

Abstract
SWI/SNF regulates growth control, differentiation and tumor suppression, yet few direct targets of this chromatin-remodeling complex have been identified in mammalian cells. We report that SWI/SNF is required for interferon (IFN)-γ induction of CIITA, the master regulator of major histocompatibility complex class II expression. Despite the presence of functional STAT1, IRF-1 and USF-1, activators implicated in CIITA expression, IFN-γ did not induce CIITA in cells lacking BRG1 and hBRM, the ATPase subunits of SWI/SNF. Reconstitution with BRG1, but not an ATPase-deficient version of this protein (K798R), rescued CIITA induction, and enhanced the rate of induction of the IFN-γ-responsive GBP-1 gene. Not ably, BRG1 inhibited the CIITA promoter in transient transfection assays, underscoring the importance of an appropriate chromosomal environment. Chroma tin immunoprecipitation revealed that BRG1 interacts directly with the endogenous CIITA promoter in an IFN-γ-inducible fashion, while in vivo DNase I footprinting and restriction enzyme accessibility assays showed that chromatin remodeling at this locus requires functional BRG1. These data provide the first link between a cytokine pathway and SWI/SNF, and suggest a novel role for this chromatin-remodeling complex in immune surveillance.
Keywords: BRG1/CIITA/chromatin remodeling/JAK–STAT/SWI/SNF
Introduction
Eukarytotic cells have a number of complexes that counteract the repressive effect of chromatin (reviewed in Workman and Kingston, 1998; Strahl and Allis, 2000; Fry and Peterson, 2001). SWI/SNF is an ∼2 MDa 10–15 subunit chromatin-remodeling complex that is conserved from yeast to humans. It is the founding member of a class of chromatin-remodeling machines that use the energy from ATP hydrolysis to facilitate transcription (reviewed in Muchardt and Yaniv, 1999; Flaus and Owen-Hughes, 2001; Fry and Peterson, 2001). Two closely related human SWI/SNF ATPase subunits have been described, BRG1 and hBRM, that are homologous to DNA helicases (Khavari et al., 1993; Muchardt and Yaniv, 1993). They are found in different complexes, but associate with a very similar group of SWI/SNF subunits (Wang et al., 1996a,b; Sif et al., 2001). These complexes do not evict nucleosomes from DNA, but can facilitate nucleosome sliding or strand exchange, and alter the structure of DNA wound around the histone octamer (see Flaus and Owen-Hughes, 2001; Fry and Peterson, 2001; Liu et al., 2001; and references therein). These alterations in DNA topology may facilitate the access of DNA-binding proteins, such as activators and/or the general transcription machinery, eventually leading to gene expression.
Gene array studies have implicated SWI/SNF in the expression of ∼5% of yeast genes (Holstege et al., 1998; Sudarsanam et al., 2000), suggesting that the complex may also have a major role in regulating transcription in higher eukaryotes. This assumption is supported by data implicating SWI/SNF in a variety of biological processes (reviewed in Muchardt and Yaniv, 1999; Fry and Peterson, 2001). For example, homozygous inactivation of BRG1, or another SWI/SNF subunit INI1/hSNF5, causes early embryonic lethality in mice, and heterozygotes are prone to a variety of tumors (Bultman et al., 2000; Klochendler-Yeivin et al., 2000; Roberts et al., 2000; Guidi et al., 2001). Components of SWI/SNF interact with a variety of activators (Fryer and Archer, 1998; Cheng et al., 1999; Kowenz-Leutz and Leutz, 1999; Dilworth et al., 2000; DiRenzo et al., 2000; Barker et al., 2001; Sullivan et al., 2001), consistent with the notion that the complex is recruited to many mammalian promoters. Nevertheless, few direct targets have been confirmed in mammalian cells. Identification of these genes is essential to clarify how SWI/SNF facilitates transcription in vivo, and to determine when it acts in the cascade of events leading to gene induction. Rapidly inducible genes are particularly useful in this regard (Agalioti et al., 2000; Dilworth et al., 2000; DiRenzo et al., 2000).
Interferons (IFNs) are cytokines that play a central role in the immune response. IFN-α and -β are produced by virus-infected cells, while IFN-γ is secreted by activated T cells and natural killer cells. One of the major roles of IFN-γ is to induce major histocompatibility complex (MHC) class II expression on the cell surface (reviewed in Boehm et al., 1997). MHC class II molecules present antigens to CD4+ T helper cells. They are either expressed constitutively on the surface of professional antigen-presenting cells (APCs), such as B lymphocytes and dendritic cells, or can be up-regulated in response to IFN-γ in non-APCs. Both constitutive and IFN-γ-induced MHC II gene expression require class II transactivator (CIITA) (Steimle et al., 1993, 1994). Correlating with the expression of MHC class II, CIITA is constitutively expressed in APCs, and can be induced by IFN-γ in other cell types (Steimle et al., 1993, 1994; Chin et al., 1994). With few exceptions, MHC class II genes are silent in the absence of CIITA (Chang et al., 1996; Williams et al., 1998). Further more, mutations in CIITA cause bare lymphocyte syndrome, a severe immune disorder characterized by a lack of MHC class II expression (Reith and Mach, 2001). Thus, CIITA is viewed as the ‘master switch’ for MHC class II induction.
IFN-γ induction of CIITA expression occurs via the JAK–STAT pathway. Ligand-induced dimerization of the IFN-γ receptor activates the JAK1 and JAK2 tyrosine kinases, leading to phosphorylation of STAT1 (signal transducer and activator of transcription). Once phosphorylated, STAT1 homodimerizes and translocates to the nucleus, where it contributes to the induction of several genes, including CIITA and interferon regulatory factor 1 (IRF-1) (reviewed in Boehm et al., 1997; Reith and Mach, 2001). The CIITA promoter (see Figure 3B) contains an IFN-γ-activated sequence (GAS), an E-box and an IRF-element (IRF-E). STAT1 is recruited to the GAS sequence, the constitutively expressed basic helix–loop– helix (bHLH) dimer USF-1 to the E-box, and a heterodimer of inducible IRF-1 and constitutively expressed IRF-2 to the IRF-E (Muhlethaler-Mottet et al., 1998; Piskurich et al., 1999; Xi et al., 2001). There is also an NF-GMa element, which may negatively regulate expression, and an NF-κB site, which is not conserved in mice (Muhlethaler-Mottet et al., 1997). STAT1, USF-1 and IRF-1/2 have all been implicated in CIITA induc tion (Lee and Benveniste, 1996; Hobart et al., 1997; Muhlethaler-Mottet et al., 1998; Piskurich et al., 1999; Xi et al., 2001), but it is not clear whether they are sufficient for induction of the chromosomal gene. Here we demonstrate an essential role for the SWI/SNF chromatin-remodeling complex in CIITA induction. This finding represents the first link between a cytokine and the ATP-dependent class of chromatin-remodeling complexes, implicates SWI/SNF in the immune response and provides novel insight into its role as a tumor suppressor.
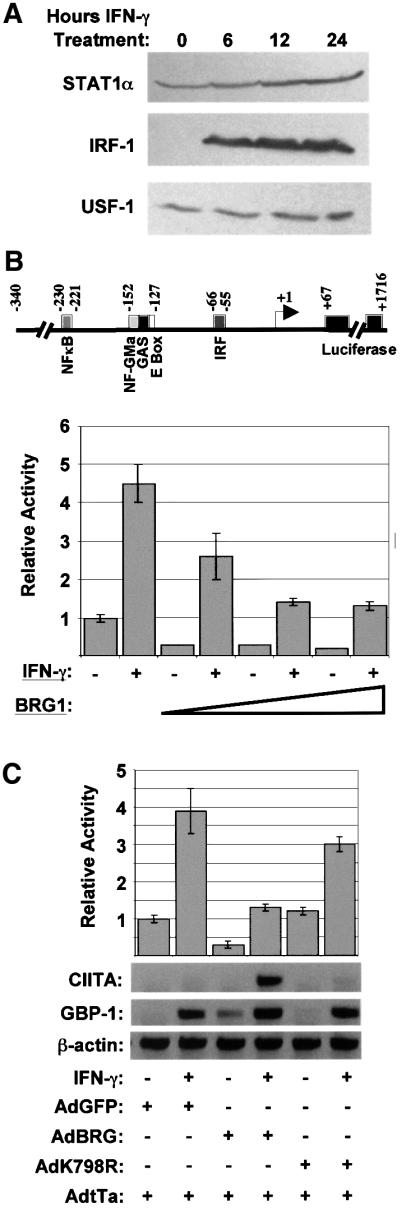
Fig. 3. The effect of BRG1 on the CIITA promoter is chromatin dependent. (A) STAT1, IRF-1 and USF-1 expression in SW13 cells. Cells were treated with IFN-γ for 0, 6, 12 or 24 h, and lysate analyzed by western blot using antibodies to STAT1, IRF-1 and USF-1. (B) IFN-γ activation of a transiently transfected CIITA reporter vector does not require BRG1. SW13 cells were transfected in duplicate with 0.8 µg of phCIITAPIV-LUC reporter plasmid (shown schematically) and increasing amounts (1, 3 and 5 µg) of a vector expressing BRG1 (pBJ5-BRG1). The empty expression vector pBJ5 was used to fill samples to ensure equimolar amounts of total plasmid. Samples were left untreated, or exposed to IFN-γ for 24 h. Baseline activity (relative activity = 1) is that obtained in untreated cells transfected with pBJ5. The results shown are the average and range of two experiments each performed in duplicate. (C) BRG1 has opposite effects on the endogenous versus transiently transfected CIITA promoter. SW13 cells were infected with AdtTA plus control AdGFP (lanes 1 and 2), AdBRG1 (lanes 3 and 4) or AdK798R (lanes 5 and 6) adenoviruses, transfected the next day with 1 µg of phCIITAPIV-LUC and then left untreated or exposed to IFN-γ for 24 h. Protein and RNA were prepared for luciferase (graph) and RT–PCR (lower panel) assays, respectively. The luciferase assays are the average and range of two experiments, each performed in duplicate. The RT–PCR is representative of two separate experiments (and reproduces the conclusions from Figure 2).
Results
BRG1 is required for IFN-γ induction of CIITA
To investigate the role of SWI/SNF in MHC class II induction, we utilized a BRG1/hBRM-deficient cell line (SW13) derived from human small-cell carcinoma of the adrenal cortex (Muchardt and Yaniv, 1993; Dunaief et al., 1994; Wang et al., 1996a). MHC class II proteins were detected on the surface of Epstein–Barr virus (EBV)-transformed lymphocytes, which constitutively express these molecules, and on IFN-γ-treated MTRB-1 cells (Figure 1A). MTRB-1 cells are BRG1 positive (Figure 1C). In contrast, IFN-γ failed to induce surface expression of MHC class II in SW13 cells (Figure 1A).

Fig. 1. IFN-γ does not induce MHC class II and CIITA in SW13 cells. (A) Lack of MHC class II induction in SW13 cells. Flow cytometry using a PE-conjugated antibody against human MHC class II DR antigen was performed on MTRB-1 control cells, or SW13 BRG1-deficient cells that were untreated (shaded curve), or exposed to IFN-γ for 24 h (solid line). The dotted line represents cells analyzed with an isotype control antibody. Untreated EBV-transformed B cells, which express MHC class II constitutively, were also analyzed. (B) Lack of CIITA induction. SW13 (lanes 1 and 2), MTRB-1 (lanes 3 and 4) or S4 (lanes 5 and 6) cells were left untreated (lanes 1, 3 and 5) or exposed to IFN-γ for 24 h (lanes 2, 4 and 6) and mRNA levels of CIITA, GBP-1 or β-actin assessed by RT–PCR. (C) Expression of BRG1. Western analysis of 50 µg of total cell lysate shows that BRG1 is expressed in control lines (MTRB-1 and S4), but not in SW13 cells.
As described above, up-regulation of the CIITA coactivator precedes MHC class II induction, so we next determined whether the defect in SW13 cells lay upstream or downstream of CIITA. At 24 h after IFN-γ treatment, CIITA message was detected by RT–PCR in two control lines but not in SW13 cells (Figure 1B). Both control lines express BRG1 (Figure 1C). An actin control showed that the RNA isolated from all three cell types was intact (Figure 1B). Furthermore, induction of guanylate-binding protein 1 (GBP-1), a classic IFN-γ-responsive gene, was detected in SW13 cells (Figure 1B, lanes 1 and 2), suggesting that the JAK–STAT pathway is intact in this line. However, as shown below, the rate of GBP-1 induction is reduced in these cells. A constitutive level of GBP-1 was detected in MTRB-1 and S4 cells, which was elevated upon IFN-γ treatment (Figure 1B, lanes 3–6).
To date, no link has been made between the JAK–STAT pathway and the SWI/SNF complex. Reconstitution of SW13 cells with BRG1 restores SWI/SNF activity, so to test its role in CIITA induction directly, we infected SW13 cells with AdBRG1, an adenovirus that expresses BRG1 (Murphy et al., 1999). Co-infection with an activator virus, AdtTa, induces BRG1 expression, which is blocked in the presence of tetracyline (Murphy et al., 1999). SW13 cells were infected with AdBRG1 and AdtTa viruses and maintained in the presence or absence of tetracycline. After 24 h, cells were treated with IFN-γ for 6, 12 or 24 h, total RNA extracted and RT–PCR performed using primers that amplify the CIITA, GBP-1 and β-actin cDNAs (Figure 2). As seen previously (Figure 1), IFN-γ did not induce CIITA in uninfected cells (Figure 2, lanes 1 and 2), or in cells infected with AdtTa alone (lanes 3 and 4). CIITA induction, however, was rescued specifically in cells infected with AdBRG1 + AdtTa (Figure 2, lanes 5 and 6) and was detectable as early as 6 h after IFN-γ treatment, similar to the time course of induction in other cell types (Steimle et al., 1994). CIITA induction was inhibited in the presence of tetracyline (lanes 7 and 8), which blocks BRG1 expression (Murphy et al., 1999). AdK798R virus, which expresses an ATPase-deficient form of BRG1 (Murphy et al., 1999), exhibited much reduced activity relative to wild-type virus, even in the absence of tetracyline (Figure 2, lanes 9–12). Consistent with the experiment in Figure 1B, GBP-1 message was detected after 24 h of IFN-γ treatment even in the absence of functional BRG1 (Figure 2, lanes 2, 4, 8, 10 and 12). Notably, however, the GBP-1 message was induced more rapidly in the presence of BRG1 and could be detected as early as 6 h (Figure 2, lane 6). This result indicates that SWI/SNF may participate in the IFN-γ induction of genes other than CIITA.

Fig. 2. BRG1 rescues CIITA induction in SW13 cells. SW13 cells were left uninfected (lanes 1 and 2), or infected with AdtTa virus alone (lanes 3 and 4) or together with AdBRG1 (lanes 5–8) or AdK798R (lanes 9–12) in the absence (lanes 1–6) or presence of tetracyline (lanes 7–12). After 20 h, cells were exposed to no cytokine (lanes 1, 3, 5, 7, 9 and 11) or IFN-γ (lanes 2, 4, 6, 8, 10 and 12) for an additional 6, 12 or 24 h. RT–PCR was used to measure CIITA, GBP-1 or β-actin RNA levels. The experiment was performed three times. Representative samples are shown.
BRG1 dependency of the CIITA promoter requires chromatin
Four alternative promoters (I–IV) situated upstream of four distinct first exons control human CIITA expression (Muhlethaler-Mottet et al., 1997). Promoters I and III drive constitutive expression in dendritic and B cells, respectively, promoter II has unknown function, and promoter IV mediates IFN-γ-inducible expression (Muhlethaler-Mottet et al., 1997). Promoter IV contains binding sites for the STAT1 homodimer, USF-1 homodimer and IRF-1/2 heterodimer, all of which have been implicated in CIITA induction. (Muhlethaler-Mottet et al., 1997, 1998; Dong et al., 1999; Piskurich et al., 1999; Xi et al., 1999; O’Keefe et al., 2001). Western analysis confirmed the constitutive expression of STAT1 and USF-1 in SW13 cells, and IRF-1 was induced to maximal levels by 6 h (Figure 3A). These levels were unaffected by infection with adenoviral vectors expressing BRG1 or the K798R mutant (data not shown). This result shows that IRF-1, unlike CIITA and GBP-1, is induced in a BRG1-independent manner, and that all the activators implicated in CIITA induction are present in IFN-γ-treated SW13 cells.
Interaction of the glucocorticoid receptor (GR) with SWI/SNF is required for induction of chromosomally integrated, but not transiently transfected mouse mammary tumor virus (MMTV) promoter (Fryer and Archer, 1998). Thus we tested whether BRG1 is required for IFN-γ induction of CIITA promoter IV in a transient transfection assay. phCIITAPIV-LUC (Figure 3B) contains the GAS, E-box and IRF elements necessary for induction in such an experiment (Muhlethaler-Mottet et al., 1998). When SW13 cells were transfected with this reporter, IFN-γ induced Luc activity 4-fold (Figure 3B), suggesting that the factors necessary for induction of promoter IV in an extra-chromosomal context are present in SW13 cells, despite the absence of a functional SWI/SNF complex. When a BRG1 expression vector was included in the assay, the fold induction was relatively unaffected but, remarkably, the absolute levels of both basal and induced promoter activity were significantly reduced (Figure 3B). We modified this assay so that expression from the endogenous chromosomal and transiently transfected CIITA promoter could be analyzed from the same cells. SW13 cells were reconstituted with BRG1 using the adenovirus system, then transfected with phCIITAPIV-LUC. Protein and RNA were prepared from untreated or IFN-γ-treated cells for luciferase and RT–PCR assays, respectively. While BRG1 rescued expression of the endo genous gene, it inhibited both background and induced levels of the phCIITAPIV-LUC reporter (Figure 3C). The K798R mutation virtually ablated regulation (Figure 3C), indicating that both activation of the endogenous gene and repression of the transiently transfected reporter require ATP hydrolysis. Thus, BRG1 is required specifically for induction of the chromosomal CIITA gene, consistent with the known role of BRG1 in chromatin remodeling, and its effect on promoter activity is reversed in a transient transfection assay.
IFN-γ induces BRG1 recruitment to the CIITA promoter
Although BRG1 influences the expression of several mammalian genes (de La Serna et al., 2000, 2001; Liu et al., 2001), direct evidence has not been obtained that BRG1 is recruited to most of these loci. To determine if BRG1 associates with the CIITA promoter directly, we employed the chromatin immunoprecipitation (ChIP) assay. A HeLa cell line derivative was used for these experiments as these cells contain functional SWI/SNF and are IFN-γ responsive. Following treatment with IFN-γ for 24 h, cells were treated with formaldehyde, lysed and the cross-linked chromatin sheared by sonication. DNA– protein complexes were immunoprecipitated with a rabbit anti-BRG1 antibody or a control rabbit anti-GAL4 antibody. The cross-links were then reversed, and the purified DNA amplified with primers specific for the CIITA promoter or an irrelevant control locus (PITX1 intron). These assays revealed that BRG1 was recruited specifically to the CIITA promoter region (Figure 4, lane 5). Moreover, this association was IFN-γ dependent as BRG1 was not associated with the CIITA promoter in untreated cells (Figure 4, lane 5).
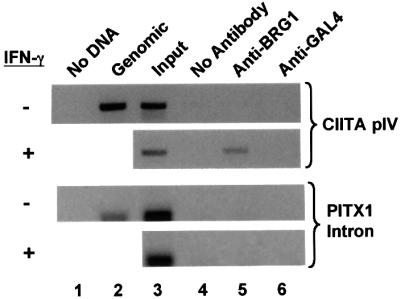
Fig. 4. IFN-γ-inducible BRG1 recruitment to the CIITA promoter. HeLa-Ini-11 cells were left untreated or exposed to IFN-γ for 24 h, formaldehyde cross-linked and the chromatin sonicated and used in ChIPs. Primers for CIITA promoter IV or the irrelevant PITX1 intron were used to amplify no template (lane 1), control human genomic DNA (lane 2), input DNA (lane 3) or DNA isolated following ChIP with no antibody (lane 4), anti-BRG1 (lane 5) or anti-GAL4 (lane 6). The CIITA primers amplify from –352 to +160 relative to the transcription start site.
BRG1-dependent chromatin remodeling at the CIITA promoter
IFN-γ induction of CIITA is associated with increased DNase I accessibility at promoter IV (Piskurich et al., 1999). Having shown that BRG1 is required for CIITA induction, and that it is recruited to the promoter in an IFN-γ-dependent fashion, we hypothesized that chromatin remodeling at promoter IV would require BRG1. Thus, in vivo DNase I footprinting was performed on a 400 bp region that contained the CIITA promoter. SW13 cells were infected with adenovirus expressing BRG1 or the inactive mutant, K798R, together with AdtTa. The infected cells were treated with IFN-γ for 24 h, after which the cell nuclei were isolated and digested with a limiting concentration of DNase I. Following purification of the DNA, ligation-mediated PCR (LM-PCR) was used to reveal areas of the promoter that were accessible to digestion (Figure 5A). In IFN-γ-treated cells expressing the K798R mutant, virtually no PCR products were detected even at the highest concentration of DNase I (Figure 5A, lanes 11–13), indicating that the locus remained inaccessible. In contrast, several amplified products were obtained from IFN-γ-treated cells infected with AdBRG (Figure 5A, lanes 5–7). The DNase I breakpoints were clustered around regions of the promoter that are known to bind critical activators, as well as the transcription initiation site.

Fig. 5. BRG1-dependent chromatin remodeling at the CIITA promoter. (A) DNase I accessibility requires BRG1. SW13 cells were infected with AdtTa plus AdBRG (lanes 2–7), or AdK798R (lanes 8–13). After 20 h, cells were left untreated (lanes 2–4 and 8–10) or exposed to IFN-γ for 24 h (lanes 5–7 and 11–13). Nuclei were harvested and treated with 12.8 (lanes 2, 5, 8 and 11), 25.6 (lanes 3, 6, 9 and 12) or 51.2 (lanes 4, 7, 10 and 13) ng/µl DNase I for 3 min at 37°C. DNA was isolated and analyzed by LM-PCR. (B) HaeIII accessibility requires BRG1. Uninfected SW13 cells (lanes 2 and 3) or cells infected with AdtTa and AdBRG (lanes 4 and 5) were treated for 24 h with IFN-γ (lanes 2–5). Nuclei were isolated and incubated in vivo with HaeIII (lanes 2 and 4) or no enzyme (lanes 3 and 5) at 37°C for 10 min (lanes 2 and 4). DNA was extracted, digested to completion in vitro with AvrII and analyzed by LM-PCR. The intensity of each HaeIII band in lanes 2 and 4 was normalized to that of the full-length primer extension product to the AvrII site. The ratio of these values gave the fold increase in HaeIII accessibility. Arrows on the left and right indicate the HaeIII and AvrII sites, respectively. The marker in both (A) and (B) (lane 1) is MspI-digested pBR322 DNA. The position of elements in the CIITA promoter is indicated to the right of each figure. Major (+1) and minor (+16) start sites of transcription are indicated.
A restriction enzyme accessibility assay also demonstrated BRG1-dependent chromatin remodeling in the CIITA promoter region. SW13 cells were infected with AdBRG1 + AdtTa and exposed to IFN-γ for 24 h. Isolated nuclei were then treated with a limiting concentration of HaeIII (in vivo digest, Figure 5B), and the DNA purified and digested to completion in vitro with AvrII, which cuts upstream of HaeIII (Figure 5B). LM-PCR was performed to visualize the full-length 529 bp primer extension product to the AvrII site, and the shorter fragments generated by HaeIII digestion. The sensitivity of the CIITA promoter to HaeIII digestion was determined by calculating the ratio of the in vivo HaeIII-digested bands to the in vitro AvrII-digested band in each lane (Figure 5B). IFN-γ-treated SW13 reconstituted with BRG1 showed an increase in HaeIII accessibility compared with cells lacking BRG1. Thus, both DNase I and restriction enzyme accessibility at the CIITA promoter are BRG1 dependent.
Discussion
BRG1-dependent CIITA induction
MHC class II induction requires the CIITA coactivator (Reith and Mach, 2001). We found that IFN-γ failed to induce MHC class II expression in SW13 cells, which was traced to a defect in CIITA up-regulation. Reconstitution of these cells with BRG1, but not an ATPase-deficient version of this protein, rescued CIITA induction. The time course of induction in reconstituted cells was typical of that seen in other IFN-γ-responsive cells, consistent with a direct effect at the CIITA locus. In support of this hypothesis, a ChIP assay revealed that BRG1 associates directly with the CIITA promoter in an IFN-γ-dependent fashion. Furthermore, in vivo footprinting and restriction enzyme accessibility assays showed that chromatin remodeling at this locus requires functional BRG1. Thus, SWI/SNF appears to participate directly in chromatin remodeling at the CIITA promoter in response to IFN-γ signaling.
SWI/SNF- and cytokine-mediated gene induction
These data represent the first evidence linking SWI/SNF to a cytokine pathway, and the JAK–STAT pathway in particular. SWI/SNF has been implicated in the regulation of subsets of genes required for myeloid differentia tion, muscle differentiation and the response to stress (Kowenz-Leutz and Leutz, 1999; de La Serna et al., 2000). In agreement with this emerging generalization, our results suggest that SWI/SNF is required at a subset of IFN-γ-responsive genes, since IRF-1 induction was normal in SW13 cells, while GBP-1 and CIITA induction were partially or completely dependent on functional BRG1, respectively. To understand the full biological implications of our findings, it will be important to define which subsets of IFN-γ-regulated genes fall into these categories. Additional studies are also required to determine whether SWI/SNF plays a role in the biological and molecular effects of other cytokines, particularly those that utilize variations of the JAK–STAT pathway (reviewed in Pellegrini and Dusanter-Fourt, 1997; Bromberg and Darnell, 2000).
SWI/SNF and tumor surveillance
There is considerable evidence that the SWI/SNF complex can act as a tumor suppressor by blocking the cell cycle, promoting differentiation and possibly mediating apoptosis (see Cheng et al., 1999; Muchardt and Yaniv, 1999; Fry and Peterson, 2001; and references therein). Mutations in SWI/SNF subunits, or defects in their expression, have been reported in a variety of tumors and tumor cell lines (Muchardt and Yaniv, 1993; Wang et al., 1996a; Versteege et al., 1998; Sevenet et al., 1999a,b; Strobeck et al., 2000; Yuge et al., 2000; Schmitz et al., 2001). In addition, heterozygous BRG1 or hSNF5/INI1 mice are predisposed to a variety of tumors (Bultman et al., 2000; Klochendler-Yeivin et al., 2000; Roberts et al., 2000). Our data suggest that SWI/SNF may also contribute to tumor suppression by rendering cells sensitive to tumor surveillance. Indeed, ectopic MHC class II expression has been used as a strategy to generate effective tumor vaccines (reviewed in Ostrand-Rosenberg et al., 1999). Therapeutic strategies that restore or mimic SWI/SNF function may, therefore, benefit cancer patients at multiple levels. Intriguingly, SWI/SNF is not the only putative tumor suppressor involved in MHC class II induction, as we and others have implicated RB in this pathway (Lu et al., 1994; Zhu et al., 1999).
Effects of SWI/SNF on transiently transfected versus chromosomal targets
Chromatin does not form properly on transiently transfected plasmids, and the ability of BRG1/hBRM to affect promoters in transient transfections is variable (Smith and Hager, 1997). For example, BRG1 potentiates CSF1 promoter activity, and GR-mediated induction of the MMTV promoter in a chromosomal context, but not in transient transfection assays (Fryer and Archer, 1998; Liu et al., 2001). In contrast, hBRM does potentiate GR induction of various artificial reporter vectors in transient assays (Muchardt and Yaniv, 1993; Singh et al., 1995). In addition, BRG1/hBRM enhance RB repression of cell cycle gene promoters in transient assays (Trouche et al., 1997; Murphy et al., 1999; Strobeck et al., 2000; Zhang et al., 2000). In our study, BRG1 was not only unnecessary for CIITA promoter IV induction in a transient assay, but actually inhibited activity. It seems unlikely that this effect is due to sequestration of critical factors by high levels of BRG1, since repression required ATPase activity. Thus, in the absence of appropriate chromatin cues, BRG1 may form an inhibitory complex on the promoter. Whatever the explanation, it is clear that results based on transient transfections should be interpreted with caution, and additional assays performed to determine how SWI/SNF affects the chromosomal gene, and whether this effect is direct.
Mechanism of SWI/SNF recruitment
Our ChIP data show that BRG1 is recruited directly to the CIITA promoter in an IFN-γ-dependent fashion. SWI/SNF can be recruited to promoters by specific activators (Fryer and Archer, 1998; Cheng et al., 1999; Kowenz-Leutz and Leutz, 1999; Neely et al., 1999; Yudkovsky et al., 1999; Dilworth et al., 2000; DiRenzo et al., 2000; Kadam et al., 2000; Barker et al., 2001; Sullivan et al., 2001). Activators implicated in CIITA induction, such as STAT1, USF-1 and IRF-1/2, have not, as yet, been shown to associate with SWI/SNF. In transient transfection assays, BRG1 was not necessary for induction, but inhibited both basal and induced CIITA promoter IV activity to a similar degree. This result suggests that BRG1 may affect the activity of a constitutively active factor that regulates the CIITA promoter, such as USF-1, IRF-2 or part of the general transcription machinery.
Alternatively, as in the case of the IFN-β promoter (Agalioti et al., 2000), the formation of an enhanceosome by multiple activators could create a novel interaction surface at the CIITA promoter that facilitates SWI/SNF binding. Histone acetylation also promotes SWI/SNF binding at the IFN-β promoter (Agalioti et al., 2000), probably through interaction of the BRG1 bromodomain with acetyl-lysine groups (reviewed in Horn and Peterson, 2001). In this regard, it is of interest that STAT1 interacts with CBP, a known histone acetyl transferase (Zhang et al., 1996; Horvai et al., 1997). Finally, SWI/SNF might be targeted to the CIITA promoter by virtue of its ability to interact with RNA polymerase II holoenzyme (Wilson et al., 1996; Cho et al., 1998; Neish et al., 1998). However, the chromatin accessibility assays (Figure 5) are more consistent with SWI/SNF binding at a stage prior to polymerase recruitment.
Analysis of CIITA induction has focused on human promoter IV. However, the ‘B-cell-specific’ promoter III, located ∼2 kb upstream, is also partially IFN-γ inducible (Piskurich et al., 1998, 1999). This promoter is silent in non-professional APCs, but constitutive DNase I-hypersensitive sites are present both around the transcription start site and ∼5 kb upstream, and access at these sites is enhanced slightly following IFN-γ treatment (Piskurich et al., 1999). It will be interesting, therefore, to determine if these sites influence the induction of promoter IV, and whether chromatin accessibility requires BRG1. Analysis of CIITA induction, therefore, provides an opportunity to expand emerging insights into the order in which activators and their cofactors, including SWI/SNF, are recruited to inducible promoters, as well as the influence of long-range effects.
Materials and methods
Cell lines and IFN-γ treatment
SW13 cells, the breast cancer lines MTRB-1, S4 (full name MDA-468-S4) (Lu et al., 1994), EBV-transformed lymphocytes and HeLa-Ini-11 cells (Sif et al., 1998) were grown in α-minimal essential medium (MEM)/10% fetal bovine serum (FBS). Human IFN-γ (BioSource International PHC4834) was used at 250 U/ml in ChIP assays, and at 300 U/ml in all other assays.
Flow cytometry
SW13 and MTRB-1 cells were plated in duplicate at 5 × 105 cells per well on a 24-well plate and treated with IFN-γ for 24 h. Trypsinized cells were washed in phosphate-buffered saline (PBS)/5% FBS, then stained with phycoerythrin (PE)-conjugated anti-HLA-DR antibody (PharMingen 34235X), or PE-conjugated anti-IgG2a isotype control (PharMingen 03025A) for 1 h at 4°C. Cells were washed five times with PFA (PBS, 2% FBS, 0.1% azide), and resuspended in 500 µl of PFA/ 1 µg/ml propidium iodide. A total of 10 000 cells were analyzed by flow cytometry using Cell Quest software (Becton Dickinson).
RNA extraction and RT–PCR
RNA was isolated from confluent 60 or 100 mm plates using TRIzol reagent (Gibco-BRL). A 2.5 µg aliquot of RNA was diluted in 20 µl of diethylpyrocarbonate (DEPC)-treated water, heated to 90°C for 5 min then combined with 50 µl of first strand master mix [10 µg Pd(N)6 salt, 1× First Strand buffer (Gibco-BRL), 1 mM dNTPs, 10 mM dithiothreitol (DTT), 50 U of Superscript II reverse transcriptase (Gibco-BRL)], and incubated at 37°C for 1 h, then for 10 min at 95°C. One-tenth of first strand cDNA was amplified for 30 cycles using the following primer pairs: sense o-hCA-3 (TGTGCAGACTCAGAGGACGAGAAGTTC), antisense o-hCA-4 (5′-CCCGAATTCCCATCTCAGGCTGATCCGTGAATCC-3′); sense o-hGBP-1 (5′-AACACTAATGGGCGACTGAT-3′), antisense o-hGBP-2 (5′-CTTCAGGGAGTATGTCAGGT-3′); sense o-ACT-1 (5′-CCCAGATCATGTTTGAGACC-3′), antisense o-ACT-2 (5′-AGTCATAGTCCGCCTAGAAG-3′).
Western blotting
A confluent 100 mm plate of SW13 cells was lysed in 50 mM Tris–HCl pH 7.4, 150 mM KCl, 15 mM NaCl, 30 mM MgCl2, 10 mM EGTA, 0.5% NP-40 and protease inhibitors. A 50 µg aliquot of lysate was analyzed by western blot as described (Bremner et al., 1995) with antibodies against IRF-1 (C-20; Santa Cruz sc-497), USF-1 (C-20; Santa Cruz sc-229), Stat1α p91 (C-111; Santa Cruz sc-417) and BRG-1/BRM (B36320; Transduction Labs).
Plasmids
phCIITAPIV-LUC: a 378 bp human CIITA pIV fragment was amplified using o-hCA-12 (5′-CCCGGTACCGGAGAGAAACAGAGACCCAC-3′) and hCA-13 (5′-GGCAAGCTTCCTCTCCCTCCCGCCAGCTC-3′), digested with KpnI and HindIII and ligated to KpnI–HindIII-digested pGL2-Basic. Clones were sequence verified. pBJ5-BRG1 and pBJ5 are described in Dunaief et al. (1994) and Khavari et al. (1993).
Luciferase/β-gal assays
SW13 cells were transfected by the calcium phosphate method and 0.3 µg of CMV-β-gal included to normalize for transfection efficiency (Bremner et al., 1995). For infection-then-transfection (Figure 3C), SW13 cells were infected as described below and transfected 24 h later. Cells were lysed in 1× reporter lysis buffer (Promega). Luc assays used 20 µl of lysate and 100 µl of luciferase assay reagent (10 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 270 µM co-enzyme A, 470 µM luciferin, 530 µM ATP in gly-gly buffer, pH 7.8).
Chromatin immunoprecipitation
HeLa Ini-11 cells were IFN-γ induced for 24 h, cross-linked with 1% formaldehyde at room temperature for 10 min, washed twice with ice-cold PBS, collected in 1 ml of PBS (3 × 107 cells per tube) and centrifuged in a bench-top microfuge (Desaga; Sarstedt-Gruppe) for 5 min at 5000 r.p.m. Cells were resuspended in 1 ml of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8) plus protease inhibitors (aprotinin, leupeptin and pepstatin), incubated on ice for 10 min and sonicated to an average size of 500 bp (Vibra Cell, Sonics and Materials Inc., Danbury). A 100 µl aliquot of sonicated chromatin (3 × 106 cell equivalents) was used per immunoprecipitation. Chromatin was diluted in 1 ml of buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl pH 8) and pre-cleared with 2 µg of sheared salmon sperm DNA and protein A–Sepharose (Sigma) (45 µl of 50% slurry in 10 mM Tris–HCl pH 8, 1 mM EDTA) for 2 h at 4°C. Immunoprecipitation (IP) was performed overnight at 4°C with no antibody, anti-BRG1 (R.Kingston) or anti-yeast GAL4 antibody (Upstate Biotechnology 06-262). A 45 µl aliquot of protein A–Sepharose, 2 µg of salmon sperm DNA and 45 µl of yeast tRNA were added per IP and incubated for 1 h. Precipitates were washed sequentially for 10 min in 1× TSEI (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8, 150 mM NaCl), 4× TSEII (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8, 500 mM NaCl), 1× buffer III (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl pH 8) and 3× TE (10 mM Tris–HCl pH 8, 1 mM EDTA). Samples were extracted twice with 250 µl of elution buffer (1% SDS, 0.1 M NaHCO3), heated at 65°C overnight to reverse cross-links, and DNA fragments purified with a QIAEX II Gel Extraction Kit. A 3 µl aliquot from a total of 50 µl was used in the PCR. CIITA promoter IV-specific primers were 5′-TTGGACTGAGTTGGAGAG-3′ and 5′-GTGACCTTGAGCAAGTAG-3′.
Virus infections
AdBRG, AdK798R and AdtTa were prepared as described (Murphy et al., 1999). Plates (100 mm) of SW13 cells at 80% confluence were infected in a final volume of 1.5 ml for 1 h with occasional rocking. The amount of virus was such that the level of BRG1 or K798R was equivalent, and similar to BRG1 levels in HeLa-Ini-11 cells (data not shown). After infection, virus was removed and 9 ml of medium added. Where indicated, 2.5 µg/ml tetracycline was added 4 h post-infection. IFN-γ was added 20 h post-infection and cells harvested 24 h later.
DNase I footprinting
SW13 cells infected with AdBRG1 or AdK798R were treated with IFN-γ for 24 h. A total of 1 × 107 SW13 cells were trypsinized from 100 mm plates and washed twice in PBS, and once in 1× RSB [reticulocyte standard buffer: 10 mM Tris–HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mm phenylmethylsulfonyl fluoride (PMSF)]. The cell pellet was resuspended in 10 ml of 50% glycerol, 0.5% NP-40, 1 mM PMSF, 1× RSB solution, lysed by pipeting 10 times, incubated on ice for 5–10 min, and nuclei visualized with trypan blue. Nuclei were spun at 4000 r.p.m. for 10 min at 4°C, washed in 10 ml of 1× RSB/0.1 mM PMSF, spun at 2500 r.p.m. for 10 min at 4°C then resuspended in 0.3 ml of 1× RSB. DNase I stock (4 mg/ml; Sigma Molecular Biology Grade D5793) was diluted in 1× RSB to a final concentration of 80 ng/ml. A 1/1000 volume of 1 M CaCl2 was added and 100 µl of nuclei digested with 12.8, 25.6 or 51.2 ng/µl DNase for 3 min at 37°C. A 100 µl aliquot of 2× STOP solution (0.6 M NaCl, 20 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% SDS) was added, then 10 µl of proteinase K (25 mg/ml), and samples were incubated at 55°C overnight. A 200 µl aliquot of 1× STOP solution was added, and samples were phenol/chloroform extracted and treated with RNase (Sigma Molecular Biology Grade) at 37°C overnight. Samples were phenol/chloroform extracted again, ethanol precipitated and resuspended in 100 µl of 10 mM Tris–HCl pH 8.0 and used in LM-PCR (see below).
Restriction enzyme accessibility assay
Nuclei were isolated as described above except restriction enzyme digestion buffer (10 mM Tris–HCl pH 7.4, 50 mM NaCl, 10 mM MgCl2, 0.2 mM EDTA, 0.2 mM EGTA, 0.15 mM spermine, 0.5 mM spermidine, 1 mM β-mercaptoethanol) was used in place of 1× RSB. Nuclei were suspended in 300 µl of 1× NEB2 (New England Biolabs), 50 µl combined with 20 U of HaeIII (New England Biolabs), and digested at 37°C for 10 min. Reactions were stopped with 50 µl of 2× proteinase K buffer (100 mM Tris–HCl pH 7.5, 200 mM NaCl, 2 mM EDTA, 1% SDS) at 55°C for 1 h. Samples were then digested overnight at 55°C with 3 µl (25 mg/ml) of proteinase K (Sigma Molecular Biology Grade). DNA was extracted as described above and resuspended in 100 µl of 10 mM Tris–HCl pH 8.0. A 1 µg aliquot was digested with AvrII, and one-quarter of the digest used for LM-PCR (see below).
Ligation-mediated PCR
LM-PCR was performed using these CIITA primers: o-hCA-22 (5′- ACCTTAGGGGTTACAGAGGAGACTT-3′); o-hCA-23 (5′-GACTTTGGTCACCTACCGCTGTTCC-3′); and o-hCA-24 (5′-TTTGGTCACCTACCGCTGTTCCCCGGGCTC-3′). Linker primers (o-LMPCR-1, 5′-GCGGTGACCCGGGAGATCTGAATTC-3′; and o-LMPCR-2, 5′-GAATTCAGATC-3′) were annealed for the 20 µM linker solution described below. A 1 µg aliquot of digested DNA was diluted in 10 µl of 10 mM Tris–HCl pH 8.0. A 4 µl aliquot of mixture A [3 µl of sequenase buffer 1 (125 mM Tris–HCl pH 7.5, 400 mM NaCl, 25 mM MgCl2) and 1 µl of o-hCA-22 (0.3 pmol/µl)] was added and samples incubated at 95°C for 5 min, and 50°C for 30 min, and placed on ice. A 9.5 µl aliquot of mixture B [9 µl of sequenase buffer 2 (40 mM Tris–HCl pH 7.5, 5 mM MgCl2, 20 mM DTT, 0.1 mM dNTPs) and 0.5 µl of Sequenase T7 DNA polymerase v. 2.0 (United States Biochemicals) (8 U/µl)] was added, and incubated at 37°C for 10 min, and 68°C for 10 min, and placed back on ice. A 48 µl aliquot of mixture C was added [20 µl of ligase buffer 1 (80 mM Tris–HCl pH 7.5, 180 µg/µl bovine serum albumin (BSA), 30 mM MgCl2), 20 µl of ligase buffer 2 (12 mM ATP, 70 mM DTT), 5 µl of 20 µM Linker oligo solution and 3 µl of T4 DNA ligase (2 U/µl)], and samples incubated at 20°C overnight. Samples were ethanol precipitated, resuspended in 20 µl of 10 mM Tris–HCl pH 8.0 and amplified with o-hCA-23 and o-LMPCR-1 for one cycle of 94°C, 3 min; 23 cycles of 94°C, 1 min/64°C, 1 min/72°C, 1 min; and one cycle of 72°C, 10 min. 32P-end-labeled o-hCA-24 (2 pmol) was added to 15% of the PCR, and seven cycles (94°C, 1 min/64°C, 1 min/72°C, 1 min) of primer extension performed. STOP mixture [24 µl of TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA), 1 µl of 5 mg/ml tRNA (Gibco-BRL) and 5 µl of 3 M NaOAc] was added, and each reaction was chloroform extracted, ethanol precipitated, and DNA resuspended in 12 µl of sequencing loading buffer. Samples were run on an 8% sequencing gel, the gel dried, exposed overnight and visualized on a phosphoimager (Bio-Rad). Densitometry was performed using Bio-Rad imaging software.
Acknowledgments
Acknowledgements
We are indebted to D.Engel for the adenoviruses, R.Kingston for anti-Brg1 antibody and HeLa-Ini-11 line, S.Goff for BRG1 plasmid, G.Beresford and J.DiRenzo for advice on ChIP assays, J.Ellis for advice on DNase I assays, and Ken Zaret for advice on LM-PCR. S.P. was supported by the KM Hunter/MRC Doctoral Research Award and the Frank Fletcher Memorial Fund, R.K. by a Fight for Sight fellowship, and S.P. and E.K. by the Vision Science Research Program, University of Toronto. This work was funded by a grant from the National Cancer Institute of Canada with funds from the Canadian Cancer Society.
References
- Agalioti T., Lomvardas,S., Parekh,B., Yie,J., Maniatis,T. and Thanos,D. (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell, 103, 667–678. [DOI] [PubMed] [Google Scholar]
- Barker N., Hurlstone,A., Musisi,H., Miles,A., Bienz,M. and Clevers,H. (2001) The chromatin remodelling factor Brg-1 interacts with β-catenin to promote target gene activation. EMBO J., 20, 4935–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U., Klamp,T., Groot,M. and Howard,J.C. (1997) Cellular responses to interferon-γ. Annu. Rev. Immunol., 15, 749–795. [DOI] [PubMed] [Google Scholar]
- Bremner R., Cohen,B.L., Sopta,M., Hamel,P.A., Ingles,C.J., Gallie,B.L. and Philips,R.A. (1995) Direct transcriptional repression by pRB and its reversal by specific cyclins. Mol. Cell. Biol., 15, 3256–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg J. and Darnell,J.E.,Jr (2000) The role of STATs in transcriptional control and their impact on cellular function. Oncogene, 19, 2468–2473. [DOI] [PubMed] [Google Scholar]
- Bultman S. et al. (2000) A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell, 6, 1287–1295. [DOI] [PubMed] [Google Scholar]
- Chang C.-H., Guerder,S., Hong,S.-C., van Ewijk,W. and Flavell,R.A. (1996) Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity, 4, 167–178. [DOI] [PubMed] [Google Scholar]
- Cheng S.W., Davies,K.P., Yung,E., Beltran,R.J., Yu,J. and Kalpana,G.V. (1999) c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nature Genet., 22, 102–105. [DOI] [PubMed] [Google Scholar]
- Chin K.C., Mao,C., Skinner,C., Riley,J.L., Wright,K.L., Moreno,C.S., Stark,G.R., Boss,J.M. and Ting,J.P. (1994) Molecular analysis of G1B and G3A IFN γ mutants reveals that defects in CIITA or RFX result in defective class II MHC and Ii gene induction. Immunity, 1, 687–697. [DOI] [PubMed] [Google Scholar]
- Cho H., Orphanides,G., Sun,X., Yang,X.J., Ogryzko,V., Lees,E., Nakatani,Y. and Reinberg,D. (1998) A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell. Biol., 18, 5355–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Serna I.L., Carlson,K.A., Hill,D.A., Guidi,C.J., Stephenson,R.O., Sif,S., Kingston,R.E. and Imbalzano,A.N. (2000) Mammalian SWI/SNF complexes contribute to activation of the hsp70 gene. Mol. Cell. Biol., 20, 2839–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Serna I.L., Carlson,K.A. and Imbalzano,A.N. (2001) Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nature Genet., 27, 187–190. [DOI] [PubMed] [Google Scholar]
- Dilworth F.J., Fromental-Ramain,C., Yamamoto,K. and Chambon,P. (2000) ATP-driven chromatin remodeling activity and histone acetyltransferases act sequentially during transactivation by RAR/RXR in vitro. Mol. Cell, 6, 1049–1058. [DOI] [PubMed] [Google Scholar]
- DiRenzo J., Shang,Y., Phelan,M., Sif,S., Myers,M., Kingston,R. and Brown,M. (2000) BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol. Cell. Biol., 20, 7541–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y., Rohn,W.M. and Benveniste,E.N. (1999) IFN-γ regulation of the type IV class II transactivator promoter in astrocytes. J. Immunol., 162, 4731–4739. [PubMed] [Google Scholar]
- Dunaief J.L., Strober,B.E., Guha,S., Khavari,P.A., Alin,K., Luban,J., Begemann,M., Crabtree,G.R. and Goff,S.P. (1994) The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell, 79, 119–130. [DOI] [PubMed] [Google Scholar]
- Flaus A. and Owen-Hughes,T. (2001) Mechanisms for ATP-dependent chromatin remodelling. Curr. Opin. Genet. Dev., 11, 148–154. [DOI] [PubMed] [Google Scholar]
- Fry C.J. and Peterson,C.L. (2001) Chromatin remodeling enzymes: who’s on first? Curr. Biol., 11, R185–R197. [DOI] [PubMed] [Google Scholar]
- Fryer C.J. and Archer,T.K. (1998) Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature, 393, 88–91. [DOI] [PubMed] [Google Scholar]
- Guidi C.J., Sands,A.T., Zambrowicz,B.P., Turner,T.K., Demers,D.A., Webster,W., Smith,T.W., Imbalzano,A.N. and Jones,S.N. (2001) Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol., 21, 3598–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobart M., Ramassar,V., Goes,N., Urmson,J. and Halloran,P.F. (1997) IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J. Immunol., 158, 4260–4269. [PubMed] [Google Scholar]
- Holstege F.C., Jennings,E.G., Wyrick,J.J., Lee,T.I., Hengartner,C.J., Green,M.R., Golub,T.R., Lander,E.S. and Young,R.A. (1998) Dissecting the regulatory circuitry of a eukaryotic genome. Cell, 95, 717–728. [DOI] [PubMed] [Google Scholar]
- Horn P.J. and Peterson,C.L. (2001) The bromodomain: a regulator of ATP-dependent chromatin remodeling? Frontiers Biosci., 6, D1019–D1023. [DOI] [PubMed] [Google Scholar]
- Horvai A.E., Xu,L., Korzus,E., Brard,G., Kalafus,D., Mullen,T.M., Rose,D.W., Rosenfeld,M.G. and Glass,C.K. (1997) Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl Acad. Sci. USA, 94, 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam S., McAlpine,G.S., Phelan,M.L., Kingston,R.E., Jones,K.A. and Emerson,B.M. (2000) Functional selectivity of recombinant mammalian SWI/SNF subunits. Genes Dev., 14, 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavari P.A., Peterson,C., Tamkun,J.W., Mendel,D.B. and Crabtree,G.R. (1993) BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature, 366, 170–174. [DOI] [PubMed] [Google Scholar]
- Klochendler-Yeivin A., Fiette,L., Barra,J., Muchardt,C., Babinet,C. and Yaniv,M. (2000) The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep., 1, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowenz-Leutz E. and Leutz,A. (1999) A C/EBPβ isoform recruits the SWI/SNF complex to activate myeloid genes. Mol. Cell, 4, 735–743. [DOI] [PubMed] [Google Scholar]
- Lee Y.J. and Benveniste,E.N. (1996) Stat1α expression is involved in IFN-γ induction of the class II transactivator and class II MHC genes. J. Immunol., 157, 1559–1568. [PubMed] [Google Scholar]
- Liu R., Liu,H., Chen,X., Kirby,M., Brown,P.O. and Zhao,K. (2001) Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell, 106, 309–318. [DOI] [PubMed] [Google Scholar]
- Lu Y., Ussery,G.D., Muncaster,M.M., Gallie,B.L. and Blanck,G. (1994) Evidence for retinoblastoma protein (RB) dependent and independent IFN-γ responses: RB coordinately rescues IFN-γ induction of MHC class II gene transcription in noninducible breast carcinoma cells. Oncogene, 9, 1015–1019. [PubMed] [Google Scholar]
- Muchardt C. and Yaniv,M. (1993) A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J., 12, 4279–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C. and Yaniv,M. (1999) The mammalian SWI/SNF complex and the control of cell growth. Semin. Cell Dev. Biol., 10, 189–195. [DOI] [PubMed] [Google Scholar]
- Muhlethaler-Mottet A., Otten,L.A., Steimle,V. and Mach,B. (1997) Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J., 16, 2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlethaler-Mottet A., Di Berardino,W., Otten,L.A. and Mach,B. (1998) Activation of the MHC class II transactivator CIITA by interferon-γ requires cooperative interaction between Stat1 and USF-1. Immunity, 8, 157–166. [DOI] [PubMed] [Google Scholar]
- Murphy D.J., Hardy,S. and Engel,D.A. (1999) Human SWI/SNF component BRG1 represses transcription of the c-fos gene. Mol. Cell. Biol., 19, 2724–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely K.E., Hassan,A.H., Wallberg,A.E., Steger,D.J., Cairns,B.R., Wright,A.P. and Workman,J.L. (1999) Activation domain-mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Mol. Cell, 4, 649–655. [DOI] [PubMed] [Google Scholar]
- Neish A.S., Anderson,S.F., Schlegel,B.P., Wei,W. and Parvin,J.D. (1998) Factors associated with the mammalian RNA polymerase II holoenzyme. Nucleic Acids Res., 26, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe G.M., Nguyen,V.T., Ping Tang,L.L. and Benveniste,E.N. (2001) IFN-γ regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J. Immunol., 166, 2260–2269. [DOI] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S., Pulaski,B.A., Clements,V.K., Qi,L., Pipeling,M.R. and Hanyok,L.A. (1999) Cell-based vaccines for the stimulation of immunity to metastatic cancers. Immunol. Rev., 170, 101–114. [DOI] [PubMed] [Google Scholar]
- Pellegrini S. and Dusanter-Fourt,I. (1997) The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). Eur. J. Biochem., 248, 615–633. [DOI] [PubMed] [Google Scholar]
- Piskurich J.F., Wang,Y., Linhoff,M.W., White,L.C. and Ting,J.P. (1998) Identification of distinct regions of 5′ flanking DNA that mediate constitutive, IFN-γ, STAT1 and TGF-β-regulated expression of the class II transactivator gene. J. Immunol., 160, 233–240. [PubMed] [Google Scholar]
- Piskurich J.F., Linhoff,M.W., Wang,Y. and Ting,J.P. (1999) Two distinct γ interferon-inducible promoters of the major histo compatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1 and transforming growth factor β. Mol. Cell. Biol., 19, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith W. and Mach,B. (2001) The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol., 19, 331–373. [DOI] [PubMed] [Google Scholar]
- Roberts C.W., Galusha,S.A., McMenamin,M.E., Fletcher,C.D. and Orkin,S.H. (2000) Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl Acad. Sci. USA, 97, 13796–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz U., Mueller,W., Weber,M., Sevenet,N., Delattre,O. and von Deimling,A. (2001) INI1 mutations in meningiomas at a potential hotspot in exon 9. Br. J. Cancer, 84, 199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenet N., Lellouch-Tubiana,A., Schofield,D., Hoang-Xuan,K., Gessler,M., Birnbaum,D., Jeanpierre,C., Jouvet,A. and Delattre,O. (1999a) Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype–phenotype correlations. Hum. Mol. Genet., 8, 2359–2368. [DOI] [PubMed] [Google Scholar]
- Sevenet N., Sheridan,E., Amram,D., Schneider,P., Handgretinger,R. and Delattre,O. (1999b) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet., 65, 1342–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sif S., Stukenberg,P.T., Kirschner,M.W. and Kingston,R.E. (1998) Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes Dev., 12, 2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sif S., Saurin,A.J., Imbalzano,A.N. and Kingston,R.E. (2001) Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genes Dev., 15, 603–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., Coe,J. and Hong,W. (1995) A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature, 374, 562–565. [DOI] [PubMed] [Google Scholar]
- Smith C.L. and Hager,G.L. (1997) Transcriptional regulation of mammalian genes in vivo. A tale of two templates. J. Biol. Chem., 272, 27493–27496. [DOI] [PubMed] [Google Scholar]
- Steimle V., Otten,L.A., Zufferey,M. and Mach,B. (1993) Comple mentation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell, 75, 135–146. [PubMed] [Google Scholar]
- Steimle V., Siegrist,C.A., Mottet,A., Lisowska-Grospierre,B. and Mach,B. (1994) Regulation of MHC class II expression by interferon-γ mediated by the transactivator gene CIITA. Science, 265, 106–109. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Strobeck M.W., Knudsen,K.E., Fribourg,A.F., DeCristofaro,M.F., Weissman,B.E., Imbalzano,A.N. and Knudsen,E.S. (2000) BRG-1 is required for RB-mediated cell cycle arrest. Proc. Natl Acad. Sci. USA, 97, 7748–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsanam P., Iyer,V.R., Brown,P.O. and Winston,F. (2000) Whole-genome expression analysis of snf/swi mutants of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 97, 3364–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan E.K., Weirich,C.S., Guyon,J.R., Sif,S. and Kingston,R.E. (2001) Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol. Cell. Biol., 21, 5826–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouche D., Le Chalony,C., Muchardt,C., Yaniv,M. and Kouzarides,T. (1997) RB and hbrm cooperate to repress the activation functions of E2F1. Proc. Natl Acad. Sci. USA, 94, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I., Sevenet,N., Lange,J., Rousseau-Merck,M.F., Ambros,P., Handgretinger,R., Aurias,A. and Delattre,O. (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature, 394, 203–206. [DOI] [PubMed] [Google Scholar]
- Wang W. et al. (1996a) Purification and biochemical heterogeneity of the mammalian SWI/SNF complex. EMBO J., 15, 5370–5382. [PMC free article] [PubMed] [Google Scholar]
- Wang W., Xue,Y., Zhou,S., Kuo,A., Cairns,B.R. and Crabtree,G.R. (1996b) Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev., 10, 2117–2130. [DOI] [PubMed] [Google Scholar]
- Williams G.S., Malin,M., Vremec,D., Chang,C.H., Boyd,R., Benoist,C. and Mathis,D. (1998) Mice lacking the transcription factor CIITA—a second look. Int. Immunol., 10, 1957–1967. [DOI] [PubMed] [Google Scholar]
- Wilson C.J., Chao,D.M., Imbalzano,A.N., Schnitzler,G.R., Kingston,R.E. and Young,R.A. (1996) RNA polymerase II holoenzyme contains SWI/SNF regulators involved in chromatin remodeling. Cell, 84, 235–244. [DOI] [PubMed] [Google Scholar]
- Workman J.L. and Kingston,R.E. (1998) Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem., 67, 545–579. [DOI] [PubMed] [Google Scholar]
- Xi H., Eason,D.D., Ghosh,D., Dovhey,S., Wright,K.L. and Blanck,G. (1999) Co-occupancy of the interferon regulatory element of the class II transactivator (CIITA) type IV promoter by interferon regulatory factors 1 and 2. Oncogene, 18, 5889–5903. [DOI] [PubMed] [Google Scholar]
- Xi H., Goodwin,B., Shepherd,A.T. and Blanck,G. (2001) Impaired class II transactivator expression in mice lacking interferon regulatory factor-2. Oncogene, 20, 4219–4227. [DOI] [PubMed] [Google Scholar]
- Yudkovsky N., Logie,C., Hahn,S. and Peterson,C.L. (1999) Recruitment of the SWI/SNF chromatin remodeling complex by transcriptional activators. Genes Dev., 13, 2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuge M., Nagai,H., Uchida,T., Murate,T., Hayashi,Y., Hotta,T., Saito,H. and Kinoshita,T. (2000) HSNF5/INI1 gene mutations in lymphoid malignancy. Cancer Genet. Cytogenet., 122, 37–42. [DOI] [PubMed] [Google Scholar]
- Zhang J.J., Vinkemeier,U., Gu,W., Chakravarti,D., Horvath,C.M. and Darnell,J.E.,Jr (1996) Two contact regions between Stat1 and CBP/p300 in interferon γ signaling. Proc. Natl Acad. Sci. USA, 93, 15092–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.S., Gavin,M., Dahiya,A., Postigo,A.A., Ma,D., Luo,R.X., Harbour,J.W. and Dean,D.C. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell, 101, 79–89. [DOI] [PubMed] [Google Scholar]
- Zhu X., Pattenden,S. and Bremner,R. (1999) pRB is required for interferon-γ-induction of the MHC class II Aβ gene. Oncogene, 18, 4940–4947. [DOI] [PubMed] [Google Scholar]