

Abstract
The major human AP endonuclease APE1 (HAP1, APEX, Ref1) initiates the repair of abasic sites generated either spontaneously, from attack of bases by free radicals, or during the course of the repair of damaged bases. APE1 therefore plays a central role in the base excision repair (BER) pathway. We report here that XRCC1, another essential protein involved in the maintenance of genome stability, physically interacts with APE1 and stimulates its enzymatic activities. A truncated form of APE1, lacking the first 35 amino acids, although catalytically proficient, loses the affinity for XRCC1 and is not stimulated by XRCC1. Chinese ovary cell lines mutated in XRCC1 have a diminished capacity to initiate the repair of AP sites. This defect is compensated by the expression of XRCC1. XRCC1, acting as both a scaffold and a modulator of the different activities involved in BER, would provide a physical link between the incision and sealing steps of the AP site repair process. The interaction described extends the coordinating role of XRCC1 to the initial step of the repair of DNA abasic sites.
Keywords: abasic sites/AP endonuclease/APE1/base excision repair/XRCC1
Introduction
Abasic (AP) sites are among the most common DNA lesions that arise in DNA and can have deleterious consequences for the cell. If left unrepaired, AP sites, which are generated spontaneously at an estimated rate of 104/cell/day (Lindahl and Nyberg, 1972) and are found at high levels in mammalian cells (Nakamura and Swenberg, 1999), are potentially genotoxic as well as mutagenic (Loeb, 1985). The loss of DNA bases to form AP sites in cellular DNA occurs spontaneously as a result of the inherent lability of the N-glycosyl bond to hydrolytic cleavage. AP sites are also generated in the DNA by the action of endogenous factors such as reactive oxygen species generated by the normal metabolism of the cell or following exposure of cells to ionizing radiation or other oxidizing agents. Moreover, the AP site is the product of DNA glycosylases and is the first DNA intermediate in the process of base excision repair (BER) (reviewed by McCullough et al., 1999). The second enzyme of this pathway, an AP endonuclease, initiates the repair of AP sites (Demple et al., 1991; Robson and Hickson, 1991). In human cells, the most abundant AP endonuclease is the APE1 protein. This DNA repair enzyme was discovered independently as HAP1, APE and APEX (as recommended by the Human Gene Map Nomenclature Committee, the APE1 acronym will be used here) and also as an activator of oxidized transcription factors (Ref1) (Xanthoudakis et al., 1992). This second function relies in a domain that is dispensable for the DNA repair activity (Walker et al., 1993; Xanthoudakis et al., 1994). As a BER enzyme, APE1 catalyzes the hydrolytic cleavage of the phosphodiester bond immediately 5′ to the AP site and hence initiates the repair of the AP site (reviewed by Demple and Harrison, 1994). APE1 has a broad specificity for AP sites and can act as a phosphodiesterase, a 3′ phosphatase and an RNase H (Chen et al., 1991; Seki et al., 1991; Winters et al., 1992; Barzilay et al., 1995; Wilson et al., 1995; Suh et al., 1997). However, the physiological relevance of these additional activities is unclear since they have specific activities between two and four orders of magnitude lower than that of the AP endonuclease activity. Following the cleavage of the AP site by APE1, the 5′-terminal sugar phosphate (dRP) that remains is removed by a dRP lyase activity associated with DNA polymerase β (Polβ). Polβ adds one nucleotide to the 3′ end of the nick. A DNA ligase subsequently seals the remaining nick, the whole process resulting then in the replacement of the AP site by the incorporation of a single nucleotide (Kubota et al., 1996). This pathway is called short-patch BER. Alternatively, after the cleavage of the AP site by APE1, the nicked DNA can be directed towards the long-patch repair pathway. In this pathway, the DNA polymerase adds several nucleotides and displaces the dRP residue as part of a ‘flap’ subsequently removed by FEN1 (Matsumoto et al., 1994; Frosina et al., 1996; Klungland and Lindahl, 1997).
The length of the replaced patch is influenced in vitro by the absence or presence of another DNA repair protein, XRCC1. This protein has been implicated in the BER of AP sites through the short-patch branch (Kubota et al., 1996). XRCC1 was first identified as a gene capable of complementing the hypersensitivity of Chinese hamster ovary (CHO) mutant cell lines to a variety of DNA-damaging agents (Thompson et al., 1990). Since then, although no enzymatic activity has been attributed to the protein, the role of XRCC1 in single strand break repair (SSBR) has been established by the discovery of its interactions with DNA ligase III (LIGIII) (Caldecott et al., 1994), poly(ADP-ribose) polymerase (PARP-1) (Caldecott et al., 1996; Masson et al., 1998), Polβ (Caldecott et al., 1996; Kubota et al., 1996) and, more recently, polynucleotide kinase (PNK) (Whitehouse et al., 2001). The multiple partners of XRCC1 in SSBR suggest a scaffolding and coordinating role for this protein during SSBR. XRCC1 is essential for the stabilization of LIGIII and can stimulate the activity of PNK, thereby accelerating the SSBR process. During in vitro BER, the presence of XRCC1 is required to limit the number of nucleotides replaced by Polβ to one, suggesting a role distinct from that of stabilization of the ligase. The interaction of XRCC1 with Polβ could, therefore, define another role for XRCC1 in BER. A recent study has shown that the N-terminal domain of XRCC1 has affinity for a nicked DNA molecule, which is the product of AP endonuclease action (Marintchev et al., 1999). The same study demonstrated that the interaction with the DNA can result in a ternary complex composed of XRCC1, Polβ and a gapped DNA in a way that the two proteins almost completely surround the damaged site in the DNA, protecting it from the cellular milieu. Furthermore, photoaffinity labeling by the product of APE1 of the PARP-1 (Lavrik et al., 2001), involved in the later steps of long-patch BER (Dantzer et al., 2000; Prasad et al., 2001), suggests that the interaction of XRCC1 with PARP could also play a role in BER. Despite these observations, however, the biochemical role of XRCC1 in the coordination of BER remains unclear.
Because the AP endonuclease activity is indispensable in all the BER pathways, we investigated the possible protein partners of APE1 to obtain a better understanding of the coordination displayed by BER. In the present study, we show the physical and functional interactions of APE1 with XRCC1, hence extending the view of a highly orchestrated mechanism for BER in human cells.
Results
XRCC1 protein interacts directly with the major human AP endonuclease
A search for protein partners for APE1 was set up using the yeast two-hybrid approach. Plasmid pEG202-HAP1 was used as bait to probe a human cDNA library in plasmid pJG45. After selection on plates containing X-gal, positive clones were analyzed by sequencing. Three clones carried human XRCC1 sequences. XRCC1 has been implicated in the BER pathway by its interactions with Polβ, PARP and LIGIIIα. In addition to that, the hypersensitivity to alkylating agents observed in XRCC1-defective cells supports the idea that XRCC1 plays a specific and critical role in the repair of damaged bases. However, no evidence for the participation of XRCC1 in the initial steps of BER has been reported. A plasmid encoding XRCC1, pXRCC1, was analyzed further. The β-galactosidase levels of the EGY48 strain harboring plasmids pEG202-HAP1 and pXRCC1 is presented in Figure 1A, along with several positive and negative controls. Neither XRCC1-expressing plasmid alone, nor in combination with pLexA-Max- or HM12-expressing constructs, conferred significant β-galactosidase activities. When both pEG202-HAP1 and pXRCC1 were present in the strain, 23- and 11-fold increases in activity were observed with respect to the individual pEG202-HAP1 and pXRCC1 plasmids, respectively. These β-galactosidase activity levels are similar to those obtained with the RAD51L3–XRCC2 couple.

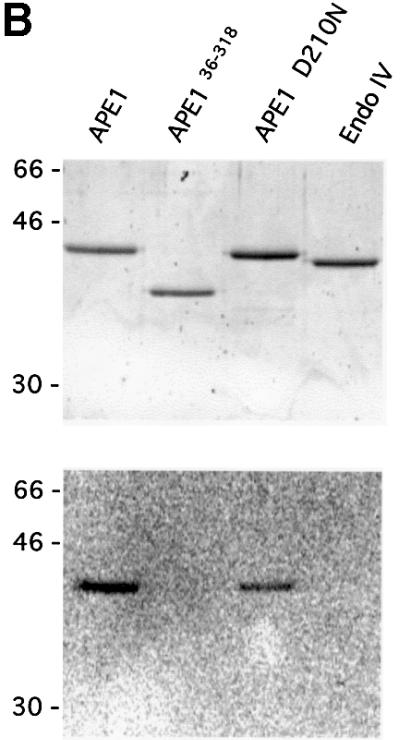
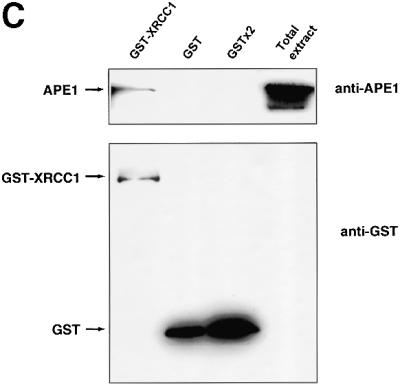
Fig. 1. Human AP endonuclease APE1 and XRCC1 proteins physically interact. (A) Two-hybrid system experiments; for details refer to Materials and methods. (B) Far-western analysis. Upper panel: Coomassie blue-stained gel showing the expected size of the loaded AP endonucleases. Lower panel: 200 ng of full-length APE1 (lane 1), APE136–318 (lane 2), APE1 D210N (lane 3) and EndoIV (lane 4) were loaded on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose membrane and incubated with 35S-labeled XRCC1. (C) GST pull-down assay in human cell extracts after transfection with either GST- or GST–XRCC1-expressing vectors. GST–XRCC1141–572, GST and associated endogenous proteins were purified selectively from total cell extracts and analyzed by western blotting as described in Materials and methods with either anti-APE1 (upper panel) or anti-GST (lower panel) antibodies. The last lane contains 20 µg of total extract from control untransfected HeLa cells.
In order to confirm the direct interaction between APE1 and XRCC1, far-western experiments were carried out. Purified recombinant proteins APE1, two mutant APE1 forms and endonuclease IV (EndoIV), a bacterial protein structurally unrelated to APE1 but with the same enzymatic function, were fractionated by SDS–PAGE and either stained with Coomassie blue (Figure 1B, upper panel) or transferred to a membrane. The membrane was subsequently incubated in the presence of a [35S]XRCC1 probe. Figure 1B (lower panel) shows that the XRCC1 probe detected both the wild-type and the catalytically inactive D210N human AP endonuclease APE1 proteins. Using similar amounts of protein, no affinity of XRCC1 was observed for the endonuclease EndoIV. Interestingly, no interaction was detected when recombinant APE136–318, a form of APE1 protein lacking the first 35 amino acids, was probed. This suggested that the interaction domain for XRCC1 on APE1 includes residues 1–35. We showed previously that truncated APE136–318 retains full ‘redox’ and endonuclease repair activities (Walker et al., 1993). Moreover, APE136–318 has the same three-dimensional structure as the wild-type protein (Gorman et al., 1997; Beernink et al., 2001).
To determine whether APE1 and XRCC1 interact in mammalian cells, we expressed an XRCC1 fragment, spanning residues 141–572, fused in-frame to GST. The GST–XRCC1 fusion or GST alone (as a control) were expressed in HeLa cells and subsequently affinity purified on glutathione–Sepharose beads, together with the associated proteins. The proteins bound were analyzed by western blots using antibodies against APE1 or GST (Figure 1C). In accordance with the experiments using purified proteins, APE1 was found associated with the GST–XRCC1 fusion protein expressed in HeLa cells, while no signal for APE1 was detected when only the GST protein was overexpressed (upper panel). Because the GST–XRCC1 fusion construct coded only for amino acids 141–572, the pull-down experiment also shows that neither the N- nor the C-terminal domains of XRCC1 are necessary for the interaction with APE1. The same membranes were incubated in the presence of antibodies against GST to confirm the expression of the GST proteins (lower panel). Taken together, these results demonstrate that, in mammalian cells, APE1 endonuclease interacts physically with XRCC1, extending the range of XRCC1 interactions to a protein involved in the initial steps of BER.
XRCC1 promotes the AP endonuclease and 3′-dRPase activities of APE1
The affinity of XRCC1 for APE1 described above prompted the search for a functional interaction between these two proteins. Since no catalytic activity has been assigned to XRCC1, the possibility that it might affect the enzymatic activity of APE1 was investigated. Using recombinant human proteins, the APE1 AP endonuclease activity was tested in the presence or absence of XRCC1. Although XRCC1 is dispensable for the cleavage of a duplex oligonucleotide harboring a unique abasic site by the AP endonuclease, the presence of purified XRCC1 protein strongly stimulated the APE1 activity in vitro (Figure 2A). The addition of XRCC1 to the reaction mixture caused a >5-fold increase in APE1-catalyzed strand cleavage at the AP site. Identical results were obtained with a DNA substrate containing a synthetic abasic site analog, a tetrahydrofuran (THF) residue, instead of a regular AP site (Figure 2B). When the XRCC1 preparation was boiled for 10 min prior to inclusion in the reaction mixture, no detectable stimulation of APE1 activity was observed for either substrate (data not shown). In the absence of APE1, the XRCC1 protein preparation showed no detectable activity on these substrates, confirming that the extra activity conferred by XRCC1 reflected stimulation of APE1 rather than contamination of the XRCC1 preparation by an AP endonuclease.
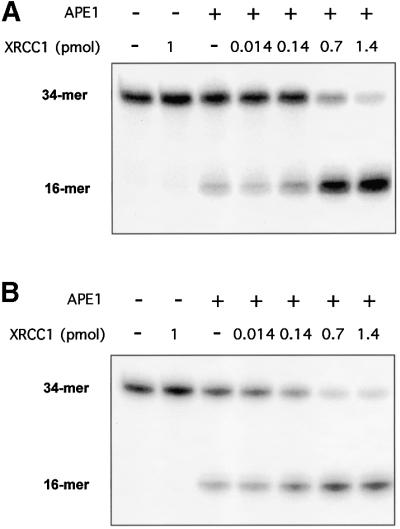

Fig. 2. Effect of XRCC1 on the AP endonuclease activity of APE1. AP endonuclease activity on a 34mer (A) [AP:C] or (B) [F:C] duplex 32P-labeled at the 5′ end on the lesion-containing strand was measured as described in Materials and methods. A limiting amount (0.03 fmol) of APE1 protein was incubated for 30 min at 37°C with the double-stranded oligonucleotide substrate (50 fmol) and increasing amounts of XRCC1 protein. The products of the reactions were separated by denaturing 20% PAGE. The positions in the gel of the 34mer substrate and the 16mer product of the reaction are indicated. (C) Quantification of the data from (A) and (B).
When the repair of a modified base is initiated by a bi-functional DNA glycosylase such as OGG1 or NTH, a possible DNA intermediate in BER is a nick with a 3′-unsaturated aldehyde moiety. This potentially toxic intermediate can also be processed by AP endonuclease, although the corresponding 3′-dRPase activity of APE1 is relatively inefficient (Demple et al., 1991). We therefore investigated whether XRCC1 also stimulated the 3′-phosphodiesterase activity of APE1 on substrates carrying a 3′-phosphoribose residue. Figure 3 shows that the presence of XRCC1 increases the efficiency of APE1 for the removal of a 3′-unsaturated aldehyde residue present in a nick. The lower band present in all lanes including the control corresponds to the labeled DNA fragment carrying a 3′-phosphate. The detection of the 3′-phosphatase activity of APE1 requires a much higher (10- to 100-fold) enzyme concentration (Demple et al., 1991).

Fig. 3. Effect of XRCC1 upon the 3′-dRPase activity of APE1. The 3′-phosphodiesterase activity of APE1 was tested on a 34mer DNA duplex containing a nick with a 3′-unsaturated aldehyde residue and labeled at the 5′ end of the lesion-carrying strand. Limiting amounts of APE1 were incubated in the presence or absence of XRCC1 (0.7 pmol) and the activity was determined by the appearance of a band corresponding to the labeled fragment carrying a 3′-OH at the expense of the substrate carrying a 3′-unsaturated aldehyde residue (dRP). The lower band corresponds to the labeled fragment carrying a 3′-phosphate.
Stimulation of APE1 by XRCC1 requires the N-terminal region of APE1
The effect of XRCC1 on the AP endonuclease activity of Escherichia coli EndoIV was tested in order to analyze the specificity of the APE1 stimulation by XRCC1. EndoIV, although structurally unrelated to the human AP endonuclease, can cleave DNA at abasic sites yielding products identical to those generated by APE1. When limiting amounts of bacterial EndoIV were used, there was no detectable stimulation by XRCC1 using as substrate oligonucleotides harboring either a regular AP site or a THF residue (Figure 4).

Fig. 4. Effect of XRCC1 upon the in vitro activities of the bacterial EndIV and the interaction mutant APE136–318. Reactions were carried out with (A) 34mer [AP:C] or (B) [F:C] duplexes as described in Figure 2 except that only one amount (0.7 pmol) of XRCC1 protein was assayed for each AP endonuclease. The products of the reactions were separated by denaturing 20% PAGE. The positions in the gel of the substrate (34mer) and the product (16mer) of the reaction are indicated.
The specificity of the stimulatory effect of XRCC1 on APE1 AP endonuclease activity most probably relies on a structural direct interaction between the two proteins. Because it was shown above that XRCC1 has a strong affinity for APE1, we analyzed the effect of XRCC1 on APE136–318, which does not interact with XRCC1 but was shown previously to retain full catalytic capacity. Equivalent amounts of AP endonuclease units from APE1 and the interaction mutant APE136–318 were tested in the presence of purified XRCC1, for their capacity to cleave a DNA duplex containing an AP site. Figure 4 shows that XRCC1 is devoid of any stimulatory capacity on the truncated version of APE1, consistent with the lack of interaction between XRCC1 and APE136–318. This result suggests strongly that the physical interaction between APE1 and XRCC1 is required for the stimulation of the repair activity of the AP endonuclease.
The stimulation of HAP1 by XRCC1 could be due to an enhancement of the binding of the enzyme to its substrate. The determination of the kinetic constants for the cleavage reaction of HAP1 shows that the addition of XRCC1 shifts the KM from 20 nM in its absence to 10 nM, without affecting the Vmax. This suggests that XRCC1 increases the affinity of HAP1 for its substrate.
Whole-cell extracts from XRCC1-defective cell lines have a diminished AP repair capacity
XRCC1-defective cell lines were isolated initially for their sensitivity to DNA-damaging agents (Thompson et al., 1980). Subsequent studies have shown that these cell lines have DNA repair defects that are responsible for their hypersensitivity to ionizing radiation and alkylating agents (Thompson and West, 2000). Because of the interactions with other proteins, XRCC1 has been postulated to participate not only in SSBR but also in BER. We wished then to examine whether, in cells, XRCC1 stimulates APE1 activity, a key step in BER. The capacities of three different CHO cell lines with mutations in XRCC1 (Shen et al., 1998) to cleave DNA at abasic sites were compared with that of cell line AA8 carrying a wild-type XRCC1. One limitation in this type of assay system utilizing whole-cell extracts is that AP sites are not only substrates for an AP endonuclease activity but also for the AP lyase activity of the bi-functional DNA glycosylases. For this reason, whole-cell extracts from the different cell lines were incubated with a 34mer oligonucleotide harboring a single THF residue. This DNA substrate cannot be cleaved by the bi-functional DNA glycosylases’ AP lyase activity. A reduction in the cleavage capacity with respect to the AA8 extract was found consistently in all three extracts derived from XRCC1-defective cell lines (Figure 5A). The levels of APE1 protein were determined in those same extracts by western blot. Interestingly, cell lines EM9, EMC11 and EMC12, while having a reduced capacity for cleaving AP sites when compared with AA8, each have between 1.5- and 2-fold higher levels of APE1 than has AA8 (Figure 5B). These results indicate that XRCC1-deficient cells have significantly lower AP endonuclease specific activity than has the parental AA8 cell line.

Fig. 5. Comparison of the AP endonuclease activity in XRCC1-proficient or deficient whole-cell extracts. XRCC1-proficient (AA8) or XRCC1-deficient (EM9, EMC11 and EMC12) CHO cell lines were used to prepare extracts. Reactions were carried out as described in Materials and methods with increasing amounts of total extract protein as indicated. An oligonucleotide harboring a single THF site was used as substrate. (A) Plot of the percentage of DNA substrate cleaved by the AP endonuclease activities of the four CHO cell line extracts versus total amount of protein used in the reaction. Each point represents the average and standard deviation from two independent experiments. (B) APE1 protein levels in 50 µg of each cell extract were determined by western blotting.
The XRCC1– cell lines used have been obtained by non-specific mutagenesis. This opens up the possibility that secondary mutations affect the phenotype being studied. We therefore repeated the above experiments using a pair of isogenic cell lines. Whole-cell extracts were prepared from XRCC1 mutant EM9 cells carrying either the empty vector D2E (EM9) or the expression vector encoding human XRCC1 (EM9-X). It has been demonstrated previously that the resistance to DNA damage and the SSBR proficiency are restored in EM9-X cells (Caldecott et al., 1992; Taylor et al., 2000). Consistent with the results shown in Figure 5, EM9 cell extracts were less able to cleave DNA at abasic sites than were extracts from EM9-X cells (Figure 6A and B). Furthermore, the addition of recombinant XRCC1 to the EM9 cell extracts stimulated the level of AP endonuclease activity. In contrast, when XRCC1 was added to EM9-X cell extracts, no further stimulation was observed on the APE1 specific activity. Figure 6B displays the western blots showing the levels of XRCC1 and APE1 proteins in the EM9 and EM9-X cells. As expected, there is no detectable XRCC1 in EM9 cells while the levels of APE1 are similar in both cell lines (Figure 6C). Taken together, these data indicate that cells lacking XRCC1 have a reduced specific activity for the major AP endonuclease activity that initiates the repair of abasic sites and that the stimulation by XRCC1 reflects a complementation of the genetic defect.


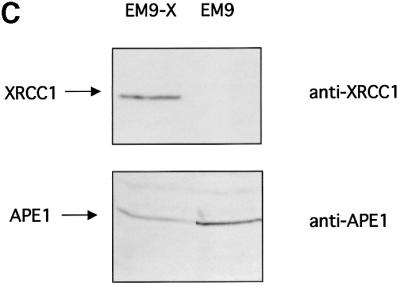
Fig. 6. Endogenous XRCC1 stimulates AP endonuclease activity in whole-cell extracts. (A) Reactions were carried out on an oligonucleotide harboring a single THF site as described in Materials and methods with increasing amounts of total protein as indicated. The products of the reactions were separated by denaturing 20% PAGE. The positions in the gel of the substrate and the product of the reaction are indicated in the figure. XRCC1-deficient (EM9) or XRCC1-proficient (EM9-X) cell lines were used to prepare the extracts. Where indicated, standard reactions were supplemented with 1.4 pmol of purified human XRCC1 (+XRCC1). (B) Plot of the percentage of DNA substrate cleaved by the AP endonuclease activities versus total amount of protein used in the reaction. Each point represents the average and standard deviation from three independent experiments. (C) XRCC1 and APE1 contents were determined by western blot analysis on 30 µg of each protein extract.
Discussion
The repair of abasic sites is essential for the preservation of the genomic stability in all cells. The physical and functional interaction between the major human AP endonuclease and the XRCC1 protein shown here provides the first evidence of a role for XRCC1 in the incision steps of the DNA repair process. XRCC1 mutant cells are particularly sensitive to exposure to alkylating agents (Thompson and West, 2000). Since the repair of the alkylated bases is done mostly by the BER, the interaction between XRCC1 and APE1, an enzyme essential for all the BER pathways described (Lindahl and Wood, 1999; Hoeijmakers, 2001), could explain the similarity of the sensitivity to genotoxic agents between mutant cells in either of those genes (for a review see Thompson and West, 2000). The role of this association of XRCC1 with APE1 could be to facilitate the processing of the abasic sites by APE1 in order to accelerate ‘normal’ repair and therefore avoiding the formation of ‘dirty’ DNA ends, blocked 5′ or 3′ extremities unsuitable for the action of the DNA polymerases, that could have deleterious consequences for the genome. Interestingly, the features of XRCC1–/– embryos (Tebbs et al., 1999) closely resemble those associated with knockout of the APE1 gene (Xanthoudakis et al., 1996; Ludwig et al., 1998). This similarity opens up yet another intriguing possibility that derives from the second function known for APE1, i.e. its role as a regulator of gene expression (Xanthoudakis et al., 1992). It is tempting to speculate that the binding of XRCC1 to APE1 is important for this second function of APE1 and therefore that in the XRCC1–/– embryos, the transcription activation role of APE1 is impaired. Although this hypothesis cannot be excluded, the known functions of XRCC1 and APE1 in DNA repair together with the data presented in this work, as well as the recent finding of mutations and polymorphisms in XRCC1 or APE1 associated with human diseases (Hu et al., 2001; Lee et al., 2001; Matullo et al., 2001) and the phenotype of the heterozygous APEX knockout mice (Meira et al., 2001), further underline the importance of these two proteins in the maintenance of genetic stability.
The experiments presented here suggest a new level of coordination in the BER pathway orchestrated by the XRCC1 protein. This protein, for which no enzymatic activity has been assigned, plays a central role in DNA SSBR (Caldecott, 2001). Its action is associated with its capacity to interact with other proteins involved in SSBR (Caldecott, 2001). At the recognition step, XRCC1 has been shown to interact with PARP-1 and to modulate its activity negatively (Caldecott et al., 1996; Masson et al., 1998). The role of this interaction between XRCC1 and the primary sensor of DNA breaks is still unclear. By its capacity to associate with DNA nicks, XRCC1 could have a sensor function by itself. In the case where the DNA ends generated by the break are not suitable for the DNA polymerase action (‘dirty’ ends), XRCC1 could recruit PNK (Whitehouse et al., 2001) to ‘clean’ the ends. Furthermore, the presence of XRCC1 stimulates both the DNA phosphatase and DNA kinase activities of PNK. The stimulation of the processing of a 3′-blocking lesion presented here suggests a similar role for the APE1–XRCC1 interaction. The role of XRCC1 in the strand healing process is related to its interactions with Polβ (Caldecott et al., 1996; Kubota et al., 1996), and the ability to form a ternary complex with gapped DNA (Marintchev et al., 1999). Finally, XRCC1 is involved in the ligation step by its interaction with and stabilization of LIGIII (Caldecott et al., 1994, 1995).
In BER, XRCC1 is known to be involved in the strand sealing of the nicked AP site left by the AP endonuclease. As in the case of SSBR, by its interactions with Polβ and LIGIII, XRCC1 seems to function as a molecular scaffold and a coordinator of the later steps of BER required for the repair of the SSB left by APE1 (Cappelli et al., 1997). Furthermore, the presence of XRCC1 directs the BER towards its short-patch branch (Kubota et al., 1996). However, it has been pointed out that the defect in the ligation step may not fully account for the sensitivity of XRCC1– cells to killing by alkylating agents (Thompson and West, 2000). Here, we show that XRCC1 is also involved in the processing of abasic sites that lead to single-strand breaks in the BER pathway. In a role similar to the one it plays in SSBR, by interacting with APE1 and stimulating its AP endonuclease activity, XRCC1 prepares the DNA substrate for the DNA polymerase activities. This is particularly important in the case of the repair of an oxidized base through BER. In those cases, the repair is initiated by the excision of the modified base by either hNTH or hOGG1. Both of these DNA glycosylases have an associated AP lyase activity that can cleave the abasic site leaving a 3′α,β-unsaturated aldehyde on the 3′ DNA end. This residue needs to be removed to allow the re-synthesis of the strand. Moreover, it has been proposed that this kind of DNA intermediate can be toxic for the cell (Sobol et al., 2000). The activation of the APE1 AP endonuclease activity by XRCC1 could dissociate the DNA glycosylase further from the AP lyase activity (Hill et al., 2001; Vidal et al., 2001) in such a way as to avoid the formation of 3′-blocked DNA ends. Moreover, the XRCC1-induced stimulation of APE1 3′-dRPase activity, a limiting activity in mammalian cell extracts (Izumi et al., 2000), allows a more efficient processing of 3′-blocking intermediates left by the AP lyase activity of either NTH or OGG1. It had been shown that Polβ can also increase AP endonuclease activity by releasing APE1 from product inhibition (Masuda et al., 1998). APE1 also facilitates the loading of the DNA polymerase on the DNA at the cleaved AP site, accelerating the excision of 5′-dRP residues (Bennett et al., 1997). The capacity of XRCC1 to interact with both APE1 and Polβ suggests the possibility that once the DNA is nicked by APE1, XRCC1 mediates the positioning of the polymerase to perform its lyase activity, thus improving the processing of the nick generated by APE1 (Sobol et al., 2000).
The results presented here show that the first 35 amino acids of APE1 are essential for the interaction with XRCC1. The first 42 amino acids have been shown to be disordered in the APE1 crystals (Mol et al., 2000; Beernink et al., 2001). Indeed, except for its N-terminal extension, APE1 is a globular protein (Gorman et al., 1997). This suggests that the N-terminal tail will be readily available to interact with other proteins, perhaps being structured by the contact with XRCC1. XRCC1 has been shown to interact physically with at least four enzymes involved in BER. It is unlikely that all these interactions are simultaneous even though for three of them distinct XRCC1 domains are responsible. Indeed Polβ interacts with the N-terminal domain (Marintchev et al., 1999), PARP with the central BRCT domain (Masson et al., 1998) and LIGIII with the second BRCT domain (Taylor et al., 1998; Dulic et al., 2001). The GST pull-down experiments described here indicate that the interaction with APE1 does not require the N-terminal or the BRCT2 domains of XRCC1. Furthermore, the stimulation capacity is retained by XRCC1 lacking the BRCT2 domain (data not shown). It remains to be established whether the BRCT1 domain or the two remaining ‘hinge’ regions of XRCC1 (Callebaut et al., 1997) are responsible for the interaction with APE1. The sequential binding of XRCC1 with the different BER partners, including DNA intermediates, would be consistent with the proposed model, for both BER and SSBR, by which the DNA–protein complexes assembled for one step of the repair process are recognized by the next protein or protein complex in the pathway (Rice, 1999; Mol et al., 2000; Thompson and West, 2000; Wilson and Kunkel, 2000; Caldecott, 2001). XRCC1 could then be the scaffold for this ‘handing over’ mechanism that would avoid the formation and exposure to the cellular milieu of potentially toxic DNA repair intermediates (Figure 7).
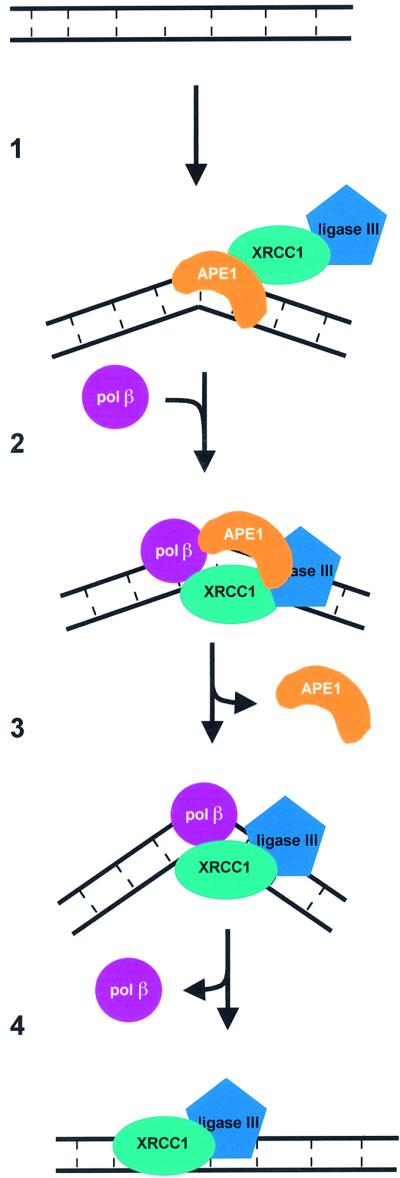
Fig. 7. A model for mammalian short-patch BER of abasic sites. (1) APE1, associated with the XRCC1–LIGIII complex, binds to an AP site present in double-stranded DNA, creating a bend in the DNA. APE1 incises the strand carrying the lesion at the phosphodiester bond located 5′ of the AP site. (2) Polβ is recruited by its interactions with APE1 and XRCC1. (3) APE1 is released; the XRCC1–Polβ complex further bends the DNA before the incorporation of a nucleotide and the elimination of the phosphoribose residue by the polymerase. (4) The remaining nick is sealed by the XRCC1–LIGIII complex.
In conclusion, the functional and physical interactions between APE1 and XRCC1 shown here create a new link between the incision and sealing steps of the BER. This observation implies a role for XRCC1 in every step in the repair of an AP site (Figure 7), suggesting that it is through this scaffolding protein that the proposed coordination of the repair process is achieved (Rice, 1999; Mol et al., 2000; Thompson and West, 2000; Wilson and Kunkel, 2000).
Materials and methods
Oligonucleotide substrates, enzymes and cell lines
The 34mer oligodeoxyribonucleotides containing either a uracil residue (U) (Genosys), a tetrahydrofuranyl residue (F) (Eurogentec) or an 8-oxoguanine were labeled at the 5′ end using [γ-32P]ATP (3000 Ci/mmol; Amersham) and T4 polynucleotide kinase (New England Biolabs). In all cases, the 32P-labeled strands were hybridized with a complementary sequence, containing a cytosine (C) opposite to the lesion, yielding the duplexes U:C, F:C and 8-oxoG:C, respectively. The duplex DNA containing an AP site (AP:C) was prepared as follows: a reaction mixture (80 µl) containing 1 pmol of the 32P-labeled [U:C] duplex was incubated for 30 min at 37°C in reaction buffer (20 mM Tris–HCl pH 8.0, 75 mM NaCl, 1 mM MgCl2) with 2 U of uracil-DNA glycosylase (UDG). The reaction was stopped by addition of 4 U of uracil glycosylase inhibitor (UGI) and further incubation for 10 min at 37°C. For the preparation of a nicked DNA with a 3′-unsaturated aldehyde residue, the 8-oxoG-containing duplex was treated with an excess of hOGG1 for 1 h at 37°C. The reaction mixture was extracted with a phenol–chloroform mix and the DNA was precipitated from the aqueous phase by ethanol precipitation. After drying, the DNA was resuspended in H2O and re-hybrized by heating at 90°C for 5 min and allowing to cool to room temperature. The recombinant APE1 and mutant derivatives APE1 D210N and APE136–318 were purified as described (Rothwell et al., 2000). EndoIV enzyme was from our laboratory stocks. UDG, UGI and restriction enzymes were from New England Biolabs. The CHO cell line AA8 and XRCC1– cells EM9, EMC11 and EMC12 (Shen et al., 1998) were obtained from E.Sage (Institut Curie, Paris). EM9-V and EM9-X (Taylor et al., 2000) were a kind gift from K.Caldecott (University of Manchester). All mammalian cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum.
Two-hybrid analysis
Two-hybrid analysis was performed essentially by the method previously described (Watt et al., 1995; Wu et al., 2001). Briefly, the full-length APE1 cDNA was amplified using PCR with 5′ and 3′ primers that contained EcoRI and XhoI sites, respectively, for direct cloning into pEG202 (kindly provided by Dr R.Brent). The HeLa cell activation library in pJG45 was kindly provided by Dr R.Brent. Screening of this library using pEG202/APE1 as bait was performed in the EGY48 strain. Positive clones were detected by growth on plates containing 2% galactose, 1% raffinose and 40 µg/ml X-gal by their blue coloration. Plasmid DNA from these clones was extracted and retransformed into EGY48 containing pEG202/APE1 to confirm that the interaction was observed a second time. Quantification of β-galactosidase activity was performed as described (Ausubel et al., 1994) using extracts (normalized for total protein) of yeast cell cultures treated with 2% galactose for 4 h. The negative control plasmids derived from pEG202 (pLexA-MAX and pHM12) have been described previously (Watt et al., 1995). The positive control bait and prey plasmids carrying XRCC2 and RAD51L3 have also been described previously (Braybrooke et al., 2000).
XRCC1 expression and purification
The pET16b expression vector containing the cDNA encoding the full-length XRCC1 protein was kindly donated by K.Caldecott. His-tagged recombinant XRCC1 was expressed in E.coli BL21 and affinity purified on a nickel column (immobilized metal ion adsorption chromatography; Pharmacia, Uppsala, Sweden) as previously described (Caldecott et al., 1995).
In vitro transcription/translation of XRCC1
In vitro transcription/translation of XRCC1 was performed using a TNT-coupled lysate system (Promega) according to the manufacturer’s instructions. The reaction containing [35S]methionine and the pBS-XRCC1 expression vector (a gift from G.de Murcia, ESBS, Strasbourg) was incubated for 90 min at 30°C. Reaction products were analyzed directly by SDS–PAGE and used in far-western assays.
Far-western blot analysis
The same amounts of purified APE1, APE1 derivative mutant proteins and EndoIV (∼200 ng) were separated on 12% SDS–PAGE (29:1 acrylamide:bisacrylamide) and electrotransferred for 1 h and 90 V at 4°C to nitrocellulose membranes (Amersham). The immobilized proteins were then denaturated by incubation (20 min at 4°C) in a buffer containing 6 M guanidine–HCl, 25 mM HEPES–KOH pH 7.7, 25 mM NaCl, 5 mM MgCl2 and 1 mM dithiothreitol (DTT). Renaturation was then carried out by successive incubations in the same buffer as above containing decreasing concentrations of guanidine-HCl, the final incubation being carried out in the absence of denaturing agent. Membranes were then blocked for 1 h at 4°C in 25 mM HEPES–KOH pH 7.7, 25 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.05% NP-40 and 5% skimmed milk and incubated overnight with 50 µl of the in vitro transcribed/translated 35S-labeled XRCC1 at 4°C in 5 ml of hybridization buffer (20 mM HEPES–KOH pH 7.7, 0.1 mM EDTA 2.5 mM MgCl2, 1 mM DTT, 0.05% NP-40 and 1% skimmed milk). The membranes were washed three times at 4°C with hybridization buffer without the probe, dried briefly and scanned with a Storm PhosphorImager (Molecular Dynamics).
GST pull-down assay from transfected human cells
Approximately 106 HeLa cells were transfected using cationic liposomes (Transfast, Promega) with 10 µg of either pBC-NLS, a mammalian expression vector expressing GST fused to a nuclear localization signal, or with pBC expressing the human XRCC1 cDNA (nucleotides 420–1716; both plasmids kindly provided by G.de Murcia) (Masson et al., 1998). After 36 h of incubation at 37°C, cells were harvested and resuspended in 150 µl of lysis buffer [20 mM Tris–HCl pH 7.5, 250 mM NaCl, 20% glycerol, 5 mM DTT and 1 mM protease inhibitor phenylmethylsulfonyl fluoride (PMSF)]. Cellular debris was cleared by centrifugation at 10 000 r.p.m. for 20 min at 4°C. Supernatants were then diluted in 1 ml of binding buffer (50 mM Tris–HCl pH 7.5, 0.1% NP-40, 150 mM NaCl and 1 mM PMSF) and incubated with glutathione– Sepharose beads for 30 min at room temperature on a rotator. Beads were washed three times with wash buffer (50 mM Tris–HCl pH 7.5, 0.5% NP-40, 250 mM NaCl and 1 mM PMSF). Bound proteins were separated on SDS–12% PAGE, transferred to nitrocellulose membranes and analyzed by western blot analysis with either rabbit polyclonal anti-APE1 (our laboratory) or goat polyclonal anti-GST antibodies (Pharmacia).
CHO cell extracts
Total cell extracts were obtained by sonication as described (Hollenbach et al., 1999). To determine the level of XRCC1 by western blot, a rabbit polyclonal antibody against the His-tagged protein (kindly provided by G.de Murcia) was used (Masson et al., 1998). In the case of APE1, rabbit polyclonal anti-APE1 antibody IHIC13 from our laboratory was used.
Cleavage assays
In a standard reaction (16 µl final volume), 50 fmol of the labeled duplexes carrying either AP, THF or 3′-unsaturated aldehyde residues was incubated in reaction buffer [25 mM Tris–HCl pH 8.0, 1 mM MgCl2, 0.4 mg/ml bovine serum albumin (BSA)] with various amounts of APE1 and XRCC1 or cell extract. Reactions were carried out at 37°C for 30 min and stopped by adding 6 µl of formamide dye, followed by heating for 5 min at 95°C before loading in the gels. When using the AP:C duplex as substrate, reactions were stopped by the addition of NaBH4 (50 mM final concentration). The products of the reactions were resolved by denaturing 20% PAGE (19:1 acrylamide:bisacrylamide). Gels were scanned and band intensities quantified using a Storm PhosphorImager (Molecular Dynamics).
Acknowledgments
Acknowledgements
Our special thanks to Patricia Auffret van der Kemp for setting up the preparation of DNA substrates and the dRPase experiments. We thank Dominic Rothwell for the preparation of APE1 proteins, Claudine Dhérin for her technical assistance, Keith Caldecott for the XRCC1 plasmid constructs and the EM9-V and EM9-X cell lines, Gilbert de Murcia and Josiane Menissier de Murcia for the anti-XRCC1 antibody, the GST constructs and their help in setting up the GST pull-down experiments, and Valérie Schreiber for her advice on the XRCC1 purification protocol. We also thank Jean-Baptiste Charbonnier for fruitful discussions and critical reading of the manuscript. A.E.V. was supported by a postdoctoral fellowship from the Ministerio de Educacion y Cultura (Spain). This work was funded by the Commissariat à l’Energie Atomique (CEA), Centre National pour la Recherche Scientifique (CNRS), the Association pour la Recherche sur le Cancer (ARC) and Electricité de France (EDF).
References
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) Current Protocols in Molecular Biology. Wiley, New York.
- Barzilay G., Mol,C.D., Robson,C.N., Walker,L.J., Cunningham,R.P., Tainer,J.A. and Hickson,I.D. (1995) Identification of critical active-site residues in the multifunctional human DNA repair enzyme HAP1. Nature Struct. Biol., 2, 561–568. [DOI] [PubMed] [Google Scholar]
- Beernink P.T., Segelke,B.W., Hadi,M.Z., Erzberger,J.P., Wilson,D.M.,III and Rupp,B. (2001) Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: implications for the catalytic mechanism. J. Mol. Biol., 307, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Bennett R.A., Wilson,D.M.,III, Wong,D. and Demple,B. (1997) Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc. Natl Acad. Sci. USA, 94, 7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braybrooke J.P., Spink,K.G., Thacker,J. and Hickson,I.D. (2000) The RAD51 family member, RAD51L3, is a DNA-stimulated ATPase that forms a complex with XRCC2. J. Biol. Chem., 275, 29100–29106. [DOI] [PubMed] [Google Scholar]
- Caldecott K.W. (2001) Mammalian DNA single-strand break repair: an X-ra(y)ted affair. BioEssays, 23, 447–455. [DOI] [PubMed] [Google Scholar]
- Caldecott K.W., Tucker,J.D. and Thompson,L.H. (1992) Construction of human XRCC1 minigenes that fully correct the CHO DNA repair mutant EM9. Nucleic Acids Res., 20, 4575–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott K.W., McKeown,C.K., Tucker,J.D., Ljungquist,S. and Thompson,L.H. (1994) An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol., 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott K.W., Tucker,J.D., Stanker,L.H. and Thompson,L.H. (1995) Characterization of the XRCC1–DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res., 23, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott K.W., Aoufouchi,S., Johnson,P. and Shall,S. (1996) XRCC1 polypeptide interacts with DNA polymerase β and possibly poly(ADP-ribose) polymerase and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res., 24, 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I., Labesse,G., Durand,P., Poupon,A., Canard,L., Chomilier,J., Henrissat,B. and Mornon,J.P. (1997) Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell. Mol. Life Sci., 53, 621–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelli E., Taylor,R., Cevasco,M., Abbondandolo,A., Caldecott,K. and Frosina,G. (1997) Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem., 272, 23970–23975. [DOI] [PubMed] [Google Scholar]
- Chen D.S., Herman,T. and Demple,B. (1991) Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res., 19, 5907–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer F., de La Rubia,G., Menissier-De Murcia,J., Hostomsky,Z., de Murcia,G. and Schreiber,V. (2000) Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry, 39, 7559–7569. [DOI] [PubMed] [Google Scholar]
- Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- Demple B., Herman,T. and Chen,D.S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic A., Bates,P.A., Zhang,X., Martin,S.R., Freemont,P.S., Lindahl,T. and Barnes,D.E. (2001) BRCT domain interactions in the heterodimeric DNA repair protein XRCC1–DNA ligase III. Biochemistry, 40, 5906–5913. [DOI] [PubMed] [Google Scholar]
- Frosina G., Fortini,P., Rossi,O., Carrozzino,F., Raspaglio,G., Cox,L.S., Lane,D.P., Abbondandolo,A. and Dogliotti,E. (1996) Two pathways for base excision repair in mammalian cells. J. Biol. Chem., 271, 9573–9578. [DOI] [PubMed] [Google Scholar]
- Gorman M.A., Morera,S., Rothwell,D.G., de La Fortelle,E., Mol,C.D., Tainer,J.A., Hickson,I.D. and Freemont,P.S. (1997) The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J., 16, 6548–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J.W., Hazra,T.K., Izumi,T. and Mitra,S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res., 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers J.H. (2001) Genome maintenance mechanisms for preventing cancer. Nature, 411, 366–374. [DOI] [PubMed] [Google Scholar]
- Hollenbach S., Dhenaut,A., Eckert,I., Radicella,J.P. and Epe,B. (1999) Overexpression of Ogg1 in mammalian cells: effects on induced and spontaneous oxidative DNA damage and mutagenesis. Carcinogenesis, 20, 1863–1868. [DOI] [PubMed] [Google Scholar]
- Hu J.J., Smith,T.R., Miller,M.S., Mohrenweiser,H.W., Golden,A. and Case,L.D. (2001) Amino acid substitution variants of APE1 and XRCC1 genes associated with ionizing radiation sensitivity. Carcinogenesis, 22, 917–922. [DOI] [PubMed] [Google Scholar]
- Izumi T., Hazra,T.K., Boldogh,I., Tomkinson,A.E., Park,M.S., Ikeda,S. and Mitra,S. (2000) Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis, 21, 1329–1334. [PubMed] [Google Scholar]
- Klungland A. and Lindahl,T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J., 16, 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y., Nash,R.A., Klungland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Lavrik O.I., Prasad,R., Sobol,R.W., Horton,J.K., Ackerman,E.J. and Wilson,S.H. (2001) Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem., 276, 25541–25548. [DOI] [PubMed] [Google Scholar]
- Lee J.M., Lee,Y.C., Yang,S.Y., Yang,P.W., Luh,S.P., Lee,C.J., Chen,C.J. and Wu,M.T. (2001) Genetic polymorphisms of XRCC1 and risk of the esophageal cancer. Int. J. Cancer, 95, 240–246. [DOI] [PubMed] [Google Scholar]
- Lindahl T. and Nyberg,B. (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry, 11, 3610–3618. [DOI] [PubMed] [Google Scholar]
- Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- Loeb L.A. (1985) Apurinic sites as mutagenic intermediates. Cell, 40, 483–484. [DOI] [PubMed] [Google Scholar]
- Ludwig D.L., MacInnes,M.A., Takiguchi,Y., Purtymun,P.E., Henrie,M., Flannery,M., Meneses,J., Pedersen,R.A. and Chen,D.J. (1998) A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat. Res., 409, 17–29. [DOI] [PubMed] [Google Scholar]
- Marintchev A., Mullen,M.A., Maciejewski,M.W., Pan,B., Gryk,M.R. and Mullen,G.P. (1999) Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nature Struct. Biol., 6, 884–893. [DOI] [PubMed] [Google Scholar]
- Masson M., Niedergang,C., Schreiber,V., Muller,S., Menissier-de Murcia,J. and de Murcia,G. (1998) XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol., 18, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y., Bennett,R.A. and Demple,B. (1998) Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. J. Biol. Chem., 273, 30360–30365. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y., Kim,K. and Bogenhagen,D.F. (1994) Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol. Cell. Biol., 14, 6187–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matullo G., Guarrera,S., Carturan,S., Peluso,M., Malaveille,C., Davico,L., Piazza,A. and Vineis,P. (2001) DNA repair gene polymorphisms, bulky DNA adducts in white blood cells and bladder cancer in a case–control study. Int. J. Cancer, 92, 562–567. [DOI] [PubMed] [Google Scholar]
- McCullough A.K., Dodson,M.L. and Lloyd,R.S. (1999) Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem., 68, 255–285. [DOI] [PubMed] [Google Scholar]
- Meira L.B., Devaraj,S., Kisby,G.E., Burns,D.K., Daniel,R.L., Hammer,R.E., Grundy,S., Jialal,I. and Friedberg,E.C. (2001) Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res., 61, 5552–5557. [PubMed] [Google Scholar]
- Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] [published erratum appears in Nature 2000 Mar 30;404(6777)]. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- Nakamura J. and Swenberg,J.A. (1999) Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res., 59, 2522–2526. [PubMed] [Google Scholar]
- Prasad R., Lavrik,O.I., Kim,S.J., Kedar,P., Yang,X.P., Vande Berg,B.J. and Wilson,S.H. (2001) DNA polymerase β-mediated long patch base excision repair: poly(ADP-ribose) polymerase-1 stimulates strand displacement DNA synthesis. J. Biol. Chem., 276, 32411–32414. [DOI] [PubMed] [Google Scholar]
- Rice P.A. (1999) Holding damaged DNA together. Nature Struct. Biol., 6, 805–806. [DOI] [PubMed] [Google Scholar]
- Robson C.N. and Hickson,I.D. (1991) Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E.coli xth (exonuclease III) mutants. Nucleic Acids Res., 19, 5519–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell D.G., Hang,B., Gorman,M.A., Freemont,P.S., Singer,B. and Hickson,I.D. (2000) Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilises substrate binding. Nucleic Acids Res., 28, 2207–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki S., Akiyama,K., Watanabe,S., Hatsushika,M., Ikeda,S. and Tsutsui,K. (1991) cDNA and deduced amino acid sequence of a mouse DNA repair enzyme (APEX nuclease) with significant homology to Escherichia coli exonuclease III. J. Biol. Chem., 266, 20797–20802. [PubMed] [Google Scholar]
- Shen M.R., Zdzienicka,M.Z., Mohrenweiser,H., Thompson,L.H. and Thelen,M.P. (1998) Mutations in hamster single-strand break repair gene XRCC1 causing defective DNA repair. Nucleic Acids Res., 26, 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobol R.W., Prasad,R., Evenski,A., Baker,A., Yang,X.P., Horton,J.K. and Wilson,S.H. (2000) The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature, 405, 807–810. [DOI] [PubMed] [Google Scholar]
- Suh D., Wilson,D.M.,III and Povirk,L.F. (1997) 3′-Phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Res., 25, 2495–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R.M., Wickstead,B., Cronin,S. and Caldecott,K.W. (1998) Role of a BRCT domain in the interaction of DNA ligase III-α with the DNA repair protein XRCC1. Curr. Biol., 8, 877–880. [DOI] [PubMed] [Google Scholar]
- Taylor R.M., Moore,D.J., Whitehouse,J., Johnson,P. and Caldecott,K.W. (2000) A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Mol. Cell. Biol., 20, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbs R.S., Flannery,M.L., Meneses,J.J., Hartmann,A., Tucker,J.D., Thompson,L.H., Cleaver,J.E. and Pedersen,R.A. (1999) Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol., 208, 513–529. [DOI] [PubMed] [Google Scholar]
- Thompson L.H. and West,M.G. (2000) XRCC1 keeps DNA from getting stranded. Mutat. Res., 459, 1–18. [DOI] [PubMed] [Google Scholar]
- Thompson L.H., Rubin,J.S., Cleaver,J.E., Whitmore,G.F. and Brookman,K. (1980) A screening method for isolating DNA repair-deficient mutants of CHO cells. Somat. Cell Genet., 6, 391–405. [DOI] [PubMed] [Google Scholar]
- Thompson L.H., Brookman,K.W., Jones,N.J., Allen,S.A. and Carrano,A.V. (1990) Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell. Biol., 10, 6160–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal A.E., Hickson,I.D., Boiteux,S. and Radicella,J.P. (2001) Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res., 29, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L.J., Robson,C.N., Black,E., Gillespie,D. and Hickson,I.D. (1993) Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol., 13, 5370–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt P.M., Louis,E.J., Borts,R.H. and Hickson,I.D. (1995) Sgs1: a eukaryotic homolog of E.coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell, 81, 253–260. [DOI] [PubMed] [Google Scholar]
- Whitehouse C.J., Taylor,R.M., Thistlethwaite,A., Zhang,H., Karimi-Busheri,F., Lasko,D.D., Weinfeld,M. and Caldecott,K.W. (2001) XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell, 104, 107–117. [DOI] [PubMed] [Google Scholar]
- Wilson D.M. III, Takeshita,M., Grollman,A.P. and Demple,B. (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem., 270, 16002–16007. [DOI] [PubMed] [Google Scholar]
- Wilson S.H. and Kunkel,T.A. (2000) Passing the baton in base excision repair. Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]
- Winters T.A., Weinfeld,M. and Jorgensen,T.J. (1992) Human HeLa cell enzymes that remove phosphoglycolate 3′-end groups from DNA. Nucleic Acids Res., 20, 2573–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Davies,S.L., Levitt,N.C. and Hickson,I.D. (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem., 276, 19375–19381. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S., Miao,G., Wang,F., Pan,Y.C. and Curran,T. (1992) Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J., 11, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S., Miao,G.G. and Curran,T. (1994) The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl Acad. Sci. USA, 91, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S., Smeyne,R.J., Wallace,J.D. and Curran,T. (1996) The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl Acad. Sci. USA, 93, 8919–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]