

Abstract
Retinitis pigmentosa (RP), the group of hereditary conditions involving death of retinal photoreceptors, represents the most prevalent cause of visual handicap among working populations in developed countries. Here we provide an overview of the molecular pathologies associated with such disorders, from which it becomes clearly apparent that RP is one of the most genetically heterogeneous of hereditary conditions for which molecular pathologies have so far been elucidated. While heterogeneity of such magnitude would appear to represent a major impediment to the development of therapeutics, mutation-independent approaches to therapy are being developed to effectively by-pass such diversity in genetic aetiology. The implications of such technologies in terms of therapeutic intervention in RP, and indeed other genetically heterogeneous conditions, will be addressed.
Keywords: gene therapy/inherited eye disease/photo receptors/retinitis pigmentosa/ribozymes
The mammalian retina
The mammalian retina comprises six classes of neuron. Input neurons are the rod and cone photoreceptors, the nuclear bodies of which are located within an outer nuclear layer (ONL; Figure 1). Light energy absorbed by the photoreactive pigments is converted to graded electrical potentials by G-protein coupled signal transduction cascades (Stryer, 1988) (Figure 2). Photoreceptors synapse with second order retinal neurons at the outer plexiform layer (OPL; Figure 1), the nuclear bodies of such cells being organized into an inner nuclear layer (INL; Figure 1). Further synapsing between such cells and the output neurons of the retina, ganglion cells, occurs in the inner plexiform layer (IPL; Figure 1). The almost two orders of magnitude reduction in the number of output, as compared with input, retinal neurons reflects the remarkable level of processing undertaken by this neuronal tissue.
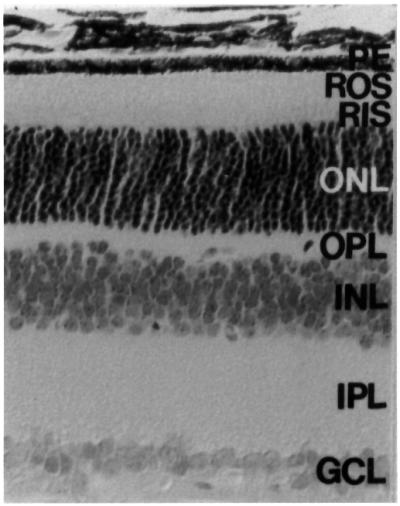
Fig. 1. Retinal histology from a wild-type mouse retina. The various layers of the retina are identified: retinal pigment epithelium (RE), rod outer segments (ROS), inner segments (RIS), outer nuclear layer (ONL), outer plexiform layer (OPL), inner nuclear layer (INL), inner plexiform layer and ganglion cell layer (GCL).

Fig. 2. Graphical summary of key components of the visual transduction cascade. Rhodopsin is indicated by RHO, transducin by T and phosphodiesterase by PDE.
Clinical and genetic aspects of retinitis pigmentosa (RP) and Leber congenital amaurosis
As with all neurons, those of the retina are susceptible to degeneration. RP involves loss of rod photoreceptor function and viability. Rod electroretinogram amplitudes, a measure of retina function, are reduced or non-recordable (Figure 3). As rod cell death progresses, cone cell viability is compromised, complete loss of vision often being the end result. As the disease progresses, pigmentary deposits may be observed on the surface of the retina (Figure 4). Where registers of visual impairment are available, RP represents the most prevalent cause of visual handicap among those of working age, while Leber congenital amaurosis (LCA), a congenital form of RP, is the most prevalent cause of hereditary visual handicap in children (Boughman and Fishman, 1983). Fifty per cent of cases of RP are sporadic, the remainder showing dominant, recessive or X-linked hereditary patterns. RP also occurs in syndromes, including Usher and Bardet–Biedl, the former involving sensorineural deafness and vestibular dysfunction, the latter, obesity and mental retardation.

Fig. 3. Rod-isolated, combined rod and cone responses to the maximal intensity flash presented in the dark-adapted state and single flash light-adapted cone responses are shown from (A) a normal individual, (B) a patient with moderately advanced RP and (C) a patient with advanced RP.

Fig. 4. Photographs of a normal (left panel) and RP retina (right panel). Features typical of RP, such as marked pigment epithelial thinning, optic disc pallor, retinal vascular attenuation and the classical ‘bone spicule’ intraretinal pigmentary deposits, are clearly evident in the RP retina.
A first step towards the identification of RP-causing genes was made in 1984 and involved the localization of a gene for X-linked RP to Xp (Bhattacharya et al., 1984). In 1989, the first dominant RP (adRP) gene was localized to 3q; this gene was found to be tightly linked to that encoding the photoreceptor pigment rhodopsin (McWilliam et al., 1989; Farrar et al., 1990). Mutations within the rhodopsin gene were subsequently documented (Dryja et al., 1990, 1991), and to date up to 150 mutations have been characterized (www.sph.uth.tmc.edu/RetNet/disease.htm; www.retina-international.com). A second locus for adRP was identified on 6p (Farrar et al., 1991). Further investigation revealed the presence of mutations within the gene encoding RDS-peripherin, a component of the photoreceptor outer segment disc membranes (Farrar et al., 1991; Kajiwara et al., 1991). These findings represented merely the beginning of the molecular story for retinal degeneration. Notably, mutations in 21 genes have now been identified in RP, seven in LCA, four in Usher syndrome and three in Bardet–Biedl syndrome (www.sph.uth.tmc.edu/RetNet/disease.htm; www.retina-international.com). Additionally, a further 24 disease loci have been mapped for RP or for syndromes incorporating RP; however, these genes remain to be isolated. In essence, the many linkage studies undertaken largely during the 1990s have thus revealed immense genetic heterogeneity and a complex underlying molecular pathology in retinal degeneration. Comprehensive referencing has not been possible in the following section, owing to limitations on article length, therefore for detailed referencing please refer to www.sph.uth.tmc.edu/RetNet/disease.htm and www.retina-international.com.
It is perhaps worthy of note in the context of providing an overview of this field that there is a great functional diversity in the types of gene that have been implicated in retinal degeneration (Table I). Of those genes implicated in RP, some encode proteins of visual transduction, including rhodopsin, the α and β subunits of rod cyclic GMP phosphodiesterase, the α subunit of the rod cyclic GMP-gated channel and arrestin. An additional protein implicated in RP, and possibly involved in visual transduction, is a member of the protein kinase C family, PRKCG (Al-Maghtheh et al., 1998). Mutations within two structural components of photoreceptor outer segment disc membranes, RDS-peripherin and Rom1, have also been identified in RP (Kajiwara et al., 1994). Defects in two other structural components of the photoreceptors and of the hair cells of the inner ear, harmonin (Verpy et al., 2000) and myosin VIIa (Weil et al., 1995), have been identified in Usher syndrome types 1A and 1B. Mutations within a number of genes encoding proteins of the retina extracelluar matrix or those involved in cell adhesion have also been identified in RP, Usher syndrome or LCA, including CRB1, a homologue of the Drosophila crumbs protein (den Hollander et al., 2001), ‘usherin’ in Usher type IIa (Eudy et al., 1998), a cadherin-like protein in Usher type 1D (Bolz et al., 2001) and a protein with homology to cofactor C, with probable involvement in the folding of β tubulin in X-linked RP (RP2). Mutations within five genes encoding proteins of the retinoid cycle, or involved in transport of retinal proteins, have also been implicated in RP or LCA (Table I). CRBP is the intracellular carrier of retinal. Light-induced isomerization of 11-cis retinal to the all-trans form is the initial step in both rod and cone visual transduction (Burstedt et al., 1999). RPE65 in the retinal pigment epithelium (RPE) is involved in the metabolism of all-trans-retinyl esters to 11-cis-retinal (Gu et al., 1997; Morimura et al., 1998). ABCR is a transmembrane protein of the outer segment disc membranes, one function being to transport retinal into and out of the outer segment discs (Sun and Nathans, 1997). RGR bears sequence homology to rhodopsin and is expressed in RPE and in Muller cells (Morimura et al., 1999). This protein binds all-trans retinol. LRAT is involved in the derivation of 11-cis retinal from vitamin A in the RPE (Ruiz et al., 2001). Mutations within these genes disrupt the cycling of retinal between the photoreceptors and the RPE, resulting in dysfunction of photoreceptor cells.
Table I. Summary of genes implicated in retinal degeneration highlighting functional diversity.
| Retinal degeneration | Gene | Function |
|---|---|---|
| RP | rhodopsin | visual transduction |
| RP | α subunits of rod cyclic GMP phosphodiesterase | visual transduction |
| RP | β subunits of rod cyclic GMP phosphodiesterase | visual transduction |
| RP | α subunit of rod cyclic GMP-gated channel | visual transduction |
| RP | arrestin | visual transduction |
| RP | PRKCG | visual transduction |
| RP | RDS-peripherin | photoreceptor structure |
| RP | ROM-1 | photoreceptor structure |
| Usher syndrome | harmonin | photoreceptor structure |
| Usher syndrome | myosin VIIa | photoreceptor structure |
| Usher syndrome | usherin | extracellular matrix |
| Usher syndrome | cadherin-like protein | extracellular matrix |
| RP/LCA | CRB1 | retinal development |
| RP | RP2 (homology with) cofactor C-β-tubulin | protein folding |
| RP | RPGR | protein transport |
| RP | CRBP | retinoid cycle |
| RP/LCA | RPE65 | retinoid cycle |
| RP | ABCR | retinoid cycle |
| RP | RGR | retinoid cycle |
| RP | LRAT | retinoid cycle |
| RP | MERTK | disc sheddding |
| RP/LCA | CRX | transcription factor |
| RP | NRL | transcription factor |
| RP/LCA | TULP1 | transcription factor |
| RP | PRPC8 | pre-mRNA slicing |
| RP | PRP31 | pre-mRNA slicing |
| LCA | AIPL1 | protein folding/trafficking |
| LCA | RPGRIP | photoreceptor cilia |
| LCA | retGC-1 | cGMP channel |
| BB | BBS2 | to be established |
| BB | BBS4 | to be established |
| BB | BBS6 | to be established |
A unique property of photoreceptors is the circadian shedding of the outer segment disc membranes and their phagocytosis by the RPE. Mutations within Mertk, encoding an epithelial expressed receptor tyrosine kinase, have been identified in the RCS rat (exhibiting aberrant disk phagocytosis) and in recessive RP in man (Gal et al., 2000), indicating that genes encoding RPE-expressed proteins involved in phagocytosis of outer segment disc membranes are involved in disease pathology.
Three transcription factors have also been implicated in the aetiology of retinal degeneration. These are CRX, NRL and TULP1. In the eye, CRX may be involved in regulation of expression of photoreceptor-specific genes (Chen et al., 1997). Some mutations in CRX may interrupt nuclear trafficking of the protein in photoreceptor cells (Fei and Hughes, 2000). NRL is expressed in developing tissue but is confined to the retina in adult (Rehemtulla et al., 1996). Mutations in NRL may affect levels of rhodopsin synthesis. NRL and CRX can act individually or synergistically to mediate photoreceptor specific-expression of rhodopsin by binding to the promoter region of that gene (Mitton et al., 2000). Mutations in CRX may alter promoter binding or NRL interaction (Mitton et al., 2000). TULP1 is a homologue of the murine tub gene (Kleyn et al., 1996). Available evidence suggests that TULP1 is a transcription factor (Hagstrom et al., 1998), mutations possibly causing deregulation of transcription of genes involved in photoreceptor viability (Banerjee et al., 1998).
A recent interesting finding has been the involvement in the pathogenesis of RP of genes and gene products involved in pre-mRNA splicing, e.g. PRPC8, PRP31 and HPRP3 (McKie et al., 2001; Vithana et al., 2001; Charkarova et al., 2002). Interestingly, although these genes are expressed in all tissues, it only causes a pathology in photoreceptor neurons—once again highlighting the functional diversity of the genes and gene products now implicated in human retinal degeneration.
Seven proteins have been implicated in LCA (Table I). The functions of RPE65, CRX, TULP1 and CRB1 have been outlined in the context of RP (above). AIPL1 bears 50% amino acid identity to human aryl hydrocarbon receptor-interacting protein (AIP) and may be involved in protein folding or trafficking. RPGRIP is probably the physiological binding partner of RPGR, localizing to the cilia of photoreceptors (Meindl et al., 1996; Dryja et al., 2001). This protein probably recruits RPGR to the cilia (Hong et al., 2000). Mutations in the retinal-specific guanylate cyclase gene (retGC-1) have also been implicated in LCA, resulting in impaired production of cGMP and closure of cGMP-gated channels.
Three genes have been identified in Bardet–Biedl syndrome (BBS2, BBS4 and BBS6; Slavotinek et al., 2000; Mykytyn et al., 2001; Nishimura et al., 2001). One of these genes shows homology to a prokaryotic chaperonin from Thermoplasma acidophilum (Slavotinek et al., 2000).
It is not difficult to envisage how photoreceptor cell death may be brought about by the presence of mutations within genes encoding some of these proteins. Defects in disc shedding, the integrity of cytoskeletal proteins, cell adhesion or in extracellular matrices of the retina could readily be perceived to result in such dysfunction as to bring about disorganization or death of photoreceptor cells. In other instances, the mechanisms of photoreceptor cell death remain more cryptic. Mutations within rhodopsin, NRL, CRX or components of the retinoid cycle may result in functionally and/or structurally abnormal rhodopsin, or in aberrant levels of the normal protein. In animal models, it has been established that structurally aberrant rhodopsin, or abnormal levels of protein, bring about rod cell dysfunction. For example, mice carrying a targeted disruption of the rhodopsin gene (rho–/– mice) present with a rapid photoreceptor degeneration (Humphries et al., 1997). In those cases where structurally abnormal protein is produced, evidence from cell culture experiments indicates that mutated proteins may not be transported from the endoplasmic reticulum to the outer segment disc membranes. Build-up of structurally aberrant protein in the nuclear compartment of photoreceptors is likely to be toxic to the cell. Moreover, aberrant folding of mutant opsins into outer segment disc membranes is likely to have a disruptive effect on such membranes and hence cell structure. However, in RP patients, photoreceptors may continue to function for many years, before succumbing to the toxic effects of mutant protein or the absence of wild-type protein. Such evidence as there is, derived almost exclusively from the study of small animal models, indicates that a common pathway of cell death is apoptosis (Portera-Cailliau et al., 1994). However, only in one case of human RP (the only case in which it has been examined) has apoptosis of retinal cells been documented; nevertheless, this process ‘as a final common pathway of cell death’ is widely accepted as the likely mode of cell death in all or most degenerative retinopathies. Accepting this hypothesis to be the case, it is clear that events leading from the presence or absence of mutant protein to triggering of the apoptotic death cascade remain to be elucidated. It is also clear that knowledge of such events may reveal a new family of targets for therapeutic intervention.
Therapeutic intervention at the genetic level: overcoming the problem of dominant mutational heterogeneity
Notwithstanding the recent successes in delivery of potentially therapeutic genes to retinal tissues in cases of recessive disease in animal models of retinal degeneration (Bennett et al., 1996; Takahashi et al., 1999; Ali et al., 2000; Acland et al., 2001), the mutational heterogeneity encountered in dominant disease represents a challenging problem with respect to the development of economically viable therapeutics. Therapeutic interventions that overcome such mutational heterogeneity could be targeted to the primary genetic defect, or indeed to modulating secondary effects associated with the disease pathology, such as apoptosis. It may be that a combined approach targeting both the primary genetic lesion and reprogramming cellular processes so as to minimize secondary pathologies may represent optimal therapeutic strategies. Mutation-independent therapeutic approaches targeting the primary defect or secondary effects are outlined below.
Ribozymes are attractive agents for suppression of mutated transcripts since they cleave target mRNAs (Haseloff and Gerlach, 1988; Pley et al., 1994), and a growing number of examples of in vivo efficacies of such agents is available (Lewin et al., 1998). Hammerhead ribozymes cleave mRNA only at NUX sites (N, any nucleoside; U, uracil; X, any nucleoside other than G). Hence, only mutations causing a sequence variation at ribozyme cleavage sites are targetable by these agents. Moreover, ribozymes are limited to targets in open loop structures of transcripts. Such limitations, considering a requirement to suppress >100 dominant mutations within the rhodopsin gene alone, would appear to exclude such agents as tools for suppression of the majority of mutations. Even if such constraints were lifted by modifications in ribozyme design, the clinical validation of suppression effectors targeting large numbers of individual mutations would not be economically viable. However, such limitations pertain only as long as mutation-specific approaches are used. In an alternative approach, Millington-Ward et al. (1997) developed a series of mutation-independent, suppression-replacement strategies for dominant mutations. The strategies (Figure 5) have been utilized in the evaluation of ribozymes targeting transcripts derived from the rhodopsin, RDS-peripherin, collagen 1A1 and 1A2 genes (Millington-Ward et al., 1997, 1999; O’Neill et al., 2000).

Fig. 5. Graphical representation of the principle of mutation-independent suppression utilizing the degeneracy of the genetic code. A ribozyme is designed to cleave a target transcript at a degenerate site. In parallel, a replacement gene that encodes wild-type protein but has been subtly modified at degenerate sites such that the ribozyme cannot cleave transcripts from the replacement gene is supplied.
The first of these methods involves the use of untranslated regions (UTRs)—both mutant and wild-type alleles will be suppressed. If suppression is targeted to UTRs, then replacement genes can be generated with modified UTRs. Suppression is entirely mutation independent and in essence involves a gene-exchange strategy. Modifications introduced into replacement genes will have no effect on the encoded protein but will protect transcripts (from replacement genes) from suppression. Sequence modifications can be as subtle as a single base change at the ribozyme cleavage site. A second strategy exploits the degeneracy of the genetic code, i.e. a single amino acid may be encoded by more than one codon (‘wobble’)—suppression is targeted to degenerate sites in mRNA. Again, both mutant and normal alleles are suppressed. In conjunction with suppression, a replacement gene is modified, exploiting the degeneracy of the genetic code such that transcripts from replacement genes are protected from suppression but encode the normal protein. A third approach involves the use of intragenic polymorphisms that occur frequently in human DNA. Suppression of a mutant allele is directed to a neutral polymorphism rather than to a rare mutation and therefore the suppression therapeutic will be relevant to a much larger proportion of patients, since the therapeutic is mutation independent (Millington-Ward et al., 1997, 1999; O’Neill et al., 2000). The technologies have been evaluated in vitro and in cell culture. Subsequent to demonstration of the ‘generic’ nature of such approaches in animals (a mouse model expressing normal and mutant human rhodopsins on a murine null-opsin background has been generated; Olsson et al., 1992; McNally et al., 1999), there would be a clear rationale to put in place the elements required to proceed from animal studies toward phase 1 human clinical trials. It is of interest to note that, in this model, wild-type human rhodopsin is sufficiently conserved between mouse and humans to rescue the disease pathology present in rho–/– mice, therefore validating the use of rho–/– mice to test the future delivery of the human gene (McNally et al., 1999). While described in the context of inherited retinopathies, such approaches are applicable to targeting any mutation with a dominant-negative effect. Some 1500 human autosomal dominant Mendelian diseases have been described. Also, many dominantly acting mutations underlie the cause of multifactorial diseases such as p53- or Ras-related cancers. Table II lists a sample of such conditions, in which mutation-independent approaches would have direct utility. Therapeutic intervention at the level of the primary genetic defect in diseases displaying such extensive genetic heterogeneity is thus becoming a realistic prospect. It is also notable that the strategies outlined above are not limited to hammerhead ribozymes, but could involve the use of many of the molecular tools for suppression now becoming available. Such suppression tools include antisense DNA/RNA, peptide nucleic acids, other forms of ribozymes including hairpin ribozymes, trans-splicing ribozymes, minizymes and maxizymes, and triple helix DNA, to name a few (Sullenger and Cech, 1994; Sioud et al., 1997; Kuwabara et al., 1998; Praseuth et al., 1999; Phylactou, 2000).
Table II. Possible disease targets for gene exchange, mutational diversity of targets, estimated disease prevalence and causative gene.
| Disease | Mutations | Prevalence | Gene |
|---|---|---|---|
| Cancers | |||
| p53 cancers | 100 | 1 in 6 | p53 |
| p21 cancers | multiple mutations | 1 in 20 | HRAS, KRAS, NRAS |
| FAP cancers | 350 | 1 in 30 | APC |
| neurofibromatosis | multiple mutations | 1 in 4000 | NF1 and NF2 |
| Neurological disorders | |||
| RP | 120 | 1 in 12 000 | rhodopsin |
| Alzheimer’s disease | 100+ | 1 in 15 | presenillins/APP |
| Bone/connective tissue disorders | |||
| OI | 150 | 1 in 24 000 | collagen 1a1 |
| OI | 80 | 1 in 24 000 | collagen 1a2 |
| Ehlers–Danlos syndrome IV | 122 | 1 in 30 000 | collagen 3a1 |
| Ehlers–Danlos syndrome VII | 9 | 1 in 50 000 | collagen 1A2 |
| EB | 50 | 1 in 30 000 | keratins |
| congenital cataract | multiple mutations | 1 in 3000 | crystalline |
| Marfan syndrome | 200 | 1 in 25 000 | fibrillin 1 |
An alternative, or indeed possibly complementary, approach for therapy would be modulation of secondary effects associated with disease pathology. In this regard, a number of studies have provided preliminary evidence that inhibition of key players which orchestrate apoptosis, such as the caspase family of serine proteases, may provide protection against photoreceptor cell apoptosis. Photoreceptor apoptosis has been clearly linked to the pathological processes active in animal models with retinal degeneration (simulating the human disease; Chang et al., 1993). For example, peptide decoys preventing caspase activation have been shown to protect in part against photoreceptor apoptosis both in cell culture and in animal models (Liu et al., 1999). Furthermore, the anti-apoptotic baculoviral gene p35 has also been shown to protect against apoptosis in a variety of tissues including photoreceptors (Bump et al., 1995; Davidson and Steller, 1998). For example, Davidson and Steller demonstrated functional rescue of degenerating photoreceptors in Drosophila using the p35 gene. Indeed, the repertoire of potential anti-apoptotic genes and molecules is extensive, and many other candidates such as c-fos and Bcl-2 have been tested in animal models of retinal degeneration with varying degrees of success (Chen et al., 1996; Bennett et al., 1998).
An additional strategy for therapy of retinopathies, which may prove to be of value individually or in combination with those outlined above, involves the delivery of appropriate neurotrophic factors to photoreceptor cells in an attempt to improve survival rates for these specialized neurons. Various neurotrophic factors have been utilized to attempt to achieve this goal, including bFGF, BDNF, GDNF and CNTF (Cayouette and Gravel, 1997; Akimoto et al., 1999; Liang et al., 2001). For example, injection of adenoviral vectors and adeno-associated viral vectors carrying CNTF have been shown to provide a benefit in terms of photoreceptor survival in rodent models of retinal degeneration (Cayouette and Gravel, 1997; Liang et al., 2001). In addition to the use of known neurotrophic factors, studies are being undertaken to identify novel neurotrophic factors with better potency in terms of increasing photoreceptor cell longevity. Such studies may reveal novel and powerful therapeutic molecules for photoreceptor degeneration. In the context of developing therapies for retinal degeneration, it is worth highlighting that many studies have been going on in parallel with those outlined above, to explore viral and non-viral methods of achieving efficient delivery of therapeutic genes to retinal tissues. However, given the large size of this field, it could form the basis of an additional review topic and therefore due to space constraints has not been covered here.
In conclusion, despite the high degree of genetic heterogeneity inherent in disorders such as dominantly inherited RP, there are novel therapeutic approaches in research and development which may effectively circumvent such heterogeneity and thereby provide rationale therapeutics at both the biological and economic levels for this group of debilitating conditions.
Acknowledgments
Acknowledgements
We acknowledge the support of The Wellcome Trust, HEA (Ireland), HRB (Ireland), Fighting Blindness (Ireland), Foundation Fighting Blindness (USA), British Retinitis Pigmentosa Society and The European Union 5th Framework Programme and the EU Human Potential Programme Contract HPRN-CT-2000-00098.
References
- Acland G.M. et al. (2001) Gene therapy restores vision in a canine model of childhood blindness. Nature Genet., 28, 92–95. [DOI] [PubMed] [Google Scholar]
- Akimoto M., Miyatake,S.-I., Kogishi,J.-I., Hangai,M., Okazaki,K., Takahashi,J.C., Saiki,M., Iwaki,M. and Honda,Y. (1999) Adenovirally expressed basic fibroblast growth factor rescues photoreceptor cells in RCS rats. Invest. Ophthalmol. Vis. Sci., 40, 273–279. [PubMed] [Google Scholar]
- Ali R.R. et al. (2000) Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nature Genet., 25, 306–310. [DOI] [PubMed] [Google Scholar]
- Al-Maghtheh M., Vithana,E.N., Inglehearn,C.F., Moore,T., Bird,A.C. and Bhattacharya,S.S. (1998) Segregation of a PRKCG mutation in two RP11 families. Am. J. Hum. Genet., 62, 1248–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P. et al. (1998) TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nature Genet., 18, 177–179. [DOI] [PubMed] [Google Scholar]
- Bennett J., Tanabe,T., Sun,D., Zeng,Y., Kjeldbye,H., Gouras,P. and Maguire,A.M. (1996) Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nature Med., 2, 649–654. [DOI] [PubMed] [Google Scholar]
- Bennett J., Zeng,Y., Bajwa,R., Klatt,L., Li,Y. and Maguire,A.M. (1998) Adenovirus-mediated delivery of rhodopsin-promoted bcl-2 results in a delay in photoreceptor cell death in the rd/rd mouse. Gene Ther., 5, 1156–1164. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.S. et al. (1984) Close genetic linkage between X-linked retinitis pigmentosa and a restriction fragment length polymorphism identified with recombinant DNA probe L1.28. Nature, 309, 253–255. [DOI] [PubMed] [Google Scholar]
- Bolz H. et al. (2001) Mutations of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nature Genet., 27, 108–112. [DOI] [PubMed] [Google Scholar]
- Boughman J.A. and Fishman,G.S. (1983) A genetic analysis of retinitis pigmentosa. Br. J. Ophthalmol., 67, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bump N.J. et al. (1995) Inhibition of ICE family proteases by baculovirus anti-apoptotic protein p35. Science, 269, 1885–1888. [DOI] [PubMed] [Google Scholar]
- Burstedt M.S., Sandgren,O., Holmgren,G. and Forsman-Semb,K. (1999) Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chomosome 15q26. Invest. Ophthalmol. Vis. Sci., 40, 995–1000. [PubMed] [Google Scholar]
- Cayouette M. and Gravel,C. (1997) Adenovirus-mediated transfer of ciliary neurotrophic factor can prevent photoreceptor degeneration in the retinal degeneration (rd) mouse. Hum. Gene Ther., 8, 423–430. [DOI] [PubMed] [Google Scholar]
- Chakarova C.F. et al. (2002) Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa.. Hum. Mol. Genet., 11, 87–92. [DOI] [PubMed] [Google Scholar]
- Chang G.Q., Hao,Y. and Wong,F. (1993) Apoptosis: final common pathways of photoreceptor death in rd, rds and rhodopsin mutant mice. Neuron, 11, 595–605. [DOI] [PubMed] [Google Scholar]
- Chen J., Flannery,J.G., LaVail,M.M., Steinberg,R.H., Xu,J. and Simon,M.I. (1996) Bcl-2 over expression reduces apoptotic photoreceptor cell death in three different retinal degenerations. Proc. Natl Acad. Sci. USA, 93, 7042–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson F.F. and Steller,H. (1998) Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature, 391, 587–590. [DOI] [PubMed] [Google Scholar]
- den Hollander A.I. et al. (2001) Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am. J. Hum. Genet., 69, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja T.P., McGee,T.L., Reichel,E., Hahn,L.B., Cowley,G.S., Yandell,D.W., Sandberg,M.A. and Berson,E.L. (1990) A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature, 343, 364–366. [DOI] [PubMed] [Google Scholar]
- Dryja T.P., Hahn,L.B., Cowley,G.S., McGee,T.L. and Berson,E.L. (1991) Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc. Natl Acad. Sci. USA, 88, 9370–9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja T.P., Adams,S.M., Grimsby,J.L., McGee,T.L., Hong,D.H., Li,T., Andreasson,S. and Berson,E.L. (2001) Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet., 68, 1295–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eudy J.D. et al. (1998) Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science, 280, 1753–1757. [DOI] [PubMed] [Google Scholar]
- Farrar G.J. et al. (1990) Autosomal dominant retinitis pigmentosa: linkage to rhodopsin and evidence for genetic heterogeneity. Genomics, 8, 35–40. [DOI] [PubMed] [Google Scholar]
- Farrar G.J., Jordan,S.A., Kenna,P., Humphries,M.M., Kumar-Singh,R., McWilliam,P., Allamand,V., Sharp,E. and Humphries,P. (1991) Autosomal dominant retinitis pigmentosa: localisation of a disease gene (RP6) to the short arm of chromosome 6. Genomics, 11, 870–874. [DOI] [PubMed] [Google Scholar]
- Fei Y. and Hughes,T.E. (2000) Nuclear trafficking of photoreceptor protein Crx: the targeting sequence and pathologic implications. Invest. Ophthalmol. Vis. Sci., 41, 2849–2856. [PubMed] [Google Scholar]
- Gal A., Li,Y., Thompson,D.A., Weir,J., Orth,U., Jacobson,S.G., Apfelstedt-Sylla,E. and Vollrath,D. (2000) Mutations in MERKTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nature Genet., 26, 270–271. [DOI] [PubMed] [Google Scholar]
- Gu S.M. et al. (1997) Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nature Genet., 17, 194–197. [DOI] [PubMed] [Google Scholar]
- Hagstrom S.A., North,M.A., Nishina,P.L., Berson,E.L. and Dryja,T.P. (1998) Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nature Genet., 18, 174–176. [DOI] [PubMed] [Google Scholar]
- Haseloff J. and Gerlach,W.L. (1988) Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature, 334, 585–591. [DOI] [PubMed] [Google Scholar]
- Hong D.H., Pawlyk,B.S., Shang,J., Sandberg,M.A., Berson,E.L. and Li,T. (2000) A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl Acad. Sci. USA, 97, 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries M.M. et al. (1997) Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nature Genet., 15, 216–219. [DOI] [PubMed] [Google Scholar]
- Kajiwara K., Hahn,L.B., Mukai,S., Travis,G.H., Berson,E.L. and Dryja,T.P. (1991) Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature, 354, 478–480. [DOI] [PubMed] [Google Scholar]
- Kajiwara K., Berson,E.L. and Dryja,T.P. (1994) Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 264, 1604–1608. [DOI] [PubMed] [Google Scholar]
- Kleyn P.W. et al. (1996) Identification and characterization of the mouse obesity gene tubby: a member of a novel gene family. Cell, 85, 281–290. [DOI] [PubMed] [Google Scholar]
- Kuwabara T., Warashina,M., Tanabe,T., Tani,K., Asano,S. and Taira,K. (1998) A novel allosterically trans-activated ribozyme, the maxizyme, with exceptional specificity in vitro and in vivo. Mol. Cell, 2, 617–627. [DOI] [PubMed] [Google Scholar]
- Lewin A.S., Drenser,K.A., Hauswirth,W.W., Nishikawa,S., Yasumura,D., Flannery,J.G. and LaVail,M.M. (1998) Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nature Med., 4, 967–971. [DOI] [PubMed] [Google Scholar]
- Liang F.Q., Dejneka,N.S., Cohen,D.R., Krasnoperova,N.V., Lem,J., Maguire,A.M., Dudus,L., Fisher,K.J. and Bennett,J. (2001) AAV-mediated delivery of ciliary neurotrophic factor prolongs photoreceptor survival in the rhodopsin knockout mouse. Mol. Ther., 3, 241–248. [DOI] [PubMed] [Google Scholar]
- Liu C., Li,Y., Peng,M., Laties,A.M. and Wen,R. (1999) Activation of caspase-3 in the retina of transgenic rats with the rhodopsin mutation S334ter during photoreceptor degeneration. J. Neurosci., 19, 4778–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie A.B. et al. (2001) Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum. Mol. Genet., 10, 1555–1562. [DOI] [PubMed] [Google Scholar]
- McNally N., Kenna,P., Humphries,M.M., Hobson,A.H., Khan,N.W., Bush,R.A., Sieving,P.A., Humphries,P. and Farrar,G.J. (1999) Structural and functional rescue of murine rod photoreceptors by human rhodopsin transgene. Hum. Mol. Genet., 8, 1309–1312. [DOI] [PubMed] [Google Scholar]
- McWilliam P. et al. (1989) Autosomal dominant retinitis pigmentosa: localisation of an ADRP gene to the long arm of chromosome 3. Genomics, 5, 612–619. [DOI] [PubMed] [Google Scholar]
- Meindl A. et al. (1996) A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nature Genet., 13, 35–42. [DOI] [PubMed] [Google Scholar]
- Millington-Ward S. et al. (1997) Strategems in vitro for gene therapies directed to dominant mutations. Hum. Mol. Genet., 6, 1415–1426. [DOI] [PubMed] [Google Scholar]
- Millington-Ward S., O’Neill,B., Kiang,A.S., Humphries,P., Kenna,P.F. and Farrar,G.J. (1999) A mutation-independent therapeutic strategem for osteogenesis imperfecta. Antisense Nucleic Acid Drug Dev., 9, 537–542. [DOI] [PubMed] [Google Scholar]
- Mitton K.P., Swain,P.K., Chen,S., Xu,S., Zack,D.J. and Swaroop,A. (2000) The leucine zipper of NRL interacts with the CRX homeodomain. A possible mechanism of transcriptional synergy in rhodopsin regulation. J. Biol. Chem., 275, 29794–29799. [DOI] [PubMed] [Google Scholar]
- Morimura H., Fishman,G.A., Grover,S.A., Fulton,A.B., Berson,E.L. and Dryja,T.P. (1998) Mutations in the RPE-65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc. Natl Acad. Sci. USA, 95, 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura H., Saindelle-Ribeaudeau,F., Berson,E.L. and Dryja,T.P. (1999) Mutations in RGR, encode a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nature Genet., 23, 393–394. [DOI] [PubMed] [Google Scholar]
- Mykytyn K. et al. (2001) Identification of the gene that when mutated causes the human obesity syndrome BBS4. Nature Genet., 28, 188–191. [DOI] [PubMed] [Google Scholar]
- Nishimura D.Y. et al. (2001) Positional cloning of a novel gene on chromosome 16q causing Bardet–Biedl syndrome (BBS2). Hum. Mol. Genet., 10, 865–874. [DOI] [PubMed] [Google Scholar]
- O’Neill B., Millington-Ward,S., O’Reilly,M., Tuohy,G., Kiang,A.S., Kenna,P.F., Humphries,P. and Farrar,G.J. (2000) Ribozyme based therapeutic approaches for autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci., 41, 2863–2869. [PubMed] [Google Scholar]
- Olsson J.E. et al. (1992) Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron, 9, 815–830. [DOI] [PubMed] [Google Scholar]
- Phylactou L.A. (2000) Ribozyme and peptide-nucleic acid-based gene therapy. Adv. Drug Deliv. Rev., 44, 97–108. [DOI] [PubMed] [Google Scholar]
- Pley H.W., Flaherty,K.M. and McKay,D.B. (1994) Three-dimensional structure of a hammerhead ribozyme. Nature, 372, 68–74. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C., Sung,C.H., Nathans,J. and Adler,R. (1994) Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl Acad. Sci. USA, 91, 974–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praseuth D., Guieysse,A.l. and Helene,C. (1999) Triple helix formation and the antigene strategy for control of gene expression. Biochim. Biophys. Acta, 1489, 181–206. [DOI] [PubMed] [Google Scholar]
- Rehemtulla A., Warwar,R., Kumar,R., Ji,X., Zack,D.J. and Swaroop,A. (1996) The basic motif-leucine zipper transcription factor Nrl can positively regulate rhodopsin gene expression. Proc. Natl Acad. Sci. USA, 93, 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A., Kuehn,M.H., Andorf,J.L., Stone,E., Hageman,G.S. and Bok,D. (2001) Genomic organization and mutation analysis of the gene encoding lecithin retinal acyltransferase in human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci., 42, 31–37. [PubMed] [Google Scholar]
- Sioud M., Opstad,A., Hendry,P., Lockett,T.J., Jennings,P.A. and McCall,M.J. (1997) A minimized hammerhead ribozyme with activity against interleukin-2 in human cells. Biochem. Biophys. Res. Commun., 231, 397–402. [DOI] [PubMed] [Google Scholar]
- Slavotinek A.M. et al. (2000) Mutations in MKKS cause Bardet–Biedl syndrome. Nature Genet., 26, 15–16. [DOI] [PubMed] [Google Scholar]
- Sullenger B.A. and Cehch,T.R. (1994) Ribozyme-mediated repair of defective mRNA by targeted trans-splicing. Nature, 371, 619–622. [DOI] [PubMed] [Google Scholar]
- Stryer L. (1988) Molecular basis of visual excitation. Cold Spring Harb. Symp. Quant. Biol., 53, 283–294. [DOI] [PubMed] [Google Scholar]
- Sun H. and Nathans,J. (1997) Stargardt’s ABCR is localized to the disc membrane of retinal rod outer segments. Nature Genet., 17, 15–16. [DOI] [PubMed] [Google Scholar]
- Takahashi M., Miyoshi,H., Verma,I.M. and Gage,F.H. (1999) Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J. Virol., 73, 7812–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpy E. et al. (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells underlies usher syndrome type 1C. Nature Genet., 26, 51–55. [DOI] [PubMed] [Google Scholar]
- Vithana E.N. et al. (2001) A human homolog of yeast pre-mRNA splicing gene RPR31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol. Cell, 8, 375–381. [DOI] [PubMed] [Google Scholar]
- Weil D. et al. (1995) Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature, 374, 60–61. [DOI] [PubMed] [Google Scholar]