

Abstract
Bifunctional DNA glycosylases employ an active site lysine or the N-terminus to form a Schiff base with an abasic (AP) site base excision repair intermediate. For 8-oxoguanine DNA glycosylase 1 (OGG1), cleaving this reversible structure is the rate-determining step in the initiation of 8-oxoguanine (8-oxoG) repair in DNA. Evolution has led OGG1 to use a product-assisted catalysis approach, where the excised 8-oxoG acts as a Brønsted base for cleavage of a Schiff base intermediate. However, the physicochemical properties of 8-oxoG significantly limit the inherent enzymatic turnover leading to a weak, cellularly absent, AP lyase activity. We hypothesized that chemical synthesis of purine analogues enables access to complex structures that are suitable as product-like catalysts. Herein, the nucleobase landscape is profiled for its potential to increase OGG1 Schiff base cleavage. 8-Substituted 6-thioguanines emerge as potent and selective scaffolds enabling OGG1 to cleave AP sites opposite any canonical nucleobase by β-elimination. This effectively broadens the enzymatic substrate scope of OGG1, shaping a complete, artificial AP-lyase function. In addition, a second class of compounds, 6-substituted pyrazolo-[3,4-d]-pyrimidines, stimulate OGG1 function at high pH, while thioguanines govern enzymatic control at acidic pH. This enables up to 20-fold increased enzyme turnover and a de novo OGG1 β-elimination in conditions commonly not tolerated. The tool compounds employed here are non-toxic in cells and stimulate the repair of AP sites through a natural, APE1 dependent pathway, as opposed to previously reported β,δ-lyase stimulator TH10785.
OGG1 lyase-activators are organocatalytic switches that enhance OGG1 initiated DNA repair by stimulating rudimentary activities through direct catalytic involvement, establishing de novo enzymatic functions on a widened substrate scope.
Oxidative damage in the form of 8-oxoG is the most common DNA lesion in human cells. An accumulation of 8-oxoG, its oxidation products or subsequent mutations caused through their presence leads to deterioration of cellular health and ultimately to neurodegenerative1,2 and cardiovascular3,4 diseases, as well as cancer.5,6 OGG1 is the enzyme responsible for the removal of 8-oxoG through its glycosylase function and literature suggests that stimulative targeting of OGG1 function may be a viable therapeutic strategy.7,8
Targeting OGG1 to improve 8-oxoG repair could be achieved using small molecule activation, increased enzyme levels or by upregulation of post-translational modifications that enhance enzyme activity. Interestingly, enhancement of OGG1 activity has been demonstrated through HDAC1-mediated deacetylation9 or through allosteric activation.10 Furthermore, we have previously reported on the activation of OGG1 through its weak AP-lyase activity by a small molecule (TH10785, Fig. 1).11
Fig. 1. Screening for OGG1 organocatalytic switches based on substrate similarity: a number of OGG1 organocatalytic switches have previously been reported. As an 8-oxoG analogue, 8-bromoguanine is a known OGG1 lyase-activator catalyzing the inherent β-elimination activity of OGG1. Based on substrate similarity, we screened an in-house library of diverse modified nucleobases and discovered 8-methylpurines as potent OGG1 AP lyase activators (3–5). Additional chemotypes covered thioguanine analogues including FDA-approved drugs (1, 2), as well as 6-amino-substituted purines (6–9) but not adenines or 9-substituted nucleobases. Assay details in Materials and methods.

Currently, it is assumed that the weak intrinsic AP-lyase activity of OGG1 is controlled by product-assisted catalysis, a distinct mode of action compared to classic allosteric regulation (Scheme S1).12 After OGG1 has extruded and recognized 8-oxoG, the base lesion is removed employing the efficient glycosylase function of the enzyme and forming a Schiff base between OGG1 residue Lys249 and the resulting apurinic site (AP site). Then, the previously excised 8-oxoG may act as a weak chemical base and abstract a proton from the Schiff base intermediate. In vitro, this process results in a weak AP-lyase activity through β-elimination, which leads to 3′-DNA strand incision and removal of OGG1 from the product. In cells, this effect is negligible and OGG1 is thus a monofunctional glycosylase.13 Instead, recruited apurinic/apyrimidinic endonuclease 1 (APE1) blocks rebinding of AP sites through OGG1 by its nuclease function, effectively preventing duration of Schiff base complexes.11,14,15
Since the recruitment and binding to the AP site by APE1 requires time,16 release of OGG1 from the Schiff base or alternatively its cleavage is the rate-determining step in the initiation of base excision repair (BER).17 Activating the AP-lyase activity of OGG1 through small molecules therefore leads to increased repair of AP sites, potentially by avoidance of APE1 recruitment. Accelerated release of OGG1 from the Schiff base would then (a) immediately stimulate AP site repair and (b) increase the capacity for 8-oxoG repair as an effect of increased OGG1 release.11,18
In agreement with the product-assisted catalysis mechanism postulated by Fromme et al.,12 we reported OGG1 lyase-activators that establish proton-abstraction events to foster an artificial β,δ-elimination generating a gap flanked by 5′P and 3′P ends. With the AP lyase activity being stimulated, this novel elimination overwrites the dominant glycosylase function of OGG1 in cells, rendering it a β,δ-AP lyase that creates products whose repair pathways are independent of APE1.18
Since small molecule activators typically bind allosteric sites and activate an already present function, we termed the bifunctional compounds organocatalytic switches (ORCAs) since they:
(a) contain a heterocyclic nitrogen center with Brønsted base-like character,
(b) bear a structural handle with active site affinity,11
(c) partake in the biochemical reaction,
(d) are not changed in the process, and
(e) as a result, alter, i.e. switch, the protein function
As a relatively new concept in the literature, several critical points remain to be addressed. The first question raised was whether there are additional chemical series that activate OGG1, especially if compounds derived from the natural substrate 8-oxoG could act as ORCAs. Indeed, we confirmed 8-bromoguanine (8-BrG), a more soluble analogue of 8-oxoG, to activate OGG1, however only by stimulation of the residual β-elimination function (Fig. 1). Secondly, while β,δ-elimination additionally requires bifunctional polynucleotide phosphatase/kinase (PNKP1) function for end cleansing, controlling OGG1 incision through β-elimination leads to dependance on APE1 only.19 At the current, this distinct initiation of base excision repair is incompletely understood in the literature. To more deeply investigate the potential and structure–activity relationships (SAR) of β-elimination activators, we sought to establish a series of purine-based ORCAs of OGG1 based on the chemotype of 8-BrG.
Here, we screened a library of nucleoside and nucleobase analogues and discovered two chemical series of nucleobase-based molecules that activate OGG1 on AP sites. Chemical modification has revealed a detailed SAR and enzymatic assays complimented by co-crystal structures suggest active site binding and not allosteric regulation as the mode of action. We observed that very potent ORCAs of this series control OGG1 function opposite any canonical nucleobase and that different scaffolds can be employed to govern enzymatic function in a pH range distinct from naïve OGG1. The compounds developed enable OGG1-mediated repair of AP sites dependent on APE1 and are powerful tools to investigate increased loading of this canonical pathway within base excision repair.
Results
Screening of in-house library
We started by screening a 500-compound in-house library enriched for nucleosides and nucleobases, which was built up from compounds available through the National Cancer Institute's Developmental Therapeutics Platform (NCI DTP). Additionally, we manually added compounds that were synthesized during our campaigns for nucleobase binding proteins, such as the NUDIX proteins and alternative DNA glycosylases to OGG1.20–24 Previously, we introduced the concentration of half-maximal activation (AC50), which reflects the activity of 10 nM OGG1 with a given compound concentration that reaches 50% assay turnover compared to 10 nM OGG1 with 2 nM APE1. Consequently, the AC50 may be altered depending on assay conditions and the amount of APE1 used. Thus, as a quantitative readout to compare compound activity with APE1 function, we determined AC50 for all screened compounds on the substrate 8-oxoA:C through kinetic readout instead of a single point read for inhibitors. 8-oxoA was incorporated instead of 8-oxoG, since the latter is prone to further oxidation and could therefore jeopardize the reproducibility of results.25 For details of this screening assay (Z′ = 0.853), the workflow and data handling, please see Fig. S1.25
Among the primary hits were the FDA approved drugs thioguanine 1 and azathioprine 2 (Fig. 1 and Fig. S2), 8-substituted thioguanines 3–5 and compounds combining the purine scaffold with amines in the 6-position (6–9). Interestingly, a number of 8-monosubstituted thioguanines, but not adenines, were found to be particularly active. In addition, we found that N9-modified analogues or nucleoside structures were inactive in the assay, suggesting the necessity of an unsubstituted nitrogen in that position of the molecule (Fig. S2).
Investigation of thioguanine analogues
Based on the promising screening results for the 6-thioguanine chemotype, we initially directed our attention towards hits within this series. First, we confirmed purity and identity, and reordered solid material from NCI DTP for the determination of AC50. Then we generated a number of analogues of 8-substituted 6-thioguanines with larger substituents (10 and S23–S26, Table 1, Table S1A). Interestingly, only small substituents were tolerated in the 8-position, as observed for compounds 3 and 10. We observed a decrease in potency upon extension of the synthetic system via propyl (10), methylamino (S25) and inactivity for a more extended N-Boc substituent (S26). Next, we explored the apparent necessity of an unsubstituted 6-thio modification by generating analogues with thioether or sulfone modifications (S27–S39, Table S1B). Again, small substituents such as ethyl (S28), iso-propyl (S29) and propene (S31) gave better results, while extended systems (S14, S15 and S36–39) were inactive below 100 μM. Finally, a combination of small substituents in both the 6-thioether as well as the 8-position failed to further improve the activity of this chemical series (S33–S35). These studies suggest a highly specific SAR for thioguanines, allowing only minor modifications to the core system.
Table 1. Optimization towards potent and selective OGG1 stimulators: assay details in Materials and methods. AC50 in μM, CI95 confidence interval 95% in μM.
| # | Structure | AC50 (CI 95) [μM] |
|---|---|---|
| 3 |
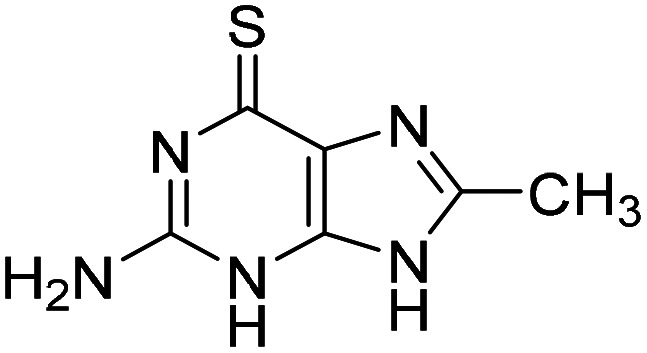
|
0.32 (0.27–0.39) |
| 10 |
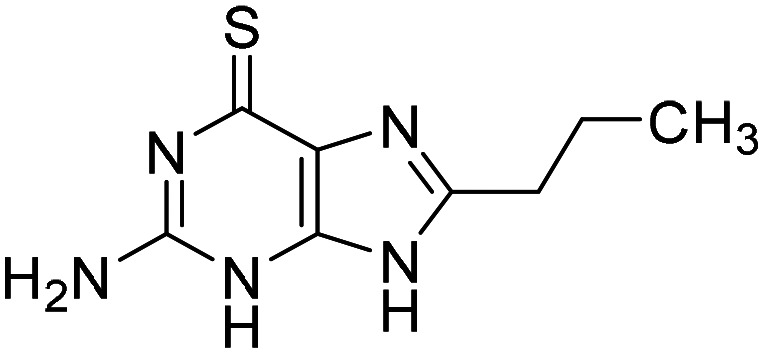
|
0.55 (0.26–1.20) |
| 11 |
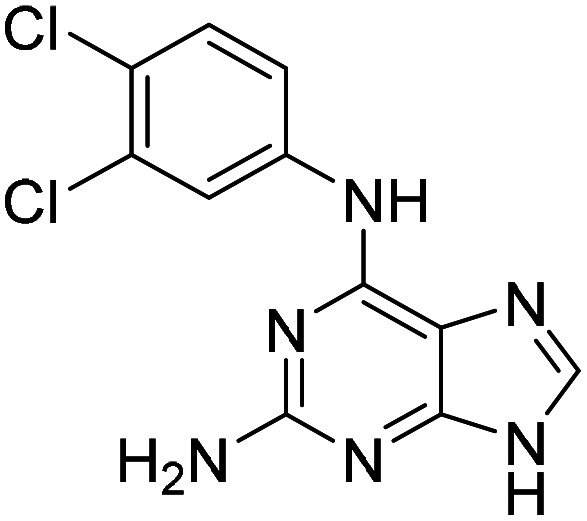
|
12.5a |
| 12 |
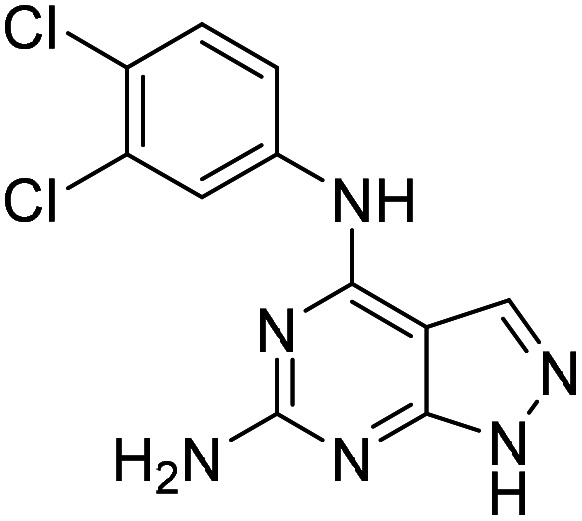
|
13.1 (2.3–75.1) |
| 13 |

|
>100 |
| 14 |
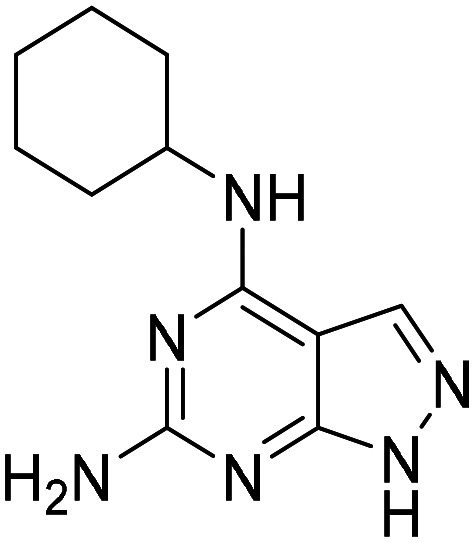
|
11.7 (5.5–24.9) |
| 15 |

|
28.2 (10.0–79.8) |
Compound only reaches AC35 due to a bell-shape activity curve.
Investigation of 6-amino substituted purines
To follow up on the primary hits with a 6-amino substituted purine core (6–9, Table 1 and Table S2), we confirmed identity, reordered solid material from NCI DTP and determined AC50 as before. Previously, we had shown that OGG1 inhibitor modifications25 combined with the quinazoline core of TH10785 would still yield activating molecules.11 This investigation was performed to show that the combination of affinity to the target and reactivity on the substrate within one small molecule was both possible and necessary to activate OGG1 on AP sites. To build on the screening results for 6–9 and to assess whether these earlier observations could be further corroborated in a scaffold hopping approach, we generated matched pairs of different amines with the core structure of hit molecule 6 (Table S2). Using a mix of acidic and alkaline coupling conditions, a number of derivatives were synthesized (11 and S40–S45). As a readout for activity, the biochemical assay was performed which showed that with the exception of 6 and 11, 6-aniline modified analogues are not significantly activating OGG1 below 100 μM. Thereby, 11, bearing a 3,4-dichloroaniline substituent, exhibited a bell-shaped activity curve peaking at a concentration of 12.5 μM but below an AC50 value (AC35, Fig. S3). Previously, we observed similar behavior for TH10785 and concluded that this curve reflects a competition with the 8-oxoA substrate in higher concentrations.11
Further, we generated matched pair compounds for some of the thioguanines synthesized previously and observed improved activity of the amine analogues (S36vs.13 and S38vs.6, Tables S1B and S2). If these molecules indeed bind the active site, this comparison suggested that the hit scaffold requires a secondary amine, namely an R1R2–NH, between the nucleobase core (R1) and the 6-modification (R2) to be able to activate OGG1. In contrast, a panel of additional sp2-richer amines (S18–20, Fig. S2, S42 and S43, Table S2) indicated a preference for small substituted tertiary amine rings. Previously, we had observed that the N-methyl version of TH10785, TH11735, was still able to induce activation of OGG1, albeit with more modest turnover.11
6-Substituted pyrazolo-[3,4-d]-pyrimidine are activators of the OGG1 AP lyase activity
Having identified the 3,4-dichloroaniline derivative (11) as the most promising member among the 6-substituted purines, we then investigated the scope of accepted nucleobase analogues, replacing the guanine alike core with adenine, uracil and other heterocycles (12 and S46–S49, Table S3A). Interestingly, while 9-methylated analogue S49 and uracil derivative S48 remained inactive, the 6-amino-pyrazolo-[3,4-d]-pyrimidine derivative 12 surpassed purine analogue 11 in the biochemical assay, with an AC50 similar to the peak effect of 11. In addition, 6-unsubstituted pyrazolo-[3,4-d]-pyrimidine S47 activated OGG1, while adenine analogue S46 remained inactive. Following this finding, we assembled a number of matched pairs based on previously synthesized 6-aminoaryl-imidazo-[3,4-d]-pyrimidines (13, 14 and S50–S56, Table S3B) and observed that all members of the pyrazolo-[3,4-d]-pyrimidine series surpassed the activity of their imidazo counterparts (11vs.12, 13vs.14).
Organocatalytic switches bind the active site of OGG1
To investigate whether the identified molecules bind to the active site during OGG1 catalysis, we evaluated the biochemical activity of 3 and 14 on OGG1 mutant variants in the fluorescence-based assay. While incision through wtOGG1 and Ser326Cys were enhanced, active site mutants Phe319Ala, Cys253Tyr and Lys249Trp were not affected by incubation with the compounds, suggesting that the activity of the compounds is exerted from within the active site (Fig. 2). This finding was corroborated by solving the X-ray co-crystal structures of mouse OGG1 in complex with 3, 10, 14 and 15 confirming active site binding (Fig. 3A–D and Fig. S4). The binding poses of compounds 14 and 15, interacting both with Phe319 and Gly42, confirmed the selectivity observed within the SAR. An overlay with the 8-oxoG-bound human OGG1 (PDB ID: 1HU0)12 indicated no significant rearrangements in the core protein structure for all structures solved. Identical placement of 3, 10 and 8-oxoG (Fig. 3C and D, Fig. S4) suggests a binding mode that allows for enhanced product-assisted like catalysis. The heterocyclic ring systems of 14 and 15 are observed to be shifted outwards due to their more spacious 6-amino substitution and are also flipped compared to one another, which may explain the lower activity observed for this series in all assays (Fig. S4). In addition, this observation supports the finding during SAR optimization of the thioguanine series, where 6-substitution led to a reduced activity compared to 6-unsubstituted analogues.
Fig. 2. Assessment of OGG1 mutants confirms activation of variants with changes outside but not within the active site: left: 3 and right: 14 are enhancing wtOGG1 and the S326C mutant, but not F319A, C253Y and K249W. 10 nM 8-oxo:A was used as a substrate and was incubated with 10 nM hOGG1. Compounds were assayed with the respective mutants and the initial slope was measured in the fluorophore-quencher assay. Assay details in Materials and methods. Structures of compounds 3 and 14 depicted in Table 1. Placement of relevant amino acids shown in Fig. S4 and S5.

Fig. 3. Organocatalytic switches bind the active site of OGG1 and closely overlap with 8-oxoG position: (A) superposition of mouse OGG1 bound 15 (green) and mouse OGG1 bound 14 (blue) and human OGG1 bound DNA-8-oxoG (magenta, PDB ID: 1HU0) monomers. DNA from the 8-oxoG complex is coloured light orange. Ligands are depicted as sticks; C atoms are coloured green (15), blue (14) or magenta (8-oxoG), O atoms red, and N atoms dark blue; (B) Close up comparison of ligand binding between the structures in (A). Amino acids which contribute to ligand positioning are depicted as thin sticks; (C) superposition of mouse OGG1 bound 3 (green) and mouse OGG1 bound 10 (blue) and human OGG1 bound DNA-8-oxoG (magenta, PDB ID: 1HU0) monomers. DNA from the 8-oxoG complex is coloured light orange. Ligands are depicted as sticks; C atoms are coloured green (3), blue (10) or magenta (8-oxoG), O atoms red, N atoms dark blue and S atoms gold; (D) close up comparison of ligand binding between the structures in (C). Amino acids which contribute to ligand positioning are depicted as thin sticks. Structures of compounds 10, 14 and 15 depicted in Table 1.

Nucleobase based organocatalytic switches increase β-lyase activity of OGG1 depending on pH
To determine whether the synthesized structures were activating a β,δ-elimination in OGG1 similar to previously reported structures TH10785 and analogues,11 we first elucidated the exact nature of the observed biochemical activity, i.e. β- or β,δ-AP-lyase. Therefore, we assessed the reaction of compound and protein on the substrate using a 32P-radiolabelled substrate (see Materials and methods) and further resolution of the products by PAGE. Compounds 13 and 3 (Table 1) – as members of two different nucleobase series – were chosen for evaluation in this way. Both compounds 3 and 13 were confirmed as activators of the inherent β-lyase functionality of OGG1 after 2 and 4 minutes respectively (Fig. 4). The corresponding reaction product of β-elimination, 3′phospho unsaturated aldehyde (3′-PUA), was rapidly formed by OGG1 in the presence of 13 and 3. This product was also observed with TH10785, although in this case OGG1 rendered an additional band with a higher electrophoretic mobility and corresponding to a β,δ-elimination 3′-P product, as previously reported.11 As expected, the phosphatase activity of T4PNK had no effect on the 3′-PUA product common to 3, 13, and TH10785 (Fig. S6). For TH10785, the enzyme removed the phosphate group from the 3′P product, generating a product with slightly reduced electrophoretic mobility, confirming its identity as the β,δ-elimination product. Extended incubation periods of 30 minutes or longer gave the β,δ-elimination product in the cases of 3 and 13, possibly through unspecific cleavage since DMSO and the OGG1 inhibitor TH5487 showed a similar response (Fig. S6). Although devoid of β,δ-AP-lyase activity, the intensity of the substrate and product bands confirmed 3 was a quantitatively stronger activator than TH10785 and 13.
Fig. 4. Effect of compounds 3 and 13 on the AP-lyase activity of hOGG1 on 8oxoG:C-containing DNA. Upper panel: PAGE gel after autoradiographic readout. The assay was performed using 1 nM of the [32P]5′-labeled uridin-containing substrate, previously treated with 0.2 U of E. coli UDG to obtain a natural AP site, incubated with 10 nM hOGG1, 20 mM EDTA and either 10% DMSO or 6.25 μM TH10785, 20 μM 13 and 20 μM 3. After incubation for the indicated times at 37 °C, reactions were stopped and samples further analyzed by 7 M urea-20% PAGE and autoradiography. C1: control of no UDG-treated DNA incubated for 8 min at 37 °C; C2: control of UDG treated DNA incubated for 8 min at 37 °C; C3: alkaline degradation of the UDG treated DNA. Lower panel: bar chart of the AP lyase activity (n = 3; means ± SEM). Significance of the results was determined with a two-tailed paired t-test. *P < 0.05; P < 0.01; ***P < 0.001. 3′-PUA – 3′ phospho unsaturated aldehyde. Structures of compounds 3 and 13 depicted in Table 1.

Due to the necessity of employing a nucleophilic lysine residue and cleavage of a Schiff base via proton abstraction, OGG1 functions optimally in a pH close to 8, where both glycosylase and AP lyase processes are ensured to progress in a reasonable time frame. Since we previously observed a pH preference for TH10785, all AC50 measurements during screening and optimization were performed at a pH of 7.5. This ensured that compounds were profiled in a way that also included their possible activity in slightly more acidic or more alkaline conditions but mainly their putative activity in a cellular system. Due to this dynamic interplay between OGG1 modulator and enzyme, we next assessed the artificially controlled AP-lyase activity across a pH range of 6.7 to 8.4. For this, we measured the initial rate of the reaction in the fluorescence-based biochemical assay using the 8-oxoA substrate and compounds TH10785, 3 and 14. We chose the most potent concentrations of each compound, according to their activity profile. As indicated in Fig. 5, 14 pronounced the already increased OGG1 enzymatic turnover at higher pH close to or over 8. The highest fold increase in function was however observed at lower pH. Like 3 (5 μM) and TH10785 (6.25 μM), 14 (100 μM) also allowed OGG1 to cleave AP sites even in a slightly acidic reaction buffer of pH 6.7, installing a de novo function. Interestingly, the effect of 3 was the strongest at low pH. Finally, TH10785 stimulated OGG1 function the most around a pH of 7.5. A weakened effect was seen for high pH, where OGG1 function was unchanged in the presence of TH10785.
Fig. 5. Compound pKa governs pH range of enzymatic activity: left: 3 activates OGG1 at a pH closer to 7 and below; middle: TH10785 follows a bell-shaped curve with a maximum at pH 7.5; right: 14 has a high pKa and thus controls OGG1 function at a pH above 8. 10 nM 8-oxoA was used as substrate and was incubated with 10 nM hOGG1. Compounds were used at most effective concentation as indicated and v0 was measured within the initial linear slope of the reaction in the flourophore-quencher assay. Structures of compounds 3 and 14 depicted in Table 1, structure of TH10785 depicted in Fig. 1.

To investigate the effects of compound binding to OGG1 further, we assessed protein stability using the melting temperature at different pH and in the presence of the compounds using nano-DSF (Table S4). The presence of the employed compounds led to a general increase in protein stability, as well as an increase in stability at extreme pH similar to compound pKa, which may partly explain the observed enhancement of enzymatic activity in the biochemical assay through binding of the compounds to OGG1.
Lastly, to assess whether proton transfer reactions have an influence on compound activity, we applied D2O as solvent, investigating a potential solvent isotope effect. In the fluorescence-based assay we observed challenged incision efficacy evidenced by the slower rate of the reaction compared to conditions using H2O (Fig. S7). These investigations draw a complex picture of protein stability, compound binding, proton abstraction and transfer, as well as substrate and leaving group solvation during elimination events.
Strongly activated OGG1 cleaves AP sites opposite all canonical nucleobases
Since OGG1 has a preference for 8-oxoG opposite cytosine,27 we hypothesized that a substantially increased AP lyase function could overwrite this cytosine selectivity. Thus, we investigated the reaction of OGG1 under saturating concentrations of substrates 8-oxoA:C and AP:A, AP:C, AP:G and AP:T, generated from their respective uracil containing precursors using UNG2. Using 3 and TH10785 to evaluate a potential influence of β- or β,δ-elimination capabilities, we found that both compounds pronounced the inherent preference for cytosine (Fig. 6, Fig. S8A and B). Further, both compounds exhibited a competition effect for the 8-oxoA substrate, where higher concentrations lead to reduced rates. This effect was less prominent for AP site substrates, where the rate is plateauing instead. In contrast to TH10785, 3 was able to stimulate a significant OGG1 AP lyase function on all substrates, suggesting that compound potency rather than type of AP lyase function governs the reaction on AP-sites. We further rule out unspecific acceleration of strand cleavage by compound 3 in absence of OGG1 by control experiments on AP:C substrate alone (Fig. S8C).
Fig. 6. Reaction velocity for TH10785 and 3 against saturating concentration of AP site substrates and 8-oxoA:C: left: 3 activates OGG1 on AP sites opposite any canonical nucleobase; right: TH10785 activates OGG1 on AP sites in the same manner and no incision dependant efficiency is observed; v0 of reaction was measured. 10 nM 8-oxo:A was used as substrate and was incubated with 10 nM hOGG1. U:X substrate was used to generate AP sites opposite canonical nucleobases using 1 nM UNG2. v0 was measured within the initial linear slope of the reaction in the fluorophore-quencher assay. Details in Materials and methods. Structures of compound 3 and TH10785 depicted in Fig. 1.

Organocatalytic switches are selective, non-toxic and stimulate the repair of AP sites in cells
To rule out any unwanted effects of the molecules regarding potential toxicity or off-target effects, we extensively profiled TH10785, 3 and 14 using a functional panel of enzymes consisting of DNA glycosylases,21 NUDIX family members20,28 and a set of protein kinases which was probed using thermal stability.29 Limited inhibition below 50% was observed within the family of DNA glycosylase at 100 μM, indicating over 100-fold selectivity for 3 and TH10785. All other tests returned negative for off-targets (Tables S6, S7 and Fig. S9, S10).
Further, considering that some thioguanines are approved cytotoxic drugs we sought to exclude an effect on cell viability. Cultivation of an immortalized cell line, BJ-TERT, and an oncogene driven cell line, BJ-Ras,23,30 over three days in the presence of a dose response of the compounds confirmed the absence of any toxicity between these two cell lines (Table S8).
Using these selective and non-cytotoxic scaffolds, we investigated whether either of the OGG1 functions were indeed improved in a cellular setting. Determining thermal stabilization using DSF revealed that 10 (1.5 K) and 14 (0.5 K) stabilized OGG1 more than 3 (0.5 K) and 13 (0.1 K). Thus, we applied 10 and 14 moving forward and induced DNA damage in U2OS cells using KBrO3 and profiled 8-oxoG, AP sites and γH2AX as a general marker for DNA damage. We observed increased levels of nuclear 8-oxoG over 6 hours post exposure (Fig. 7A). This effect was rescued by the OGG1 organocatalytic switches. Cellular efficacy followed the biochemical activity for ORCAs of the β-elimination, as 10 was superior to 14, with TH10785 being most efficient as a stimulator of β,δ-elimination. This indeed indicated an increased cellular repair of 8-oxoG lesions in DNA, possibly through more free protein after accelerated repair.11 However, considering the compounds redirect OGG1 function towards resolving AP sites, we expected a more pronounced effect for analogues 10 and 14 when assessing the levels of this particular type of DNA damage. Using the aldehyde reactive probe31 and fluorescence-activated cell sorting we indeed observed reduced numbers of AP sites for compound 10, performing at the level of TH10785 (Fig. 7B). 14 was found to be inactive at these concentrations, possibly due to a suboptimal concentration in relation to its biochemical activity. Lastly, γH2AX levels were unaltered between the different compounds (Fig. S11), indicating sustainable DNA repair upon faster initiation of BER. Collectively, these studies confirm enhanced, compound-mediated OGG1 activity in canonical repair of oxidative DNA damage in vitro and in cells. The effects observed suggest OGG1 is rendered an AP lyase in presence of potent OGG1-ORCAs.
Fig. 7. Organocatalytic switches stimulate removal of 8-oxoG and AP sites: (A) effect for compounds TH10785, 14 and 10 at 10 μM in a cellular setting. Quantification of nuclear 8-oxoG levels across different time points in U2OS cells exposed to organocatalytic switches or DMSO, under oxidative stress conditions (20 mM of KBrO3 for 1 h). Each bar represents the mean ± SEM. Data are the average of three independent experiments. For each experiment, 25 fields and around 1000 cells were captured per condition. Statistical significance was calculated using two-way ANOVA for multiple comparisons. ns, non-significant; *P < 0.05; ****P < 0.0001; (B) a comparative analysis of ARP-STREP_PE signal induction over DMSO, reported in percentage, is shown for compounds TH10785, 14 and 10 at 10 μM. Each bar represents the mean ± SD. Data are the average of five independent experiments with at least three biological replicates each. Statistical significance was calculated using an unpaired two-tailed Student's t-test. ns, non-significant; ***P < 0.001; ****P < 0.0001. Structures of compounds 10 and 14 depicted in Table 1, structure of TH10785 depicted in Fig. 1.

Discussion
Removal of 8-oxoG from DNA is necessary to maintain cellular health and genomic integrity. At the same time, the presence of 8-oxoG is crucial to recruit repair enzymes and transcription factors to remodel cellular responses in inflammation,32,33 after physical activity and in human disease.34,35 Both scenarios benefit from a tight control of human BER, in which OGG1 recognizes and removes 8-oxoG as the initiating enzyme. While bifunctional enzymes with similar substrate scope exist in lower organisms,36 OGG1 appears to have a negligible AP lyase function which cleaves the reaction product, an AP site. Whether this occurred as a result of an evolutionary advantage has not been thoroughly investigated. However, literature indicates that an AP lyase function is at the center of transcriptional processes, since both OGG1 and the succeeding enzyme APE1 are implicated in the recruitment of transcription factors to genomic regions rich in potential substrates.17,37
Still, APE1 is recruited to relieve OGG1 from the AP site. Due to the required unbinding of the AP site from OGG1, rebinding of APE1 and incision through APE1, significant rearrangements are accomplished within the repair complex. Furthermore, when bound to an AP site via a reversible Schiff base, OGG1 remains unavailable for additional excision events. In scenarios, such as neurodegenerative and cardiometabolic diseases as well as rapid aging, this delay of repair may cause an accumulation of 8-oxoG as well as AP sites. Mutations and excessive DNA damage may be the result, threatening genomic integrity. Thus, improving OGG1 function by enhancing a weak AP lyase activity has recently gained traction with first applications in patient-derived models of liver fibrosis.38,39
The discovery of TH10785, an organocatalytic switch, has been reported that improves AP site cleavage of OGG1 independent of APE1.11 The small molecule binds the active site and removes an activated proton from the Schiff base intermediate. This effectively shapes an β,δ-elimination reaction that resembles the AP lyase function of DNA glycosylases from lower organisms, such as Fpg from bacteria.36
At the same time, human BER has evolved to accomplish different DNA-end cleansing depending on the elimination reaction that was performed. β-Eliminations generate a 3′-PUA which is still a substrate of APE1. β,δ-Eliminations, however, require PNKP1 before the reaction product converges again with the stream resulting from β-elimination. Thus, TH10785 rewires the initiation of base excision repair to become independent of APE1 and to rely on PNKP1 instead.
OGG1 can employ the excised 8-oxoG for a weak β-elimination and we have shown that 8-bromo guanine indeed activates this reaction further. Thus, we hypothesized that complex nucleobase space may present (a) additional more potent analogues and (b) offer insight in the mechanistic differences of OGG1 β- and β,δ-eliminations. Therefore, we profiled the nucleobase chemical space for organocatalytic switches of OGG1 using a fluorophore-quencher assay and discovered two distinct selective chemical series, including 6-thioguanines and 6-aminopurines. An early indication of active site binding was observed in the inactivity of 9-substituted analogues during the screen. Assuming an orientation similar to 8-oxoG during catalysis, these analogues would not be able to abstract protons from the intermediate Schiff base.
Investigation of the SAR revealed that 6-thioguanine tolerated only minor groups in the 8-position, with an AC50 of up to 0.32 μM, which was later supported by obtained co-crystal structures. 6-Aminoguanine derivatives on the other hand were optimized to bear substituted aniline substituents, reaching moderate μM activity while their thioguanine counterparts displayed only weak (compound S36) or no activation (compound S38, Table S1). This apparent requirement for H-donor-bonding in addition to a π-stacking with the bicyclic hetero-aryl system agreed with the current knowledge on organocatalytic switch binding, requiring both interactions with OGG1 active site amino acids Gly42 and Phe319. Especially the adaptation of the aniline substitution pattern from previously published OGG1 inhibitors increased the potency of these 6-amino purines. Finally, investigating the space of alternative scaffolds we observed that members of a pyrazolo-[3,4-d]-pyrimidine series surpassed the activity of the corresponding imidazo-pyrimidine series. Taken together, these results suggest that previously optimized OGG1 inhibitor chemical space can be utilized to optimize the affinity handle of a series of OGG1 organocatalytic switches.25 At the same time, the polar and nitrogen-rich scaffold of nucleobases appears suitable to stimulate proton abstraction during OGG1 catalysis.
In an attempt to rationalize the observed SAR, we then assessed key molecules in the catalysis of OGG1 mutants. As before,11 we only saw a stimulation of mutants outside of the active site. This confirmed an orthosteric binding of the small molecules. The exact orientation of compounds 3 and 10 which was further elucidated using co-crystal structures was in line with the 8-oxoG binding mode (PDB ID: 1HU0), enabling for a mechanism like the product-assisted catalysis. All four obtained co-crystal structures confirmed the established SAR, with the small molecules engaging key amino acid residues Gly42 and Phe319. Especially, the thioguanine series showed excellent similarity to the placement of 8-oxoG during catalysis. This overlapping in binding mode confirmed the observed SAR and the limited toleration of substituents in 8-position. At the same time, the co-crystal structures of 14 and 15, offered deeper insight into the activity of nucleobase analogues. The spacious aniline derivatives led to a outwards shifted position of the entire molecule, as visible by the position of active site residue Cys253 (Fig. S4). Together, the crystal structures suggest, that a combination of 6- and 8-substitution would be too sterically demanding for the active site, explaining the lower activity of these analogues.
Assessment in the fluorophore-quencher assay allowed to compare compounds to a pathway stimulated by APE1, but does not provide the identity of the reaction product. We therefore used a 32P-labelled DNA substrate and resolved the reaction of OGG1 with 3, 13 and TH10785 by PAGE. As before, TH10785 induced β,δ-elimination while 3 and 13 catalyzed an β-elimination in accordance with their previously established AC50. Although, these findings confirmed the earlier observation of Fromme et al. that guanine analogues are assisting in catalysis driven by product-alike properties, the absence of a β,δ-elimination catalyzed by potent analogues such as 3, suggest that TH10785 acts through a distinct mechanism. Previously, it was assumed that a cascade β,δ-elimination occurs through the stimulation of an initiating β-elimination by TH10785. The fact, that potent molecules identified in this study fail to control the second elimination, warrants further investigation.
Since proton abstraction and the lysine attack are both processes that thrive in more basic conditions, we hypothesized that compound pKa and pH environment play a role in OGG1 activity. 3 promoted an AP lyase activity in slightly acidic conditions, while 14 was more active at higher pH over 8. As observed earlier, TH10785 performed best around pH 7.5. These findings pointed towards the presence of a basic nitrogen or a changed protonation state of the enzyme binding site. With respect to the former, the observed activity enhancement was in accordance with the calculated26 pKa of the nitrogen bases within the used compounds, TH10785 (1N: 6.55 ± 1.13), 3 (9N: 2.59 ± 2.22) and 14 (9N: 13.53 ± 2.00) demonstrating that compounds 3, 14 and TH10785 are more potent in catalyzing reactions that are closer to their individual pKa. Since the AC50 is dependent on pH, compound pKa can have important ramifications on attenuating activity in different biological compartments.
Since a number of processes during glycosylase and AP lyase function require proton abstraction, we investigated protein stabilization by the compounds as well as a solvent effect using D2O as solvent. While stabilization was indeed pH dependent and followed the general trend of compound pKa, the use of D2O indicated significant slower incision events in the fluorophore-quencher assay. The active center nitrogen of the compounds experiences a vivid proton or deuterium exchange in solution. Thus, this result points towards challenged proton abstraction from the α-carbon of the Schiff base intermediate. Altogether, these investigations draw a complex picture of protein stability, compound binding, proton abstraction and transfer, as well as substrate and leaving group solvation during elimination events. The question of whether pKa is the driver of the elimination reaction or favors protein-Schiff base binding remains elusive. Given, that all structures used in this study bear multiple nitrogen atoms with a variety of pKa, we see the need to identify a minimum structure of organocatalytic switches. Modulating the pKa of a minimalist structure would then allow to assess whether active nitrogen pKa is an additional descriptor for the development of ORCAs.
Another evolutionary selection that OGG1 underwent, is the selectivity for an 8-oxoG:C pair as opposed to for example the selectivity of MUTYH for 8-oxoG:A. With the AP lyase activity stimulated, we thus asked the question whether OGG1 continues to discriminate against the other canonical nucleobases. Generating AP sites opposite adenine, cytosine, guanine and thymine and assessing the burst phase of the reaction of OGG1 with 3 and TH10785, we observed a continued selectivity for cytosine independent of incision mode. Still, incision events were markedly stimulated for all substrates used, enabling cleavage of all AP sites in double stranded DNA. The reduction of reaction rates in the presence of higher concentrations of compound 3 or TH10785 strongly occurs for 8-oxoA, but less for AP site containing substrates. These observations indicated competition with substrates that require extended space within the active site which is obstructed when a compound binds prior to the excision of the damaged base.11 Additionally, OGG1-independent acceleration of substrate cleavage by the compounds 3 or 14 was ruled out by enzyme-free control experiments.
These properties prompted us to evaluate the effects of the compounds in cells. Since OGG1 is able to incise AP sites in the presence of organocatalytic switches as a new substrate in addition to the removal of 8-oxoG, we set out to assess the levels of both substrates after a burst in oxidative DNA damage. We determined the levels of 8-oxoG and AP sites using immunofluorescence using an anti-8-oxoG-antibody and quantification of AP sites through an aldehyde reactive probe and FACS, respectively. As a proxy of target engagement, we used DSF to prioritize one compound per series, i.e.10 and 14. We observed both reduced levels of 8-oxoG and AP sites using compound 10 confirming an acceleration of initiation of base excision repair. No such effect was observed for 14, which was likely connected to the fact that the concentration used is close to the compound's AC50. In addition, it may be possible that compounds are less active in certain cellular environments, depending on their individual pKa. Future research will need to address the possibility with a chemical series that allows for robust pKa modulation, as mentioned above.
The presented molecules activate the canonical but rudimentary β-elimination activity of OGG1 from within the active site of the protein. Active site affinity and a reactive center in the form of a basic nitrogen are combined in one molecule, as evidenced by studies involving OGG1 mutants, substrate scope, generated products and enzymatic activity covering a range of pH environments. Co-crystal structures of a number of analogues further confirm the product-assisted hypothesis of Fromme et al. and paint a detailed picture of functional enhancement of enzymatic activity on Schiff bases.
In conclusion, we demonstrate that a chemical space beyond TH10785 exists that increases the repair of OGG1. Importantly, we show that these OGG1 ORCAs may act through a distinct mechanism of action and in contrast to TH10785 primarily stimulate the β-elimination during OGG1 catalysis. At the current time, it remains a challenge to explain why some molecules assist in β-elimination and why others stimulate a β,δ-elimination. Future research will have to show whether distinct chemical series can stimulate a cascade event or more readily abstract the γ-proton for an additional elimination reaction.
Still, OGG1-ORCAs are the first chemical entities that rewrite an enzymatic function in cells by partaking in the biochemical reaction, allowing for increased DNA damage repair. Considering the widespread implications of OGG1 function within a number of diseases, including neurodegeneration,1,2 obesity3,4 and inflammation,7,32 these small molecules are novel, powerful tools to unravel disease biology, and offer the potential for further development into promising drug candidates.38,40
Author contributions
Chemical synthesis: ECH, NDDE, OW, FO, AD, MEA, KN, MV, JS, MK and MM; biochemistry: ECH, NDDE, LM, AdP, PC, KZ, EW, AK, HD, ASJ, MdV and MM; structural biology: ERS, SK and PS; computational chemistry: EJH, NDDE and MM; cell biology: CBB, IA, ML and MM; supervision: OW, ASJ, MS, PS, MdV, SKn, LS, TH and MM; resources: TH, PS, MdV, CBB, LS, SKn and MM; writing and editing: ECH, NDDE, AE, OW and MM; conceptualization: ECH, NDDE and MM.
Conflicts of interest
OW and TH are listed as inventors on a U.S. patent no. WO2019166639 A1, covering OGG1 inhibitors. The patent is fully owned by a non-profit public foundation, the Helleday Foundation, and TH is a member of the foundation board. MS is an employee of Oxcia, a company developing OGG1 inhibitors. EW, OW, EJH, IA, ASJ, TH and MS are shareholders of Oxcia. The remaining authors declare no competing financial interests.
Supplementary Material
Acknowledgments
This work was funded by the Swedish Research Council (2018-03406 PS), the Alfred Österlund foundation (PS), the Crafoord foundation (20190532 PS), the Helleday Foundation (FO, LM), the Swedish Cancer Society (CAN2021/1490 TH), the Åke-Olsson foundation for haematological research (2020-00306 MM), a Novo Nordisk Pioneer Innovator Grant (NNF23OC0085944 MM), Karolinska Institutet Research Foundation Grants (2020-02186 and 2022-01776 MM), a Childhood Cancer Research Postdoctoral grant (ML), the Åke Wiberg Foundation (M23-0043 MM), Grant PID2020-115978GB-I00 (MdV) funded by MCIN/AEI/10.13039/501100011033, a Health Research Fund, Carlos III Health Institute, co-funded by European Regional Development (FEDER) funds (PI20-00335 LSG) and the Research Talent Attraction Grants (2023-T1/SAL-GL-29292 CBB). This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement no 875510 (MM, EJH, EW, IA, ASJ, AK, SKn, FO, KZ, MK, NDE). The JU receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA and Ontario Institute for Cancer Research, Royal Institution for the Advancement of Learning McGill University, Kungliga Tekniska Högskolan, Diamond Light Source Limited. This communication reflects the views of the authors and the JU is not liable for any use that may be made of the information contained herein. We would like to thank the Chemical Biology Consortium Sweden (CBCS), the Protein science facility (PSF) at Karolinska and the Structural Genomics Consortium (SGC) at Karolinska and Goethe-Universität Frankfurt for support. We are thankful to Athina Pliakou, Mari Kullman Magnusson, Kristina Edfeldt, Therese Pham, Opher Gileadi, and Michael Sundström for administrative support. We thank MAXIV Laboratory (Sweden, proposal MX20200204), Diamond Light Source (United Kingdom, proposals MX15806 and MX21625), and their scientists from the BioMAX and I03 beamlines for their support during data collection.
Data availability
The data supporting this article, description of the materials and methods, spectral data and compound purity data have been included as part of the supplementary information (SI). See DOI: https://doi.org/10.1039/d4cb00323c.
References
- Oka S. Leon J. Sakumi K. Abolhassani N. Sheng Z. Tsuchimoto D. LaFerla F. M. Nakabeppu Y. MTH1 and OGG1 maintain a low level of 8-oxoguanine in Alzheimer's brain, and prevent the progression of Alzheimer's pathogenesis. Sci. Rep. 2021;11:5819. doi: 10.1038/s41598-021-84640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukae J. Takanashi M. Kubo S. Nishioka K. Nakabeppu Y. Mori H. Mizuno Y. Hattori N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson's disease and related neurodegenerative disorders. Acta Neuropathol. 2005;109:256–262. doi: 10.1007/s00401-004-0937-9. [DOI] [PubMed] [Google Scholar]
- Komakula S. S. B. Tumova J. Kumaraswamy D. Burchat N. Vartanian V. Ye H. Dobrzyn A. Lloyd R. S. Sampath H. The DNA Repair Protein OGG1 Protects Against Obesity by Altering Mitochondrial Energetics in White Adipose Tissue. Sci. Rep. 2018;8:14886. doi: 10.1038/s41598-018-33151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath H. Vartanian V. Rollins M. R. Sakumi K. Nakabeppu Y. Lloyd R. S. 8-Oxoguanine DNA Glycosylase (OGG1) Deficiency Increases Susceptibility to Obesity and Metabolic Dysfunction. PLoS One. 2012;7:e51697. doi: 10.1371/journal.pone.0051697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero J. M. Benítez-Buelga C. Rajagopal V. Zhenjun Z. Torres-Ruiz R. Müller S. Hanna B. M. F. Loseva O. Wallner O. Michel M. Rodríguez-Perales S. Gad H. Visnes T. Helleday T. Benítez J. Osorio A. Small molecule inhibitor of OGG1 blocks oxidative DNA damage repair at telomeres and potentiates methotrexate anticancer effects. Sci. Rep. 2021;11:3490. doi: 10.1038/s41598-021-82917-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visnes T. Benítez-Buelga C. Cázares-Körner A. Sanjiv K. Hanna B. M. F. Mortusewicz O. Rajagopal V. Albers J. J. Hagey D. W. Bekkhus T. Eshtad S. Baquero J. M. Masuyer G. Wallner O. Müller S. Pham T. Göktürk C. Rasti A. Suman S. Torres-Ruiz R. Sarno A. Wiita E. Homan E. J. Karsten S. Marimuthu K. Michel M. Koolmeister T. Scobie M. Loseva O. Almlöf I. Unterlass J. E. Pettke A. Boström J. Pandey M. Gad H. Herr P. Jemth A.-S. El Andaloussi S. Kalderén C. Rodriguez-Perales S. Benítez J. Krokan H. E. Altun M. Stenmark P. Berglund U. W. Helleday T. Targeting OGG1 arrests cancer cell proliferation by inducing replication stress. Nucleic Acids Res. 2020;48:12234–12251. doi: 10.1093/nar/gkaa1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M. Chu X. Duan Sahbaz B. Gray S. Pekhale K. Park J.-H. Croteau D. L. Bohr V. A. Mitochondrial OGG1 expression reduces age-associated neuroinflammation by regulating cytosolic mitochondrial DNA. Free Radical Biol. Med. 2023;203:34–44. doi: 10.1016/j.freeradbiomed.2023.03.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptiste B. A. Katchur S. R. Fivenson E. M. Croteau D. L. Rumsey W. L. Bohr V. A. Enhanced mitochondrial DNA repair of the common disease-associated variant, Ser326Cys, of hOGG1 through small molecule intervention. Free Radical Biol. Med. 2018;124:149–162. doi: 10.1016/j.freeradbiomed.2018.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao P.-C. Patnaik D. Watson L. A. Gao F. Pan L. Wang J. Adaikkan C. Penney J. Cam H. P. Huang W.-C. Pantano L. Lee A. Nott A. Phan T. X. Gjoneska E. Elmsaouri S. Haggarty S. J. Tsai L.-H. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer's disease. Nat. Commun. 2020;11:2484. doi: 10.1038/s41467-020-16361-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G. Katchur S. R. Jiang Y. Briand J. Schaber M. Kreatsoulas C. Schwartz B. Thrall S. Davis A. M. Duvall S. Kaufman B. A. Rumsey W. L. Small molecule-mediated allosteric activation of the base excision repair enzyme 8-oxoguanine DNA glycosylase and its impact on mitochondrial function. Sci. Rep. 2022;12:14685. doi: 10.1038/s41598-022-18878-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M. Benítez-Buelga C. Calvo P. A. Hanna B. M. F. Mortusewicz O. Masuyer G. Davies J. Wallner O. Sanjiv K. Albers J. J. Castañeda-Zegarra S. Jemth A.-S. Visnes T. Sastre-Perona A. Danda A. N. Homan E. J. Marimuthu K. Zhenjun Z. Chi C. N. Sarno A. Wiita E. von Nicolai C. Komor A. J. Rajagopal V. Müller S. Hank E. C. Varga M. Scaletti E. R. Pandey M. Karsten S. Haslene-Hox H. Loevenich S. Marttila P. Rasti A. Mamonov K. Ortis F. Schömberg F. Loseva O. Stewart J. D’Arcy-Evans N. Koolmeister T. Henriksson M. Michel D. de Ory A. Acero L. Calvete O. Scobie M. Hertweck C. Vilotijevic I. Kalderén C. Osorio A. Perona R. Stolz A. Stenmark P. Berglund U. W. de Vega M. Helleday T. Small-molecule activation of OGG1 increases oxidative DNA damage repair by gaining a new function. Science. 2022;376:1471–1476. doi: 10.1126/science.abf8980. [DOI] [PubMed] [Google Scholar]
- Fromme J. C. Bruner S. D. Yang W. Karplus M. Verdine G. L. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Mol. Biol. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- Dalhus B. Forsbring M. Helle I. H. Vik E. S. Forstrøm R. J. Backe P. H. Alseth I. Bjørås M. Separation-of-Function Mutants Unravel the Dual-Reaction Mode of Human 8-Oxoguanine DNA Glycosylase. Structure. 2011;19:117–127. doi: 10.1016/j.str.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Visnes T. Grube M. Fekry Hanna B. M. Benitez-Buelga C. Cázares-Körner A. Helleday T. Targeting BER enzymes in cancer therapy. DNA Repair. 2018;71:118–126. doi: 10.1016/j.dnarep.2018.08.015. [DOI] [PubMed] [Google Scholar]
- Hill J. W. Hazra T. K. Izumi T. Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorenko V. S. Nevinsky G. A. Zharkov D. O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair. 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Sampath H. Lloyd R. S. Roles of OGG1 in transcriptional regulation and maintenance of metabolic homeostasis. DNA Repair. 2019;81:102667. doi: 10.1016/j.dnarep.2019.102667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitéz-Buelga C. Helleday T. Michel M. Synthetic switches of OGG1 control initiation of base excision repair and offer new treatment strategies. Clin. Transl. Med. 2022;12:e1035. doi: 10.1002/ctm2.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan H. E. Bjørås M. Base Excision Repair. Cold Spring Harbor Perspect. Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M. Homan E. J. Wiita E. Pedersen K. Almlöf I. Gustavsson A.-L. Lundbäck T. Helleday T. Warpman Berglund U. In silico Druggability Assessment of the NUDIX Hydrolase Protein Family as a Workflow for Target Prioritization. Front. Chem. 2020;8:443. doi: 10.3389/fchem.2020.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M. Visnes T. Homan E. J. Seashore-Ludlow B. Hedenström M. Wiita E. Vallin K. Paulin C. B. J. Zhang J. Wallner O. Scobie M. Schmidt A. Jenmalm-Jensen A. Warpman Berglund U. Helleday T. Computational and Experimental Druggability Assessment of Human DNA Glycosylases. ACS Omega. 2019;4:11642–11656. doi: 10.1021/acsomega.9b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page B. D. G. Valerie N. C. K. Wright R. H. G. Wallner O. Isaksson R. Carter M. Rudd S. G. Loseva O. Jemth A.-S. Almlöf I. Font-Mateu J. Llona-Minguez S. Baranczewski P. Jeppsson F. Homan E. Almqvist H. Axelsson H. Regmi S. Gustavsson A.-L. Lundbäck T. Scobie M. Strömberg K. Stenmark P. Beato M. Helleday T. Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells. Nat. Commun. 2018;9:250. doi: 10.1038/s41467-017-02293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H. Koolmeister T. Jemth A.-S. Eshtad S. Jacques S. A. Ström C. E. Svensson L. M. Schultz N. Lundbäck T. Einarsdottir B. O. Saleh A. Göktürk C. Baranczewski P. Svensson R. Berntsson R. P.-A. Gustafsson R. Strömberg K. Sanjiv K. Jacques-Cordonnier M.-C. Desroses M. Gustavsson A.-L. Olofsson R. Johansson F. Homan E. J. Loseva O. Bräutigam L. Johansson L. Höglund A. Hagenkort A. Pham T. Altun M. Gaugaz F. Z. Vikingsson S. Evers B. Henriksson M. Vallin K. S. A. Wallner O. A. Hammarström L. G. J. Wiita E. Almlöf I. Kalderén C. Axelsson H. Djureinovic T. Puigvert J. C. Häggblad M. Jeppsson F. Martens U. Lundin C. Lundgren B. Granelli I. Jensen A. J. Artursson P. Nilsson J. A. Stenmark P. Scobie M. Berglund U. W. Helleday T. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508:215–221. doi: 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- Zhang S. M. Desroses M. Hagenkort A. Valerie N. C. K. Rehling D. Carter M. Wallner O. Koolmeister T. Throup A. Jemth A.-S. Almlöf I. Loseva O. Lundbäck T. Axelsson H. Regmi S. Sarno A. Krämer A. Pudelko L. Bräutigam L. Rasti A. Göttmann M. Wiita E. Kutzner J. Schaller T. Kalderén C. Cázares-Körner A. Page B. D. G. Krimpenfort R. Eshtad S. Altun M. Rudd S. G. Knapp S. Scobie M. Homan E. J. Berglund U. W. Stenmark P. Helleday T. Development of a chemical probe against NUDT15. Nat. Chem. Biol. 2020;16:1120–1128. doi: 10.1038/s41589-020-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner O. Cázares-Körner A. Scaletti E. R. Masuyer G. Bekkhus T. Visnes T. Mamonov K. Ortis F. Lundbäck T. Volkova M. Koolmeister T. Wiita E. Loseva O. Pandey M. Homan E. Benítez-Buelga C. Davies J. Scobie M. Berglund U. W. Kalderén C. Stenmark P. Helleday T. Michel M. Optimization of N-Piperidinyl-Benzimidazolone Derivatives as Potent and Selective Inhibitors of 8-Oxo Guanine DNA Glycosylase 1. ChemMedChem. 2022;18(1):e202200310. doi: 10.1002/cmdc.202200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochevarov A. D. Harder E. Hughes T. F. Greenwood J. R. Braden D. A. Philipp D. M. Rinaldo D. Halls M. D. Zhang J. Friesner R. A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013;113:2110–2142. [Google Scholar]
- Bruner S. D. Norman D. P. Verdine G. L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- Carreras-Puigvert J. Zitnik M. Jemth A.-S. Carter M. Unterlass J. E. Hallström B. Loseva O. Karem Z. Calderón-Montaño J. M. Lindskog C. Edqvist P.-H. Matuszewski D. J. Blal H. A. Berntsson R. P. A. Häggblad M. Martens U. Studham M. Lundgren B. Wählby C. Sonnhammer E. L. L. Lundberg E. Stenmark P. Zupan B. Helleday T. A comprehensive structural, biochemical and biological profiling of the human NUDIX hydrolase family. Nat. Commun. 2017;8:1541. doi: 10.1038/s41467-017-01642-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O. Niesen F. H. Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 2012;795:109–118. doi: 10.1007/978-1-61779-337-0_7. [DOI] [PubMed] [Google Scholar]
- Huber K. V. M. Salah E. Radic B. Gridling M. Elkins J. M. Stukalov A. Jemth A.-S. Göktürk C. Sanjiv K. Strömberg K. Pham T. Berglund U. W. Colinge J. Bennett K. L. Loizou J. I. Helleday T. Knapp S. Superti-Furga G. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014;508:222–227. doi: 10.1038/nature13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo K. Ide H. Wallace S. S. Kow Y. W. A novel sensitive and specific assay for abasic sites, the most commonly produced DNA lesion. Biochemistry. 1992;31:3703–3708. doi: 10.1021/bi00129a020. [DOI] [PubMed] [Google Scholar]
- Visnes T. Cázares-Körner A. Hao W. Wallner O. Masuyer G. Loseva O. Mortusewicz O. Wiita E. Sarno A. Manoilov A. Astorga-Wells J. Jemth A.-S. Pan L. Sanjiv K. Karsten S. Gokturk C. Grube M. Homan E. J. Hanna B. M. F. Paulin C. B. J. Pham T. Rasti A. Berglund U. W. von Nicolai C. Benitez-Buelga C. Koolmeister T. Ivanic D. Iliev P. Scobie M. Krokan H. E. Baranczewski P. Artursson P. Altun M. Jensen A. J. Kalderén C. Ba X. Zubarev R. A. Stenmark P. Boldogh I. Helleday T. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science. 2018;362:834–839. doi: 10.1126/science.aar8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba X. Bacsi A. Luo J. Aguilera-Aguirre L. Zeng X. Radak Z. Brasier A. R. Boldogh I. 8-Oxoguanine DNA Glycosylase-1 Augments Proinflammatory Gene Expression by Facilitating the Recruitment of Site-Specific Transcription Factors. J. Immunol. 2014;192:2384–2394. doi: 10.4049/jimmunol.1302472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote K. Rienks M. Schmidt L. Theofilatos K. Yasmin Ozols M. Eckersley A. Shah A. Figg N. Finigan A. O’Shaughnessy K. Wilkinson I. Mayr M. Bennett M. Oxidative DNA damage promotes vascular ageing associated with changes in extracellular matrix-regulating proteins. Cardiovasc. Res. 2024:cvae091. doi: 10.1093/cvr/cvae091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl F. Liang C. Böttcher-Loschinski R. Stoll A. Flamann C. Richter S. Lischer C. Völkl S. Jacobs B. Böttcher M. Jitschin R. Bruns H. Fischer T. Holler E. Rösler W. Dandekar T. Mackensen A. Mougiakakos D. Oxidative DNA damage in reconstituting T cells is associated with relapse and inferior survival after allo-SCT. Blood. 2023;141:1626–1639. doi: 10.1182/blood.2022017267. [DOI] [PubMed] [Google Scholar]
- Sowlati-Hashjin S. Wetmore S. D. Quantum mechanical study of the β- and δ-lyase reactions during the base excision repair process: application to FPG. Phys. Chem. Chem. Phys. 2015;17:24696–24706. doi: 10.1039/c5cp04250j. [DOI] [PubMed] [Google Scholar]
- Bhakat K. K. Mantha A. K. Mitra S. Transcriptional Regulatory Functions of Mammalian AP-Endonuclease (APE1/Ref-1), an Essential Multifunctional Protein. Antioxid. Redox Signaling. 2009;11:621–637. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youhanna S. Kemas A. M. Wright S. C. Zhong Y. Klumpp B. Klein K. Motso A. Michel M. Ziegler N. Shang M. Sabatier P. Kannt A. Sheng H. Oliva-Vilarnau N. Büttner F. A. Seashore-Ludlow B. Schreiner J. Windbergs M. Cornillet M. Björkström N. K. Hülsmeier A. J. Hornemann T. Olsen J. V. Wang Y. Gramignoli R. Sundström M. Lauschke V. M. Chemogenomic Screening in a Patient-Derived 3D Fatty Liver Disease Model Reveals the CHRM1-TRPM8 Axis as a Novel Module for Targeted Intervention. Adv. Sci. 2024:2407572. doi: 10.1002/advs.202407572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehler M. Zhou K. Kemas A. M. Del Prado A. Hutchinson E. S. Nairn E. H. Varga M. Plattner Y. Zhong Y. Purewal-Sidhu O. Haslam J. Wiita E. Gildie H. Singerova K. Szaruga Z. Almlöf I. Hormann F. M. Liu K.-C. Wallner O. Ortis F. Homan E. J. Gileadi O. Rudd S. G. Stenmark P. De Vega M. Helleday T. D’Arcy-Evans N. D. Lauschke V. M. Michel M. Organocatalytic switches of DNA glycosylase OGG1 catalyze a highly efficient AP-lyase function. Chem. – Eur. J. 2025:e202500382. doi: 10.1002/chem.202500382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visnes T. Zhou K. Kemas A. M. Campopiano D. Lauschke V. M. Michel M. Chemical Switching: A Concept Inspired by Strategies from Biocatalysis and Organocatalysis. ChemBioChem. 2025;26(11):e202500220. doi: 10.1002/cbic.202500220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article, description of the materials and methods, spectral data and compound purity data have been included as part of the supplementary information (SI). See DOI: https://doi.org/10.1039/d4cb00323c.