLa nuclear extract (NE) depleted for U2AF activity. Two different approaches were used to deplete the extract for U2AF: chromatographic depletion, exploiting the affinity of U2AF65 for oligo(dT)-cellulose (Valcárcel et al., 1997), or immunodepletion, using a U2AF65-specific antibody (Gama-Carvalho et al., 1997). The chromatographic depletion was performed by passing NE over an oligo(dT)-cellulose column in 1 M KCl buffer. Under these conditions U2AF65 and the associated U2AF35 bind strongly to the oligo(dT) column, leading to an equimolar depletion of both U2AF subunits. The column was subsequently washed in 2.5 M KCl buffer followed by elution of the U2AF activity with 2 M guanidine buffer. Finally, the guanidine eluate and the U2AF-depleted NE [oligo(dT)ΔNE] were dialysed against 100 mM KCl buffer (buffer D). Alternatively, immunodepletion of U2AF from NE was performed by incubating NE with a U2AF65-specific monoclonal antibody (αMC3), leading to equimolar depletion of both U2AF subunits (αMC3ΔNE). A mock immunodepletion was performed using an unrelated antibody (αMyc) to obtain a control extract (mockΔNE). The U2AF65 protein content in oligo(dT)ΔNE and αMC3ΔNE was estimated to be ∼1 and 4–10%, respectively, of the normal level based on comparative western blot analysis (Figure 2A).
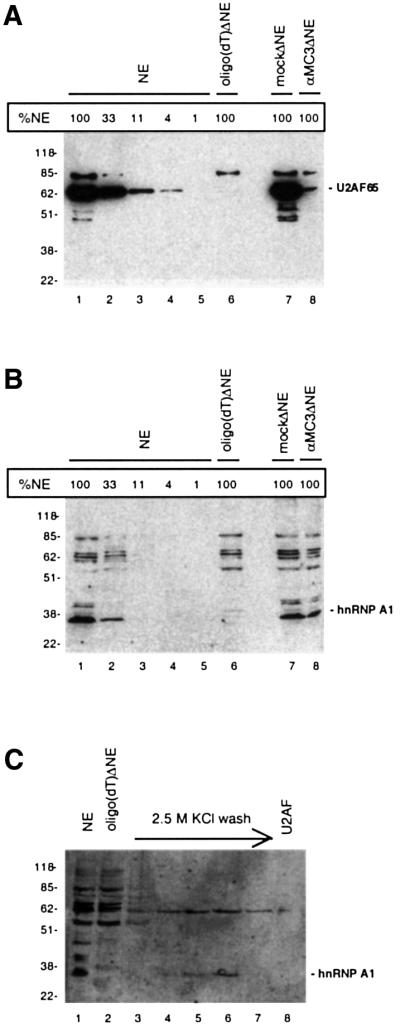
Fig. 2. Monitoring the depletion efficiency of U2AF65 (A) and hnRNP A1 [(B) and (C)] by western blotting. (A) Western blot of non-depleted extract (NE; lane 1), oligo(dT)-depleted extract [oligo(dT)ΔNE; lane 6], immunodepleted extract (αMC3ΔNE; lane 7), mock immunodepleted extract (mockΔNE; lane 8), and serial dilutions of non-depleted NE used for estimating the degree of depletion (lanes 2–5), using U2AF65-specific antibody (αMC3). Ponceau staining of the filter verified that equal amounts of extracts were loaded and transferred to the filter (data not shown). (B) Western blot of the same filter as in (A) probed with hnRNP A1-specific antibody (αA1). (C) Immunodetection of hnRNP A1 in the 2.5 M KCl buffer wash of the oligo(dT) column (lanes 3–7) and the eluted U2AF activity (lane 8). The samples were loaded next to samples containing NE (lane 1) and oligo(dT)ΔNE (lane 2). Bands above or below the marked bands probably derive from unspecific binding of the antibody. A size marker (kDa) is indicated to the left, and the positions of U2AF65 and hnRNP A1 are marked.
The tat pre-mRNA or an artificial pre-mRNA optimized for constitutive splicing, PIP7.A (Figure 1B), was incubated under splicing conditions in non-depleted, oligo(dT)-depleted and immunodepleted extracts, in the absence or presence of U2AF activity eluted from the oligo(dT) column (Figure 3A). Splicing of PIP7.A was completely abolished by either of the depletion methods (Figure 3A, lanes 2 and 5), and splicing was fully restored by complementing both of the depleted extracts with U2AF activity (Figure 3A, lanes 3 and 6). Splicing of tat pre-mRNA was very inefficient in non-depleted NE and addition of extra U2AF activity did not activate splicing (Figure 3B, lanes 1 and 2). Surprisingly, tat pre-mRNA was very efficiently spliced in U2AF-supplemented oligo(dT)ΔNE (Figure 3B, lane 4). It may primarily be the 65 kDa subunit of U2AF that is responsible for the activation of splicing, because the stimulatory effect was also observed when using recombinant U2AF65 protein to complement the oligo(dT)ΔNE (data not shown). In contrast, using immunodepleted NE gave a very different result: no splicing was observed, either with or without addition of U2AF activity (Figure 3B, lanes 5–8). These results indicate that the oligo(dT) depletion of NE, as well as removing U2AF, also removes an inhibitory factor that is usually responsible for inhibition of tat splicing, and that this factor is not removed by the immunodepletion approach. Moreover, since the inhibitory activity is absent in the eluted U2AF fraction, the putative factor is most likely to be removed during the 2.5 M KCl buffer wash of the oligo(dT) column or inactivated by the 2 M guanidine during the elution of U2AF.

Fig. 3. In vitro splicing of PIP7.A (A) and tat (B) in different NES. The extracts are denoted: NE (non-depleted); mockΔNE (mock immunodepleted); oligo(dT)ΔNE [oligo(dT)-chromatography depleted]; and αMC3ΔNE (U2AF65 immunodepleted). U2AF denotes the addition of U2AF activity recovered from the oligo(dT) column. The identities of the splicing products are indicated.
Interestingly, we observed that the level of tat pre-mRNA splicing increased significantly in oligo(dT)ΔNE, despite the fact that this extract only contains ∼1% of the normal level of U2AF and no extra SR proteins (Figures 3B, lane 3, and 4, lane 2). These observations suggest that in the absence of hnRNP A1, splicing of the tat intron can proceed at very low U2AF concentrations, conditions that are unable to support splicing of optimized splicing constructs, e.g. PIP7.A (Figure 3A, lane 2).
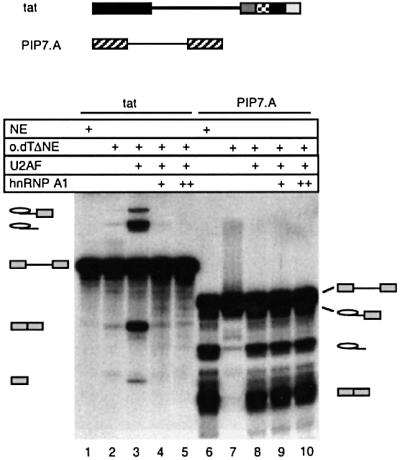
Fig. 4. hnRNP A1 specifically inhibits tat splicing. Splicing reactions containing tat (lanes 1–5) or PIP7.A (lanes 6–10) were performed in non-depleted extract (NE; lanes 1 and 6) or in oligo(dT)-depleted extract [oligo(dT)ΔNE; lanes 2–5 and 7–10]. The depleted extract was reconstituted with U2AF activity in lanes 3–6 and 8–10, and 17.5 ng/µl (+) or 35 ng/µl (++) (final concentration) GST–hnRNP A1 was added in lanes 4 and 9, and lanes 5 and 10, respectively. The identities of the splicing products are indicated.
The inhibition of tat splicing is mediated by hnRNP A1
To determine whether hnRNP A1 was the putative inhibitory factor that had been removed in the depletion process, a western blot was performed using a polyclonal hnRNP A1 antibody (αA1). It showed that ∼90% of the hnRNP A1 was depleted in oligo(dT)ΔNE (Figure 2B), whereas co-depletion of hnRNP A1 was not observed during immunodepletion of U2AF from NE (Figure 2B, lanes 7 and 8). Clearly, the co-depletion of hnRNP A1 was a side-effect of the oligo(dT) depletion procedure, suggesting that hnRNP A1 binds to oligo(dT)-cellulose, potentially via interactions with other proteins. To determine the fate of the hnRNP A1 protein in the depletion process, we tested the wash fractions and the U2AF eluate by western blotting using the hnRNP A1-specific antibody (Figure 2C). The hnRNP A1 protein was not detectable in the dialysed guanidine fraction containing the U2AF activity (Figure 2C, lane 8), but was clearly visible in the 2.5 M KCl buffer wash fractions (Figure 2C, lanes 5 and 6). This demonstrated that hnRNP A1 was eluted from the column during the 2.5 M KCl buffer wash and therefore not present in the U2AF-supplemented oligo(dT)ΔNE.
To confirm that hnRNP A1 was responsible for the inhibition, the oligo(dT)-depleted extract was reconstituted both with eluted U2AF and recombinant hnRNP A1 proteins and the processing of the tat and PIP7.A pre-mRNAs was tested (Figure 4). Increasing the concentration of hnRNP A1 completely inhibited tat splicing (Figure 4, lanes 4 and 5). These results confirm that the inhibitory factor(s) removed during the oligo(dT) depletion is hnRNP A1, and that the inhibition is specific to the tat pre-mRNA, since splicing of PIP7.A was unaffected by the hnRNP A1 addition (Figure 4, lanes 9 and 10).
The observation that splicing of tat in U2AF-supplemented oligo(dT)ΔNE proceeded efficiently without addition of recombinant SF2/ASF raised the question of whether the SF2/ASF-responsive enhancer, ESE3, is necessary for splicing under these conditions. To address this question, the entire region containing ESE3 and ESS3 was replaced with a sequence of similar length from the first part of the dsx exon 4 (tat-dsx-exon; Figure 1B), which has no detectable ESE or ESS activity in HeLa NE (Lynch and Maniatis, 1995). Splicing of this pre-mRNA did not occur in U2AF-supplemented oligo(dT)ΔNE (data not shown). This demonstrated that tat splicing is also dependent on the ESE3 activity in the absence of the inhibitory effect of hnRNP A1.
Binding of hnRNP A1 to an ISS near tat branch point –26 and to the ESS3 inhibits splicing
The binding of hnRNP A1 has previously been investigated by SELEX, and a high-affinity binding motif, UAGGG(A/U), which binds hnRNP A1 with KD ∼1 nM, has been defined (Burd and Dreyfuss, 1994). A search for putative hnRNP A1 binding sites in the vicinity of the 3′ splice site of the tat pre-mRNA revealed five putative motifs that each matched the consensus sequence with five out of six nucleotides (Figure 1C). Four of the sites are partially overlapping and located within the region –22 to –44 nucleotides (nt) upstream of SA7, and the last site coincides with ESS3 (Figure 1C). The function of these sites as binding platforms for hnRNP A1 proteins was demonstrated by gel shift analysis (Figure 5), which showed that hnRNP A1 formed two complexes with an RNA fragment containing the intronic hnRNP A1 binding motifs (UVtat–AvaI; Figure 5A, lanes 1–4), but failed to bind the same RNA, where the hnRNP A1 motifs were deleted (UVtatΔA1–AvaI; Figure 5A, lanes 9–12) or scrambled (UVtat-sA1–AvaI; Figure 5A, lanes 5–8). hnRNP A1 also formed two complexes with the 3′ exon alone in the gel-shift assay (UVtat-exon; Figure 5B, lanes 1–5). Deleting the ESS3 lowered the binding affinity for hnRNP A1 (UVtat-exon–Sau3A; Figure 5B, lanes 6–10), although two complexes were still detectable at the highest titration point. Even at this titration point hnRNP A1 binding was specific, since no binding was observed to the control substrate derived from PIP7.A (UVPIP; Figure 5B, lanes 11–15).

Fig. 5. Gel mobility-shift assay mapping the hnRNP A1 binding sites in the intron (A) and in the exon part of UVtat (B). The RNA substrates, indicated above the autoradiograms, were incubated in 10 µl reactions containing 0, 30, 100 and 300 ng of GST–hnRNP A1 in (A) and 0, 10, 30, 100 and 300 ng of GST–hnRNP A1 in (B). The migration of the free RNA probes and the complexes is indicated. The asterisk marks the position of RNA dimers of some of the substrates.
To investigate the role of the hnRNP A1 binding sites, the splicing efficiencies of pre-mRNAs, containing deletion or scrambling of the intronic binding sites and/or deletion of the ESS3 element, were tested in the absence or presence of recombinant SF2/ASF (Figures 1B and 6A). The data revealed that scrambling or deleting the intronic hnRNP A1 binding sites (tat-sA1, Figure 6A, lanes 5–6; and tatΔA1, lanes 9–10), respectively, had a positive effect on tat splicing, showing that these motifs function as an ISS. Deleting the ESS3 (Figure 6A, tatΔ4, lanes 3–4) had a small but highly reproducible positive effect on splicing. The inhibition was strongly abrogated when both the ISS and ESS3 were removed in tat-sA1Δ4 (lanes 7–8) and tat-ΔA1Δ4 (lanes 11–12), particularly in the absence of extra SF2/ASF, suggesting that the inhibitory effects of the ISS and ESS3 are more than additive. The increase in splicing observed when deleting or scrambling the ISS is not a consequence of elimination of the coinciding bp –26 sequence, since elimination of this branch point by a single point mutation does not lead to an increase in splicing (data not shown).

Fig. 6. In vitro splicing analysis determining the effect of ISS and ESS3 mutations. (A) Splicing of tat pre-mRNA in NE in the absence and presence of 32 ng/µl recombinant SF2/ASF (lanes 1 and 2) as compared with tat-derived pre-mRNAs with deleted ESS3 (tatΔ4; lanes 3 and 4), scrambled ISS (tat-sA1; lanes 5 and 6), deleted ISS (tatΔA1; lanes 9 and 10), scrambled ISS and deleted ESS3 (tat-sA1Δ4; lanes 7 and 8), and deleted ISS and ESS3 (tatΔA1Δ4; lanes 11 and 12). (B) Splicing of the chimeric PIPtat pre-mRNA (lanes 1–5), as compared with PIPtat-derived pre-mRNAs with deleted ESS3 (PIPtatΔ4; lanes 6–10), and scrambled ISS and deleted ESS3 (PIPtat-sA1Δ4; lanes 11–15). (C) Splicing of PIPtat pre-mRNA (lanes 1–5) and PIPtat with scrambled ISS (PIPtat-sA1; lanes 6–10). The source of NE was: non-depleted NE, oligo(dT)ΔNE or oligo(dT)ΔNE supplemented with U2AF alone or together with 12.5 ng/µl (+) or 37.5 ng/µl (++) (final concentration) GST–hnRNP A1 as indicated above the lanes. The asterisk indicates a degradation product that co-migrates with the exon–exon product of PIPtat. The identities of the splicing products are indicated.
To eliminate potential contribution from hnRNP A1 binding sites in the 5′ half of the tat pre-mRNA to the inhibition of splicing, we also analysed the 3′ splice site region from tat pre-mRNA in the context of the 5′ splice site from PIP7.A (PIPtat; Figure 1B). The chimeric pre-mRNAs were analysed in non-depleted NE, oligo(dT)ΔNE or oligo(dT)ΔNE supplemented with U2AF and hnRNP A1, as indicated in Figure 6B. In contrast to what was observed for the tat transcript, the splicing of PIPtat was clearly detectable in the absence of extra SF2/ASF and the ability to splice in the absence of U2AF was eliminated, suggesting a role of the tat 5′-exon for these characteristics. However, the splicing of PIPtat was efficiently inhibited by hnRNP A1 (Figure 6B and C, lanes 1–5). The inhibitory effect of hnRNP A1 was reduced when ESS3 that harbours the exonic binding site was deleted (PIPtatΔ4; Figure 6B, lanes 6–10). Likewise, scrambling of the ISS led to a reduced response to hnRNP A1 (PIPtat-sA1; Figure 6C, lanes 6–10). When scrambling of the ISS was combined with deletion of the ESS3 the inhibition of splicing by hnRNP A1 was almost eliminated, leading to efficient splicing also at the highest hnRNP A1 titration point (PIPtat-sA1Δ4; Figure 6B, lanes 11–15). We can conclude that both the ISS and ESS3 function as hnRNP A1-responsive elements.
hnRNP A1 inhibits U2 snRNP binding, but not U2AF65 binding
SF2/ASF-mediated activation of tat splicing correlates with an increased level of U2AF65 binding to the suboptimal polypyrimidine tract of SA7 (Tange and Kjems, 2001). To investigate the effect of hnRNP A1 on the association of splicing factors with the 3′ splice region of tat, a UV-crosslinking assay was performed (Figure 7B, lanes 1–3). The RNA substrate, spanning the 3′ splice site region and 3′ exon of the tat pre-mRNA (UVtat; Figure 1C), was randomly labelled and incubated under splicing conditions in the absence or presence of recombinant SF2/ASF and hnRNP A1. The in vitro splicing efficiency of the corresponding tat pre-mRNA was assayed under the same conditions in parallel (Figure 7A, lanes 1–3).

Fig. 7. Analysing the effect of hnRNP A1 on spliceosome assembly. (A) In vitro splicing of tat pre-mRNA in NE (lane 1), NE supplemented with 32 ng/µl SF2/ASF (lane 2) or NE supplemented with both 32 ng/µl SF2/ASF and 20 ng/µl hnRNP A1 (lane 3). The identities of the splicing products are indicated. (B) Autoradiogram of SDS–polyacrylamide gel showing UV-crosslinking of proteins to UVtat. Bands corresponding to U2AF65 and GST–hnRNP A1 are indicated, and a protein size marker (in kDa) is shown to the left. The experimental conditions are the same as in (A) except that the samples were incubated for 30 min. (C) Native gel electrophoresis showing complex A3′ formation on UVtat and derivatives with scrambling/deletion of the ISS and/or the ESS3. The substrates were incubated for 25 min in non-depleted NE (lanes 1, 4, 7 and 10), oligo(dT)ΔNE supplemented with U2AF (lanes 2, 5, 8 and 11) and oligo(dT)ΔNE supplemented with U2AF and 15 ng/µl hnRNP A1 (lanes 3, 6, 9 and 12).
Three major crosslinked bands at 65, 42 and 35 kDa were observed that all originated primarily from crosslinking of proteins to the intron part of UVtat (data not shown) and based on immunoprecipitation data the cross-linked protein with an apparent mol. wt of 65 kDa in Figure 7B was identified as U2AF65 (data not shown). The 42 kDa band most likely represents hnRNP C1/C2, while part of the 35 kDa band in all probability is hnRNP A1 (Figure 5A, and see below). Addition of SF2/ASF clearly increased U2AF65 crosslinking to UVtat (Figure 7B, lanes 1 and 2), which correlated with the activation of splicing shown in Figure 7A (lanes 1 and 2). When both SF2/ASF and hnRNP A1 were added to the reaction, a 62 kDa band corresponding to the size of GST–hnRNP A1 appeared (Figure 7B, lane 3), and splicing became strongly inhibited (Figure 7A, lane 3). Notably, the binding of U2AF65 remained unaffected upon addition of recombinant hnRNP A1 (Figure 7B, lane 3), implying that hnRNP A1 inhibits splicing at a step subsequent to the association of U2AF65 protein. Moreover, it was found that the crosslinking efficiency of recombinant hnRNP A1 was independent of addition of SF2/ASF (data not shown). These observations strongly suggest that the activation and inhibition of splicing by SF2/ASF and hnRNP A1, respectively, can occur at functionally distinct steps in the spliceosome assembly process.
Next we analysed the effect on U2 snRNP binding. The spliceosomal complex A3′, which represents stable binding of U2 snRNP to a 3′ splice site substrate in NE in the presence of ATP, can be visualized by native gel electrophoresis (Konarska and Sharp, 1986). We tested the influence of hnRNP A1 on complex A3′ formation on UVtat (Figure 7C). In non-depleted NE only complex H formation could be observed (Figure 7C, lane 1). Removal of hnRNP A1 led to efficient complex A3′ formation (Figure 7C, lane 2) and reconstituting the reaction with recombinant hnRNP A1, in an amount sufficient to inhibit tat splicing, restored the strong inhibition of complex A3′ formation, leading to formation of a more distinct complex H (Figure 7C, lane 3). The strong inhibition of complex A3′ formation by hnRNP A1 was largely unaffected by deletion of the ESS3 (Figure 7C, lanes 4–6), whereas scrambling of the ISS rendered complex A3′ formation essentially resistant to hnRNP A1 (Figure 7C, lanes 7–9). Mutation of both the ISS and ESS3 increased the resistance of complex A3′ formation to hnRNP A1 further (Figure 7C, lanes 10–12). In conclusion, hnRNP A1 inhibits complex A3′ formation on UVtat, primarily via the ISS. The ESS3 also mediates an inhibitory effect on A3′ formation although this effect is weaker and primarily seen when the ISS also is mutated.
The ISS and ESS3 inhibit the assembly of complex A
To investigate the effect of the ISS and ESS3 on spliceosome assembly, native electrophoresis was performed of the splicing complexes formed on chimeric pre-mRNAs (Figure 8). In non-depleted extract there was only a weak spliceosome assembly activity on PIPtat (lane 1). In contrast, PIPtat-sA1Δ4 assembled efficiently into complex A and B (lane 5). As expected, spliceosome assembly in U2AF-supplemented oligo(dT)ΔNE that lacks hnRNP A1 occurred efficiently on both chimeric pre-mRNAs (lanes 3 and 7). However, when recombinant hnRNP A1 was added to the reaction it led to an almost complete inhibition of complex A, B and C assembly on PIPtat (lane 4), while complex formation on PIPtat-sA1Δ4 was largely unaffected by this addition (lane 8). Complex formation on PIPtat-sA1 was also tested in this assay and found to be intermediate between PIPtat and PIPtat-sA1Δ4 (data not shown). Taken together, this shows that the effect of the ISS and ESS3 is to inhibit spliceosome assembly before or at the step of complex A assembly.

Fig. 8. Native gel electrophoresis analysing the assembly of spliceosomes on tat pre-mRNA-derived constructs in response to hnRNP A1 (15 ng/µl) and inactivation of the ISS and ESS3 elements. The pre-mRNAs, PIPtat, PIPtat-sA1Δ4 and, as control, PIP7.A, were incubated for 20 min under the indicated conditions before analysing the complexes by native polyacrylamide gel electrophoresis. The positions of complexes H, A, B and C are indicated.
Discussion
We have analysed the splicing of the second tat intron in HIV-1, which corresponds to the joining of SD4 and SA7. This process is relatively inefficient both in vivo and in vitro, mainly due to inefficient 3′ splice site (SA7) utilization. To obtain moderate tat splicing activity in vitro, an enhancer (ESE3) in the 3′ exon (third tat exon), as well as elevated concentrations of SF2/ASF, is required (Krainer et al., 1990; Fu, 1993; Amendt et al., 1995; Dyhr-Mikkelsen and Kjems, 1995; Staffa and Cochrane, 1995; Mayeda et al., 1999; Tange and Kjems, 2001). The inefficiency of splicing is not based solely on intrinsic weakness of the 3′ splice site (SA7), but also originates from negative regulation by several cis-acting inhibitory signals located both upstream and downstream of the splice site. When these inhibitory elements were mutated or deleted, splicing occurred without addition of SF2/ASF. At least three regions contribute to the negative regulation of the 3′ splice site: a novel ISS element covering nucleotides –22 to –44 upstream of the 3′ splice site (this study), the previously described ESS3 in the third tat exon (Amendt et al., 1995; Staffa and Cochrane, 1995; Tange and Kjems, 2001) and a less well defined ESS that overlaps with the ESE3 (Tange and Kjems, 2001).
By a depletion–reconstitution procedure we have identified hnRNP A1 as a potent inhibitor of tat splicing and as a mediator of the inhibitory effect of the ISS and ESS3. Mutating these elements rendered splicing of a tat-derived chimeric pre-mRNA insensitive to hnRNP A1 addition. The ISS and ESS3 contain four and one motifs, respectively, which closely resemble the hnRNP A1 high-affinity binding site UAGGG(A/U) (Burd and Dreyfuss, 1994) (Figure 1C), and gel-shift analysis demonstrated that hnRNP A1 binds specifically to these sites. Deletion of these sequences, individually or in combinations, confirmed that the binding motifs coincided with functional silencing activity, suggesting that direct binding of hnRNP A1 to the ISS and ESS3 is a prerequisite for inhibition of splicing. The splicing of several other pre-mRNAs, including PIP7.A and ESE-dependent pre-mRNAs, such as IgM and dsx, was largely insensitive to hnRNP A1 addition, showing that the inhibition of tat splicing is specific (data not shown).
hnRNP A1 has been implicated in regulation of splicing in several systems (see Introduction). The mechanism of action in most of these systems has not been resolved, but several lines of evidence suggest that hnRNP A1 may inhibit the association of basal splicing factors with pre-mRNA and/or counteract the positive effects of other splicing factors, especially SR proteins. For the ISS reported here, we suggest a steric blocking model (Figure 9). The positional overlap between the ISS element and the branch point –26, as well as the anchoring sites for U2 snRNP at both branch points –26 and –16 (T.Ø.Tange, C.K.Damgaard and J.Kjems, in preparation), suggest that hnRNP A1 binding physically blocks the association of branch point binding factors such as SF1/mBBP and/or U2 snRNP (Gozani et al., 1996; Berglund et al., 1997). Consistent with this view we observed that hnRNP A1 blocked U2 snRNP binding to the branch point in an ISS-dependent manner. This model bears some resemblance to the mechanism suggested for repression of the adenovirus late 3′ splice site IIIa where SR proteins bound to a purine-rich intronic element (3RE) near the 3′ splice site IIIa interfere with U2 snRNP binding, possibly by steric blocking (Kanopka et al., 1996). Notably, hnRNP A1 did not affect SF2/ASF-mediated stabilization of U2AF65 binding, suggesting that hnRNP A1 and SF2/ASF can function at different steps during spliceosome assembly on the tat pre-mRNA.

Fig. 9. A putative model for regulating splicing of the 3′ splice site in tat pre-mRNA. Binding of hnRNP A1 (A1) to the ISS may physically block the association of factors with the branch point sequences, e.g. SF1/mBBP and/or U2 snRNP, but without affecting U2AF65 binding to the polypyrimidine tract. SF2/ASF binds the ESE3 and stimulates binding of U2AF65 while the ESS3 has a negative effect on U2AF65 binding (Tange and Kjems, 2001). In addition, our data imply that ESS3 also affects spliceosome assembly after U2AF binding via an hnRNP A1-dependent mechanism.
It has been shown that hnRNP A1, and the related proteins hnRNP A1b, A2 and B1, bind HIV-1 ESS2 and repress splicing of the first tat intron (SD1 to SA3) (Caputi et al., 1999) and heterologous introns (Del Gatto-Konczak et al., 1999). Several lines of evidence suggest that ESS3 functions similarly: (i) the sequences of ESS2 and ESS3 are homologous (Amendt et al., 1995); (ii) hnRNP A1 binds to both ESS elements (Caputi et al., 1999; this study); and (iii) both ESS elements appear to inhibit spliceosome assembly before or at the step of U2 snRNP binding (Amendt et al., 1995; Si et al., 1998; Tange and Kjems, 2001; this study). Our spliceosome assembly data show that the ESS3 has a significantly weaker inhibitory effect on U2 snRNP binding than the ISS and due to the distance between the ESS3 and the tat branch points its mechanism is unlikely to be a direct physical block of U2 snRNP binding as suggested for the ISS. One possibility is that the ESS3 indirectly inhibits the association of U2AF65 with the tat polypyrimidine tract by counteracting the stimulatory effect of the upstream ESE3 on U2AF65 binding (Tange and Kjems, 2001). Additionally, the ESS3 probably has a negative effect on spliceosome assembly at a post-U2AF65 binding step (U2 snRNP binding), as suggested by its ability to function in the absence of ESE3 in a heterologous context (Si et al., 1998; Tange and Kjems, 2001). Although the ISS and ESS3 can function independently as silencer elements, our results indicate that mutation of both elements has a positive effect on splicing that is more than additive. Since hnRNP A1 has been shown to dimerize (Blanchette and Chabot, 1999), it is possible that hnRNP A1 associated with the ESS3 can stabilize the binding of hnRNP A1 to the ISS through dimerization.
While studying splicing of tat, we discovered that splicing of tat pre-mRNA was stimulated in NE depleted for both hnRNP A1 and U2AF. It is unlikely that the remaining ∼1% U2AF in the depleted NE is enough to support splicing of the tat intron because the optimized PIP7.A pre-mRNA and the ESE-dependent pre-mRNAs, IgM and dsxPRE, exhibited negligible levels of splicing under these conditions (data not shown). Instead, the ESE3 element and possibly also elements near the 5′ splice site may activate splicing via a U2AF-independent pathway that exists in parallel to the normal U2AF-dependent pathway, which is also dependent on ESE3.
HIV-1 virus is faced with the problem that many of its mRNAs harbour introns, which act as nuclear retention signals. To overrule this retention mechanism the virus expresses the Rev protein, which binds to the Rev response element (RRE) in the tat intron and activates nuclear export of viral RNA. It has been shown that hnRNP A1 binds the instability element (INS) in the p17gag gene and stimulates the Rev export function in a co-operative fashion (Najera et al., 1999). It is therefore tempting to speculate that the ESS2, ISS and ESS3 elements also play a role in the Rev-mediated export of unspliced and partially spliced HIV-1 mRNA at late stages of infection by recruiting hnRNP A1 as a nuclear export factor.
Materials and methods
Constructs
The following plasmids have been described previously: pPIP7.A (Jamison et al., 1992), pTAT4 (Dyhr-Mikkelsen and Kjems, 1995), pIgM (Watakabe et al., 1993), pdsxPRE and pdsxΔE (Lynch and Maniatis, 1995), GST–hnRNP A1 (Blanchette and Chabot, 1999) and His-tagged SF2/ASF (Ge et al., 1991). The pBS-tat, which covers the HIV-1 isolate HXB3 (DDBJ/EMBL/GenBank accession No. M14100) nt 5811–6158 and 8141–8485 (DDBJ/EMBL/GenBank accession No. K03455; HXB2 numbering), was generated by PCR using pTAT4 as template and primers tagged with EcoRI and HindIII sites. The resulting EcoRI–HindIII-cleaved fragment was inserted between the EcoRI and HindIII sites in the multiple cloning site of the pBS+ vector (Stratagene). This construct was used as parental plasmid for constructing point mutation/deletion mutants: pBS-tatΔ4, pBS-tat-sA1, pBS-tat-sA1Δ4, pBS-tatΔA1, pBS-tatΔA1Δ4, using PCR and standard cloning techniques. (Sambrook et al., 1989; see Figure 1 for sequences of mutagenized regions). To generate pBS-tat-dsx-exon, a fragment, covering the first 65 nt of the Drosophila double sex gene (dsx) exon 4, was made by PCR using pdsxΔE as template and primers tagged with AvaI and HindIII. The fragment was inserted between the AvaI (HIV-1 nt 8393) and HindIII sites of pBS-tat. To construct pBS-UVtat, a PCR fragment, covering the last 65 nt of the tat intron and the first 108 nt of the third tat exon, was generated with tags containing EcoRI and HindIII sites and inserted between the EcoRI and HindIII sites of pBS+. pBS-UVtat-exon was constructed by inserting a PCR fragment tagged with HindIII and BamHI sites, and covering the first 101 nt of the third tat exon, between the HindIII and BamHI sites of pBS+. pUVPIP was constructed by deleting the sequence between the EcoRI and XhoI sites in pPIP7.A by fill-in blunt-end ligation. The PIP7.A/tat chimeric constructs pPIPtat, pPIPtatΔ4, pPIPtat-sA1, pPIPtat-sA1Δ4, pPIPtatΔA1 and pPIPtatΔA1Δ4 were made by replacing the SalI and HindIII fragment in pPIP7.A with PCR fragments tagged with SalI and HindIII sites, and covering the last 65 nt of the tat intron and the first 108 nt of the third tat exon without and with point mutations/deletions. All constructs were verified by sequencing.
Protein expression and U2AF-depletion
His-tagged SF2/ASF was expressed and purified as described in Tange and Kjems (2001). SF2/ASF was dialysed against SF2/ASF buffer [20 mM HEPES–KOH pH 7.9, 0.5 M guanidine, 42 mM (NH4)2SO4, 15% glycerol, 0.2 mM EDTA, 0.5 mM dithiothreitol (DTT)] and stored at –80°C. GST–hnRNP A1 was expressed and purified essentially as described in Blanchette and Chabot (1999) and kept in GST–hnRNP A1 buffer (20 mM HEPES–KOH pH 7.9, 150 mM KCl, 20% glycerol, 1 mM DTT, 0.2 mM EDTA). Protein concentrations were measured using a Bradford protein assay kit (Bio-Rad) and/or estimated from Coomassie Blue-stained SDS–polyacrylamide gels. Oligo(dT) depletion was performed as described in Valcárcel et al. (1997) and immunodepletion of U2AF from NE was performed as described in Gama-Carvalho et al. (1997). Total protein concentration was measured using a Bradford protein assay kit (Bio-Rad). Generally, an ∼10 and ∼30% loss of total protein concentration was observed in the oligo(dT) depletion and immunodepletion, respectively, which was adjusted for in the assays.
In vitro transcription and splicing
Internally labelled RNAs were prepared by standard in vitro run-off transcription with [α-32P]UTP (Amersham Pharmacia) as described in Tange and Kjems (2001). All templates for tat-, PIPtat- and UVtat-derived transcripts were linearized with HindIII or inside the HIV-1 sequence with AvaI or Sau3A (denoted with a –AvaI and –Sau3A suffix), and transcription was with T7 RNA polymerase except that the template for UVtat-exon was linearized with BamHI and T3 RNA polymerase (USB) was used for transcription.
In vitro splicing was performed in 10 µl reactions, except for reactions shown in Figures 6A and 7A and B, which were in 25 µl volume, and as described in Tange and Kjems (2001). Splicing reactions were prepared by mixing: 3 µl oligo(dT)ΔNE or NE, 1 µl buffer D or U2AF, 0.5 µl SF2/ASF buffer, 0.5 µl H2O, 1 µl GST–hnRNP A1 buffer or GST–hnRNP A1, 1 µl 10× splice buffer [20–25 mM MgCl2, 5 mM ATP, 200 mM creatine phosphate, 3.5 mM DTT, 3.2 U/µl RNasin (Promega)], 2 µl 15% polyvinyl alcohol, 1 µl labelled pre-mRNA (5 fmol). Reactions with tat-derived pre-mRNAs contained 2.5 mM MgCl2, while reactions with PIP7.A and PIPtat-derived pre-mRNAs contained 2 mM MgCl2. The reactions were incubated at 30°C for 2 h followed by phenol/chloroform extraction, precipitation and gel electrophoresis.
Western blotting
Proteins were separated on a 10% SDS–polyacrylamide gel and blotted onto nitrocellulose filter using a semidry blotter apparatus (Bio-Rad). The primary antibody was monoclonal αMC3 (1:1000) for U2AF65 detection and rabbit polyclonal αA1 (1:100) for hnRNP A1 detection. The secondary antibody was horseradish peroxidase-coupled, anti-mouse IgG (1:5000; Amersham Pharmacia) for αMC3 detection, and peroxidase-coupled, anti-rabbit IgG (1:5000; Amersham Pharmacia) for αA1 detection. The secondary antibodies were detected by enhanced chemiluminescence (Amersham Pharmacia).
Gel-shift and UV-crosslinking assays
RNA–hnRNP A1 complexes were prepared by mixing 1 µl (0–300 ng) GST–hnRNP A1 in GST–hnRNP A1 buffer, 5 µl 2× binding buffer (20 mM HEPES–KOH pH 7.6, 200 mM KCl, 4 mM MgCl2, 1 mM EDTA, 2 mM DTT, 20% glycerol, 100 ng/µl Escherichia coli bulk tRNA), 0.1 µl RNasin (40 U/µl), 2.9 µl H2O and 1 µl labelled RNA (5 fmol). The reactions were mixed and incubated for 15 min at room temperature and run on a native 4% polyacrylamide gel containing 1× TBE.
To investigate spliceosome assembly, splicing reactions with 5 fmol PIPtat-derived pre-mRNAs or UVtat-derived RNAs were incubated for 20–30 min at 30°C. The reactions were stopped with heparin (5 µg/µl final concentration; Sigma). An aliquot of each reaction was separated on a native 4% acrylamide:bisacrylamide (80:1) and 0.5% agarose gel containing 50 mM Tris base–50 mM glycine.
UV-crosslinking was performed as described in Tange and Kjems (2001).
Acknowledgments
Acknowledgements
We are grateful to the following people for kindly providing plasmid constructs and antibodies: Jim Manley for SF2/ASF plasmid, Benoit Chabot for hnRNP A1 plasmid and Silvano Riva for αA1 antibody. We also thank Rita Rosendahl for excellent technical assistance and Jakob Nilsson and Mette K.Lund for critical reading of the manuscript. The work was supported in part by grants from the Danish Medical and Natural Research Councils, and the Karen Elise Jensen Foundation NOVO’s Found, the Danish AIDS Foundation and the Carlsberg Foundation. T.Ø.T. and C.K.D. were supported by Århus University and T.Ø.T. by EMBO.
References
- Amendt B.A., Hesslein,D., Chang,L.J. and Stoltzfus,C.M. (1994) Presence of negative and positive cis-acting RNA splicing elements within and flanking the first tat coding exon of human immunodeficiency virus type 1. Mol. Cell. Biol., 14, 3960–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendt B.A., Si,Z.H. and Stoltzfus,C.M. (1995) Presence of exon splicing silencers within human immunodeficiency virus type 1 tat exon 2 and tat-rev exon 3: evidence for inhibition mediated by cellular factors. Mol. Cell. Biol., 15, 4606–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J.A., Chua,K., Abovich,N., Reed,R. and Rosbash,M. (1997) The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell, 89, 781–787. [DOI] [PubMed] [Google Scholar]
- Blanchette M. and Chabot,B. (1999) Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J., 18, 1939–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe B.J. (2000) Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci., 25, 106–110. [DOI] [PubMed] [Google Scholar]
- Burd C.G. and Dreyfuss,G. (1994) RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J., 13, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C.B., Tuschl,T. and Sharp,P.A. (1999) Splicing of precursors to mRNAs by the spliceosome. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World. Cold Spring Habor Laboratory Press, Cold Spring Harbor, NY, pp. 525–560.
- Caputi M., Mayeda,A., Krainer,A.R. and Zahler,A.M. (1999) hnRNP A/B proteins are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J., 18, 4060–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.D., Kobayashi,R. and Helfman,D.M. (1999) Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat β-tropomyosin gene. Genes Dev., 13, 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gatto F. and Breathnach,R. (1995) Exon and intron sequences, respectively, repress and activate splicing of a fibroblast growth factor receptor 2 alternative exon. Mol. Cell. Biol., 15, 4825–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gatto-Konczak F., Olive,M., Gesnel,M.C. and Breathnach,R. (1999) hnRNP A1 recruited to an exon in vivo can function as an exon splicing silencer. Mol. Cell. Biol., 19, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyhr-Mikkelsen H. and Kjems,J. (1995) Inefficient spliceosome assembly and abnormal branch site selection in splicing of an HIV-1 transcript in vitro. J. Biol. Chem., 270, 24060–24066. [DOI] [PubMed] [Google Scholar]
- Eperon I.C., Makarova,O.V., Mayeda,A., Munroe,S.H., Caceres,J.F., Hayward,D.G. and Krainer,A.R. (2000) Selection of alternative 5′ splice sites: role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol. Cell. Biol., 20, 8303–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.D. (1993) Specific commitment of different pre-mRNAs to splicing by single SR proteins. Nature, 365, 82–85. [DOI] [PubMed] [Google Scholar]
- Fu X.D., Mayeda,A., Maniatis,T. and Krainer,A.R. (1992) General splicing factors SF2 and SC35 have equivalent activities in vitro and both affect alternative 5′ and 3′ splice site selection. Proc. Natl Acad. Sci. USA, 89, 11224–11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Carvalho M., Krauss,R.D., Chiang,L., Valcárcel,J., Green,M.R. and Carmo-Fonseca,M. (1997) Targeting of U2AF65 to sites of active splicing in the nucleus. J. Cell Biol., 137, 975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H., Zuo,P. and Manley,J.L. (1991) Primary structure of the human splicing factor ASF reveals similarities with Drosophila regulators. Cell, 66, 373–382. [DOI] [PubMed] [Google Scholar]
- Gontarek R.R., McNally,M.T. and Beemon,K. (1993) Mutation of an RSV intronic element abolishes both U11/U12 snRNP binding and negative regulation of splicing. Genes Dev., 7, 1926–1936. [DOI] [PubMed] [Google Scholar]
- Gooding C., Roberts,G.C. and Smith,C.W. (1998) Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated α-tropomyosin exon. RNA, 4, 85–100. [PMC free article] [PubMed] [Google Scholar]
- Gozani O., Feld,R. and Reed,R. (1996) Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev., 10, 233–243. [DOI] [PubMed] [Google Scholar]
- Gozani O., Potashkin,J. and Reed,R. (1998) A potential role for U2AF–SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol. Cell. Biol., 18, 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B.R. (2000) Sorting out the complexity of SR protein functions. RNA, 6, 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquenet S., Ropers,D., Bilodeau,P.S., Damier,L., Mougin,A., Stoltzfus,C.M. and Branlant,C. (2001) Conserved stem–loop structures in the HIV-1 RNA region containing the A3 3′ splice site and its cis-regulatory element: possible involvement in RNA splicing. Nucleic Acids Res., 29, 464–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison S.F., Crow,A. and Garcia-Blanco,M.A. (1992) The spliceosome assembly pathway in mammalian extracts. Mol. Cell. Biol., 12, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammler S. et al. (2001) The sequence complementarity between HIV-1 5′ splice site SD4 and U1 snRNA determines the steady-state level of an unstable env pre-mRNA. RNA, 7, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan J.L. and Green,M.R. (1999) Pre-mRNA splicing of IgM exons M1 and M2 is directed by a juxtaposed splicing enhancer and inhibitor. Genes Dev., 13, 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanopka A., Muhlemann,O. and Akusjarvi,G. (1996) Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature, 381, 535–538. [DOI] [PubMed] [Google Scholar]
- Konarska M.M. and Sharp,P.A. (1986) Electrophoretic separation of complexes involved in the splicing of precursors to mRNAs. Cell, 46, 845–855. [DOI] [PubMed] [Google Scholar]
- Krainer A.R., Conway,G.C. and Kozak,D. (1990) Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev., 4, 1158–1171. [DOI] [PubMed] [Google Scholar]
- Krecic A.M. and Swanson,M.S. (1999) hnRNP complexes: composition, structure and function. Curr. Opin. Cell Biol., 11, 363–371. [DOI] [PubMed] [Google Scholar]
- Liu H.X., Zhang,M. and Krainer,A.R. (1998) Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev., 12, 1998–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch K.W. and Maniatis,T. (1995) Synergistic interactions between two distinct elements of a regulated splicing enhancer. Genes Dev., 9, 284–293. [DOI] [PubMed] [Google Scholar]
- Mayeda A., Helfman,D.M. and Krainer,A.R. (1993) Modulation of exon skipping and inclusion by heterogeneous nuclear ribonucleoprotein A1 and pre-mRNA splicing factor SF2/ASF. Mol. Cell. Biol., 13, 2993–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A., Screaton,G.R., Chandler,S.D., Fu,X.D. and Krainer,A.R. (1999) Substrate specificities of SR proteins in constitutive splicing are determined by their RNA recognition motifs and composite pre-mRNA exonic elements. Mol. Cell. Biol., 19, 1853–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally M.T. and Beemon,K. (1992) Intronic sequences and 3′ splice sites control Rous sarcoma virus RNA splicing. J. Virol., 66, 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J. (2000) Intron recognition comes of AGe. Nature Struct. Biol., 7, 14–16. [DOI] [PubMed] [Google Scholar]
- Najera I., Krieg,M. and Karn,J. (1999) Synergistic stimulation of HIV-1 rev-dependent export of unspliced mRNA to the cytoplasm by hnRNP A1. J. Mol. Biol., 285, 1951–1964. [DOI] [PubMed] [Google Scholar]
- O’Reilly M.M., McNally,M.T. and Beemon,K.L. (1995) Two strong 5′ splice sites and competing, suboptimal 3′ splice sites involved in alternative splicing of human immunodeficiency virus type 1 RNA. Virology, 213, 373–385. [DOI] [PubMed] [Google Scholar]
- Ramchatesingh J., Zahler,A.M., Neugebauer,K.M., Roth,M.B. and Cooper,T.A. (1995) A subset of SR proteins activates splicing of the cardiac troponin T alternative exon by direct interactions with an exonic enhancer. Mol. Cell. Biol., 15, 4898–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schaal T.D. and Maniatis,T. (1999) Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein-specific enhancer sequences. Mol. Cell. Biol., 19, 1705–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Z., Amendt,B. and Stoltzfus,C. (1997) Splicing efficiency of human immunodeficiency virus type 1 tat RNA is determined by both a suboptimal 3′ splice site and a 10 nucleotide exon splicing silencer element located within tat exon 2. Nucleic Acids Res., 25, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Z.H., Rauch,D. and Stoltzfus,C.M. (1998) The exon splicing silencer in human immunodeficiency virus type 1 Tat exon 3 is bipartite and acts early in spliceosome assembly. Mol. Cell. Biol., 18, 5404–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.W. and Valcárcel,J. (2000) Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci., 25, 381–388. [DOI] [PubMed] [Google Scholar]
- Staffa A. and Cochrane,A. (1994) The tat/rev intron of human immunodeficiency virus type 1 is inefficiently spliced because of suboptimal signals in the 3′ splice site. J. Virol., 68, 3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staffa A. and Cochrane,A. (1995) Identification of positive and negative splicing regulatory elements within the terminal tat-rev exon of human immunodeficiency virus type 1. Mol. Cell. Biol., 15, 4597–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Mayeda,A., Hampson,R.K., Krainer,A.R. and Rottman,F.M. (1993) General splicing factor SF2/ASF promotes alternative splicing by binding to an exonic splicing enhancer. Genes Dev., 7, 2598–2608. [DOI] [PubMed] [Google Scholar]
- Tange T. and Kjems,J. (2001) SF2/ASF binds to a splicing enhancer in the third HIV-1 tat exon and stimulates U2AF binding independently of the RS domain. J. Mol. Biol., 312, 649–662. [DOI] [PubMed] [Google Scholar]
- Valcárcel J., Singh,R., Zamore,P.D. and Green,M.R. (1993) The protein Sex-lethal antagonizes the splicing factor U2AF to regulate alternative splicing of transformer pre-mRNA. Nature, 362, 171–175. [DOI] [PubMed] [Google Scholar]
- Valcárcel J., Gaur,R.K., Singh,R. and Green,M.R. (1996) Interaction of U2AF65 RS region with pre-mRNA of branch point and promotion base pairing with U2 snRNA. Science, 273, 1706–1709. [DOI] [PubMed] [Google Scholar]
- Valcárcel J., Martínez,C. and Green,M.R. (1997) Functional analysis of splicing factors and regulators. In Richter,J.D. (ed.), mRNA Formation and Function. Academic Press, San Diego, CA, pp. 31–53.
- Wang Z., Hoffmann,H.M. and Grabowski,P.J. (1995) Intrinsic U2AF binding is modulated by exon enhancer signals in parallel with changes in splicing activity. RNA, 1, 21–35. [PMC free article] [PubMed] [Google Scholar]
- Watakabe A., Tanaka,K. and Shimura,Y. (1993) The role of exon sequences in splice site selection. Genes Dev., 7, 407–418. [DOI] [PubMed] [Google Scholar]
- Wentz M.P., Moore,B.E., Cloyd,M.W., Berget,S.M. and Donehower, L.A. (1997) A naturally arising mutation of a potential silencer of exon splicing in human immunodeficiency virus type 1 induces dominant aberrant splicing and arrests virus production. J. Virol., 71, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.Y. and Maniatis,T. (1993) Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell, 75, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Zamore P.D. and Green,M.R. (1989) Identification, purification and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc. Natl Acad. Sci. USA, 86, 9243–9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo P. and Maniatis,T. (1996) The splicing factor U2AF35 mediates critical protein–protein interactions in constitutive and enhancer-dependent splicing. Genes Dev., 10, 1356–1368. [DOI] [PubMed] [Google Scholar]