

Abstract
The oncogenic latent membrane protein 1 (LMP1) of the Epstein–Barr virus recruits tumor necrosis factor-receptor (TNFR)-associated factors (TRAFs), the TNFR-associated death domain protein (TRADD) and JAK3 to induce intracellular signaling pathways. LMP1 serves as the prototype of a TRADD-binding receptor that transforms cells but does not induce apoptosis. Here we show that TRAF6 critically mediates LMP1 signaling to p38 mitogen-activated protein kinase (MAPK) via a MAPK kinase 6-dependent pathway. In addition, NF-κB but not c-Jun N-terminal kinase 1 (JNK1) induction by LMP1 involves TRAF6. The PxQxT motif of the LMP1 C-terminal activator region 1 (CTAR1) and tyrosine 384 of CTAR2 together are essential for full p38 MAPK activation and for TRAF6 recruitment to the LMP1 signaling complex. Dominant-negative TRADD blocks p38 MAPK activation by LMP1. The data suggest that entry of TRAF6 into the LMP1 complex is mediated by TRADD and TRAF2. In TRAF6-knockout fibroblasts, significant induction of p38 MAPK by LMP1 is dependent on the ectopic expression of TRAF6. We describe a novel role of TRAF6 as an essential signaling mediator of a transforming oncogene, downstream of TRADD and TRAF2.
Keywords: LMP1/p38 MAPK/signal transduction/TRADD/TRAF6
Introduction
The latent membrane protein 1 (LMP1) is the primary oncogene of the Epstein–Barr virus (EBV), a γ herpes virus classified as a human DNA tumor virus. Persistent latent infection with EBV is widespread in human populations, and can result in the development of malignancies such as Hodgkin’s lymphoma, Burkitt’s lymphoma, AIDS-associated lymphoproliferative diseases and nasopharyngeal carcinoma (for a review see Klein, 1994; Farrell, 1995; Kieff, 1996). LMP1 is critically involved in the effective immortalization and proliferation of B-cells latently infected by EBV (Kaye et al., 1993, 1995; Kilger et al., 1998). LMP1 has attracted great interest since it is the only EBV protein with the capacity to transform rodent fibroblast cell lines in culture (Wang et al., 1985; Baichwal and Sugden, 1988; Moorthy and Thorley-Lawson, 1993). Recently, LMP1 has been proven to constitute a genuine viral oncogene because it induces lymphomas in mice carrying LMP1 as a transgene (Kulwichit et al., 1998).
LMP1 mimics some of the phenotypic effects of an activated CD40 receptor in B cells such as induction of prolonged cell survival, proliferation and DNA synthesis (Zimber-Strobl et al., 1996; Kilger et al., 1998; Busch and Bishop, 1999; Uchida et al., 1999). However, in CD40-knockout mice LMP1 failed to systemically substitute for CD40 function (Uchida et al., 1999). Although LMP1 and CD40 share some properties, this observation demonstrates that profound differences in function exist between the two molecules. This conclusion is further supported by the observation that LMP1, but not CD40, signals through the tumor necrosis factor receptor (TNFR)-associated death domain (TRADD) adapter protein (Izumi and Kieff, 1997; Kieser et al., 1999). Emerging evidence suggests that LMP1 transforms cells by simultaneously antagonizing apoptosis, suppressing cellular senescence and mediating proliferative, growth factor-like effects (Henderson et al., 1991; Laherty et al., 1992; Kaye et al., 1995; Kenney et al., 1998; Yang et al., 2000).
LMP1 is a transmembrane protein of 386 amino acids consisting of a short N-terminal cytoplasmic domain of 24 amino acids, six transmembrane-spanning domains of 162 amino acids and a C-terminal cytoplasmic domain of 200 amino acids (Liebowitz et al., 1986; Kieff, 1996) (see Figure 8). LMP1 acts as a constitutively active receptor-like molecule independently of the binding of a ligand. The six transmembrane domains mediate spontaneous autoaggregation of LMP1 molecules within the plasma membrane, a prerequisite for LMP1 function (Floettmann and Rowe, 1997; Gires et al., 1997). Notably, clustering of LMP1 molecules is both necessary and sufficient for LMP1 activity. The short N-terminal domain of LMP1 is essential for correct insertion of LMP1 into the membrane, for association with cytoskeletal elements, and for the regulation of LMP1 degradation and stability (Liebowitz et al., 1987; Izumi et al., 1994; Aviel et al., 2000).

Fig. 8. Model of TRAF6 function in LMP1 signal transduction. For a detailed explanation refer to the text.
Two subdomains of the LMP1 C-terminus, the so-called C-terminal activator regions (CTARs) 1 and 2, are critical for growth transformation (Moorthy and Thorley-Lawson, 1993; Huen et al., 1995; Kaye et al., 1995; Mitchell and Sugden, 1995; Izumi et al., 1997). Extensive molecular analysis has revealed that CTARs serve as a platform for the binding of signaling molecules leading to the activation of intracellular signal transduction pathways (reviewed in Farrell, 1998). Both CTAR1 (amino acids 194–231) and CTAR2 (amino acids 332–386) participate in the induction of the transcription factor NF-κB (Huen et al., 1995; Mitchell and Sugden, 1995). The PxQxT motif of CTAR1 (P204xQ206xT208) provides a docking site for TNFR-associated factors (TRAFs) that can also be found in other TRAF-interacting receptors like CD30 or CD40 (reviewed in Arch et al., 1998; Inoue et al., 2000). So far, TRAF1, 2, 3 and 5 have been shown to bind to PxQxT of LMP1 (Mosialos et al., 1995; Devergne et al., 1996, 1998; Brodeur et al., 1997; Sandberg et al., 1997). CTAR1 signaling to NF-κB is mediated primarily by TRAF2 (Devergne et al., 1996; Kaye et al., 1996). Co-expression of TRAF1 further increases LMP1’s ability to induce NF-κB (Devergne et al., 1996). LMP1 is a strong NF-κB inducer in 293 human embryonic kidney (HEK 293) cells, in which TRAF1 expression is barely detectable, suggesting that TRAF1 is not essential for NF-κB signaling of LMP1 (Devergne et al., 1998). TRAF3 rather counteracts NF-κB activation by LMP1 (Devergne et al., 1996), and a role of TRAF5 in LMP1 signaling has not been shown so far. In contrast to CTAR1, CTAR2 does not bind TRAFs directly but triggers NF-κB through direct interaction with the TRADD protein (Izumi and Kieff, 1997; Kieser et al., 1999). TRADD serves as a bridging protein between LMP1 and TRAF2 transmitting the signal to the NF-κB-inducing kinase (NIK)/I-κB kinase (IKK)/NF-κB pathway, where CTAR1 and CTAR2 signals to NF-κB converge (Sylla et al., 1998). Tyrosines 384 and 385 of CTAR2 are essential for TRADD binding (Izumi and Kieff, 1997). In contrast to its interaction with TNF-receptor 1 and other known TRADD-binding receptors, TRADD binding to LMP1 is independent of the TRADD death domain (Kieser et al., 1999). This is especially interesting because LMP1 is the only known receptor-like molecule signaling through TRADD that does not possess a death domain and that does not induce apoptosis (Izumi et al., 1999). Thus, the study of LMP1 signaling will shed new light on the functions of TNFR1- and TRADD-induced signaling pathways.
In contrast to NF-κB, activation of the c-Jun N-terminal kinase 1 (JNK1)/activator protein 1 (AP1) pathway by LMP1 is triggered solely at CTAR2 and appears to be independent of TRAF2 (Kieser et al., 1997, 1999). The JNK1-activating region of LMP1 comprises amino acids 379–384, a region that is identical to the NF-κB-inducing sequence of CTAR2 and that overlaps with the TRADD-binding domain of LMP1 (Floettmann and Rowe, 1997; Izumi and Kieff, 1997; Eliopoulos et al., 1999a; Kieser et al., 1999). In addition, receptor-interacting protein (RIP) binds to this motif directly, although its role in LMP1 function is not yet known (Izumi et al., 1999). The JAK3/STAT signaling cascade, known to be involved in the control of cell proliferation, is triggered by the CTAR3 domain, which is located between CTAR1 and CTAR2 (Gires et al., 1999).
Recently, it has been shown that the p38 mitogen-activated protein kinase (MAPK) pathway is also induced by LMP1. p38 MAPK mediates cytokine induction by LMP1 (Eliopoulos et al., 1999b; Vockerodt et al., 2001). TRAF2 is involved in p38 MAPK activation by LMP1, but signaling downstream of TRAF2 is unknown (Eliopoulos et al., 1999b). This led us to search for signaling molecules transmitting LMP1 signals to p38 MAPK. Here we show that TRAF6 is an essential and specific mediator of LMP1 signal transduction to p38 MAPK downstream of TRADD and TRAF2 by interacting with the active LMP1 signaling complex at the plasma membrane. Thus, our results provide new clues to the molecular and biological functions of TRADD and TRAF6 within the cell.
Results
LMP1 induces p38 MAPK through a TRAF6-dependent pathway
TRAF6 was a prime candidate for mediating LMP1 signaling to p38 MAPK. This working hypothesis was based on two facts: LMP1 is known to engage TRAF molecules for signaling (see Introduction) and TRAF6 effectively induces p38 MAPK upon overexpression (Baud et al., 1999). To investigate whether TRAF6 is involved in the signaling pathway from LMP1 to p38 MAPK, we tested the ability of a dominant-negative mutant of human TRAF6, TRAF6(300–524), to block LMP1 activation of p38 MAPK in transient transfection assays in HEK 293 cells. TRAF6(300–524) lacks the typical RING finger domain. With the exception of TRAF1, all known TRAF molecules contain an N-terminal RING finger, the deletion of which generates a TRAF mutant that does not mediate downstream signaling but still interacts with the known TRAF-binding proteins (Arch et al., 1998; Inoue et al., 2000). A hemagglutinin (HA)-tagged p38 MAPK (HA- p38 MAPK) was co-transfected together with LMP1 and TRAF6(300–524) into HEK 293 cells. Twenty-four hours post-transfection, HA-p38 MAPK was immunoprecipitated from cleared cell lysates and assayed for its activity in in vitro kinase assays using purified glutathione S-transferase (GST)-tagged ATF2 (GST–ATF2) as a substrate (Figure 1).


Fig. 1. Dominant-negative TRAF6 efficiently blocks LMP1 signaling to p38. (A) Transient p38 MAPK assays in HEK 293 cells. Cells were transfected with 1.0 µg of HA-p38 MAPK together with 1.0 µg of wild-type (wt) LMP1 or LMP1(Δ194–386) lacking the C-terminal signaling domain and 1.0 µg of wt TRAF6 or dominant-negative TRAF6(300–524) expression vectors, as indicated. Total amounts of transfected DNAs were adjusted to 3.0 µg using pcDNA3.1 empty vector. At 24 h post-transfection, non-radioactive HA-p38 MAPK immunocomplex kinase assays were performed. Top panels, HA-p38 MAPK activity. Immunoblot analysis (IB) of in vitro GST–ATF2 phosphorylation by the precipitated HA-p38 MAPK using a phospho (P)-ATF2-specific antibody. Second panels from top, immunoblot of the immunoprecipitated (IP) HA-p38 MAPK. Third panels from top, immunoblot analysis of TRAF6 expression using the rabbit H-274 antibody. One asterisk, wt TRAF6; two asterisks, TRAF6(300–524). Bottom panels, LMP1 expression. Apparent molecular weights are given in kDa. The left and right colums are derived from two separate and independent experiments. (B) Transient p38 MAPK assays in HEK 293 cells. Transfections and kinase assays were performed essentially as described in (A) with the following alterations. As indicated, 1.0 µg of pSV-LMP1:CD40 was co-transfected. In vitro HA-p38 MAPK assays were performed in the presence of [γ-32P]ATP. Top panel, autoradiograph of GST–ATF2 phosphorylation. Middle panel, phosphoimager quantitation of GST–ATF2 phosphorylation normalized to the immunoprecipitated HA-p38 MAPK. Bottom panel, precipitated HA-p38 MAPK.
As shown in Figure 1A, co-expression of TRAF6 (300–524) entirely blocked p38 MAPK induction by LMP1 (left column), whereas wild-type TRAF6 augmented LMP1-induced p38 MAPK activity (right column). LMP1Δ194–386 lacks the complete signaling C-terminus and therefore was incorporated into the experiment as the null control. LMP1Δ194–386 yielded identical p38 MAPK base levels as the empty pHEBO vector (data not shown). TRAF6 protein and LMP1 were expressed readily as shown by immunoblot analysis of the cleared cell lysates performed prior to the immunoprecipitation of HA-p38 MAPK. The LMP1Δ194–386 mutant cannot be detected by the anti-LMP1 antibody cocktail CS1-4 since all CS1-4 epitopes are located within the deleted C-terminus of LMP1 (Rowe et al., 1987). In the experiments shown in Figure 1A, in vitro phosphorylation of GST–ATF2 by the immunoprecipitated HA-p38 MAPK was detected by immunostaining of the kinase assay blots with an anti-phospho-ATF2 antibody. For phosphoimager-based quantitation of the TRAF6(300–524) effects on LMP1 signaling to p38 MAPK, radioactive in vitro kinase assays were performed in an additional experiment yielding essentially identical results (Figure 1B). The lower p38 MAPK induction levels in the radioactive kinase assay as compared with the non-radioactive experiment are due to the lower and therefore sub-optimal total ATP concentrations in the radioactive assays. LMP1 induced p38 MAPK by a factor of 2.2-fold in the radioactive assay. TRAF6(300–524) co-expression resulted in a reduction of p38 MAPK activity to base levels (0.8-fold). In addition, the LMP1:CD40 chimera, which signals like a constitutively active CD40 receptor (Gires et al., 1997; Kieser et al., 1999), was included in the experiment as a positive control. As expected, dominant-negative TRAF6 also interfered with p38 MAPK induction by LMP1:CD40 (Figure 1B). In summary, we concluded from these data that LMP1 signaling to p38 MAPK is critically mediated by the TRAF6 adapter molecule.
MKK6 mediates LMP1 signaling to p38 MAPK
Next we set out to identify components of the LMP1→p38 MAPK signaling pathway downstream of TRAF6. The MAPKK MKK6 phosphorylates and thereby activates p38 MAPK. MKK6 has already been shown to mediate p38 MAPK activation by several extracellular and intracellular stimuli (for a review see Nebreda and Porras, 2000). Therefore, MKK6 was an obvious candidate to also be involved in LMP1 signaling to p38 MAPK. To test this scenario, a Flag-tagged kinase-negative MKK6 mutant, Flag-MKK6(Ala) (Raingeaud et al., 1996; Hoffmeyer et al., 1999), was co-transfected with LMP1 and HA-p38 MAPK into HEK 293 cells and subsequently non-radioactive HA-p38 MAPK assays were performed (Figure 2). Flag-MKK6(Ala) interfered with LMP1 induction of p38 MAPK in a dominant-negative manner, demonstrating its involvement in LMP1 signaling to p38 MAPK. Flag-MKK6(Ala) co-expression suppressed LMP1-induced p38 MAPK activity by 50%. As determined by immunoblotting analysis, LMP1 and Flag-MKK6(Ala) were equally expressed and identical amounts of HA-p38 MAPK were immunoprecipitated from all cleared lysates (Figure 2, three lower panels). Our findings clearly show that LMP1 induces p38 MAPK via a TRAF6- and MKK6-dependent signaling pathway.

Fig. 2. MKK6 mediates p38 MAPK induction by LMP1. Transient p38 MAPK assays in HEK 293 cells. As indicated, cells were transfected with 1.0 µg of HA-p38 MAPK together with 0.5 µg of wild-type LMP1 or LMP1(Δ194–386) expression vectors and 1.0 µg of MKK6(Ala), a dominant-negative Flag-tagged MKK6 mutant. Total amounts of transfected DNAs were adjusted to 2.5 µg using carrier DNA. At 24 h post-transfection, non-radioactive HA-p38 MAPK immunocomplex kinase assays were performed. Top panel, HA-p38 MAPK activity. Second panel from top, quantitation of HA-p38 MAPK activity normalized to HA-p38 MAPK IP. Third panel from top, immunoprecipitated HA-p38 MAPK. Second panel from bottom, LMP1 expression. Bottom panel, Flag-MKK6(Ala) expression. One out of three independent experiments with identical results is shown.
The PxQxT motif and tyrosine 384 of LMP1 cooperate to activate p38 MAPK
The next step was to map the domains of the LMP1 molecule that are responsible for activation of p38 MAPK. Previous studies have shown that LMP1-triggered NF-κB activity is mediated by CTAR1 and CTAR2 via a TRAF-dependent pathway (see Introduction). Since p38 MAPK is also activated via TRAF molecules, we tested whether mutation of the TRAF-interaction motif PxQxT and/or mutation of the TRADD-binding tyrosine 384 would have an impact on LMP1’s ability to induce p38 MAPK. As shown in Figure 3, mutation of the PxQxT motif to AxAxA impaired p38 MAPK induction by LMP1. p38 MAPK induction by LMP1(PQT→AAA) was reduced to 56.8 ± 14.6% of wild-type LMP1 levels, normalized to the immunoprecipitated HA-p38 MAPK protein and to LMP1 expression. Mutation of the tyrosine residue at position 384 to glycine almost completely abrogated LMP1 induction of p38 MAPK. Residual p38 MAPK activity induced by LMP1(Y384G) was 7.0 ± 1.3% of wild-type LMP1. Mutation of both the PxQxT motif of CTAR1 and tyrosine 384 of CTAR2 resulted in a LMP1 molecule, LMP1(PQT→AAA/Y384G), that was unable to induce p38 MAPK. Apparently, the PxQxT motif of CTAR1 and tyrosine 384 of CTAR2 together are essential for full induction of p38 MAPK by LMP1, the TRADD interaction site being the predominant p38 MAPK activator region. This result is reminiscent of a recently published observation showing that physical interaction and cooperativity of both CTAR1 and CTAR2 are required for the full signaling capacity of LMP1 to NF-κB (Floettmann et al., 1998).

Fig. 3. The TRAF-interaction motif of CTAR1 and tyrosine 384 of CTAR2 cooperate to induce p38 MAPK. Transient HA-p38 MAPK kinase assays in HEK 293 cells. As indicated, 1.0 µg of HA-p38 MAPK was transfected together with 1.0 µg of LMP1, LMP1(PQT→AAA) mutated in CTAR1, LMP1(Y384G) mutated in CTAR2, LMP1(PQT→AAA/Y384G) double mutant expression vectors, or LMP1Δ194–386 lacking the complete C-terminal signaling domain as a negative control. At 24 h post-transfection, non-radioactive HA-p38 MAPK immunocomplex kinase assays were performed. Blots of one representative experiment out of four is shown. Top panel, immunoblot of GST–ATF2 in vitro phosphorylated by the precipitated HA-p38 MAPK. Second panel from top, immunoprecipitated HA-p38 MAPK. Third panel from top, LMP1 expression. Bar graph, p38 MAPK activation by LMP1 and LMP1 mutants given as a percentage relative to wild-type LMP1. HA-p38 MAPK induction by wild-type LMP1 was set to 100%. Data are mean p38 MAPK activities of four independent experiments normalized to the immunoprecipitated HA-p38 MAPK and to the expression of LMP1. In all assays, LMP1(PQT→AAA/Y384G) was completely inactive with respect to p38 MAPK activation.
TRAF6 is involved in NF-κB but not in JNK1 signaling of LMP1
We show here that TRAF6 is a critical mediator of p38 MAPK induction by LMP1. Similar to p38 MAPK, NF-κB signaling is also triggered by both the PxQxT and tyrosine 384 motifs of LMP1 (Floettmann and Rowe, 1997; Devergne et al., 1998). This led us to ask whether TRAF6 might also be involved in LMP1 signaling to NF-κB. The dominant-negative TRAF6(300–524) mutant was tested for its ability to block LMP1 activation of an NF-κB reporter construct in HEK 293 cells. As shown in Figure 4A, dominant-negative TRAF6 repressed LMP1 induction of NF-κB by 40%. Wild-type TRAF6 augmented NF-κB induction by LMP1 (data not shown). In accordance with previously published data (Floettmann and Rowe, 1997; Devergne et al., 1998), mutation of the PxQxT motif or the tyrosine 384 residue reduced LMP1’s capacity to activate NF-κB, whereas LMP1(PQT→AAA/Y384G) was completely inactive. Dominant-negative TRAF6 significantly blocked NF-κB activation by both domains, the effect on CTAR1 being more pronounced than on CTAR2. NF-κB activation by LMP1(Y384G) was reduced by 80%, whereas LMP1(PQT→AAA)-induced NF-κB activity was repressed by 56%. The effect of dominant-negative TRAF6 was stronger on either of the two single domain mutants than on wild-type LMP1. It has been suggested that CTAR1 and CTAR2 do physically interact and cooperate in function (Floettmann et al., 1998). Thus, it is very likely that molecules binding to CTAR1 and CTAR2 influence each other in the wild-type situation, e.g. stabilize the holo-complex. If one of the two domains is mutated, the complex of the remaining functional domain might become more sensitive to TRAF6(300–524). To demonstrate the specificity of the TRAF6(300–524) effect on LMP1 signaling to NF-κB, appropriate control experiments were performed (Figure 4B). As expected, TRAF6(300–524) interfered with LMP1:CD40 but not with NIK signaling to NF-κB. NIK is supposed to work downstream of TRAF molecules in the signaling cascade to NF-κB and, thus, should not be affected by dominant-negative TRAF6. These findings show that LMP1 signaling to NF-κB involves TRAF6. The quality of TRAF6 function in NF-κB signaling differs from its role in p38 MAPK activation. p38 MAPK signaling by LMP1 is primarily dependent on TRAF6 (Figure 1). In contrast, TRAF6 does not appear to be a key player in NF-κB signaling of LMP1, but rather acts as a modulator. Apparently, the mechanisms leading to NF-κB or p38 MAPK induction already differ at the level of the LMP1 complex.



Fig. 4. Dominant-negative TRAF6 interferes with NF-κB but not with JNK1 induction by LMP1. (A) TRAF6(300–524) expression partially inhibits NF-κB signaling triggered by both CTAR1 and CTAR2 of LMP1. Transient NF-κB luciferase reporter assays in HEK 293 cells. Transfected expression vectors: 0.5 µg of LMP1Δ194–386, wild-type LMP1, LMP1(PQT→AAA), LMP1(Y384G), or LMP1(PQT→AAA/Y384G), 1.5 µg of TRAF6(300–524) or pcDNA3.1 empty vector, as indicated. (B) Control experiment: TRAF6(300–524) blocks LMP1:CD40 but not NIK-induced NF-κB activity. Tranfections were performed as described in (A). pSV-LMP1:CD40 (0.5 µg) or pcDNA3-NIK (0.5 µg) were co-transfected. For (A) and (B), luciferase activities were corrected for transfection efficiencies. NF-κB activities (y-axis) are given as x-fold induction calculated versus the signaling-defect LMP1Δ194–386 mutant. LMP1Δ194–386 values were set to 1. Data are mean values of three independent experiments. (C) JNK1 activation by LMP1 is independent of TRAF6. Transient HA-JNK1 assays in HEK 293 cells. Transfections were performed as described in the legend to Figure 1 with 1.0 µg pSRα-HA-JNK1 transfected instead of pCMV-HA-p38. Top panel, HA-JNK1 activity. Autoradiograph of GST–c-Jun phosphorylation. Middle panel, phosphoimager quantitation of GST–c-Jun phosphorylation normalized to the immunoprecipitated HA-JNK1. Bottom panel, immunoprecipitated HA-JNK1.
Tyrosine 384 is also essential for LMP1 activation of JNK1 (Eliopoulos et al., 1999a; Kieser et al., 1999). Thus, we examined a potential role of TRAF6 in LMP1 signaling to JNK1. As shown in Figure 4C, wild-type TRAF6 induced JNK1 upon overexpression and cooperated with LMP1 and LMP1:CD40 to induce JNK1. However, TRAF6(300–524) did not affect JNK1 activation by LMP1 or LMP1:CD40 at all under conditions that were sufficient to fully block LMP1-induced p38 MAPK activity (cf. Figure 1). Since LMP1 induces p38 MAPK via a TRAF2-dependent pathway (Eliopoulos et al., 1999b), whereas JNK1 is activated independently of TRAF2 (Kieser et al., 1999), this result was to be expected and further demonstrates the specificity of the TRAF6 effect on p38 MAPK signaling. The p38 MAPK and JNK1 pathways triggered by LMP1 apparently bifurcate upstream of TRAF2 and TRAF6.
TRAF6 co-localizes with LMP1 complexes at the plasma membrane
In the functional studies presented in this paper, an essential role of TRAF6 in signal transduction of LMP1 has been demonstrated. It was then important to show that TRAF6 interacts with LMP1 signaling complexes within the cell. To allow in situ studies of LMP1 and TRAF6 co-localization, we constructed a fusion protein consisting of an HA-tagged LMP1 C-terminally fused to enhanced green fluorescent protein (HA-LMP1–EGFP). In this fusion protein, the C-terminus of LMP1 and EGFP are separated by a spacer of five alanines. HA-LMP1–EGFP was fully functional with respect to its signaling activity (Figure 5B). HA-LMP1–EGFP was transiently co-transfected together with TRAF6 into HEK 293 cells and subsequently the subcellular localization of both proteins was analyzed by fluorescence microscopy (Figure 5A). Confocal images were generated by digital deconvolution (see Materials and methods). HA-LMP1–EGFP was detected via its green autofluorescence (Figure 5A, a) and TRAF6 via immunofluorescence in red (b). As shown in (a), HA-LMP1–EGFP molecules form clearly visible patches (arrows) within the plasma membrane. These patches are a result of the two-dimensional oligomerization of LMP1 molecules within the membrane through intermolecular interactions between their transmembrane domains (Gires et al., 1997). The nucleus of the cell appears as a darker, non-stained zone marked with ‘n’. The green staining of the cytoplasma stems from intracellular HA-LMP1–EGFP which forms light green spots within the cell. Due to the longer exposure times that were necessary to detect HA-LMP1–EGFP patches within the plasma membrane, these intracellular spots had to be overexposed and appear as an artificial green stain filling the cytoplasma. Very recently, a similar pattern of intracellular LMP1–EGFP distribution has been described by another group of investigators (Kaykas et al., 2001). An explanation for the large amounts of intracellular HA-LMP1–EGFP accumulating over time seems to be a tendency of HA-LMP1–EGFP to partially precipitate within the cell, probably at the sites of protein sythesis. Figure 5A, b shows the TRAF6 staining of the same cell shown in (a). Because TRAF6 had been overexpressed, the cytoplasm is stained intense red. Notably, TRAF6 localized to patches in the plasma membrane (arrows) that were completely overlapping with the HA-LMP1–EGFP clusters shown in (a). The overlay of (a) and (b) in (c) confirmed the co-localization of both molecules at the plasma membrane. In LMP1-negative HEK 293 cells, TRAF6 did not display the clustered pattern at the plasma membrane (data not shown). Apparently, TRAF6 is recruited by HA-LMP1–EGFP molecules into HA-LMP1–EGFP clusters at the plasma membrane.

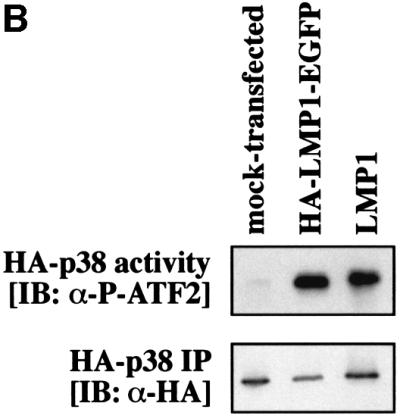


Fig. 5. TRAF6 interaction with the LMP1 signaling complex. (A) LMP1 and TRAF6 co-localize in clusters at the plasma membrane. HEK 293 cells were transfected with pcDNA3.1-TRAF6 and pSV-HA-LMP1–EGFP expressing an HA-LMP1 variant that is C-terminally fused to EGFP. The day after transfection, cells were processed for immunostaining, fluorescence microscopy and confocal imaging. Confocal images were generated from a single cell with HA-LMP1–EGFP autofluorescence in green (a), TRAF6 immunofluorescence in red (b) and the overlay of both images (c). Arrows indicate clusters of HA-LMP1–EGFP molecules within the plasma membrane (a). TRAF6 co-localizes with HA-LMP1–EGFP clusters (b and c). n, nucleus. Clustering of TRAF6 at the plasma membrane could not be detected in control transfections lacking HA-LMP1–EGFP expression (data not shown). (B) HA-LMP1–EGFP is fully functional with respect to p38 MAPK activation. Transient HA-p38 MAPK assays were performed in HEK 293 cells. Equal amounts of LMP1 and HA-LMP1–EGFP expression vectors were transfected. (C) TRAF6 rapidly translocates to induced clusters of a conditional NGFR:LMP1 chimera at the plasma membrane. HeLa cells were transiently transfected with 1 µg of pSV-NGFR:LMP1 expressing a conditional chimera of the low affinity p75 NGFR extracellular and transmembrane domains and the intracellular signaling domain of LMP1. Oligomerization and signaling activity of NGFR:LMP1 can be induced by crosslinking with specific antibodies. Twenty-four hours post-transfection, all cells were incubated with a mouse anti-NGFR antibody specific for the extracellular NGFR portion of the NGFR:LMP1 chimera. Subsequently, cells were either treated with an anti-mouse IgG secondary antibody for 15 min to crosslink NGFR:LMP1 (three lower panels), or left untreated as a negative control (two upper panels). The cells were fixed and immunofluorescence analysis was performed. Left column, NGFR:LMP1 immunofluorescence (red). Middle column, TRAF6 immunofluorescence (green). Right column, overlay of NGFR:LMP1 and TRAF6. The yellow/orange color of the induced NGFR:LMP1 clusters in the overlay images (three lower panels, right column) clearly demonstrate the co-localization of TRAF6 with activated NGFR:LMP1 at the plasma membrane. In contrast, unstimulated cells display a homogenous NGFR:LMP1 plasma membrane stain without detectable TRAF6 co-localization (upper two panels). (D) TRAF6 and TRAF6(300–524) are recruited to HA-LMP1 depending on the PxQxT motif and tyrosine 384 of LMP1. HEK 293 cells were transfected with pcDNA3.1-TRAF6 (upper graph) or pcDNA3.1-TRAF6(300–524) (lower graph) together with wild-type pSV-HA-LMP1, pSV-HA-LMP1(PQT→AAA/Y384G) or pcDNA3.1. Binding of TRAF6 to wild-type or mutated HA-LMP1 was assayed by a novel sandwich ELISA assay as described in the text. Wild-type TRAF6 was detected by the anti-TRAF6 H274 antibody, the specificity of which was shown by immunoblotting (inlay). m, mock-transfected HEK 293 cells; tr., TRAF6-transfected HEK 293 cells. Xpress-tagged TRAF6(300–524) was detected by an anti-Xpress-tag antibody. The cartoons to the right illustrate the set-up of the ELISA assays, the ‘X’ representing factors that mediate TRAF6 binding to HA-LMP1. POX, peroxidase.
To further substantiate our observations, we used a conditional chimera consisting of the human low-affinity p75 nerve growth factor-receptor (NGFR) extracellular and transmembrane domains fused to the C-terminal cytoplasmic signaling domain of LMP1, NGFR:LMP1 (Gires et al., 1997). The activity of the chimera can be triggered at will by crosslinking of NGFR:LMP1 molecules in the plasma membrane using specific antibodies directed against the extracellular part of the fusion protein. Sequential application of a mouse anti-NGFR primary antibody and an anti-mouse IgG secondary antibody to the cell culture medium results in the oligomerization and visible clustering of NGFR:LMP1 molecules within the plasma membrane and, hence, in the activation of NGFR:LMP1 signal transduction. Upon induction, NGFR: LMP1 signals like wild-type LMP1 due to its LMP1 signaling domain (Gires et al., 1997). In the non-crosslinked state, NGFR:LMP1 is signaling inactive. Here, we tested whether TRAF6 is located within membrane clusters of activated NGFR:LMP1 molecules in HeLa cells (Figure 5C). HeLa cells were transiently transfected with NGFR:LMP1. At 24 h post-transfection, an anti-NGFR antibody was applied to all cells for 1 h. After washing with medium, a subset of cells was treated with an anti-mouse IgG secondary antibody for another 15 min to crosslink the primary antibody leading to the oligomerization and activation of NGFR:LMP1, or left untreated as a negative control. Subsequent immunofluorescence analysis shows NGFR:LMP1 in red and endogenous TRAF6 in green. In some cells, in addition to a homogeneous cytoplasmic stain, TRAF6 shows a speckled distribution within the cytoplasma that did not correlate with NGFR:LMP1 transfection or antibody treatment of the cells and, thus, is not relevant for the conclusions drawn here from these experiments. The molecular basis for this distribution is unknown. As expected, NGFR:LMP1 was homogeneously distributed all over the plasma membrane in the unstimulated cells, and TRAF6 shows no detectable co-localization with NGFR:LMP1 (Figure 5C, two upper panels). Fifteen minutes after application of the crosslinking secondary antibody, the picture had changed completely (three lower panels). NGFR:LMP1 formed clearly visible clusters in the plasma membrane and, most notably, TRAF6 molecules could be found in the NGFR:LMP1 patches (compare TRAF6 stain and overlay of NGFR:LMP1 and TRAF6 stains). From these experiments, we concluded (i) that TRAF6 translocates from the cytoplasm to NGFR:LMP1 upon activation and (ii) that TRAF6 might be part of the active LMP1 signaling complex.
TRAF6 is part of the LMP1 signaling complex
The next question to be addressed was how does TRAF6 interact with the LMP1 signaling complex. The minimal TRAF6-binding motif of CD40 has been defined with the core sequence QxPxExxE/F (Pullen et al., 1998, 1999). No such motif can be found in LMP1, suggesting that TRAF6 does not bind directly to LMP1. Accordingly, TRAF6 did not co-precipitate with GST–LMP1 in pull-down assays from COS-7 or HEK 293 cell lysates (Ishida et al., 1996; Brodeur et al., 1997). We could not detect any significant TRAF6 binding to LMP1 in immunoprecipitations of HA-LMP1 from HEK 293 cells or in pull-down assays with GST–LMP1 and in vitro translated TRAF6 (data not shown). However, we found that TRAF6 has an important function in LMP1 signaling and that it co-localizes with LMP1 at the plasma membrane. Thus, it was tempting to speculate that TRAF6 binding to LMP1 might be indirect. In many cases, the detection of indirect protein–protein interactions is difficult in immunoprecipitation assays due to reduced stability of the protein holo-complex and the limited sensitivity of the detection systems. This led us to develop a two-component sandwich ELISA-based assay to analyze protein complexes and protein–protein interactions with higher sensitivity (Figure 5D). For this purpose, HEK 293 cells were transfected with the indicated expression vectors. Twenty-seven hours post-transfection, cells were lysed in a buffer containing 1% Triton X-100. As depicted in the cartoons shown in Figure 5D, HA-LMP1 or HA-LMP1(PQT→AAA/Y384) is trapped from the cell lysate by an anti-HA-tag primary antibody immobilized on 96-well ELISA plates. After washing of the plates, bound or co-bound proteins could be detected by specific secondary antibodies.
We asked whether TRAF6 could be found in protein complexes of HA-LMP1 bound to the immobilized anti-HA antibody 3F10. As shown in Figure 5D, upper panel, specific and significant binding of TRAF6 to HA-LMP1 could be detected in the sandwich ELISA assay. Using the anti-TRAF6 H274 secondary antibody, a strong signal in the ELISA was obtained upon co-transfection of TRAF6 with wild-type HA-LMP1 as compared with the co-transfection of TRAF6 with an empty vector. This result further supports our conclusions drawn from the co-localization studies and shows that TRAF6 is part of the LMP1 signaling complex. Mutation of both the PxQxT motif of CTAR1 and tyrosine 384 of CTAR2 resulted in the complete loss of TRAF6 recruitment to the complex. HA-LMP1 and HA-LMP1(PQT→AAA/Y384G) were expressed to identical levels when analyzed by immunoblotting (data not shown). PxQxT and tyrosine 384 are essential for the induction of the TRAF6-dependent p38 MAPK and NF-κB pathways. Mutation of these two sites also prevents the recruitment of TRAF6 to the LMP1 complex. This led us to conclude that TRAF6 most likely enters the LMP1 complex through factors interacting directly with the PxQxT motif and tyrosine 384.
To confirm this result, we repeated the experiment co-transfecting an Xpress-tagged TRAF6(300–524) instead of wild-type TRAF6 with essentially identical results (Figure 5D, lower panel). Moreover, the usage of the mouse anti-Xpress-tag instead of the rabbit anti-TRAF6 H274 antibody for the detection of TRAF6(300– 524) further excludes artefacts due to a non-specific binding of the antibodies. The result of this experiment also indicated that TRAF6 interaction with the LMP1 complex is independent of the TRAF6 RING finger domain.
Dominant-negative TRADD(1–194) blocks LMP1 signaling to p38 MAPK
Tyrosine 384 located within CTAR2 is the major p38 MAPK activator site of the LMP1 molecule. Tyrosine 384 is essential for direct TRADD binding to LMP1 (Izumi and Kieff, 1997). Thus, TRADD should work upstream of TRAF6 and TRAF2 in CTAR2 signaling to p38 MAPK. We have reported on a TRADD mutant lacking its death domain, TRADD(1–194), which binds directly to LMP1 and interferes with CTAR2 signaling to NF-κB in a dominant-negative fashion (Kieser et al., 1999). Figure 6 shows that myc epitope-tagged TRADD(1–194) efficiently interfered with LMP1 signaling to p38 MAPK. For the transient p38 MAPK assays, identical amounts of HA-p38 MAPK were immunoprecipitated and LMP1 and myc-TRADD(1–194) were expressed at identical levels (Figure 6, three lower panels). This result demonstrates that CTAR2 signals to p38 MAPK via a TRADD-dependent pathway. Since TRADD interacts directly with CTAR2, we concluded that TRAF6 works downstream of TRADD in p38 MAPK signaling of CTAR2.

Fig. 6. Dominant-negative TRADD(1–194) blocks p38 MAPK signaling by LMP1. Transient HA-p38 MAPK assays in HEK 293 cells. Cells were transfected with 1.0 µg of HA-p38 MAPK together with 1.0 µg of wild-type LMP1, LMP1(Δ194–386) or myc-TRADD(1–194) expression vectors, as indicated. pcDNA3-p35 (0.5 µg) was co-transfected to prevent apoptosis. Total amounts of transfected DNA were adjusted to 3.0 µg using pRK5 empty vector. Top panel, HA-p38 MAPK activity. Second panel from top, quantitation of HA-p38 MAPK activity normalized to the immunoprecipitated HA-p38 MAPK. Third panel from top, immunoprecipitated HA-p38 MAPK. Second panel from bottom, LMP1 expression. Bottom panel, immunoblot analysis of myc-TRADD(1–194) expression using the anti-myc tag antibody 9E10. One out of three independent experiments with identical results is shown.
In TRAF6–/– fibroblasts, ectopic TRAF6 expression is required for efficient p38 MAPK activation by LMP1
To provide additional evidence for the critical role of TRAF6 in LMP1 signaling to p38 MAPK, we tested the effect of ectopic TRAF6 expression on p38 MAPK induction by LMP1 in fibroblasts established from TRAF6-knockout mice (Lomaga et al., 1999). As shown in Figure 7, LMP1 alone induced p38 MAPK in TRAF6–/– cells only marginally. This was to be expected given that TRAF6 plays an important role in p38 MAPK signaling of LMP1. Residual and very weak p38 MAPK activation seen upon LMP1 transfection might reflect a partial rescue of LMP1 functions by other TRAFs. Transfection of TRAF6 alone also caused a very weak induction of p38 MAPK. This demonstrates that ectopic TRAF6 protein levels were comparable with physiological TRAF6 levels in that they do not massively induce p38 MAPK per se. For detection of TRAF6, the protein had to be immunoprecipitated from cell lysates prior to immunoblotting analysis (Figure 7, third panel from top). Notably, co-expression of both LMP1 and TRAF6 resulted in a very strong activation of p38 MAPK in the TRAF6–/– mouse fibroblasts. Restoration of TRAF6 expression to physiological levels apparently rescued LMP1 activation of p38 MAPK. In summary, this result also delivered additional and strong evidence for a pivotal role of TRAF6 in LMP1 signal transduction to p38 MAPK.

Fig. 7. Ectopic expression of TRAF6 enables LMP1 to strongly induce p38 MAPK in TRAF6-deficient mouse fibroblasts. TRAF6–/– mouse fibroblasts were transfected in 10 cm tissue culture plates with 3.3 µg of HA-p38 MAPK together with 9.9 µg of wild-type LMP1 or LMP1(Δ194–386) and 6.6 µg of TRAF6 expression vectors, as indicated. Transfected DNA levels were adjusted using 6.6 µg of pcDNA3.1 empty vector. At 24 h post-transfection, non-radioactive HA-p38 MAPK immunocomplex kinase assays were performed. Top panel, HA-p38 MAPK activity. Second panel from top, immunoprecipitated HA-p38 MAPK. Third panel from top, TRAF6 expression. TRAF6 was immunoprecipitated from cleared cell lysates using the anti-TRAF6 D10 antibody. Bottom panel, LMP1 expression. Immunoblot from total cell lysates.
Discussion
In this paper we have demonstrated that TRAF6 plays a critical role in signal transduction of the EBV oncoprotein LMP1. To date, TRAF6 has been shown to meditate signaling of TNF-R family and Toll/interleukin 1 (IL-1) receptor family members such as CD40, p75 NGFR or IL-1 receptor (for a review see Inoue et al., 2000). LMP1 is the first transforming protein shown to signal through TRAF6. Interestingly, in LMP1 signaling TRAF6 functions downstream of TRADD, an adapter protein that has mainly been implicated in the cytotoxic activity of TNF receptor 1 and death receptors (for a review see Yuan, 1997). Thus, the results presented here add a new quality to the spectrum of TRAF6 and TRADD functions within the cell and will help to understand the LMP1 signaling network resulting in immortalization and cell proliferation.
The involvement of TRAF6 is restricted to a subset of LMP1-triggered pathways demonstrating the specificity of TRAF6 function in LMP1 signaling. TRAF6 is the key mediator of p38 MAPK induction but rather acts as a modulator of the NF-κB pathway, e.g. by interacting with TRAF2. A dominant-negative TRAF6 mutant completely inhibited p38 MAPK, whereas the NF-κB pathway was only partially blocked. Most likely, other TRAF molecules like TRAF2 play the pivotal role in NF-κB induction by LMP1. TRAF2 interacts directly with NIK thereby transmitting LMP1 signals to the NF-κB pathway (Malinin et al., 1997; Sylla et al., 1998). A second possibility is that TRAF6 acts as the mediator of an alternative pathway leading from LMP1 to NF-κB: for example, in Il-1 receptor signaling TRAF6 induces NIK through the TGFβ-activating kinase (TAK1) (Ninomiya-Tsuji et al., 1999).
JNK1 induction by LMP1 is independent of TRAF6. This result is consistent with our previously published observation that the JNK1 and NF-κB pathways bifurcate upstream of TRAF2 at the level of TRADD or LMP1 itself (Kieser et al., 1999). The p38 MAPK and JNK1 pathways apparently also bifurcate upstream of TRAF2 and TRAF6. MKK6, which directly phosphorylates and thereby activates p38 MAPK, is mediating LMP1 upregulation of p38 MAPK downstream of TRAF6. The signaling molecules linking TRAF6 with MKK6 in LMP1 signaling are not known. Currently, we are carrying out experiments to identify the involved signaling mediators. In summary, it is evident that the mechanisms leading to the activation of p38 MAPK, JNK1 and NF-κB by LMP1 already differ at the receptor complex level. Figure 8 illustrates our current understanding of TRAF6 functions in LMP1 signaling.
We have shown that the TRAF-interaction motif PxQxT of CTAR1 and the tyrosine 384 residue located within CTAR2 together are essential for p38 MAPK induction, tyrosine 384 being the major p38 MAPK activating site of LMP1. This result is consistent with and refines data published recently (Eliopoulos et al., 1999b). LMP1 has no TRAF6-binding motif as it is known from CD40 (Pullen et al., 1998, 1999). TRAF6 most likely does not bind to LMP1 directly (data not shown; Ishida et al., 1996; Brodeur et al., 1997). However, we could detect TRAF6 in the LMP1 signaling complex by fluorescence microscopy and a sensitive sandwich ELISA technique. Mutation of the PxQxT motif and tyrosine 384 abolished TRAF6 recruitment to the complex. From the literature it is evident that TRAF2 binds directly to the PxQxT motif of LMP1 and TRADD to tyrosine 384 (Devergne et al., 1996, 1998; Brodeur et al., 1997; Izumi and Kieff, 1997; Sandberg et al., 1997; Kieser et al., 1999). Dominant-negative TRAF2 blocked p38 MAPK induction by LMP1 (Eliopoulos et al., 1999b). Our results presented in this paper also show that dominant-negative TRADD efficiently interferes with LMP1-triggered p38 MAPK activation. Taken together, these data strongly suggest that TRAF6 recruitment to the LMP1 signaling complex is mediated by TRAF2 and TRADD. Both adapter molecules must act upstream of TRAF6 in LMP1 signaling to p38 MAPK and NF-κB since they both block these pathways and bind directly to LMP1 via the critical PxQxT and tyrosine 384 sites.
How does TRAF6 interact with the components of the LMP1 complex and how is the holo-complex assembled? The most elegant way would be a direct interaction between TRAF6 and TRAF2. It is well documented that TRAF molecules form homo- and heterodimers by interaction via their C-terminal TRAF-C domains (Arch et al., 1998; Inoue et al., 2000). A direct interaction between TRAF6 and TRAF2 has already been shown in a yeast two-hybrid assay and in co-immunoprecipitations from mammalian cells (Cao et al., 1996). The PxQxT motif of LMP1 could recruit TRAF6 using TRAF2 as a bridging protein. As discussed, TRAF2 binds directly to PxQxT and acts upstream of TRAF6 in the signaling cascade to p38 MAPK. In the case of the major p38 MAPK inducing site, tyrosine 384, the situation is more complex. TRAFs do not bind to tyrosine 384 directly. We propose the following possible mechanism based upon data already published and presented herein. TRADD was shown to assemble the TNF-receptor 1 signaling complex by serving as a platform for the direct binding of, for example, TRAF2, RIP and FADD (Yuan, 1997). Notably, TRADD can simultaneously recruit TRAFs and FADD to TNF-receptor 1 (Yuan, 1997). In the case of LMP1, TRADD binds to LMP1 by a direct interaction of the TRADD N-terminus and tyrosine 384 of LMP1 (Izumi and Kieff, 1997; Kieser et al., 1999). TRADD, acting as a bridging protein, could then bind TRAF2/TRAF6 heteromers thereby recruiting TRAF6 to the complex. The LMP1 holo-complex could be further stabilized by interaction between TRAF molecules binding to CTAR1 and CTAR2.
p38 MAPK is preferentially activated by cellular stress and pro-inflammatory cytokines such as IL-1 and TNFα, and plays a key role in regulating apoptotic and inflammatory responses (for reviews see Nebreda and Porras, 2000; Ono and Han, 2000). More recently, it became evident that p38 MAPK is also involved in proliferative signaling as it is required for optimal CD40-induced B-cell proliferation (Craxton et al., 1998; Aicher et al., 1999). In the context of LMP1 function, which is the induction of proliferative and anti-apoptotic signals, p38 MAPK mediates induction of cytokine gene expression (Eliopoulos et al., 1999b; Vockerodt et al., 2001). The role of NF-κB in LMP1 signaling is to set survival signals within the cell by inducing anti-apoptotic genes (for a review see Cahir McFarland et al., 1999). TRAF6 is specifically involved in both pathways and, thus, in the biological functions they mediate. In summary, we have shown a novel role of TRAF6 as an important signaling mediator of LMP1. Apparently, TRADD and TRAF6 can act within one pathway mediating signaling of a transforming and anti-apoptotic oncogene. These results shed new light on the molecular and biological functions of both adapter proteins within the cell and might help to understand the oncogenic potential of LMP1.
Materials and methods
Expression plasmids and cloning
pSV-LMP1 (Kieser et al., 1997), pSV-LMP1(PQT→AAA), pSV-LMP1(PQT→AAA/Y384G), pSV-LMP1(Y384G), pSV-LMP1Δ194– 386, pRK-myc-TRADD(1–194) (Kieser et al., 1999), pSV-LMP1: CD40, pSV-NGFR:LMP1 (Gires et al., 1997), pSRα-HA-JNK1 (Minden et al., 1995), pCMV-HA-p38 (Baud et al., 1999), pcDNA3-NIK (Malinin et al., 1997), pcDNA3.1-Flag-MKK6(Ala) (Hoffmeyer et al., 1999) and pcDNA3-p35 (Seshagiri and Miller, 1997) have been described previously. pEGFP-C1 is commercially available (Clontech). All pSV-HA-LMP1 plasmids are based upon pHEBO (Sugden et al., 1985), in which N-terminally HA-tagged LMP1 variants are expressed from a subgenomic EBV fragment under the transcriptional control of the SV40 promoter/enhancer. Wild-type pSV-HA-LMP1 was cloned by assembly of fragments of HA-LMP1 single domain mutants. pSV-HA-LMP1(PQT→AAA/Y384G) was cloned by inserting a PCR fragment (primers: AK7, 5′-GGG GGG GGA GAG GGG CCC ACC GGG CCC GCG AC and AK10, 5′-CCC CCC CGG GCC TGA CTG GCC TTA GTC ATA GCC GCT TAG CTG AAC TGG GCC) of pSV-LMP1(PQT→AAA) via SfiI into 2068M (kind gift from M.Sandberg). pSV-HA-LMP1–EGFP was cloned by inserting a PCR fragment (primers: AK38, 5′-GGG GGG GGC TAA GCT ACT ATG ACG CTG CTG CTG CTG CTA TGG TGA GCA AGG GCG AGG AGC TG and AK40, 5′-CCC CCC CAG CTT AGC TTA TCT AGA TCC GGT GGA TCC CGG) of pEGFP-C1 via Bpu1102I into pSV-HA-LMP1. In the HA-LMP1–EGFP fusion protein, the C-terminus of LMP1 and the N-terminus of EGFP are separated by a spacer of five alanine residues. TRAF6 was cloned from mRNA extracted from human RAMOS B-cells by RT–PCR (5′ primer, 5′-AAG AAT TCG GCC TCC CCG CGC ACT AG and 3′ primer, 5′-GGC ACT GTT TTC TCG AGG TAG TTG TTT T). Primer sequences were derived from the published TRAF6 sequence (Cao et al., 1996). The PCR product was inserted into pBScriptIISK+ (Stratagene) via the EcoRI and XhoI restriction sites. To generate wild-type pcDNA3.1-TRAF6, the EcoRI–XhoI fragment was subcloned into pcDNA3.1 (Invitrogen). To obtain the dominant-negative TRAF6(300– 524) the sequence between XmnI and XhoI was excised and subcloned into pcDNA3.1/His containing an N-terminal Xpress-tag.
Cell culture methods
HEK 293 and HeLa cells were grown in full medium containing 10% fetal calf serum (FCS) (Gibco). TRAF6–/– mouse fibroblasts (Lomaga et al., 1999) were cultured in full medium containing 10% FCS and 100 nM sodium selenite. For transfections, cells were grown to subconfluence in 6-well plates (Nunc) or, where indicated, in 10 cm tissue culture plates (Nunc) for immunocomplex kinase assays, immunoblotting, reporter gene assays or fluorescence microscopy. HEK 293, HeLa and TRAF6–/– cells were transfected with the indicated amounts of DNA using the lipofectamine reagent (Gibco) according to the manufacturer’s protocol. Four microliters of lipofectamine were used per transfection of a 6-well and 26.5 µl per transfection of a 10 cm plate. Transfections for ELISAs were performed using the calcium phosphate precipitation method and are described separately. After transfection, cells were grown in medium containing 1% FCS for 24 h to downregulate serum-activated signaling pathways. Subsequently, immunocomplex kinase assays, immunoblotting, reporter gene assays or fluorescence microscopy were performed.
Immunocomplex kinase assays and immunoblotting
Immunocomplex kinase assays were performed as described (Kieser, 2001). Briefly, cells were treated as described in the figure legends and in cell culture methods. Subsequently, cells were lysed in TBST buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 0.5 mM β-glycerophosphate, 0.5 mM sodium pyrophosphate, 0.5 mM sodium fluoride, 0.5 mM sodium molybdate, 0.5 mM sodium orthovanadate). Cleared lysates were incubated overnight with the monoclonal rat anti-HA epitope anti body 3F10 (Roche), immobilized to GammaBind–Sepharose beads (Amersham-Pharmacia), to immunoprecipitate HA-p38 MAPK or HA-JNK1. For immunoprecipitation of TRAF6, cleared lysates were incubated with mouse anti-TRAF6 D10 antibody (Santa Cruz Biotechnology) bound to protein A–Sepharose beads (Sigma). Beads were washed twice with TBST. For radioactive kinase assays, beads were washed another two times with kinase reaction buffer I (20 mM Tris–HCl pH 7.4, 20 mM NaCl, 10 mM MgCl2, 1 µM DTT, 2 µM ATP). Radioactive in vitro kinase reactions to assay the activity of the immunoprecipitated HA-p38 MAPK or HA-JNK1 were performed in kinase reaction buffer I in the presence of 10 µCi [γ-32P]ATP (Amersham-Pharmacia) per reaction sample using purified GST–ATF2 or GST–c-Jun as substrates, respectively. For non-radioactive HA-p38 MAPK kinase assays, beads were washed another two times with kinase reaction buffer II (25 mM Tris–HCl pH 7.4, 10 mM MgCl2, 2 µM DTT, 200 µM ATP). In vitro kinase reactions were performed in kinase reaction buffer II using purified GST–ATF2 as a substrate. Kinase reactions or cell lysates were separated by SDS–PAGE and blotted onto Hybond-C membranes (Amersham-Pharmacia). Radioactive kinase reactions were analyzed by autoradiography and phosphoimager scanning, non-radioactive p38 MAPK assays were analyzed by immunostaining of kinase assay blots using the rabbit anti-phospho-ATF2(Thr 69/71) antibody (New England Biolabs) as a primary antibody. For quantitation, immunoblots were scanned and analyzed by densitometry. As indicated, the following primary antibodies were used for immunoblotting analysis: mouse anti-LMP1 CS1-4 (Dako), mouse anti-HA epitope 12CA5 (Roche), mouse anti-myc epitope 9E10 (Roche), rabbit anti-TRAF2 C20, rabbit anti-TRAF6 H274, goat anti-TRAF6 C20, rabbit anti-p38 MAPK H147 (all Santa Cruz Biotechnology) and mouse anti-Flag M2 (Sigma). Immunoblots were developed using peroxidase-coupled secondary antibodies (Dianova) and the ECL reagent (Amersham-Pharmacia).
Sandwich ELISA
HEK 293 cells were transfected in 3.5 cm dishes (TPP, Peske, Aindling, Germany) by the calcium phosphate precipitation method as described (Kingston, 1993). Briefly, cells were seeded at a density of 2.5 × 105 cells per dish, grown overnight to subconfluency and subsequently transfected with 2.5 µg of each indicated expression plasmid in a total volume of 2.5 ml. Twenty-seven hours post-transfection, cells were lysed on ice in Dulbecco’s phosphate-buffered saline containing 1% Triton X-100, 0.2% NaAzid, Aprotinin (Sigma) and Pefabloc (Roth). Lysates were cleared by centrifugation and immediately used for further analysis. For the HA-LMP1–TRAF6 complex ELISA, plastic-adsorbed rat anti-HA epitope 3F10 (Roche) as the primary antibody and rabbit anti-TRAF6 H274 as the secondary antibody were used. For the HA-LMP1– TRAF6(300–524) complex ELISA, mouse anti-Xpress (Invitrogen) was used as the secondary antibody for detection of Xpress-tagged TRAF6(300–524). Peroxidase-conjugated goat anti-rabbit Ig or mouse anti-rat Ig (Dianova) were used to develop the ELISA.
Fluorescence microscopy studies
HEK 293 cells were transfected as described above. Twenty-four hours post-transfection, cells were trypsinized and washed twice with Hanks’ buffered salts solution (HBSS) (Gibco). Cells were plated in HBSS onto poly-prep slides coated with poly-l-lysine (Sigma). After adherence, the cells were incubated for another 5 h in medium containing 1% FCS. Cells were fixed in 3% paraformaldehyde/PBS for 20 min. Subsequently, cells were permeabilized in 0.2% Triton X-100 for 5 min and blocked in 2% glycine/PBS pH 7.4 for 1 h and in 10% FCS/PBS for another 30 min. For TRAF6 immunofluorescence (Figure 5A), cells were incubated with the rabbit anti-TRAF6 H274 antibody as a primary antibody for 4 h and with Texas Red-coupled anti-rabbit IgG(H+L) (Dianova) as a secondary antibody for another 2 h. Between all incubation steps the cells were washed carefully using PBS. Subcellular localization of HA-LMP1– EGFP was analyzed by EGFP autofluorescence. For crosslinking of NGFR:LMP1 (Figure 5C), HeLa cells were transfected as described above and subsequently plated on glass slides. At 24 h post-transfection, cells were incubated with ∼1 µg/ml of an affinity-purified mouse anti-human NGFR antibody (ATCC) for 1 h and, where indicated, with 5 µg/ml of goat anti-mouse IgG+IgM(H+L) (Dianova) as a secondary antibody for another 15 min to crosslink NGFR:LMP1. Cells were fixed, permeabilized and blocked as described above. For NGFR:LMP1 immunofluorescence no additional primary antibody was applied. The anti-NGFR antibody that served for the stimulation was detected directly using Cy3-coupled goat anti-mouse IgG(H+L) (Dianova) as a secondary antibody. For TRAF6 detection, rabbit anti-TRAF6 H274 was used as a primary and FITC-coupled goat anti-rabbit IgG(H+L) (Dianova) as a secondary antibody. Fluorescence microscopy was performed using a Zeiss Axiovert 200 fluorescence microscope equipped with a high-resolution digital video camera (Hamamatsu). Red and green fluorescence were recorded separately using the respective filter sets. Confocal images were generated from three recorded layers per image applying the Openlab 2.1 deconvolution software (Improvision). Overlays of green and red fluorescence were generated from deconvolved single images using the Openlab 2.1 software.
Reporter assays
NF-κB reporter gene assays were performed using the 3× κB-Luc reporter plasmid (Mitchell and Sugden, 1995) as described (Kieser et al., 1999). Luciferase activities were standardized for transfection efficiencies by co-transfection of a β-Gal-reporter plasmid as described (Kieser et al., 1999).
Acknowledgments
Acknowledgements
We thank C.Federle and C.Schneider for expert technical assistance, Drs M.Karin, J.-H.Lin, S.Ludwig, L.K.Miller, M.Sandberg, B.Sugden, D.J.Templeton and D.Wallach for plasmids, and Dr W.Nagel for advice with immunofluorescence studies. This work was supported by grants from the Hermann-von-Helmholtz-Gesellschaft Deutscher Forschungs zentren to A.K. (HGF-Strategiefonds III: Viral Regulating Factors), from the Deutsche Forschungsgemeinschaft to H.E. (Gerhard Hess Programm and SFB 455) and to W.H. (HA135413 and SFB 455), by Public Health Service Grant CA20723, and by institutional grants. S.P. was supported by a fellowship from the Hanns Seidel Stiftung.
References
- Aicher A., Shu,G.L., Magaletti,D., Mulvania,T., Pezzutto,A., Craxton,A. and Clark,E.A. (1999) Differential role for p38 mitogen-activated protein kinase in regulating CD40-induced gene expression in dendritic cells and B cells. J. Immunol., 163, 5786–5795. [PubMed] [Google Scholar]
- Arch R.H., Gedrich,R.W. and Thompson,C.B. (1998) Tumor necrosis factor receptor-associated factors (TRAFs) — a family of adapter proteins that regulates life and death. Genes Dev., 12, 2821–2830. [DOI] [PubMed] [Google Scholar]
- Aviel S., Winberg,G., Massucci,M. and Ciechanover,A. (2000) Degradation of the Epstein–Barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway. Targeting via ubiquitination of the N-terminal residue. J. Biol. Chem., 275, 23491–23499. [DOI] [PubMed] [Google Scholar]
- Baichwal V.R. and Sugden,B. (1988) Transformation of Balb 3T3 cells by the BNLF-1 gene of Epstein–Barr virus. Oncogene, 2, 461–467. [PubMed] [Google Scholar]
- Baud V., Liu,Z.G., Bennett,B., Suzuki,N., Xia,Y. and Karin,M. (1999) Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev., 13, 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur S.R., Cheng,G., Baltimore,D. and Thorley-Lawson,D.A. (1997) Localization of the major NF-κB-activating site and the sole TRAF3 binding site of LMP-1 defines two distinct signaling motifs. J. Biol. Chem., 272, 19777–19784. [DOI] [PubMed] [Google Scholar]
- Busch L.K. and Bishop,G.A. (1999) The EBV transforming protein, latent membrane protein 1, mimics and cooperates with CD40 signaling in B lymphocytes. J. Immunol., 162, 2555–2561. [PubMed] [Google Scholar]
- Cahir McFarland E.D., Izumi,K.M. and Mosialos,G. (1999) Epstein–Barr virus transformation: involvement of latent membrane protein 1-mediated activation of NF-κB. Oncogene, 18, 6959–6964. [DOI] [PubMed] [Google Scholar]
- Cao Z., Xiong,J., Takeuchi,M., Kurama,T. and Goeddel,D.V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature, 383, 443–446. [DOI] [PubMed] [Google Scholar]
- Craxton A., Shu,G., Graves,J.D., Saklatvala,J., Krebs,E.G. and Clark,E.A. (1998) p38 MAPK is required for CD40-induced gene expression and proliferation in B lymphocytes. J. Immunol., 161, 3225–3236. [PubMed] [Google Scholar]
- Devergne O., Hatzivassiliou,E., Izumi,K.M., Kaye,K.M., Kleijnen,M.F., Kieff,E. and Mosialos,G. (1996) Association of TRAF1, TRAF2, and TRAF3 with an Epstein–Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-κB activation. Mol. Cell. Biol., 16, 7098–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devergne O., McFarland,E.C., Mosialos,G., Izumi,K.M., Ware,C.F. and Kieff,E. (1998) Role of the TRAF binding site and NF-κB activation in Epstein–Barr virus latent membrane protein 1-induced cell gene expression. J. Virol., 72, 7900–7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos A.G., Blake,S.M., Floettmann,J.E., Rowe,M. and Young,L.S. (1999a) Epstein–Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J. Virol., 73, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos A.G., Gallagher,N.J., Blake,S.M., Dawson,C.W. and Young,L.S. (1999b) Activation of the p38 mitogen-activated protein kinase pathway by Epstein–Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem., 274, 16085–16096. [DOI] [PubMed] [Google Scholar]
- Farrell P.J. (1995) Epstein–Barr virus immortalizing genes. Trends Microbiol., 3, 105–109. [DOI] [PubMed] [Google Scholar]
- Farrell P.J. (1998) Signal transduction from the Epstein–Barr virus LMP-1 transforming protein. Trends Microbiol., 6, 175–177. [DOI] [PubMed] [Google Scholar]
- Floettmann J.E. and Rowe,M. (1997) Epstein–Barr virus latent membrane protein-1 (LMP1) C-terminus activation region 2 (CTAR2) maps to the far C-terminus and requires oligomerisation for NF-κB activation. Oncogene, 15, 1851–1858. [DOI] [PubMed] [Google Scholar]
- Floettmann J.E., Eliopoulos,A.G., Jones,M., Young,L.S. and Rowe,M. (1998) Epstein–Barr virus latent membrane protein-1 (LMP1) signalling is distinct from CD40 and involves physical cooperation of its two C-terminus functional regions. Oncogene, 17, 2383–2392. [DOI] [PubMed] [Google Scholar]
- Gires O., Zimber-Strobl,U., Gonnella,R., Ueffing,M., Marschall,G., Zeidler,R., Pich,D. and Hammerschmidt,W. (1997) Latent membrane protein 1 of Epstein–Barr virus mimics a constitutively active receptor molecule. EMBO J., 16, 6131–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gires O. et al. (1999) Latent membrane protein 1 of Epstein–Barr virus interacts with JAK3 and activates STAT proteins. EMBO J., 18, 3064–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S., Rowe,M., Gregory,C., Croom-Carter,D., Wang,F., Longnecker,R., Kieff,E. and Rickinson,A. (1991) Induction of bcl-2 expression by Epstein–Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell, 65, 1107–1115. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer A., Grosse-Wilde,A., Flory,E., Neufeld,B., Kunz,M., Rapp,U.R. and Ludwig,S. (1999) Different mitogen-activated protein kinase signaling pathways cooperate to regulate tumor necrosis factor α gene expression in T lymphocytes. J. Biol. Chem., 274, 4319–4327. [DOI] [PubMed] [Google Scholar]
- Huen D.S., Henderson,S.A., Croom-Carter,D. and Rowe,M. (1995) The Epstein–Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene, 10, 549–560. [PubMed] [Google Scholar]
- Inoue J., Ishida,T., Tsukamoto,N., Kobayashi,N., Naito,A., Azuma,S. and Yamamoto,T. (2000) Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp. Cell Res., 254, 14–24. [DOI] [PubMed] [Google Scholar]
- Ishida T. et al. (1996) Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J. Biol. Chem., 271, 28745–28748. [DOI] [PubMed] [Google Scholar]
- Izumi K.M. and Kieff,E.D. (1997) The Epstein–Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-κB. Proc. Natl Acad. Sci. USA, 94, 12592–12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi K.M., Kaye,K.M. and Kieff,E.D. (1994) Epstein–Barr virus recombinant molecular genetic analysis of the LMP1 amino-terminal cytoplasmic domain reveals a probable structural role, with no component essential for primary B-lymphocyte growth transformation. J. Virol., 68, 4369–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi K.M., Kaye,K.M. and Kieff,E.D. (1997) The Epstein–Barr virus LMP1 amino acid sequence that engages tumor necrosis factor receptor associated factors is critical for primary B lymphocyte growth transformation. Proc. Natl Acad. Sci. USA, 94, 1447–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi K.M., McFarland,E.C., Ting,A.T., Riley,E.A., Seed,B. and Kieff,E.D. (1999) The Epstein–Barr virus oncoprotein latent membrane protein 1 engages the tumor necrosis factor receptor-associated proteins TRADD and receptor-interacting protein (RIP) but does not induce apoptosis or require RIP for NF-κB activation. Mol. Cell. Biol., 19, 5759–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye K.M., Izumi,K.M. and Kieff,E. (1993) Epstein–Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl Acad. Sci. USA, 90, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye K.M., Izumi,K.M., Mosialos,G. and Kieff,E. (1995) The Epstein–Barr virus LMP1 cytoplasmic carboxy terminus is essential for B-lymphocyte transformation; fibroblast cocultivation complements a critical function within the terminal 155 residues. J. Virol., 69, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye K.M., Devergne,O., Harada,J.N., Izumi,K.M., Yalamanchili,R., Kieff,E. and Mosialos,G. (1996) Tumor necrosis factor receptor associated factor 2 is a mediator of NF-κB activation by latent infection membrane protein 1, the Epstein–Barr virus transforming protein. Proc. Natl Acad. Sci. USA, 93, 11085–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaykas A., Worringer,K. and Sugden,B. (2001) CD40 and LMP-1 both signal from lipid rafts but LMP-1 assembles a distinct, more efficient signaling complex. EMBO J., 20, 2641–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney J.L., Guinness,M.E., Curiel,T. and Lacy,J. (1998) Antisense to the Epstein–Barr virus (EBV)-encoded latent membrane protein 1 (LMP-1) suppresses LMP-1 and bcl-2 expression and promotes apoptosis in EBV-immortalized B cells. Blood, 92, 1721–1727. [PubMed] [Google Scholar]
- Kieff E. (1996) Epstein–Barr virus and its replication. In Fields,B.N., Knipe,D.M., Howley,P.M., Chanock,R.M., Monath,T.P., Melnick,J.L., Roizman,B. and Straus,S.E. (eds), Virology. Lippincott-Raven, Philadelphia, PA, pp. 2343–2396.
- Kieser A. (2001) Assaying the activity of kinases regulated by LMP1. In Wilson,J.B. and May,G.W. (eds), Methods in Molecular Biology: Epstein–Barr Virus Protocols. Humana Press, Totowa, NJ, Vol. 174, pp. 325–336. [DOI] [PubMed]
- Kieser A., Kilger,E., Gires,O., Ueffing,M., Kolch,W. and Hammerschmidt,W. (1997) Epstein–Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J., 16, 6478–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser A., Kaiser,C. and Hammerschmidt,W. (1999) LMP1 signal transduction differs substantially from TNF receptor 1 signaling in the molecular functions of TRADD and TRAF2. EMBO J., 18, 2511–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilger E., Kieser,A., Baumann,M. and Hammerschmidt,W. (1998) Epstein–Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J., 17, 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston R.E. (1993) Calcium phosphate transfection. In Ausubel,F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (eds), Current Protocols in Molecular Biology. John Wiley & Sons, New York, NY, pp. 9.1.4–9.1.11.
- Klein G. (1994) Epstein–Barr virus strategy in normal and neoplastic B cells. Cell, 77, 791–793. [DOI] [PubMed] [Google Scholar]
- Kulwichit W., Edwards,R.H., Davenport,E.M., Baskar,J.F., Godfrey,V. and Raab-Traub,N. (1998) Expression of the Epstein–Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc. Natl Acad. Sci. USA, 95, 11963–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty C.D., Hu,H.M., Opipari,A.W., Wang,F. and Dixit,V.M. (1992) The Epstein–Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor κ B. J. Biol. Chem., 267, 24157–24160. [PubMed] [Google Scholar]
- Liebowitz D., Wang,D. and Kieff,E. (1986) Orientation and patching of the latent infection membrane protein encoded by Epstein–Barr virus. J. Virol., 58, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebowitz D., Kopan,R., Fuchs,E., Sample,J. and Kieff,E. (1987) An Epstein–Barr virus transforming protein associates with vimentin in lymphocytes. Mol. Cell. Biol., 7, 2299–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomaga M.A. et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev., 13, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin N.L., Boldin,M.P., Kovalenko,A.V. and Wallach,D. (1997) MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature, 385, 540–544. [DOI] [PubMed] [Google Scholar]
- Minden A., Lin,A., Claret,F.X., Abo,A. and Karin,M. (1995) Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell, 81, 1147–1157. [DOI] [PubMed] [Google Scholar]
- Mitchell T. and Sugden,B. (1995) Stimulation of NF-κB-mediated transcription by mutant derivatives of the latent membrane protein of Epstein–Barr virus. J. Virol., 69, 2968–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy R.K. and Thorley-Lawson,D.A. (1993) All three domains of the Epstein–Barr virus-encoded latent membrane protein LMP-1 are required for transformation of rat-1 fibroblasts. J. Virol., 67, 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosialos G., Birkenbach,M., Yalamanchili,R., VanArsdale,T., Ware,C. and Kieff,E. (1995) The Epstein–Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell, 80, 389–399. [DOI] [PubMed] [Google Scholar]
- Nebreda A.R. and Porras,A. (2000) p38 MAP kinases: beyond the stress response. Trends Biochem. Sci., 25, 257–260. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J., Kishimoto,K., Hiyama,A., Inoue,J., Cao,Z. and Matsumoto,K. (1999) The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature, 398, 252–256. [DOI] [PubMed] [Google Scholar]
- Ono K. and Han,J. (2000) The p38 signal transduction pathway: activation and function. Cell. Signal., 12, 1–13. [DOI] [PubMed] [Google Scholar]
- Pullen S.S., Miller,H.G., Everdeen,D.S., Dang,T.T.A., Crute,J.J. and Kehry,M.R. (1998) CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry, 37, 11836–11845. [DOI] [PubMed] [Google Scholar]
- Pullen S.S., Dang,T.T., Crute,J.J. and Kehry,M.R. (1999) CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J. Biol. Chem., 274, 14246–14254. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Whitmarsh,A.J., Barrett,T., Derijard,B. and Davis,R.J. (1996) MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol., 16, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe M., Evans,H.S., Young,L.S., Hennessy,K., Kieff,E. and Rickinson,A.B. (1987) Monoclonal antibodies to the latent membrane protein of Epstein–Barr virus reveal heterogeneity of the protein and inducible expression in virus-transformed cells. J. Gen. Virol., 68, 1575–1586. [DOI] [PubMed] [Google Scholar]
- Sandberg M., Hammerschmidt,W. and Sugden,B. (1997) Characteriz ation of LMP-1’s association with TRAF1, TRAF2, and TRAF3. J. Virol., 71, 4649–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshagiri S. and Miller,L.K. (1997) Baculovirus inhibitors of apoptosis (IAPs) block activation of Sf-caspase-1. Proc. Natl Acad. Sci. USA, 94, 13606–13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden B., Marsh,K. and Yates,J. (1985) A vector that replicates as a plasmid and can be efficiently selected in B-lymphoblasts transformed by Epstein–Barr virus. Mol. Cell. Biol., 5, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylla B.S., Hung,S.C., Davidson,D.M., Hatzivassiliou,E., Malinin,N.L., Wallach,D., Gilmore,T.D., Kieff,E. and Mosialos,G. (1998) Epstein–Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-κB through a pathway that includes the NF-κB-inducing kinase and the IκB kinases IKKα and IKKβ. Proc. Natl Acad. Sci. USA, 95, 10106–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida J., Yasui,T., Takaoka-Shichijo,Y., Muraoka,M., Kulwichit,W., Raab-Traub,N. and Kikutani,H. (1999) Mimicry of CD40 signals by Epstein–Barr virus LMP1 in B lymphocyte responses. Science, 286, 300–303. [DOI] [PubMed] [Google Scholar]
- Vockerodt M., Haier,B., Buttgereit,P., Tesch,H. and Kube,D. (2001) The Epstein–Barr virus latent membrane protein 1 induces interleukin-10 in Burkitt’s lymphoma cells but not in Hodgkin’s cells involving the p38/SAPK2 pathway. Virology, 280, 183–198. [DOI] [PubMed] [Google Scholar]
- Wang D., Liebowitz,D. and Kieff,E. (1985) An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell, 43, 831–840. [DOI] [PubMed] [Google Scholar]
- Yang X., He,Z., Xin,B. and Cao,L. (2000) LMP1 of Epstein–Barr virus suppresses cellular senescence associated with the inhibition of p16INK4a expression. Oncogene, 19, 2002–2013. [DOI] [PubMed] [Google Scholar]
- Ye H., Park,Y.C., Kreishman,M., Kieff,E. and Wu,H. (1999) The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol. Cell, 4, 321–330. [DOI] [PubMed] [Google Scholar]
- Yuan J. (1997) Transducing signals of life and death. Curr. Opin. Cell Biol., 9, 247–251. [DOI] [PubMed] [Google Scholar]
- Zimber-Strobl U., Kempkes,B., Marschall,G., Zeidler,R., Van Kooten,C., Banchereau,J., Bornkamm,G.W. and Hammerschmidt,W. (1996) Epstein–Barr virus latent membrane protein (LMP1) is not sufficient to maintain proliferation of B cells but both it and activated CD40 can prolong their survival. EMBO J., 15, 7070–7078. [PMC free article] [PubMed] [Google Scholar]