

Abstract
The SET domain proteins, SUV39 and G9a have recently been shown to be histone methyltransferases specific for lysines 9 and 27 (G9a only) of histone 3 (H3). The SET domains of the Saccharomyces cerevisiae Set1 and Drosophila trithorax proteins are closely related to each other but distinct from SUV39 and G9a. We characterized the complex associated with Set1 and Set1C and found that it is comprised of eight members, one of which, Bre2, is homologous to the trithorax-group (trxG) protein, Ash2. Set1C requires Set1 for complex integrity and mutation of Set1 and Set1C components shortens telomeres. One Set1C member, Swd2/Cpf10 is also present in cleavage polyadenylation factor (CPF). Set1C methylates lysine 4 of H3, thus adding a new specificity and a new subclass of SET domain proteins known to methyltransferases. Since methylation of H3 lysine 4 is widespread in eukaryotes, we screened the databases and found other Set1 homologues. We propose that eukaryotic Set1Cs are H3 lysine 4 methyltransferases and are related to trxG action through association with Ash2 homologues.
Keywords: chromatin/epigenetic/histone methyltransferase/telomere/trithorax-group
Introduction
Members of the trithorax group (trxG) have been identified by genetic screens of Drosophila for mutations that suppress phenotypes caused by disregulation of polycomb-group (PcG) action or mimic loss-of-function homeotic mutant phenotypes. As expected from these complex genetic screens, the trxG appears to encompass several subclasses of gene regulatory factors (Kennison, 1995). One subclass involves chromatin remodelling activity. The realization that the trxG member Brahma (BRM) is a homologue of Saccharomyces cerevisiae Swi2/Snf2 (Peterson and Herskowitz, 1992; Carlson and Laurent, 1994; Elfring et al., 1994) led to the definition of the SWI/SNF complex as a chromatin remodelling machine (Cote et al., 1994; Logie and Peterson, 1997) and the identification of another trxG member, moira, as a further component of the Drosophila SWI/SNF complex (Papoulas et al., 1998). Another trxG subclass encompasses the DNA binding proteins, zeste and GAGA factor. Although these proteins act independently, both appear to play similar roles in the stabilization of higher-order chromatin looping (Chen and Pirrotta, 1993; Katsani et al., 1999). A third subclass within the trxG (called here trxG3) that remains poorly understood includes trithorax itself, ash1 and ash2 (Shearn, 1989).
Insight into the potential molecular actions of trxG3 members came from the identification of several domains within their protein sequences (Mazo et al., 1990; Stassen et al., 1995; Adamson and Shearn, 1996; Tripoulas et al., 1996). Both trithorax (Trx) and Ash1 include a SET domain, which was identified through its occurrence in the chromatin factors, Su(var)3-9, enhancer of zeste [E(Z)] and Trx (Jones and Gelbart, 1993; Tschiersch et al., 1994). All three trxG3 members also include one or more PHD fingers (Aasland et al., 1995) and Ash2 includes a SPRY domain (Ponting et al., 1997). Of these domains, the SET domain in Su(var)3-9 homologues, in particular mammalian SUV39H1 and Schizosaccharomyces pombe Clr4, was recently identified as the first histone lysine methyltrans ferase (Rea et al., 2000), thus suggesting that other proteins containing SET domains could also be histone lysine methyltransferases. Subsequently, two other SET domain proteins, human G9a (Tachibana et al., 2001) and Drosophila Ash1 (C.Beisel, A.Imhof and F.Sauer, in preparation) have been shown to possess histone lysine methyltransferase activities. Notably each of these proteins have an additional cysteine-rich motif immediately N-terminal to their SET domains, previously referred to as a Cys-rich cluster or a preSET region. The presence of the preSET region in SUV39H1 is required for methyltransferase activity (Rea et al., 2000). Trx homologs contain a different type of preSET region, termed ATA2 (Prasad et al., 1997). Whether the absence of a SUV39-type preSET domain is the reason why Trx and E(Z) so far have tested negative in methyltransferase assays (Rea et al., 2000; C.Beisel, A.Imhof and F.Sauer, in preparation) is not yet clear.
By sequence alignments, the genome of S.cerevisiae encodes six genes with significant matches to the SET domain (now termed Set1–6; Pijnappel et al., 2001). Of these, the SET domain of Set1 is more similar to Trx SET domains than any other. Set1 does not appear to include a preSET Cys-rich region. In fact, S.cerevisiae does not appear to have a clear Su(var)3-9 homologue. set1 is not essential to yeast, however thorough analyses of its mutant phenotype revealed a variety of defects, including roles in silencing at mating-type loci and telomeres, metabolism, maintenance of telomere length (Nislow et al., 1997) and DNA repair (Corda et al., 1999; Schramke et al., 2001). Notably, expression of mammalian E(Z) homologues, either human EZH2 or murine Ezh1, in set1 strains restored the loss of gene repression at telomeres (Laible et al., 1997). To explore further Set1 function in S.cerevisiae, we characterized the Set1 complex (Set1C) and its proteomic environment using a sequential tagging and mass spectrometry approach (Rigaut et al., 1999; Shevchenko et al., 1999). We define Set1C as a complex of eight members which includes two proteins that display a SPRY domain and a PHD finger respectively. Thus it appears that Set1C incorporates an Ash2 analogue by physically associating two proteins that each carry a part of Ash2. Here we show that Set1C has histone methyltransferase activity specific for lysine 4 of histone 3 (H3) and is thus both the first histone lysine methyltransferase described in S.cerevisiae and the first from the Set1/Trx branch of SET domains. Together, these results imply that the Cys-rich preSET region is not essential for histone lysine methyltransferase activity in certain contexts and that unexpected aspects of trxG3 action also exist in S.cerevisiae.
Results
Biochemical characterization of Set1C
The protein complex associated with Set1 was identified by C-terminal tagging of Set1 with the TAP tag (Rigaut et al., 1999) and identification of co-purified proteins by MALDI mass spectrometry (Wilm et al., 1996; Shevchenko et al., 1999). All Coomassie stained bands present in Figure 1 were identified, however, only the eight that were subsequently confirmed as specific (see below) are depicted. All other bands in Figure 1 were identified as highly abundant ribosomal or heat shock proteins and have also been found in tagging experiments of unrelated proteins (results listed in Materials and methods).

Fig. 1. The composition of the Set1 complex. The affinity-purified Set1-TAP complex was separated on 7–25% SDS–PAGE and visualized by staining with Coomassie Blue. Molecular weight markers indicated on the left in kilodaltons. All bands present in this gel were identified and those subsequently determined to be specific to Set1C by repetition and further affinity purification exercises are depicted to the right. Each protein is depicted with identifiable protein domains and motifs as indicated in the key below. The thickened grey lines in Set1 and Spp1 indicate regions of further conservation to S.pombe proteins, Set1 and SPBC13G1.08c, respectively and the thickened grey line in Bre2 indicates the extent of further homology to Ash2 either side of the SPRY domain (see Figure 3C). The length of each polypeptide (aa) is noted on the right. The domains indicated are: PHD finger (Pfam:PF00628); n-SET (N-terminal SET associated domain in SET1 family; see Figure 3A); SET domain (Pfam:PF00856); postSET, C-terminal SET-associated peptide (SMART:00508); WD domain (Pfam:PF00400); RIIa, protein kinase A regulatory subunit dimerization domain (Pfam:PF02197); RRM, RNA recognition motif (Pfam:PF00076) and SPRY, domain in SPlA kinase and the ryanodine receptor (Pfam:PF00622). The SPRY domain in Bre2 is interrupted by three insertions (see Figure 3C). WD40 domains showing significant alignment scores are shown in light green, inferred alignments in white (data not shown).
Each of the seven proteins specifically co-purifying with Set1-TAP in Figure 1 were tagged and complexes were purified and identified (Figure 2). In five of the seven cases, all eight members of Set1C were specifically retrieved without any new co-purifying proteins. In the sixth case, tagging of Sdc1 yielded all members of Set1C except for Swd2, which was not identified in this experiment, possibly for technical reasons. In the last case, Swd2-TAP pulled down only a part of Set1C (Set1, Bre2, Spp1) and also, unexpectedly, co-purified the yeast cleavage polyadenylation factor (CPF) complex. The presence of Swd2 in CPF has been confirmed by independent work on CPF by B.Dichtl and W.Keller. Although all members of CPF were not identified in the experiment of Figure 2, our Swd2-TAP extracts are active for cleavage and polyadenylation (results of B.Dichtl and W.Keller, personnal communication). Swd2 is called Cpf10 by B.Dichtl and W.Keller. Notably, Swd2/Cpf10 is the only member of Set1C that is essential to yeast (SGD database; B.Dichtl and W.Keller, personal communication; our unpublished observations), probably because it provides an essential function for CPF, not Set1C. Since Swd2 was co-purified with Set1C when six of the seven other members were tagged, we conclude that it is a bona fide member and reason that either it is less stably associated with Set1C or that the TAP tag on Swd2 disturbs Set1C or both.

Fig. 2. Sequential affinity purification of Set1C. Each of the other seven members of Set1C was TAP-tagged and the purified complexes visualized by Coomassie Blue staining as indicated above each panel. The identity of each protein was established by mass spectroscopy and is indicated by numbers (see key to the right). Many unlabelled bands were also identified and were found to be highly abundant proteins (see Material and methods). The presence of the tag, after TEV cleavage, increases the size of the tagged protein by ∼10 kDa.
The stoichiometry of Set1C appeared to be uniform regardless of which member, except Swd2, was tagged. Careful inspection of Coomassie staining intensities from each of the affinity purified preparations led to a similiar estimate of Set1C stoichiometry. To arrive at this consensus, we discounted stoichiometries of the tagged proteins, since they may be over represented if the complex partially disassembled during purification. We estimate Set1C as Set1 (2); Bre2 (2); Swd1 (1); Spp1 (2); Swd2 (1); Swd3 (1); Sdc1 (<1); Shg1 (<1; relative stoichiometry in parentheses). In contrast to our experience with the Set3 complex (Set3C; Pijnappel et al., 2001), no Set1C member appeared to be clearly present as free protein, that is, in obvious (>4-fold) stoichiometric excess when purified in tagged form.
Bioinformatic analysis of Set1C members
The protein sequences of the eight members of Set1C were analyzed for matches in the databases (Figure 3). Database searches identified candidate orthologues of Set1 in S.pombe, Caenorhabditis elegans, Drosophila and humans. These five predicted proteins not only share very similar SET domains, but also two further regions of similarity, one of which includes a region with similarity to the RNA recognition motif (RRM, also known as RNP), the canonical RNA-binding domain (Figure 3A). Whereas all five protein sequences present the RNP motif, similarities between Set1 and S.pombe Set1 extend further either side of the RNP motif (denoted as a thick grey line in Figure 1). Immediately N-terminal to the SET domain, in the preSET position, these five proteins display a novel conserved region of ∼160 residues called here n-SET. Hence we conclude that Set1 orthologues are present in four diverse eukaryotes. Additionally, a predicted protein from the Arabidopsis genome shows Set1 homology in the n-SET and SET domains (Figure 3A), however, no associated RNP motif was identifiable from available sequence data.




Fig. 3. Sequence analyses of the Set1C. (A) Multiple sequence alignment of Set1 family members showing the RRM region (upper) and n-SET/SET/postSET region (lower). The alignment is colour coded in order to highlight the conserved features according to Gibson et al. (1994). The amino acid co-ordinates in each sequence are indicated after each of the two sequence blocks. The RRM domain (also called RNP) is highlighted with an orange bar. Four RRMs from human U2AF and RNPA are included for comparison. Assignment of secondary structure elements (H, helix; E, strand) is based on the known RRM structure (Burd and Dreyfuss, 1994). Below, the n-SET region is denoted by a light green bar, the SET domain by a dark green bar and the postSET motif by a yellow bar. The conserved residues characteristic for the methyltransferase catalytic core of the SET domain (Rea et al., 2000) are indicated with red dots. The position of stop codons is indicated by ‘<’. (B) Multiple alignments of selected preSET regions are shown. At the top, the preSET regions found in SUV39 and G9a families, termed preSET-s, is shown. At the bottom, the preSET region found in E(Z) and ASH1 families, preSET-e, is shown. (C) Multiple alignment of the SPRY region of Bre2 with the Ash2 family. The region of homology shown extends further N- and C-terminally than the defined SPRY domain, which, in this alignment, starts at residue 61 and ends at 283. Three insertions in Bre2 of 32, 46 and 42 residues are indicated. (D) Multiple alignment of a region in Sdc1 with Dpy30 and other sequences, including four human protein kinase A factors for reference. This region contains a motif that is related to the dimerization domain (here called RIIa) of protein kinase A regulatory subunits. The position of two α-helices in the RIIa structure (pdb:1r2a) is shown. The database sources of all proteins used in this figure and elsewhere in this paper are given in Table II.
The n-SET region is not cysteine-rich. However, the nature of the Cys-rich regions previously observed in SUV39, Ash1 and E(Z) families has not been clearly defined. Hence we examined these Cys-rich regions for significant alignments and found that they fall into two groups encompassing either SUV39 and G9a, called here preSET-s, or E(Z) and Ash1 called here preSET-e (Figure 3B). Neither show any significant similarity to n-SET. Together with the preSET region in the Trx family ATA2 (Prasad et al., 1997), we conclude that preSET regions fall into at least four distinct classes, ATA2, n-SET, preSET-s and preSET-e.
Bre2 was previously identified in a screen for mutations resistant to brefeldin, the toxin that disrupts the Golgi apparatus (Dinter and Berger, 1998). The relationship between brefeldin resistance of Δbre2 and our finding that Bre2 is entirely restricted to Set1C is unclear; however, loss of Set1 produces a plethora of cellular defects (Nislow et al., 1997; Corda et al., 1999), which may include perturbations of cytoplasmic protein translocation. Sequence analysis of Bre2 reveals a SPRY domain (Figure 3C), which is a domain originally found in splA kinase, ryanodine receptors and the trxG protein, Ash2 (Ponting et al., 1997). Amongst SPRY domains, Bre2 is most similar to that in the Ash2 family of proteins. In fact, the region of homology between Bre2 and Ash2 extends beyond the SPRY domain. The SPRY domain in Bre2 is interrupted by three large insertions relative to most other SPRY domains. These interruptions probably indicate insertions in flexible loops within the protein fold (Figure 3C). Notably, all members of the Ash2-family so far, except Bre2, carry a PHD finger.
Spp1 is one of the 14 genes in S.cerevisiae that present a PHD finger (unpublished observations). The PHD finger of Spp1 is most closely related to the PHD finger of S.pombe SPCC594.05c and then to human CGBP (data not shown), a protein that binds preferentially to unmethylated CpGs (Shin Voo et al., 2000).
Set1C includes three putative seven WD40 repeat proteins. Whereas Swd3 includes seven statistically significant WD40 repeats, Swd1 and Swd2 show only five or four, respectively, and the other repeats are inferred (not shown, white in Figure 1).
Sdc1 shows a significant match to Dpy-30 (Figure 3D), a protein required for sex-specific association of chromosome condensation factors during dosage compensation in C.elegans (Hsu et al., 1995). Sdc1, Dpy-30 and close relatives include a short motif related to the dimerization motif in the regulatory subunit of protein kinase A. This motif consists of two α-helices that form a special type of four-helix bundle during dimerization (Newlon et al., 1999).
Shg1 shows no significant similarity to any protein sequence in the databases.
Dissection of Set1C
Interactions within Set1C were evaluated by deletion of set1 in strains carrying the TAP tag fused to other Set1C members (Figure 4). In the absence of Set1, we observed that (i) Bre2-TAP and Sdc1 remain associated and no other Set1C members were found; (ii) Sdc1-TAP and Bre2 remain associated and no other Set1C members were found; (iii) Swd1-TAP and Swd3 remain associated and no other Set1C members were found and (iv) Spp1-TAP and Shg1-TAP were retrieved as free proteins with neither associating with any other Set1C member. Hence, Set1 is central to Set1C, which almost entirely disassembles in its absence. Of the variety of protein–protein interactions required for assembly of Set1C, only the interactions between Swd1 and Swd3, and Bre2 and Sdc1 have been previously mapped by two-hybrid approaches (Uetz et al., 2000; Ito et al., 2001). Notably, the stoichiometric relationship between Bre2-TAP and Sdc1 is 2:1; however, between Sdc1-TAP and Bre2 it is 1:1 (Figure 4). This implies that all Sdc1 is associated with Bre2, however, only half Bre2 is associated with Sdc1. This observation also recapitulates the relative stoichiometries of Bre2 and Sdc1 in Set1C as 2:1 and the observed lack of free protein of either. Hence we conclude that all cellular Bre2 and Sdc1 are incorporated into Set1C at a 2:1 ratio and Sdc1 stably binds Bre2.

Fig. 4. Dissection of interactions within Set1C. The structure of Set1C was examined in set1 strains carrying TAP-tagged Set1C members, as indicated above each panel, by affinity purification. All protein identities, including many unlabelled unspecific bands were established by mass spectroscopy. Numbers are the same as in Figure 2 and are 2, Bre2; 3, Swd1; 4, Spp1; 6, Swd3; 7, Sdc1; 8, Shg1.
Set1C specifically methylates lysine 4 of H3
Set1C extracts purified from either Bre2-TAP or Shg1-TAP strains showed histone methyltransferase activity when incubated with an H3 tail peptide (Figure 5A). Unexpectedly, Set1C extracts purified from the Set1-TAP strain showed no activity. Since the composition, stoichiometry and method of preparation of Set1C was the same in all three preparations, we attribute this result to inhibition of Set1C methyltransferase activity by the positioning of the protein tag at the C-terminus of Set1. As for Set1, the SET domain is very often found at the very C-terminus of SET domain proteins and the position of the stop codon can be regarded as a conserved feature (Ash1 is a notable exception). The presence of the protein tag at the C-terminus of Set1 may interfere with appropriate folding of the domain, inhibition of a dynamic aspect of enzyme activity or access of the substrate. These results also imply that full methyltransferase activity is not necessary for Set1C formation. As controls for the methyltransferase activity of Set1C, a TAP-Clr4 extract, purified by the same protocol, showed the expected enzyme activity and extracts made from Δset1 strains showed no activity (Figure 5A). To determine the specificity of Set1C, free histones were incubated with Set1C extracts and methylated products visualized by gel electrophoresis and fluorography (Figure 5B). Again, TAP-Clr4 displayed the expected specificity towards H3 and only the Bre2-TAP and Shg1-TAP extracts showed activity. In each case, only H3 was methylated with no sign of activity for the other histones. To determine which site on H3 was methylated, we incubated Set1C extracts with H3 peptides specifically mutated at either lysine 4 or lysine 9. Whereas the mutant lysine 9 peptide was methylated, the mutant lysine 4 peptide was not. As expected, incubation of these peptides with TAP-Clr4 gave the opposite result with no detectable methylation of the mutant lysine 9 peptide and strong methylation of the mutant lysine 4 peptide (not shown). Therefore, we conclude that Set1C is a histone lysine methyltransferase specific for lysine 4 of histone H3. Notably, we have so far been unable to obtain any methyltransferase activity from recombinantly expressed parts of Set1 (not shown). Our failure may merely reflect a technical problem, however, could indicate, along with the Set1-TAP result above, a sensitivity of the enzyme activity to its appropriate environment within in the complex.

Fig. 5. Set1C specifically methylates lysine 4 of H3. Histone methyltransferase activity was assayed by incubation of an H3 tail peptide (A) or free histones (B), with affinity purified extracts prepared from yeast strains carrying the TAP tag fused to Clr4 or Set1C components as indicated above each lane. The extracts were prepared from either wild-type or set1 strains as indicated. The TAP-Clr4 extract was prepared from the wild-type strain carrying a TAP-Clr4 CEN plasmid. (A) Extracts were incubated with an H3 N-terminal peptide in the presence of S-adenosyl-L-[methyl-3H]methionine and incorporated radioactivity determined by filter binding. Buffer, incubation of all reagents without any added extract. (B) Extracts were incubated with free histones in the presence of S-adenosyl-L-[methyl-3H]methionine, followed by gel electrophoresis and Coomassie Blue staining (left) and fluorography (right). (C) Set1C, isolated from a wild-type strain including Bre2-TAP, was incubated with H3 N-terminal peptides carrying either a lysine 4 to leucine (K4L) or lysine 9 to leucine (K9L) mutation.
Set1C activity in telomere length maintenance
Amongst a variety of phenotypic effects caused by the absence of set1, Nislow et al. (1997) noted that telomere lengths were shortened. We therefore examined telomere lengths in strains carrying Set1C mutations. In agreement with Nislow et al. (1997), removal of set1 significantly shortened telomere lengths by ∼100 bp (Figure 6, lanes 1 and 2). Telomeres are also shortened, although not as dramatically, in the Set1-TAP strain (lane 3). This concords with our demonstration that Set1C isolated using Set1-TAP lacks methyltransferase activity in vitro (Figure 5) and indicates that the histone methyltransferase activity of Set1C contributes to maintenance of telomere lengths. As a control, a strain containing N-terminal TAP-tagged Set1 (TAP-Set1; lane 4) did not show any reduction of telomere lengths, thus further strengthening our conclusion that C-terminal positioning of the TAP tag on Set1 selectively interferes with histone methyltransferase activity. Strains containing deletions of the Set1C components, bre2, swd1 and swd3 (lanes 5–7) also showed reduced telomere lengths. Whereas loss of either swd1 or swd3 resulted in a similar degree of shortening as loss of set1, loss of bre2 produced a milder effect. Taken together with the biochemical characterization of Set1C, these results indicate that Set1 action in maintenance of telomere lengths is mediated by Set1C and involves its methyltransferase activity. The intermediate impact on telomere lengths observed in Set1-TAP and bre2 strains also suggest that telomere maintenance by Set1C is not solely reliant on its methyltranseferase activity and another aspect of Set1C makes a contribution.
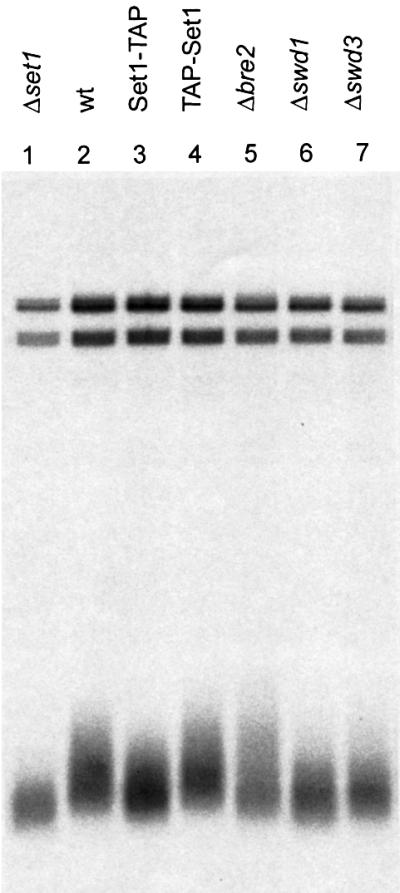
Fig. 6. Set1C activity is required for maintenance of telomere lengths. Telomeres were visualized by a Southern blotting strategy using a telomere specific probe. Genomic DNAs were isolated from the following strains: set1 (lane 1); wild type (lane 2); Set1-TAP (lane 3); TAP-Set1 (lane 4); bre2 (lane 5); swd1 (lane 6); swd3 (lane 7).
Discussion
The stable maintenance of gene expression patterns through mitotic divisions, usually termed epigenetic regulation, appears to be important during development in higher organisms. Epigenetic mechanisms provide a way for multicellular organisms to utilize genomic information in multiple overlapping patterns according to different cell lineages (Francis and Kingston, 2001). The search for a relationship between epigenetic mechanisms and development in higher eukaryotes has been led by the genetic screens in Drosophila that identified two opposing groups of candidates, PcG and trxG. PcG members are required to maintain patterns of gene repression, whereas trxG members appear to be required to maintain patterns of gene activity (Kennison, 1995; Mahmoudi and Verrijzer, 2001). Closer analyses of these phenomena have not yet delivered simple explanations of PcG and trxG epigenetic mechanism in development and recent evidence implies complex linkages to signalling, the cell cycle and the basal transcription apparatus (Gould, 1997; Jacobs et al., 1999; Voncken et al., 1999; Breiling et al., 2001; Saurin et al., 2001).
Clearer insights into epigenetic mechanisms have emerged from studies of the maintenance of chromatin states, such as those associated in yeast with centromeres (Karpen and Allshire, 1997), telomeres (Gottschling et al., 1990) mating-type silencing (Grunstein, 1998) or synthetic gene expression states in Drosophila (Cavalli and Paro 1998, 1999; Wakimoto, 1998). These studies lend support to the proposition that histone tails are the template for an epigenetic code, written in post-translational modifications involving phosphorylation, acetylation and methylation (Turner, 2000; Jenuwein and Allis, 2001). Of these modifications, the high turnover of phosphoryl and acetyl adducts on histone tails complicate simple models of inheritable chromatin states. In contrast, methyl groups on histone tails appear to be more stable (for discussion see van Holde, 1988). Consequently, histone tail methylation patterns may be primary inheritable enscriptions of epigenetic states which subsequently limit and/or direct other modifications (Rea et al., 2000; Bannister et al., 2001; Lachner et al., 2001; Nakayama et al., 2001).
To evaluate the merits of this suggestion, the specificities of histone methyltransferases and subsequent impact on patterns and hierarchies of histone tail modifications need to be unravelled. Towards this end, our identification of Set1C as the first H3 lysine 4 methyltransferase activity adds several aspects.
To the specificities of SUV39/Clr4 for H3 lysine 9 (Rea et al., 2000) and Ash1/G9a for H3 lysines 9 and 27 (Tachibana et al., 2001; C.Beisel, A.Imhof and F.Sauer, in preparation), we add the specificity for H3 lysine 4. Methylation of H3 lysine 4 appears to be widespread in eukaryotes (Strahl et al., 1999). Consequently, we anticipated that the corresponding methyltransferase may be widely conserved, so we screened the databases for Set1 homology. We found candidate orthologues in S.pombe, C.elegans and humans and also, after detailed sifting, in the Drosophila genome (Figure 3A). These predicted Set1 proteins have a similar architecture. They encompass three regions of clear homology (Figure 3A); (i) a centrally located region with homology to an RNA binding region known as the RRM motif (this may indicate a role for RNA binding, however RRM motifs also mediate protein–protein interactions); (ii) a region immediately N-terminal to the SET domain that is distinct and called the n-SET region and (iii) a very similar SET domain, which is followed by the most common SET domain peptide adduct, postSET and stop codon. Therefore we speculate that Set1 orthologues are widely distributed in eukaryotes and will be, in their respective complexes, H3 lysine 4 methyltransferases.
Set1C associates Set1 with Bre2. As for most Set1C proteins, it appears that all cellular Set1 and Bre2 are bound in Set1C in vegetatively growing haploid yeast. Bre2 shows sequence homology to Ash2 that stretches beyond their shared SPRY domains (Figure 3C). Bre2 does not include, however, a PHD finger, which is a conserved feature of Ash2 orthologues. Intriguingly, Set1C includes a PHD finger protein, Spp1. On the basis of these observations, we predict that the Ash2 complex in higher eukaryotes will include a Set1 orthologue and mediate H3 lysine 4 methyltransferase activity.
Thus, emerging and circumstantial evidence suggests that histone lysine methyltransferase activity is a common mechanistic feature of trxG3 (Trx, Ash1, Ash2) action. For Trx, lack of methyltransferase activity in vitro using recombinantly expressed protein and the absence of a cysteine-rich preSET region, have raised doubts about its ability to act as a methyltransferase (Rea et al., 2000; C.Beisel, A.Imhof and F.Sauer, in preparation). However, we show that Set1, which lacks a Cys-rich preSET region, functions as a histone lysine methyltransferase in the context of its native complex. Since the SET domain of Set1 is closely similar to that of Trx and the Trx SET domain includes all residues currently known to be important for enzyme activity, including the arginine beside the essential glutamine/histidine core (Rea et al., 2000; Figure 3A), we consider it probable that Trx acts as a methyltransferase in the context of its native complexes. Recombinantly expressed Ash1 has been recently shown to be a methyltransferase specific for H3 lysines 9 and 27 (C.Beisel, A.Imhof and F.Sauer, in preparation) and here we associate Ash2 with the Set1C H3 lysine 4 methyltransferase activity. Thus the previous genetic linkage between trxG3 members (Shearn, 1989) may reflect a common mechanistic linkage based on histone lysine methyltransferase activities of their protein complexes.
In addition to demonstrating by point mutation that the conserved core of the SUV39 SET domain was key to H3 lysine 9 methyltransferase activity, Rea et al. (2000) also showed that the Cys-rich preSET region was also required. Here we refine the classification of preSET regions by separating the Cys-rich regions of SUV39 and G9a families from those of E(Z) and Ash1 families to define the preSET-s (SUV3-9 class) and preSET-e [E(Z) class] regions (Figure 3B) and observe a novel preSET region in Set1 homologues, termed n-SET (Figure 3A). Along with the previously observed ATA2 preSET region in the Trx family (Prasad et al., 1997), it is now apparent that SET domains are N-terminally flanked by at least four preSET domains.
Many SET domains are also associated at their C-terminals with one of two short postSET peptide extensions. Most commonly, this C-terminal extension includes three cysteines and is immediately followed by a stop codon, except for the Ash1 subclass. SET domains in E(Z) homologues do not include these three cysteines, but do include other conserved residues plus a stop codon (postSET-z). Consequently, SET domain proteins can be classified by a combinatorial rule according to the (i) type of preSET region (one of four, so far; preSET-s, preSET-e, n-SET, ATA2), (ii) presence of a postSET or postSET-z C-terminal extension and (iii) presence or absence of a stop codon immediately following the postSET extension.
Interestingly, classification of SET domains by this combinatorial rule yields the same groupings as classification by homology within the SET domain itself. Figure 7 shows a tree of SET domains based solely on sequence alignments of SET domains excluding flanking regions. This analysis revealed that SET domains fall into four major branches; SUV39, Ash1, Set1/Trx and E(Z) groups. On the right of Figure 7, the distribution of N- and C-terminal associated regions and stop codons is depicted for each of the branches. A clear correspondence between the two ways to classify SET domains is evident. Boxed SET proteins in Figure 7 depict those currently shown to be associated with histone lysine methyltransferase activity. It can be seen that enzymatic activity has been found in three different branches. Furthermore, although no E(Z) member has yet been shown to associate with histone lysine methyltransferase activity, E(Z)H1 has been shown to rescue set1 defects in telomeric silencing in yeast (Laible et al., 1998). These analyses indicate that enzymatically active histone lysine methyltransferases are likely to be widespread amongst SET domain proteins and possibly all those listed in Figure 7 will have activity when tested in suitable ways. For some, this may require testing for activity using native complexes.

Fig. 7. Classification of SET domains by two different criterion yields the same groupings. At the left, a non-rooted tree of the SET domains, based on multiple sequence alignment of the SET domain, without inclusion of preSET and postSET regions, is shown. Four major groups are evident [SUV39, ASH1, SET1/TRX, E(Z)]. To the right, the distribution of flanking sequence elements is depicted. Known methyltransferases, present in three of the four major branches, are boxed.
Materials and methods
Strains
Strains used in this study are listed in Table I. Yeast transformations were performed as described (Soni et al., 1993). All haploid strains were derived from MGD353-13D (Puig et al., 1998). Gene disruptions and TAP-tag introduction were performed as described (Puig et al., 1998; Rigaut et al., 1999). Correct cassette integrations were confirmed by PCR and western blot analysis (for tagging) or PCR and genomic Southern blot (for disruptions).
Table I. Saccharomyces cerevisiae strains used in this study.
| Name | Genotype | Source |
|---|---|---|
| MGD-353-13D | Mata; ade2; arg4 (RV–); leu2-3112; trp1-289; ura3-52 | Puig et al. (1988) |
| AR2 | Mata; Set1 C-term. K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR6 | Mata; Spp1 C-term. TAP K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR11 | Mata; Swd1 C-term. TAP K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR12 | Mata; Swd3 C-term. TAP K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR13 | Mata; Swd2/Cpf10 C-term. TAP K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR16 | Mata; Sdc1 C-term. TAP K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR36 | Mata; Bre2 C-term. TAP K.l. TRP1 ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR35 | Mata; Shg1 C-term. TAP K.l. TRP ade2; arg4 (RV–); leu2-3112; ura3-52 | this study |
| AR18 | Mata; Spp1 C-term. TAP K.l. TRP1, Set1:: K.l.URA3 ade2; arg4 (RV–); leu2-3112 | this study |
| AR19 | Mata; Swd1 C-term. TAP K.l. TRP1, Set1::K.l.URA3 ade2; arg4 (RV–); leu2-3112 | this study |
| AR34 | Mata; Sdc1 C-term. TAP K.l. TRP1, Set1:: K.l.URA3 ade2; arg4 (RV–); leu2-3112 | this study |
| AR32 | Mata; Bre2 C-term. TAP K.l. TRP1, Set1:: K.l.URA3 ade2; arg4 (RV–); leu2-3112 | this study |
| AR31 | Mata; Shg1 C-term. TAP K.l. TRP1, Set1:: K.l.URA3 ade2; arg4 (RV–); leu2-3112 | this study |
| DS167 | Mata; ade2; arg4 (RV–); trp1-289; ura3-52; leu2-3112 +pYX142NT-Clr4 (LEU) | this study |
TAP purification and mass spectrometry (MS)
The extraction of yeast cells was performed as described for the yeast SWI/SNF complex (Logie and Peterson, 1999). The TAP tag consists of a calmodulin-binding peptide (CBP), a TEV protease cleavage site and two IgG-binding units of protein A as described (Rigaut et al., 1999). TAP purification was performed according to Rigaut et al. (1999) with the following modifications: 10 ml supernatant of the 43 000 g centrifugation (Logie and Peterson, 1999) was allowed to bind to 200 µl IgG–Sepharose (Pharmacia), equilibrated in buffer E (Logie and Peterson, 1999) for 2 h at 4°C using a disposable chromatography column (Bio-Rad). Two to three columns (the equivalent of 4–6 l yeast culture at OD600) were used per purification shown. The IgG–Sepharose column was washed with 35 ml buffer E lacking proteinase inhibitors, followed by 10 ml TEV cleavage buffer (Rigaut et al., 1999). TEV cleavage was performed using 10 µl (100 U) rTEV (Gibco) in 1 ml TEV cleavage buffer for 2 h at 16°C. Calmodulin–Sepharose (Stratagene) purification was as described (Rigaut et al., 1999). Purified proteins were concentrated as described by Wessel and Flügge (1984). After separation on 7–25% SDS–PAGE gradient gels, proteins were stained with Coomassie Blue, in-gel digested with trypsin and identified by MS as described (Shevchenko et al., 2000).
Histone methyltransferase assay (HMT)
HMT assays were done essentially as described previously (Strahl et al., 1999; Rea et al., 2000). Partially purified extracts were incubated in 1× methyltransferase buffer (MTB; 50 mM Tris pH 8.5, 20 mM KCl, 10 mM MgCl2 and 250 mM sucrose) in the presence of S-adenosyl-L-[methyl-3H]methionine (74 Ci/mmol, Amersham) as a methyl group donor at 1 µM final concentration and either 10 µg of free histones (Roche) or 5 µg N-terminal histone H3 peptides [wt 1–28 aa (Sigma); K4L and K9L 1–20 aa] for 1.5 h at 30°C in a total volume of 50 µl. Reactions were either spotted in duplicate on Whatman P81 paper, washed with 4 × 15 min with 50 mM NaHCO3 pH 9.0, completely dried and counted in a LSC or electrophoresed on 15% AA-SDS gels and subjected to fluorography.
Genomic Southern analysis
Yeast genomic DNA was extracted from exponentially growing cells. Two micrograms of each were digested with XhoI (NEB) and resolved on a 0.75% agarose gel in TBE buffer. The gels were capilliary blotted onto nylon (Biodyne B, Pall) and hybridized in Church/Gilbert solution at 65°C using a PCR fragment amplified from genomic DNA using the primers AGTTTAGCAGGCATCATC and CCTACTCTTTCCCACTTG to amplify the y′ repeats. The PCR fragment was labelled by random priming (Amersham) and recognises chromosomes 2, 4, 5, 6, 7, 8, 9, 10, 12, 13, 14, 15 and 16.
Sequence analysis
Database searches where performed with Blastp, Tblastn and psi-Blast (Altshul et al., 1997). Psi-Blast was only used to identify close homologues and searches were ended after three iterations. Previously characterized protein domains were identified with Pfam (Bateman et al., 2000) and SMART (Schultz et al., 2000) and with the interactive use of PairWise (version 1.6.2b; Birney et al., 1996). Multiple alignments were generated with Clustal_X (Thompson et al., 1997) and subsequently manually edited. The alignments to the RIIa and RRM motifs were performed with Clustal_X in profile mode, using corresponding seed alignments from Pfam as a reference.
Prediction of a second exon in the complete genome sequence of Drosophila melanogaster sequence (DDBJ/EMBL/GenBank accession No. AE02989) GI:10729576 was done by visual inspection and with the aid of Blast2 sequences and PairWise. The details of this prediction will be made available on the webpage http://www.uib.no/aasland/set/
A non-rooted tree of the SET domains (positions 1–138 in the alignment shown in Figure 3A) was generated with the NJ-method as implemented in Clustal_X. Positions with gaps where excluded.
Contaminanting proteins found in Figures 1, 2 and 4, listed as database entry numbers:
Figure 1
P11484, P26782, P10592, P02994, P23248, Q12109, P14126, P05753, P26783, P22203, P41805, P05737, P26786, P05740, P26781, Q12672, P04649.
Figure 2
Shg1 lane: P02994, Q12109, P00925, P00560, P14126, P49626, P00359, P05736, P05753, P29453, P22203, P26783, P05737, P53030, P26768, P07280, P26782, P02407, P05749, P04649.
Bre2 lane: P04050, P02994, Q12109, P14126, P10664, P05750, P32905, P05750, P05736, P22203, P26783, P05737, P26786, O13516, P05740, P02407, P04649.
Swd1 lane: P10592, P06634, P40150, P02994, Q01852, P23248, P26783, P22203, P26786, P07281, P02407, P53221, P04649, P06367.
Swd3 lane: P32589, P32503, P10591, P06634, P40150, P02994, Q12109, Q01852, P02365, P05750, P23248, P05753, P26783, P41805, P05737, P26786, P05740, P05756, P02406, P07280, P20407.
Swd2 lane: P10592, P11484, P02994, Q12109, P49626, P00359, P46654, P28495, P02365, P05750, P23248, P26783, P22203, P26786, P48164, P05740, P05735, P07280, P53221, P04649.
Spp1 lane: P10592, P11484, P02994, P14126, P10664, P02365, P26248, P29453, P05753, P26783, P05737, P05754, P41805, P26786, P26785, O13516, P05740, P04449, P32827, P02406, P26782, P02407, P06367, P04649, P07282.
Sdc1 lane: P16521, U43281, P46655, P10591, P40150, P02994, P14126, P10664, P39015, P00359, P05750, P46654, P23248, P05736, P05753, P25443, P22203, X89368, P05737, P41805, P05754, P40212, P05735, P38828, P05755, P26785, P05740, P47913, P24000, P41056, P07282, P05745.
Figure 4
Bre2/Set1 ko: P32589, P10591, P11484, P06634, P02994, P14126, Q01852, P10664, P00359, P26783, P41805, P26786, P05740, P07280, P02407, P26782, P04649, P06367.
Swd1/Set1 ko: P32589, P10591, P06634, P40150, P02994, Q12109, P14126, P25491, Q01852, P49626, P40531, P00359, P00358, P33442, P26783, P22203, P41805, P26786, P05740, P24000, P05735, P26781, P07281, P02407, P26782, P04456, P40213, P54780, P06367, P04649.
Spp1/Set1 ko: Q9URQ7, P02994, P00925, P14126, P10664, P00359, M26506, P05750, P32905, P23248, P05753, P26783, P22203, P41805, P26786, P05740, P47913, P26781, P04449, P02406, P07280, P02407, P26782, P04649, P39516, P05749, P07282.
Sdc1/Set1 ko: P32324, P32565, P32589, P10591, P40150, P06634, P02994, P00925, P00560, P14126, Q01852, P10664, P00359, P23248, P26783, P41805, P26786, P26782, P07280, P53221, P02407, P04649, P06367.
Table II. Source of sequence data.
| Abbreviation | Database source | Alias |
|---|---|---|
| Sc_Set1 | GI:6321911 | YHR119w |
| Sc_Bre2 | sw:YL1R_YEAST | FUN16, YLR015w |
| Sc_Swd1 | GI:6319320 | YAR003w |
| Sc_Spp1 | GI:2132201 | YPL138c |
| Sc_Swd2/Cpf10 | GI:549660 | YKL018w |
| Sc_Swd3 | GI:586308 | YBR175w, sw:YB25_YEAST |
| Sc_Sdc1 | GI:2131513 | YDR469w |
| Sc_Shg1 | GI:419867 | YBR258c |
| Mm_Suv39h2 | GI:9956936 | |
| Hs_SUV39H1 | GI:4507321 | |
| Dm_Suv39 | GI:1174492 | sw: SUV9_DROME |
| Sp_Clr4 | GI:11359019 | |
| Hs_G9a | GI:478844 | |
| Hs_WHSC1 | GI:13699811 | long isoform |
| Dm_ASH1 | GI:7511811 | |
| Sp_SET1 | GI:4704279 | SPCC306.04c |
| Dm_CG17396 | GI:7289568 | a |
| Hs_KIAA0339 | GI:6683126 | |
| Hs_KIAA1076 | GI:5689489 | b |
| Ce_C26E6.9c | GI:14550342 | |
| At_ATXR7 | GI:15488422 | c |
| Hs_MLL | GI:1170364 | sw:HRX_HUMAN |
| Dm_TRX | GI:1169582 | sw:TRX_DROME |
| Hs_EZH1 | GI:3334182 | sw:EZH1_HUMAN, ENX-2 |
| Hs_EZH2 | GI:3334180 | sw:EZH2_HUMAN |
| Dm_Ez | GI:1169582 | sw:EZ_DROME |
| Hs_RNPA | GI:133254 | sw:ROA1_HUMAN |
| Hs_U2AF | GI:267188 | sw:U2AF_HUMAN |
| Dm_CG6444 | GI:7297750 | |
| Dm_CG11591 | GI:7292426 | |
| Ce_DPY30 | GI:1706492 | sw:DP30_CAEEL |
| Hs_DPY30 | GI:14916555 | sw:DP30_HUMAN |
| Hs_NDK5 | GI:3914118 | |
| Hs_KAP1 | GI:1346362 | |
| Hs_KAP2 | GI:125198 | |
| Hs_KAP0 | GI:125193 | |
| Hs_SP17 | GI:2833264 | sw:SP17_HUMAN |
| Hs_ASH2L | GI:4210447 | |
| Hs_ASH2L2 | GI:3046995 | |
| Mm_ASH2L | GI:8392929 | |
| Dm_ASH2 | GI:7301188 | |
| Ce_Y17G7B.2a | GI:7509435 | |
| Sp_SPBC13G1.08c | GI:7491349 |
aThe N-terminal part of Dm_CG17396 is in the separate entry CG17395 (GI:7289567).
bFull-length Hs_KIAA1076 was obtained from Ensembl (www.ensembl.org) as AC079360.19.1.57549.167590.
cFull-length At_ATXR7 was obtained from DDBJ/EMBL/GenBank accession No. 3449329. The last 12 residues of this sequence were corrected due to a frame shift error.
Acknowledgments
Acknowledgements
We thank Axel Imhof, Thomas Jenuwein and Frank Sauer for discussions, Robin Allshire and Thomas Jenuwein for valuable materials and Bernhard Dichtl, Walter Keller and Frank Sauer for communication of results prior to publication.
References
- Aasland R., Gibson,T.J. and Stewart,A.F. (1995) The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci., 20, 56–59. [DOI] [PubMed] [Google Scholar]
- Adamson A.L. and Shearn,A. (1996) Molecular genetic analysis of Drosophila ash2, a member of the trithorax group required for imaginal disc pattern formation. Genetics, 144, 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A.J., Zegerman,P., Partridge,J.F., Miska,E.A., Thomas,J.O., Allshire,R.C. and Kouzarides,T. (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature, 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L.L. (2000) The Pfam protein families database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E., Thompson,J.D. and Gibson,T.J. (1996) PairWise and SearchWise: finding the optimal alignment in a simultaneous comparison of a protein profile against all DNA translation frames. Nucleic Acids Res., 24, 2730–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiling A., Turner,B.M., Bianchi,M.E. and Orlando,V. (2001) General transcription factors bind promoters repressed by Polycomb group proteins. Nature, 412, 651–655. [DOI] [PubMed] [Google Scholar]
- Burd C.G. and Dreyfuss,G. (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- Carlson M. and Laurent,B.C. (1994) The SNF/SWI family of global transcriptional activators. Curr. Opin. Cell Biol., 6, 396–402. [DOI] [PubMed] [Google Scholar]
- Cavalli G. and Paro,R. (1998) The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell, 93, 505–518. [DOI] [PubMed] [Google Scholar]
- Cavalli G. and Paro,R. (1999) Epigenetic inheritance of active chromatin after removal of the main transactivator. Science, 286, 955–958. [DOI] [PubMed] [Google Scholar]
- Chen J.D. and Pirrotta,V. (1993) Multimerization of the Drosophila zeste protein is required for efficient DNA binding. EMBO J., 12, 2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corda Y., Schramke,V., Longhese,M.P., Smokvina,T., Paciotti,V., Brevet,V., Gilson,E. and Geli,V. (1999) Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nature Genet., 21, 204–208. [DOI] [PubMed] [Google Scholar]
- Cote J., Quinn,J., Workman,J,L. and Peterson,C.L. (1994) Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science, 265, 53–60. [DOI] [PubMed] [Google Scholar]
- Dinter A. and Berger,E.G. (1998) Golgi-disturbing agents. Histochem. Cell Biol., 109, 571–590. [DOI] [PubMed] [Google Scholar]
- Elfring L.K., Deuring,R., McCallum,C.M., Peterson,C.L. and Tamkun,J.W. (1994) Identification and characterization of Drosophila relatives of the yeast transcriptional activator SNF2/SWI2. Mol. Cell. Biol., 14, 2225–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis N.J. and Kingston,R.E. (2001) Mechanisms of transcriptional memory. Nature Rev. Mol. Cell. Biol., 2, 409–421. [DOI] [PubMed] [Google Scholar]
- Gibson T.J., Hyvonen,M., Musacchio,A., Saraste,M. and Birney,E. (1994) PH domain: the first anniversary. Trends Biochem. Sci., 19, 349–353. [DOI] [PubMed] [Google Scholar]
- Gottschling D.E., Aparicio,O.M., Billington,B.L. and Zakian,V.A. (1990) Position effects at S.cerevisiae telomeres: reversible repression of Pol II transcription. Cell, 63, 751–762. [DOI] [PubMed] [Google Scholar]
- Gould A. (1997) Functions of mammalian polycomb group and trithorax group related genes. Curr. Opin. Genet. Dev., 7, 488–494. [DOI] [PubMed] [Google Scholar]
- Grunstein M. (1998) Yeast heterochromatin: regulation of its assembly and inheritance by histones. Cell, 93, 325–328. [DOI] [PubMed] [Google Scholar]
- Hsu D.R. Chuang,P.T. and Meyer,B.J. (1995) DPY-30, a nuclear protein essential early in embryogenesis for Caenorhabditis elegans dosage compensation. Development, 121, 3323–3334. [DOI] [PubMed] [Google Scholar]
- Ito T., Chiba,T., Ozawa,R., Yoshida,M., Hattori,M. and Sakaki,Y. (2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA, 98, 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.J., Kieboom,K., Marino,S., DePinho,R.A. and van Lohuizen,M. (1999) The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature, 397, 164–168. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis,C.D. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jones R.S. and Gelbart,W.M. (1993) The Drosophila polycomb-group gene enhancer of zeste contains a region with sequence similarity to trithorax. Mol. Cell. Biol., 13, 6357–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpen G.H. and Allshire,R.C. (1997) The case for epigenetic effects on centromere identity and function. Trends Genet., 13, 489–496. [DOI] [PubMed] [Google Scholar]
- Katsani K.R., Hajibagheri,M.A. and Verrijzer,C.P. (1999) Co-operative DNA binding by GAGA transcription factor requires the conserved BTB/POZ domain and reorganizes promoter topology. EMBO J., 18, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennison, J.A. (1995) The polycomb and trithorax group proteins of Drosophila: trans-regulators of homeotic gene function. Annu. Rev. Genet., 29, 289–303. [DOI] [PubMed] [Google Scholar]
- Lachner M., O’Carroll,D., Rea,S., Mechtler,K. and Jenuwein,T. (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature, 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Laible G., Wolf,A., Dorn,R., Reuter,G., Nislow,C., Lebersorger,A., Popkin,D., Pillus,L. and Jenuwein,T. (1997) Mammalian homologues of the polycomb-group gene enhancer of zeste mediate gene silencing in Drosophila heterochromatin and at S.cerevisiae telomeres. EMBO J., 16, 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logie C. and Peterson,C.L. (1997) Catalytic activity of the yeast SWI/SNF complex on reconstituted nucleosome arrays. EMBO J., 16, 6772–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logie C. and Peterson,C.L. (1999). Purification and biochemical properties of yeast SWI/SNF complex. Methods Enzymol., 304, 726–741. [DOI] [PubMed] [Google Scholar]
- Mazo A.M., Huang,D.H., Mozer,B.A. and Dawid,I.B. (1990) The trithorax gene, a trans-acting regulator of the bithorax complex in Drosophila, encodes a protein with zinc-binding domains. Proc. Natl Acad. Sci. USA, 87, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T. and Verrijzer,C.P. (2001) Chromatin silencing and activation by polycomb and trithorax group proteins. Oncogene, 20, 3055–3066. [DOI] [PubMed] [Google Scholar]
- Nakayama J., Rice,J.C., Strahl,B.D., Allis,C.D. and Grewal,S.I. (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science, 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Newlon M.G., Roy,M., Morikis,D., Hausken,Z.E., Coghlan,V., Scott,J.D. and Jennings,P.A. (1999) The molecular basis for protein kinase A anchoring revealed by solution NMR. Nature Struct. Biol., 6, 222–227. [DOI] [PubMed] [Google Scholar]
- Nislow C., Ray,E. and Pillus,L. (1997) SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol. Biol. Cell., 8, 2421–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoulas O., Beek,S.J., Moseley,S.L., McCallum,C.M., Sarte,M., Shearn,A. and Tamkun,J.W. (1998) The Drosophila trithorax group proteins BRM, ASH1 and ASH2 are subunits of distinct protein complexes. Development, 125, 3955–3966. [DOI] [PubMed] [Google Scholar]
- Peterson C.L. and Herskowitz,I. (1992) Characterization of the yeast SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell, 68, 573–583. [DOI] [PubMed] [Google Scholar]
- Pijnappel W.W.M. et al. (2001) The S.cerevisiae SET3 complex includes two histone deacetylases, HOS2 and HST1, and is a meiotic-specific repressor of the sporulation gene program, Genes Dev., 15, 2991–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C., Schultz,J. and Bork,P. (1997) SPRY domains in ryanodine receptors (Ca2+-release channels). Trends Biochem. Sci., 22, 193–194 [DOI] [PubMed] [Google Scholar]
- Prasad R. et al. (1997) Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene, 31, 549–560. [DOI] [PubMed] [Google Scholar]
- Puig O., Rutz,B., Luukkonen,B.G., Kandels-Lewis,S., Bragado-Nilsson,E. and Séraphin,B. (1998). New constructs and strategies for efficient PCR-based gene manipulations in yeast. Yeast, 14, 1139–1146. [DOI] [PubMed] [Google Scholar]
- Rea S. et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature, 406, 593–599. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko,A., Rutz,B., Wilm,M., Mann,M. and Séraphin,B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnol., 17, 1030–1032. [DOI] [PubMed] [Google Scholar]
- Saurin A.J., Shao,Z., Erdjument-Bromage,H., Tempst,P. and Kingston,R.E. (2001) A Drosophila polycomb group complex includes zeste and dTAFII proteins. Nature, 412, 655–660. [DOI] [PubMed] [Google Scholar]
- Schramke V., Neecke,H., Brevet,V., Corda,Y., Lucchini,G., Longhese,M.P., Gilson,E. and Geli,V. (2001) The set1δ mutation unveils a novel signaling pathway relayed by the Rad53-dependent hyperphosphorylation of replication protein A that leads to transcriptional activation of repair genes. Genes Dev., 15, 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J., Copley,R.R., Doerks,T., Ponting,C.P. and Bork,P. (2000) SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res., 28, 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn A. (1989) The ash-1, ash-2 and trithorax genes of Drosophila melanogaster are functionally related. Genetics, 121, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Zachariae,W. and Shevchenko,A. (1999) A strategy for the characterization of protein interaction networks by mass spectrometry. Biochem. Soc. Trans, 27, 549–554. [DOI] [PubMed] [Google Scholar]
- Shevchenko A., Loboda,A., Ens,W. and Standing,K.G. (2000). MALDI quadrupole time-of-flight mass spectrometry: a powerful tool for proteomic research. Anal. Chem., 72, 2132–2141. [DOI] [PubMed] [Google Scholar]
- Shin Voo K., Carlone,D.L., Jacobsen,B.M., Flodin,A. and Skalnik,D.G. (2000) Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax and methyl-CpG binding domain protein 1. Mol. Cell. Biol., 20, 2108–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni R., Carmichael,J.P. and Murray,J.A. (1993). Parameters affecting lithium acetate-mediated transformation of Saccharomyces cerevisiae and development of a rapid and simplified procedure. Curr. Genet., 24, 455–459. [DOI] [PubMed] [Google Scholar]
- Stec I., Wright,T.J., van Ommen,G.J., de Boer,P.A., van Haeringen,A., Moorman,A.F., Altherr,M.R. and den Dunnen,J.T. (1998). WHSC1, a 90 kb SET domain-containing gene maps in the Wolf-Hirschorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet., 7, 1071–1082. [DOI] [PubMed] [Google Scholar]
- Stassen M.J., Bailey,D., Nelson,S., Chinwalla,V. and Harte,P.J. (1995) The Drosophila trithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mech. Dev., 52, 209–223. [DOI] [PubMed] [Google Scholar]
- Strahl B.D., Ohba,R., Cook,R.G. and Allis,C.D. (1999) Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl Acad. Sci. USA, 96, 14967–14972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M., Sugimoto,K., Fukushima,T. and Shinkai,Y. (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem., 276, 25309–25317. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschiersch B., Hofmann,A., Krauss,V., Dorn,R., Korge,G. and Reuter,G. (1994) The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J., 13, 3822–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripoulas N., LaJeunesse,D., Gildea,J. and Shearn,A. (1996) The Drosophila ash1 gene product, which is localized at specific sites on polytene chromosomes, contains a SET domain and a PHD finger. Genetics, 143, 913–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B.M. (2000) Histone acetylation and an epigenetic code. BioEssays, 22, 836–845. [DOI] [PubMed] [Google Scholar]
- Uetz P. et al. (2000) A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- van Holde K.E. (1988) Chromatin. In Rich,A. (ed.), Springer Series in Molecular Biology. Springer-Verlag, New York, NY.
- Voncken J.W., Schweizer,D., Aagaard,L., Sattler,L., Jantsch,M.F. and van Lohuizen,M. (1999) Chromatin-association of the polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J. Cell Sci., 112, 4627–4639. [DOI] [PubMed] [Google Scholar]
- Wakimoto B.T. (1998) Beyond the nucleosome: epigenetic aspects of position-effect variegation in Drosophila. Cell, 93, 321–324. [DOI] [PubMed] [Google Scholar]
- Wilm M., Shevchenko,A., Houthaeve,T., Breit,S., Schweigerer,L., Fotsis,T. and Mann,M. (1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature, 379, 466–469. [DOI] [PubMed] [Google Scholar]
- Wessel D. and Flugge,U.I. (1984). A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem., 138, 141–143. [DOI] [PubMed] [Google Scholar]